

Abstract
Thyroid hormone is essential for the proper development and function of the brain. The active form of thyroid hormone is T3, which binds to nuclear receptors. Recently, a transporter specific for T3, MCT8 (monocarboxylate transporter 8) was identified. MCT8 is highly expressed in liver and brain. The gene is located in Xq13 and mutations in MCT8 are responsible for an X-linked condition, Allan--Herndon--Dudley syndrome (AHDS). This syndrome is characterized by congenital hypotonia that progresses to spasticity with severe psychomotor delays. Affected males also present with muscle hypoplasia, generalized muscle weakness, and limited speech. Importantly, these patients have elevated serum levels of free T3, low to below normal serum levels of free T4, and levels of thyroid stimulating hormone that are within the normal range. This constellation of measurements of thyroid function enables quick screening for AHDS in males presenting with mental retardation, congenital hypotonia, and generalized muscle weakness.
Keywords: Allan--Herndon--Dudley syndrome, MCT8, T3, thyroid hormone transporter
Thyroid hormone action in the brain
Thyroid hormone plays an important role in the development and proper function of multiple organs. Of particular interest for this chapter is the role of thyroid hormone in brain development.1 Thyroid hormone exists in two forms: T4 (3,3′,5,5′-tetraiodothyronine) and T3 (3,3′,5-triodothyronine). Both forms are produced and secreted by the follicular cells of the thyroid gland. The action of thyroid hormone is mediated through the binding of T3 to nuclear thyroid hormone receptors within the target cell. Therefore, the degree of biologic activity of thyroid hormone is related to the level of T3 within the cell. In most target cells for thyroid hormone, T4 is converted to T3 by the iodothyronine deiodinases -- D1 or D2. T3 can also be converted to an inactive form (T2) by another deiodinase, D3. D3 can also convert T4 into reverse T3 (rT3) (Figure 1A).2
Figure 1.


(A) Schematic representation of the thyroid hormone transport and deiodination in a target cell. Both T3 and T4 enter the cell and are acted upon by deiodinases. The active form, T3, enters the nucleus where it binds to a receptor that then activates the synthesis of specific messenger RNAs coding for specific proteins. D1 is predominantly expressed in liver, kidney, and thyroid; D2 is predominantly expressed in brain, anterior pituitary, skeletal muscle, and thyroid. (B) Schematic representation of the source and the transport of T3 in the brain. T4 enters a glial via an unknown mechanism, whereupon it is converted to T3 by the D2 deiodinase. T3 exits the cell via an unknown mechanism and is transported into a neuron via MCT8. Once inside the neuron, it either enters the nucleus, binding a receptor that initiates protein synthesis, or it is inactivated to T2 by D3 deiodinase. D1, D2, D3, deiodinases; RXR, retinoid X receptor;T4, T3 thyroid hormone; TR, thyroid hormone receptor; TRE, thyroid hormone responsive element. ? signifies that the transport/export mechanism is unknown.
For T3 to exert its biologic activity within the cell, it must either be converted from T4 or must enter the cell. Initially, it was thought that, because of their lipophilic structures, T4 and T3 would cross the plasma membrane by passive diffusion. However, became clear that the transport of these molecules across the membrane is facilitated by transporters,3 and studies were undertaken to identify such transporters. Friesema et al identified MCT8 (monocarboxylate transporter 8) as a specific thyroid hormone transporter using functional studies in Xenopus oocytes.4 Tissue distribution studies using MCT8-specific antibodies found the protein to be expressed in kidney, brain, liver, and heart. Thus, circulating T3 could be transported into the target cell by MCT8 and thereby exert its biological function.
However, the situation is not this straightforward in the brain. Fliers et al used immunohistochemistry, mRNA in-situ analysis, and enzyme studies to show the differential distribution of D2, D3, and MCT8.5 They found D3, MCT8, and thyroid hormone receptor (TR) to be expressed in neurons in the paraventricular nucleus (PVN) that release thyrotropin-releasing hormone (TRH); in agreement with earlier studies, D2 was limited to glial cells, such as astrocytes and third ventricle tanycytes.6-8 Based on their observations, Fliers et al5 proposed a model for the action of thyroid hormone (T3/T4), which is summarized in Figure 1B. T4 is taken up by glial cells via an unknown mechanism/transporter.5 Once in the glial cell, T4 is converted to T3 by D2. T3 can either enter the nucleus or exit the glial cell, again via an unknown mechanism. The circulating T3 is then taken up by a TRH-producing neuron via MCT8. Once in the neuron, it binds to a thyroid hormone receptor (TR), which then forms a complex with the retinoid X receptor (RXR). This complex binds to a T3-responsive element (TRE) and, in turn, causes a change in the transcription of specific genes and thus in the subsequent translation into proteins.
Genomic and protein structure
MCT8, also referred to as SLC16A2 (solute carrier family 16, member 2), was originally identified and isolated as a novel gene.9 Because of the presence of an N-terminal PEST domain consisting of proline/glutamic acid repeats, as well as 12 hydrophobic transmembrane domains (TMD), the gene was designated XPCT for X-linked PEST-containing transporter. Lafreniere et al showed that XPCT was expressed only from the active X chromosome in females, and so was subject to X-inactivation.9 In 2003, Friesema et al used Xenopus oocytes to show that the rat homolog of MCT8/SLC16A2 could transport thyroid hormone (T3).4
The MCT8 gene covers approximately 112.6 kb of genomic DNA. However, the transcript is only 4.3 kb in length and contains six exons (Figure 2A). The first exon is relatively large (818 bp) and has two alternative start sites. As a result, the MCT8 gene encodes for two potential proteins, of 613 and 539 amino acids, respectively. Both proteins contain the 12 TMDs and the shorter one is highly conserved in lower species. The 12 TMDs are dispersed throughout the six exons, although exon 3 encodes four TMDs and both transmembrane domains 9 and 11 are coded for by portions of two different exons (exons 4 and 5 for TMD9 and exons 5 and 6 for TMD11) (Figure 2A).
Figure 2.


(A) Genomic organization of MCT8/SLC16A2. The boxes indicate the six exons. The blue numbered areas indicate the location of the 12 transmembrane domains within the coding sequence. Location of various nonsense and missense mutations are indicated. Note the vast majority are in the transmembrane domains. ATG, transcription initiation start site; TAA, transcription stop site. (B) Structural organization of the MCT8/SLC16A2 protein. The illustration shows the 12 transmembrane domains. The two different translation start condons in the N-terminal domain are indicated by gray-shaded methionine (‘M’) residues. The currently known missense and truncating mutations are represented by various colors. Green, mutations identified by Dumitrescu et al;10 royal blue, mutations identified by Friesema et al11 and Jansen et al (unpublished data);23 light blue, mutations identified by Lenzer et al;16 red, mutations identified by Schwartz et al17 and Schwartz and Turner (unpublished data); brown, mutation identified by Holden et al;19 rose, mutation identified by Maranduba et al;20 turquoise, mutation identified by Herzovich et al.21 The F229del (F-) mutation (purple) has been observed in two unrelated families.11,17 The black circles indicate the amino acid residue that is altered. A number indicates a deletion of that many base pairs; 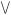 indicates an insertion of an amino acid (3 bp); – indicates the deletion of the indicated amino acid. Modified from Figure 2 of ref 23.
indicates an insertion of an amino acid (3 bp); – indicates the deletion of the indicated amino acid. Modified from Figure 2 of ref 23.
Mutations in MCT8 and Allan--Herndon--Dudley syndrome
The critical role played by MCT8 in the biological function of thyroid hormone in brain development quickly became apparent after its identification by Friesema et al in 2003.4 Two groups, independent of one another, reported patients with a severe neurological presentation and mutations in MCT8.10,11
Dumitrescu et al studied two unrelated males with global developmental delay, central hypotonia, spastic quadriplegia, dystonic movements, nystagmus, and impaired hearing.10 Of interest were the abnormally high levels of free T3, low levels of free T4 and only slightly elevated levels of thyroid stimulating hormone (TSH) in the serum. These findings suggested a defect in thyroid hormone metabolism. Dumitrescu et al excluded defects in D2 and D3 but identified two mutations in MCT8.10
Friesema et al made similar findings in five young males who presented with poor head control, involuntary hand writhing, severe mental retardation, and a lack of speech.11 Again, thyroid studies noted greatly increased serum concentrations of total T3, and total and free T4 levels at the lower end of normal; serum TSH levels varied from normal to high. The possibility of mutations in either of the two known T3 receptors or in the deiodinases was ruled out by sequencing. Thus, an analysis of MCT8 was undertaken. All five patients were found to have mutations, ranging from a large deletion of 24 kb, including exon 1, to missense mutations.
Allan--Herndon--Dudley syndrome (AHDS, OMIM #300523) was first described in 1944,12 and, as such, was one of first X-linked mental retardation (XLMR) syndromes reported. It was also one of the first XLMR conditions to be mapped on the X chromosome.13 Subsequently, other families have been described14,15 on the basis of childhood hypotonia, muscle hypoplasia, severe mental retardation, poor head control, dysarthria, and athetoid movements. Based on the overlap between the clinical features in AHDS and the patients with mutations in MCT8 ,10,11,16 Schwartz et al conducted a mutational analysis of MCT8 in six families with AHDS.17 Mutations, including truncations, in-frame deletions, and missense mutations, were found in all six families. Thus, AHDS is clearly associated with mutations in MCT8. The locations of most of the MCT8 mutations identified thus far are shown in Figure 2.
Clinical characteristics of AHDS patients
Clinical findings
The pioneer American geneticists William Allan and Nash Herndon, and their social worker, Florence Dudley, gave the initial clinical description of AHDS in 1944.12 Their report came just 1 year after the description of fragile X syndrome by Martin and Bell.18 They identified the major findings to be hypotonia (‘weakness and inability to hold head up’), muscular hypoplasia (‘atrophy of muscles of trunk and limbs’), and developmental retardation. Most patients learned to walk, but only with a clumsy and awkward gait. Most made some effort at speech, but with great difficulty (‘mumbling and gibberish’). Bowel and bladder control was delayed and imperfectly achieved.
Twenty-five families with at least 89 affected individuals have been identified and shown to have mutations in MCT8.10,11,16,17,19-22 Only one individual was affected in 13 families. This contrasts with the other families with affected individuals in multiple generations, the largest being the family report by Allan, Herndon, and Dudley, with 29 males affected in seven generations.12 Clinical details from 55 individuals (23 children and 32 adults) in 20 families form the basis for the phenotypic description given here. Clinical details for the four other families are limited.16,21
Of the 25 families, 12 were reported from Western Europe, seven from North America, four from Latin America, one from Australia and one from Japan. Two of the North American families have American--Indian ancestry. No Asian, Middle Eastern, African or African--American families have yet been identified.
Prenatal growth was normal in all for whom birth measurements were available. Birth weight ranged from 3.0 to 4.4 kg (25th to > 97th centiles) and birth length from 48.3 to 53.3 cm (15th to 90th centiles). In the few instances in which birth head circumferences were available, the measurements were in the middle centiles. The pregnancies were not complicated and decreased fetal movements have been noted in only a few instances. There is no evidence for increased prenatal or neonatal mortality.
Postnatal head and statural growth was usually normal (Figures 3, 4). About 20% of affected males had short stature (height < 3rd centile) (Figure 3) and an equal percentage had microcephaly (OFC < 3rd centile) (Figure 4). The few adults with short stature had coexisting scoliosis or contractures that compromised accurate height/length measurements. All individuals with a head circumference below the 3rd centile were children aged < 10 years. All adult head circumferences were within the normal range, distributed from the 3rd to the 90th centiles (Figure 4). The paucity of muscle bulk was reflected in the larger percentage (about two-thirds) of persons with weight below the 3rd centile for age. The low muscle bulk was evident early in childhood and persisted throughout life (Figure 5).
Figure 3.

Height plotted against age in 31 males with Allan--Herndon--Dudley syndrome. Open circles, eight height measurements < 3rd centile; gray circle, one height measurement > 97th centile; solid circles, specific height centiles.
Figure 4.

Head circumference plotted against age in 39 males with Allan--Herndon--Dudley syndrome. Open circles, eight head circumference measurements < 3rd centile; solid circles, specific head circumference centiles.
Figure 5.
VI-33 in K8005 at age 15 years (left) and 65 years (right) showing marked muscle hypoplasia.17
Craniofacial findings
There is a misconception that microcephaly was present in the original family and, hence, should be considered a part of AHDS. The misconception is based on the title of the Allan, Herndon, and Dudley article, ‘Some examples of the inheritance of mental deficiency: apparently sex linked idiocy and microcephaly’.12 In fact, the microcephaly in the title refers to a second family described in the article. In that second family, three persons with microcephaly, one female and two males, were affected in three different sibships. The authors were appropriately cautious in assigning a cause for the microcephaly, recognizing both environmental and genetic possibilities. None of the individuals in the family with ‘sex-linked idiocy’, now known as AHDS, had microcephaly, although two of the 13 birth head circumferences recorded during later follow-up of the family were between the 3rd and 10th centiles.13
Prenatal and infantile hypotonia often produce recognizable changes in facial structures. Typically, the changes affect the lower face, predominantly in the form of open mouth and tented upper lip. Ptosis, cupped ears, thickening of the soft tissue of the nose and ears, upturned earlobes, and a decrease in facial creases can also occur. Among the cases with published photographs or facial descriptions, some have the findings of facial hypotonia whereas others do not (Figures 6, 7). Five individuals have a distinctly myopathic face. By adulthood, the overall facial configuration has usually become tall and narrow; only one adult male retained the rounded face of childhood. Narrowness of the bitemporal diameter and frontal balding were noted in the original family. Bitemporal narrowing has been noted in several subsequently reported families, but frontal balding has not.
Figure 6.


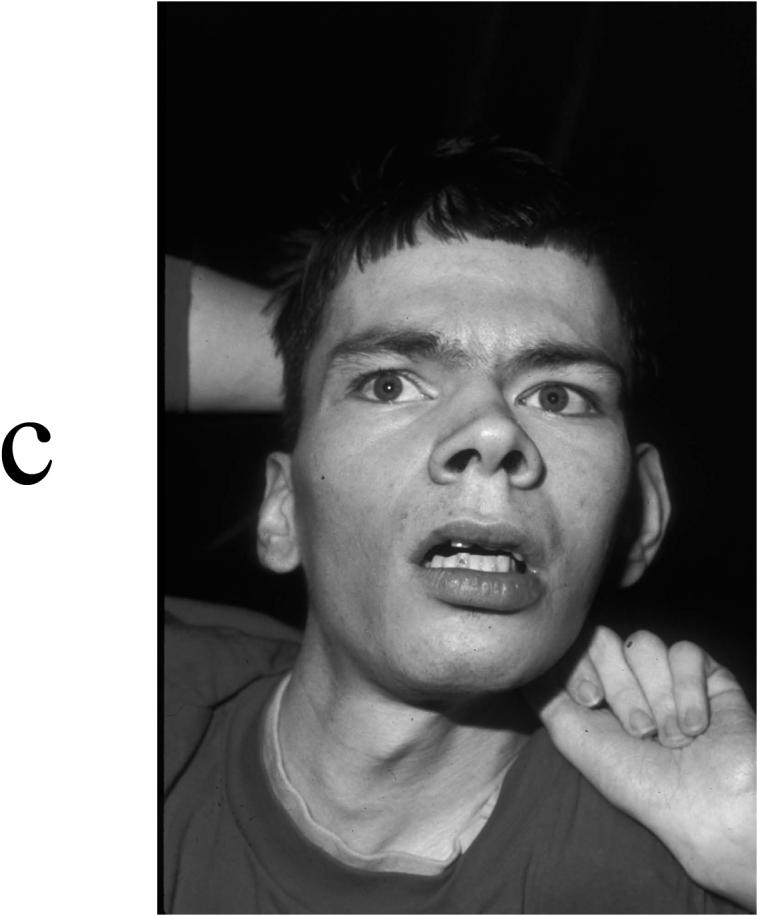

Appearance of four affected males in K8225. (A) IV-1 at age 1 year, showing a normal face. (B) III-11 at age 14 years, showing an elongated and myopathic face with prominence of the eyes. (C) III-3 at age 28 years, showing synophrys and prominence of the lower lip. (D) II-8 at age 39 years, showing an elongated myopathic face with prominence of zygomatic areas and an open mouth.
Figure 7.

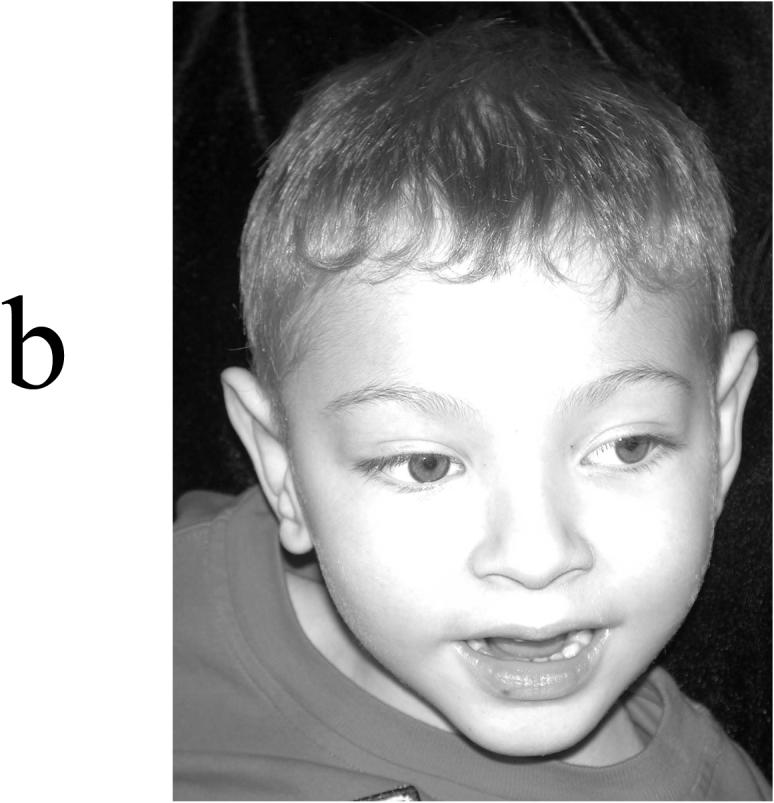
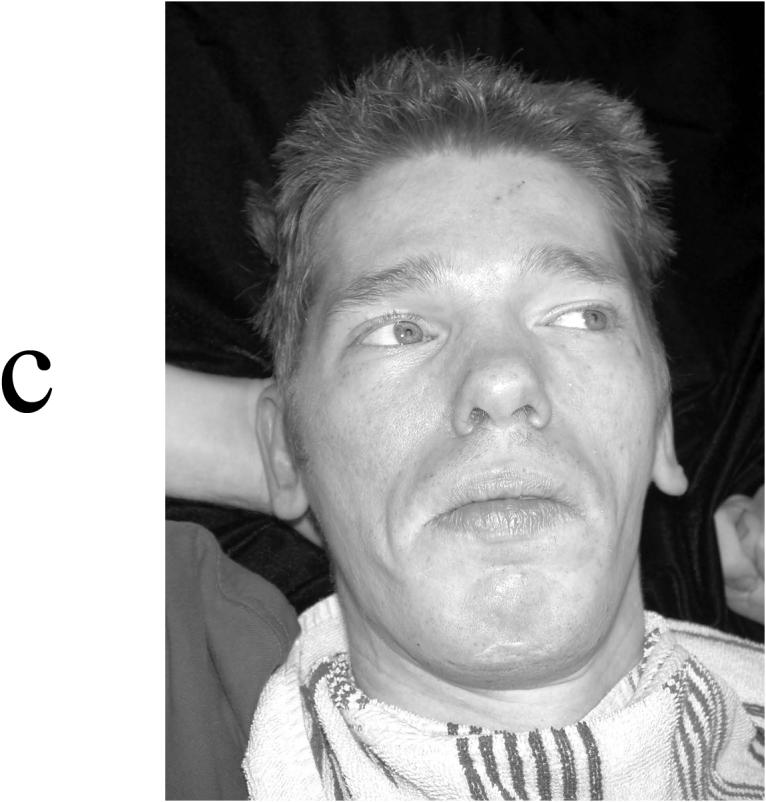
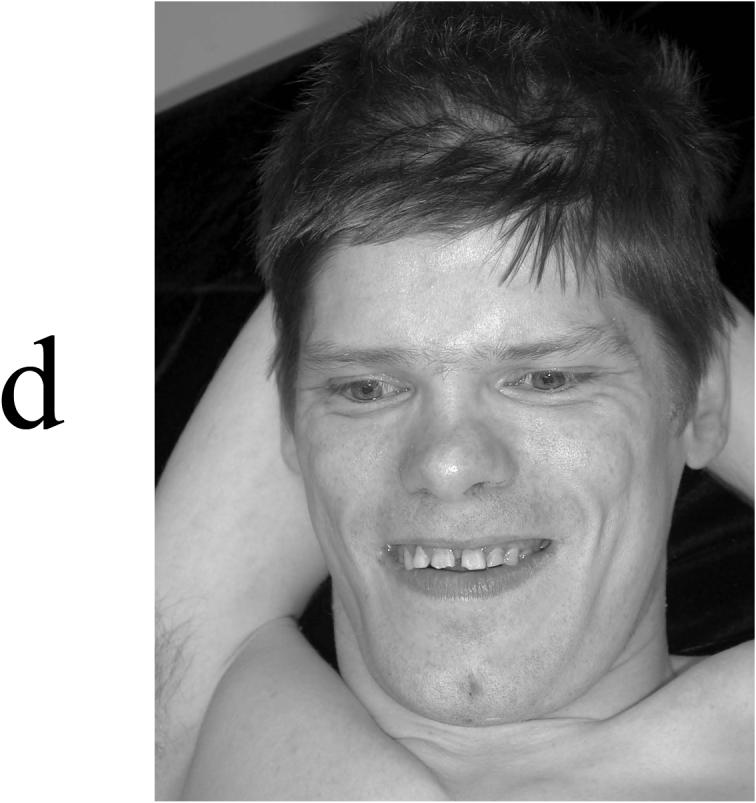
Appearance of four affected males in K9248. (A) V-3 at age 22 months, showing a cupped left ear, depressed nasal bridge, and tenting of the upper lip with short philtrum. (B) V-6 at age 5 years, showing incomplete folding of superior helices and short philtrum. (C) IV-6 at age 38 years, showing elongated face with widow's peak, flattening of midface, and square jaw. (D) IV-5 at age 40 years, showing a long face with short philtrum.
Ocular manifestations are few. Ptosis has been noted in several patients, as have prominent eyes and staring gaze. Notably absent were strabismus, nystagmus, corneal clouding, premature cataracts, and funduscopic abnormalities. Blindness of unstated cause has been noted in one individual11 and anisocoria in two individuals.14
Ear length was generous, measuring above the 97th centile in about half of the adults. This contrasts with ear size in childhood, which measured within the normal range. In some cases, the ear configuration might reflect facial hypotonia, being cupped and simply formed with flattening of the antihelix, unfolding of the superior helix, and upturned earlobes. In other individuals, the helical rim was positioned close to the cranium with prominence of the antihelix. In still others, the ears appeared entirely normal.
Malformations
Major malformations are not a manifestation of AHDS. Pectus excavatum and scoliosis are present in many, perhaps manifestations of the hypotonia and muscle hypoplasia. Long and thin everted feet are typical. The wrists might be held in ulnar deviation.
Genital development
Most affected males have no discernable abnormality of the genital system and undergo puberty at the appropriate time. Failure of one or both testes to descend was noted in four instances. One 16-year-old male with severe physical and mental disabilities had no sign of pubertal development. Testicular volume usually measures within the normal range, the exception being three cases in which the adult volume in one or both testes was below 10 cc.
Neurological manifestations
The neurological phenotype is quite complex. In the simplest view, it is a picture of congenital hypotonia and weakness, which progresses to spasticity and dystonic posturing. Weakness and hypotonia are present from birth and persist throughout life. The progression to spasticity occurs at different rates, being discernible in some children as hyperreflexia ([retention of primitive reflexes, Babinski signs and clonus), and by adulthood as hyperreflexia, contractures, Babinski signs, and clonus. Facial and distal limb dystonic movements or posturing occur, initiated and perhaps accentuated by attempts at speech or purposeful limb movements.
Layered on this neurological background are episodic and more notable involuntary movements. These so-called paroxysmal dyskinesias can occur spontaneously or be provoked by passive movement of body or limb position. In childhood, well-known provocative actions include diaper (nappy) changes, transfer from bed to chair, and other physical care maneuvers. The dyskinesias are multipatterned but are typically characterized by turning of the head, hyperextension of the neck, yawn-like opening of the mouth, stretching of the limbs, and fisting or dystonic posturing of the distal limbs. The episodes generally last less than a minute and can be terminated by flexion of the neck and trunk. The dyskinesias appear less dramatic with age, perhaps because voluntary efforts lessen the frequency or attenuate the severity of attacks.
Impairment of cognitive abilities is by far the most important of the neurological findings. It imposes a lifelong disability that is not offset by strengths in any other area of function. Cognitive impairment is evident from the outset by delay of motor and language milestones. Ultimately, all affected males will have severe impairment of cognitive and adaptive skills. IQ measurements on standardized tests have ranged from < 20 to 34 (Figure 8). No individual has been capable of independent living. Although some have considered this to be a progressive disorder, there is no evidence of progression in the severity of cognitive impairment. Involuntary movements -- athetoid and dystonic in nature -- appear to be more common and more pronounced in adult life, but are well described in infancy and childhood as well.
Figure 8.

IQ measurements (Stanford--Binet) plotted against age in 26 males with Allan--Herndon--Dudley syndrome. Open circles, nine IQ measurements < 20; gray circles, two IQ measurements < 30; solid circles, specific IQ values.
The special senses are generally normal. Impairment of vision or hearing is exceptional.
Accompanying the low muscle bulk is a generalized weakness. This is manifest by difficulty in supporting the head (one is reminded that the original family called this phenomenon ‘limber neck’) and development of motor milestones. Stevenson et al updated the pedigree some 50 years after the initial report, identifying five new affected males and noting that the muscle hypotonia progressed to spastic paraplegia by adult life.13 Most individuals who live to adulthood walk, albeit clumsily and awkwardly. With aging, this skill might be lost.
Seizures
Although about one-quarter of individuals will have seizures, they do not commonly dominate the clinical picture. Staring attacks, myoclonic jerks, and tonic--clonic convulsions have been reported. The onset is typically in infancy or early childhood and in general appears responsive to anticonvulsants.
Behaviorally, affected males are attentive, friendly, and docile. They exhibit no proneness to aggressive or destructive activities.
Although several affected males have lived to their middle 80s, overall the lifespan is compromised. In one kindred (K8900), one died before age 10 years, two died in their twenties, and one died in his thirties. In another (K9320), two of three affected males died at 4 years and 10 years. Several other males have died in childhood or early adult life, normally of recurrent infections.10,12,15
Chronic or systemic illness does not appear to be a component of the syndrome complex. Several individuals in one family had recurrent cutaneous fungal infections. One child and two adults developed diabetes mellitus. One infant of 11 months of age died of leukemia.
The typical thyroid profile in affected males is elevated serum free T3 and total T3, low or low normal free T4 and total T4 and normal TSH. Those with low circulating T4 were not frankly hypothyroid, although pale and cool skin, dry hair, and constipation were reported in some. None had prolonged neonatal jaundice, macroglossia, umbilical hernia, or myxedema. Likewise, elevated circulating T3 did not produce systemic hyperthyroidism, although the generalized muscular asthenia might be related manifestations. Carrier females do not generally show these alterations in circulating thyroid hormones.
Carriers
Female carriers of MCT8 mutations typically do not express phenotypic abnormalities; they appear to grow and develop normally. X-inactivation studies are available on a number of carriers and most do not show skewing of X-inactivation; however, two exceptions have been reported. One carrier mother was considered to have neurological residua from perinatal injury,17 another had severe mental retardation of unknown cause.
Diagnosis of AHDS Patients with MCT8 Mutations and AHDS
AHDS is an X-linked mental retardation condition. As such, only males will present with a clinical manifestation. Based on the clinical information available on patients with MCT8 mutations, a relatively short list of features that would constitute AHDS can be compiled. These features are: childhood hypotonia, severe mental retardation, hyperreflexia, muscle hypoplasia/asthenic build, and abnormal hand positioning. However, only the hypotonia and severe mental retardation are present in 100% of patients. Thus, it is suggested that males presenting with two or more of the aforementioned clinical characteristics are screened for disturbances in serum levels of the thyroid hormones, T3 and T4. The presence of abnormally high serum free T3 and low normal or low free or total T4 in such a male would be a strong indicator of AHDS and the presence of a MCT8 mutation.
Thyroid status of patients with MCT8 mutations and AHDS
As has already been mentioned, all patients with AHDS and MCT8 mutations exhibit abnormally high levels of serum free T3 levels, serum T4 levels in the low normal or below normal range, and a serum TSH concentration level in the upper normal range. Thus, despite the significantly elevated levels of serum T3, and rather low levels of T4, the serum TSH levels are elevated rather than suppressed. This would indicate that the decrease in serum T4 is not compensated for by the high levels of T3 as regards the negative feedback inhibition of TSH secretion.
The presence of the cognitive impairment observed in patients with ADHS is in agreement with the critical role MCT8 plays in supplying T3 to neurons (see Figure 2B). However, the other aspects of the ADHS phenotype (see above) need some further explanation; their presence does not suggest a disturbance in thyroid function. Recent studies in patients, and the examination of a mouse model deficient in MCT8, have provided some insights into in this perplexing problem.
Studies in ADHS patients have found increased levels of sex-hormone-binding globulin (SHBG).23 However, levels of serum T4-binding globulin are not elevated in these patients.21,23 As SHBG synthesis by the liver is stimulated by T3, the positive correlation between SHBG levels and serum T3 suggests that the liver is exposed to toxic levels of T3.24
The muscle wasting that is observed in patients with ADHS might also correlate with the increase in serum levels of T3. Herzovich et al. studied a patient with an MCT8 mutation and observed high serum levels of both lactic acid and ammonium.21 They postulated that these values reflected peripheral hyperthyroidism of skeletal muscle due to an impaired balance of ATP.25,26 A similar hypothesis was put forth by Maranduba et al to explain the low body weight observed in patients with AHDS.20
The mechanism of the pathophysiology of MCT8 deficiency was investigated by Dumitrescu et al using knockout mice.27 These mice recapitulated the abnormal thyroid findings observed in patients with a MCT8 mutation: high serum levels of T3 and low levels of T4. Furthermore, Dumitresca et al were able to show that the cerebrum was T3 deficient whereas other tissues, which did not depend exclusively on MCT8, were thyrotoxic.10 Additionally, when the mice were deprived of thyroid hormone and given L-T3, they exhibited a suppression of TSH that required higher serum levels of T3. This would indicate a resistance of the hypothalamus to T3. However, the MCT8-deficient mice did not show any neurological defects; however, neither do mice with iodine deficiency.27 It is possible that more detailed studies are needed in MCT8-deficient mice to elucidate any affect on the central nervous system.
Treatment, management and counseling for ADHS patients
Some ADHS patients have been treated with supplementation of L-T4 without success10,21 and, for the present, these patients are not amenable to treatment. They also demonstrate both ‘hyperthyroid’ and ‘hypothyroid’ tissues:21 The former due to the high levels of circulating T3, the latter to the lack of intracellular T3. Thus, trying to treat the ‘hypothyroid’ neurons by increasing circulating levels of T3 will adversely affect the tissues prone to a ‘hyperthyroid’ condition because of their ability to take up T3.
Counseling families with a male with ADHS is similar to that reserved for any family with an X-linked recessive condition. Boys will be at a 50% risk of being affected if the mother is a carrier of an MCT8 mutation. Girls will not be affected but will be at a 50% risk of being a carrier if the mother has an MCT8 mutation.
Summary
Allan--Herndon--Dudley syndrome is an X-linked condition of mental retardation associated with severe mental retardation and mutations in MCT8. Mutations in MCT8, a thyroid hormone transporter gene, results in high levels of serum free T3, rather low levels of serum T4, and a TSH concentration in the normal range. However, although these biochemical findings are suggestive of a thyroid hormone disturbance, the clinical presentation observed in AHDS is not typical of hypothyroidism. Neonatal jaundice, macroglossia, hoarseness, and myxedematous skin changes are all absent, but the degree of cognitive impairment is similar to both cretinism and to that seen in untreated congenital hypothyroidism.1
At present, no treatment is available for ADHS. L-T4 treatment has been unsuccessful in modifying serum thyroid hormone levels and neurological symptoms.10,21 Also, the severity of the disease probably indicates that the damage due to the presence of a MCT8 mutation is likely irreversible.
The alterations in free T3 and free T4 levels observed in AHDS patients do provide a simple and cost-effective method of screening males with developmental delay and congenital hypotonia for MCT8 mutations. This knowledge will be of value for carrier testing and even prenatal testing in select families.
Practice points
Males presenting with congenital hypotonia, mental retardation, and generalized muscle weakness should have thyroid hormone studies done.
Males with abnormally high free T3, but low normal free T4 and TSH levels in the normal range should be tested for mutations in MCT8.
Males with MCT8 mutations or Allan--Herndon--Dudley syndrome (AHDS) have both ‘hyperthyroid’ tissues (bone, skeletal muscle, liver) and ‘hypothyroid’ tissues (neurons).
Female carriers of MCT8 mutations do not exhibit any of the AHDS clinical features.
AHDS is an X-linked mental retardation syndrome and families with AHDS should be counseled as such.
Acknowledgements
We wish to thank the families who have participated in our study of the Allan--Herndon--Dudley syndrome. We also appreciate the cooperation of the following people who provided clinical samples: Drs Martin Bialer, Judith Martin, Jewell Ward, Nancy J. Carpenter, James Lewis, Silvana Marsa, and Javier Sanabria. Some of the data presented were collected using funding provided, in part, by a grant from NICHD (HD26202) to C.E.S. and a grant from the South Carolina Department of Disabilities and Special Needs. This chapter is dedicated to the memory of Ethan Francis Schwartz, 1996−−1998.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
The authors state that there are no conflicts of interest.
References
- 1.Morreale de Escobar G, Obregon MJ, Escobar del Rey F. Role of thyroid hormone during early brain development. Eur J Endocrinol. 2004;151(Suppl 3):U25–U37. doi: 10.1530/eje.0.151u025. [DOI] [PubMed] [Google Scholar]
- 2.Bianco AC, Salvatore D, Gereben B, et al. Biochemistry, cellular and molecular biology, and physiological roles of iodothyronine selenodeiodinases. Endocr Rev. 2002;23:38–89. doi: 10.1210/edrv.23.1.0455. [DOI] [PubMed] [Google Scholar]
- 3.Hennemann G, Docter R, Friesema EC, et al. Plasma membrane transport of thyroid hormones and its role in thyroid hormone metabolism and bioavailability. Endocr Rev. 2001;22:451–476. doi: 10.1210/edrv.22.4.0435. [DOI] [PubMed] [Google Scholar]
- 4.Friesema EC, Ganguly S, Abdalla A, et al. Identification of monocarboxylate transporter 8 as a specific thyroid hormone transporter 8 as a specific thyroid hormone transporter. J Biol Chem. 2003;278:40126–40135. doi: 10.1074/jbc.M300909200. [DOI] [PubMed] [Google Scholar]
- 5.Fliers E, Alkemade A, Wiersinga WM, Swaab DF. Hypothalamic thyroid hormone feedback in health and disease. Progress in Brain Research. 2006;153:189–207. doi: 10.1016/S0079-6123(06)53011-0. [DOI] [PubMed] [Google Scholar]
- 6.Tu HM, Kim SW, Salvatore D, et al. Regional distribution of type 2 thyroxine deiodinases messenger ribonucleic acid in rat hypothalamus and pituitary and its regulation by thyroid hormone. Endocrinology. 1997;138(8):3359–3368. doi: 10.1210/endo.138.8.5318. [DOI] [PubMed] [Google Scholar]
- 7.Guadano-Ferraz A, Obregon MJ, St Germain DL, Bernal J. The type 2 iodothyronine deiodinase is expressed primarily in glial cells in the neonatal rat brain. Proc. Natl. Acad. Sci. USA. 1997;94(19):10391–10396. doi: 10.1073/pnas.94.19.10391. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Diano S, Leonard JL, Meli R, et al. Hypothalamic type II iodothyronine deiodinase: a light and electron microscopic study. Brain Res. 2003;976(1):130–134. doi: 10.1016/s0006-8993(03)02692-1. [DOI] [PubMed] [Google Scholar]
- 9.Lafreniere RG, Carrel L, Willard HF. A novel transmembrane transporter encoged by the XPCT gene in Xq13.2. Hum Mol Genet. 1994;3:1133–1139. doi: 10.1093/hmg/3.7.1133. [DOI] [PubMed] [Google Scholar]
- 10.Dumitrescu AM, Liao XH, Best TB, et al. A novel syndrome combining thyroid and neurological abnormalities is associated with mutations in a monocarboxylate transporter gene. Am J Hum Genet. 2004;74:168–175. doi: 10.1086/380999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Friesema EC, Grueters A, Biebermann H, et al. Association between mutations in a thyroid hormone transporter and severe X-linked psychomotor retardation. Lancet. 2004;364:1435–1437. doi: 10.1016/S0140-6736(04)17226-7. [DOI] [PubMed] [Google Scholar]
- 12.Allan W, Herndon CN, Dudley FC, et al. Some examples of the inheritance of mental deficiency: apparently sex-linked idiocy and microcephaly. Am J Mental Defic. 1944;48:325–334. [Google Scholar]
- 13.Schwartz CE, Ulmer J, Brown A, et al. Allan-Herndon syndrome. II. Linkage to DNA Markers in Xq21. Am J Hum Genet. 1990;47:454–458. [PMC free article] [PubMed] [Google Scholar]
- 14.Bialer MG, Lawrence L, Stevenson RE, et al. Allan-Herndon-Dudley syndrome: clinical and linkage studies on a second family. Am J Med Genet. 1992;43:491–497. doi: 10.1002/ajmg.1320430173. [DOI] [PubMed] [Google Scholar]
- 15.Zorick TS, Kleimann S, Sertie A, et al. Fine mapping and clinical reevaluation of a Brazilian pedigree with a severe form of X-linked mental retardation associated with other neurological dysfunction. Am J Med Genet A. 2004;127:321–323. doi: 10.1002/ajmg.a.30009. [DOI] [PubMed] [Google Scholar]
- 16.Lenzner S, Rosenkranz MD, Grueters A, et al. Severe X-linked mental retardation caused by mutations in the gene for the thyroid hormone transporter MCT8 (abstract C32). European Human Genetics Conference; Munich. 2004 June 12−15.2004. [Google Scholar]
- 17.Schwartz CE, May MM, Carpenter NJ, et al. Allan-Herndon-Dudley syndrome and the monocarboxylate transporter 8 (MCT8) gene. Am J Hum Genet. 2005;77:41–53. doi: 10.1086/431313. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Martin JP, Bell J. A pedigree of mental defect showing sex linkage. J Neurol Psychiatr. 1943;6:154–157. doi: 10.1136/jnnp.6.3-4.154. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Holden KR, Zuniga OF, May MM, et al. X-linked MCT8 gene mutations: characterization of the pediatric neurologic phenotype. J Child Neurol. 2005;20(10):852–7. doi: 10.1177/08830738050200101601. [DOI] [PubMed] [Google Scholar]
- 20.Maranduba CM, Friesema EC, Kok F, et al. Decreased cellular uptake and metabolism in Allan-Herndon-Dudley syndrome (AHDS) due to a novel mutation in the MCT8 thyroid hormone transporter. J Med Genet. 2006;43(5):457–60. doi: 10.1136/jmg.2005.035840. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Herzovich V, Vaiani E, Marino R, et al. Unexpected Peripheral Markers of Thyroid Function in a Patient with a Novel Mutation of the MCT8 Thyroid Hormone Transporter Gene. Horm Res. 2006;67(1):1–6. doi: 10.1159/000095805. [DOI] [PubMed] [Google Scholar]
- 22.Kakinuma H, Itoh M, Takahashi H. A novel mutation in the monocarboxylate transporter 8 gene in a boy with putamen lesions and low free T4 levels in cerebrospinal fluid. J Petdiatr. 2005;147(4):552–4. doi: 10.1016/j.jpeds.2005.05.012. [DOI] [PubMed] [Google Scholar]
- 23.Friesema EC, Jansen J, Heuer H, et al. Mechanisms of disease: psychomotor retardation and high T3 levels caused by mutations in monocarboxylate transporter 8. Nat Clin Pract Endocrinol Metab. 2006;2(9):512–23. doi: 10.1038/ncpendmet0262. [DOI] [PubMed] [Google Scholar]
- 24.Pugeat M, Crave JC, Tourniaire J, Forest MG. Clinical utility of sex hormone-binding globulin measurement. Horm Res. 1996;45(3−5):148–55. doi: 10.1159/000184778. [DOI] [PubMed] [Google Scholar]
- 25.Doi J, Ohtsubo A, Ohtsuka A, et al. Triiodothyronine but Not Thyroxine Accelerates Myofibrillar Proteolysis via ATP Production in Cultured Muscle Cells. Bioscience, Biotechnology, and Biochemistry. 2003;67(11):2451–2454. doi: 10.1271/bbb.67.2451. [DOI] [PubMed] [Google Scholar]
- 26.Fukeri H, Taniguchi S, Ueta Y, et al. Enhanced activity of the purine nucleotide cycle of the exercising muscle in patients with hyperthyroidism. J Clin Endrocrinol Metab. 2001;86(5):2205–10. doi: 10.1210/jcem.86.5.7516. [DOI] [PubMed] [Google Scholar]
- 27.Dumitrescu AM, Liao XH, Weiss RE, et al. Tissue-specific thyroid hormone deprivation and excess in monocarboxylate transporter (mct)8-deficient mice. Endocrinology. 2006;147(9):4036–43. doi: 10.1210/en.2006-0390. [DOI] [PubMed] [Google Scholar]