

Summary
Bacteriophage T4 capsid is a prolate icosahedron composed of the major capsid protein gp23*, the vertex protein gp24*, and the portal protein gp20. Assembled on its surface are 810 molecules of the non-essential small outer capsid protein, Soc (10 kDa), and 155 molecules of the highly antigenic outer capsid protein, Hoc (39 kDa). In this study Soc, a “triplex” protein that stabilizes T4 capsid, is targeted for molecular engineering of T4 particle surface. Using a defined in vitro assembly system, anthrax toxins, protective antigen, lethal factor and their domains, fused to Soc were efficiently displayed on the capsid. Both the N- and C-termini of the 80 amino acid Soc polypeptide can be simultaneously used to display antigens. Proteins as large as 93 kDa can be stably anchored on the capsid through Soc-capsid interactions. Using both Soc and Hoc, up to 1662 anthrax toxin molecules are assembled on phage T4 capsid under controlled conditions. We infer from the binding data that a relatively high affinity capsid binding site is located in the middle of the rod-shaped Soc, with the N- and C-termini facing the two- and three-fold symmetry axes of the capsid, respectively. Soc subunits interact at these interfaces, gluing the adjacent capsid protein hexamers and generating a cage-like outer scaffold. Antigen fusion does interfere with the inter-subunit interactions, but these interactions are not essential for capsid binding and antigen display. These features make the T4-Soc platform the most robust phage display system reported to date. The study offers insights into the architectural design of bacteriophage T4 virion, one of the most stable viruses known, and how its capsid surface can be engineered for novel applications in basic molecular biology and biotechnology.
Keywords: bacteriophage T4, Soc, Hoc, virus assembly, phage display, anthrax toxins
Introduction
Bacteriophage T4 infects Escherichia coli and belongs to the family of Myoviridae. It is a large double-stranded DNA virus and packages 171 kb DNA, about 1.02 genome lengths. The head is a prolate icosahedron, 120 nm long and 86 nm wide (Figure 1).1 Attached to this is a 120 nm long contractile tail terminating in a baseplate and six long tail fibers.2 The phage T4 head is composed of three essential capsid proteins: i) 930 copies of the major capsid protein gene product (gp)a23*b (49 kDa), which form hexagonal lattice; ii) 55 copies of the vertex protein gp24* (47 kDa), which form eleven of the twelve vertices, one pentamer at each vertex; iii) 12 copies of the portal protein gp20 (61 kDa), which form a dodecameric head-tail connector vertex through which DNA enters during packaging and exits during infection.1,3
Figure 1. Schematics of the anthrax toxin-Soc fusion recombinants.
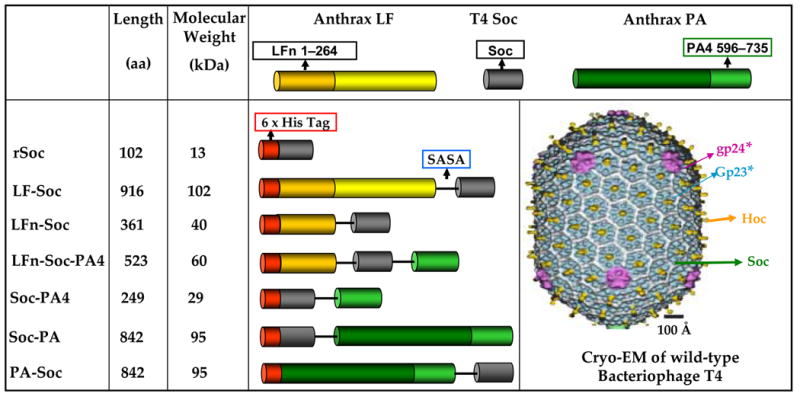
The anthrax toxin genes, PA, PA4, LFE687C, and LFn, were fused to the N- and/or C- terminus of Soc via the linker, SASA, as shown. Hexa-histidine tag was fused to the N-terminus of each gene fusion. The fusions were constructed using the PCR-based splicing by overlap extension (SOE) strategy. The numbers in the boxes represent the aa numbers of the respective proteins. The cryo-EM reconstruction of phage T4 capsid1 is shown to indicate the disposition of the capsid proteins; gp23* subunits are shown in blue, gp24* in purple, Hoc in yellow, and Soc in white.
A unique feature of T4 head is that it is decorated with two non-essential outer capsid proteins.4,5 The highly antigenic outer capsid protein (Hoc, 39 kDa), a dumbbell shaped monomer, binds at the center of each gp23* hexon, up to 155 copies per capsid (Figure 1). The small outer capsid protein (Soc, 10 kDa), a rod-shaped molecule, binds between gp23* hexons, but not between gp23* and gp24* at the hexon-penton junctions. Soc is visualized as a trimer at the trigonal points where three gp23* hexons meet (local quasi three-fold symmetry axes), and a dimer between two adjacent hexons (local quasi two-fold symmetry axes).1,6,7
Soc is the first discovered member of the viral “triplex” proteins, which decorate the outer surface of large icosahedral viruses.4 These include: i) gpD from bacteriophage λ (11 kDa; 420 copies),8,9 ii) gpDec from bacteriophage L (14 kDa; 180 copies),10,11 iii) pIX from adenovirus (14 kDa; 240 copies),12,13 and iv) triplex protein complex, vp19-(vp23)2, from human herpes virus (vp19, 50 kDa, 320 copies, and vp23, 34 kDa, 640 copies).14 No sequence similarity is evident among the triplex proteins. These proteins exhibit exquisite specificity towards the mature conformational state of the respective capsid protein. The P22 triplex proteins even discriminate subtle differences in the capsid protein conformational states at the quasi three-fold axes, the ones closest to the icosahedral two-fold axes and the true icosahedral three-fold vertices.11 Some of these proteins have been useful for the display of foreign peptides and proteins on viral capsid, for studying protein-protein interactions, and delivery of antigens and genes into targeted cells.15–18
Soc exhibits unique features to elucidate the finer details of icosahedral virus architecture, and to develop multi-component display platforms. Soc, unlike the other triplex proteins, is present at high density, 810 copies per capsid, and completely dispensable.1,4 Soc binding sites emerge after the capsid is assembled, following maturation cleavages and prohead expansion, as the DNA is being packaged inside the capsid.19 This allows for in vitro assembly of recombinant Soc, or Soc-fused antigens, on soc− phage or empty expanded capsids20 in a purified system under defined conditions. The instability of soc− phage to pH 10.6 (wild-type phage is stable to pH10.6)5 allows quantification of virus infectivity in response to structural modifications of Soc. Combined with the recently developed in vitro Hoc and complex display systems,18,21 Soc offers tremendous opportunities to engineer phage T4 outer capsid.
Here, we report the development and analysis of an in vitro Soc assemblyc system. Full-length or structural domains of anthrax toxins, protective antigen (PA, 83 kDa) and lethal factor (LFd, 89 kDa), were fused to Soc, over-expressed, purified, and assembled on soc− phage particles. Remarkably, the 80aa-Soc can efficiently anchor anthrax antigens as large as 93 kDa on the capsid surface. Up to 1662 copies of multiple anthrax toxins can be assembled on a single capsid and the copy number can be manipulated by changing the ratio of the binding components. Analysis of the binding properties of Soc-anthrax fusions suggests that the capsid binding site is located in the middle of Soc, and the N- and C- termini face the two- and three-fold symmetry axes, respectively. Inter-Soc subunit interactions at the trimeric and dimeric interfaces cement the inter-capsomer contacts, greatly stabilizing the phage T4 capsid architecture.
Results
Fusion of anthrax toxins to Soc
Recombinant native Soc (rSoc) as well as six fusions of anthrax toxins and domains to Soc were constructed. Of these, three were N-terminal fusions (LF-Soc, 102 kDa; LFn-Soc, 40 kDa; PA-Soc, 95 kDa), two were C-terminal fusions (Soc-PA domain-4 (PA4), 29 kDa; Soc-PA, 95 kDa), and one was a double fusion to both the N- and C-termini of Soc (LFn-Soc-PA4, 60 kDa) (Figure 1). With a hexa-histidine tag at the N-terminus and a four aa structureless linker (SASA) between Soc and anthrax domains, the constructs were designed to analyze the effect(s) of protein fusion on Soc-capsid interactions. For example, the 83 kDa PA was fused to both the N- and C-termini of Soc to determine whether fusion of a bulky molecule, or which fusion, affects the capsid binding site. Simultaneous fusion of anthrax domains to both the N- and C-termini is of particular interest. The recombinants were over-expressed in E. coli up to about ~20% of the total cell protein (data not shown). In some cases (rSoc and Soc-PA4), the over-expressed proteins remained in the insoluble inclusion bodies, requiring urea denaturation and renaturation followed by HisTrap column chromatography and FPLC gel filtration (see Materials and Methods). About 6 to 8 mg of anthrax domain fusion proteins, and 2 to 3 mg of full-length fusion proteins, with a purity of ~95% were obtained from one liter of induced culture (Figure 2(a)).
Figure 2. The anthrax toxin-Soc fusion recombinants retained function.
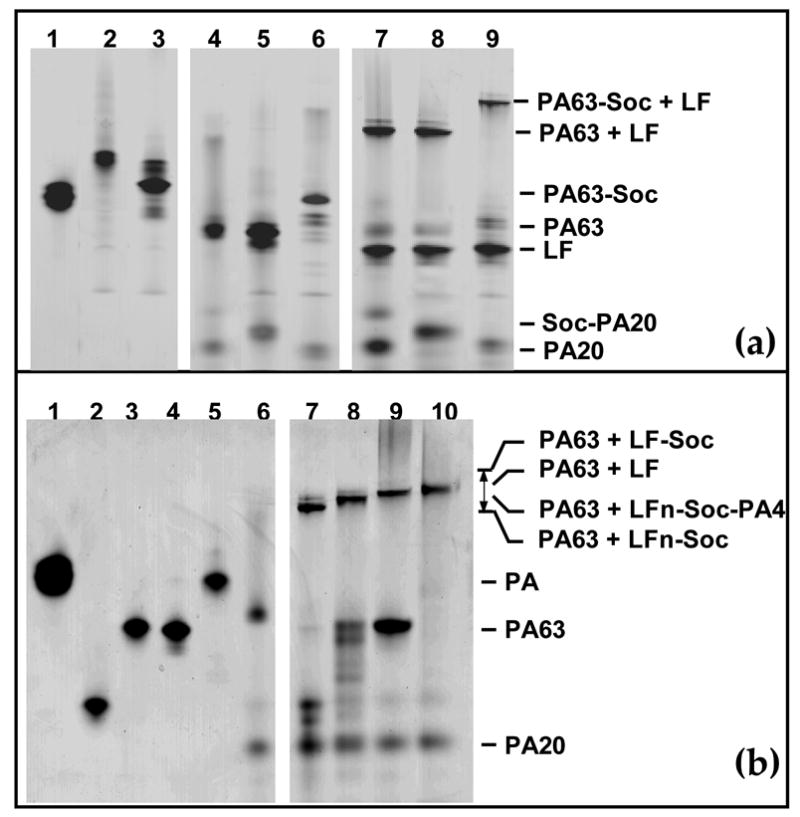
Trypsin-nicking and functional assays were performed using the purified proteins as described in Materials and Methods. The samples were subjected to 4–12% gradient native PAGE (Invitrogen) and stained with Coomassie blue. (a): Trypsin nicking of PA-Soc and Soc-PA and in vitro binding to LF. Lanes 1: PA; 2: Soc-PA; 3: PA-Soc; 4: nPA; 5: Soc-nPA; 6: nPA-Soc; 7: LF + nPA; 8: LF + Soc-nPA; 9: LF + nPA-Soc. (b): in vitro binding of LF-Soc, LFn-Soc, and LFn-Soc-PA4 to trypsin-nicked PA (PA63). Lanes 1–5, PA, LFn-Soc, LFn-Soc-PA4, LF, and LF-Soc, respectively; 6: nPA; 7: nPA + LFn-Soc; 8: nPA + LFn-Soc-PA4; 9: nPA + LF; 10: nPA + LF-Soc.
Anthrax toxin-Soc fusion proteins retain function
In the biochemical pathway for lethal toxin formation, the 83 kDa PA, following binding to the host cell receptors (CMG2 or TEM8) through PA4, is cleaved by the furin protease to PA63 and PA20.22 The PA63 heptamerizes and interacts with the N-terminal domain of LF (LFn) and/or EF. In vitro, this can be mimicked by trypsin nicking of PA and heptamerization in the presence of LF or LFn. The gel migration patterns of the (PA63)7-LF(LFn) complexes, which were well documented in literature,22–24 provided the basis to assess the function of anthrax toxin-Soc fusion proteins.
All the fusion proteins migrated as a single sharp band following denaturing (data not shown) as well as native PAGE (Figure 2, panels (a) and (b)). The latter property indicated that the fusion proteins folded into a native conformation; otherwise, these would have migrated as a broad smear. The purified Soc-PA and PA-Soc, like their native counterpart PA, were nicked to Soc-PA63 and PA63-Soc, releasing PA20 and Soc-PA20, respectively (Figure 2(a), lanes 4–6). When the nicked proteins were treated with native LF, all formed higher order complexes (lanes 7–9). Similarly, the purified LFn-Soc (Figure 2(b), lane 2), LFn-Soc-PA4 (lane 3), and LF-Soc (lane 5) interacted with trypsin-nicked PA (nPA) as well as the native LF (lane 4) [compare the high mol. wt. band in lane 9 (LF-PA63 complex) with lanes 7, 8, and 10 (LFn-Soc-, LFn-Soc-PA4-, and LF-Soc-PA63, respectively). It is significant that the double fusion construct, LFn-Soc-PA4, showed as good a binding as LFn-Soc. Not only that the Soc-fused PA63-LF complexes migrated similarly to those reported in literature,23 the positions of the complexes strictly correlated with the increase or decrease in the size of the respective fusion protein. For example the PA63-Soc-LF complex, which is about 70,000 kDa larger due to Soc fusion to LF (Figure 2(a), lane 9), migrated slower than the native PA63-LF complex (lanes 7, 8).
Anthrax toxin-Soc recombinants efficiently assemble on phage T4
The recombinant Soc (rSoc) efficiently assembled in vitro on hoc−soc− phage in the presence of excess BSA and native LF; no non-specific binding of BSA or LF was evidente (Figure 3(a), compare lanes 4 and 6 with lane 2; see Materials and Methods for experimental details). Binding reached saturation at a relatively low Soc:capsid binding site ratio of 30:1 (4.6 μM Soc; Figure 3(b)), and is of high affinity with an apparent Kd of 73 nMf (Figure 3 (e)). The Bmax as calculated from the saturation binding curve was 746 per capsid, which means that >92% of the binding sites were filled.
Figure 3. Quantitative analyses and binding kinetics of rSoc, LFn-Soc and Soc-PA4.
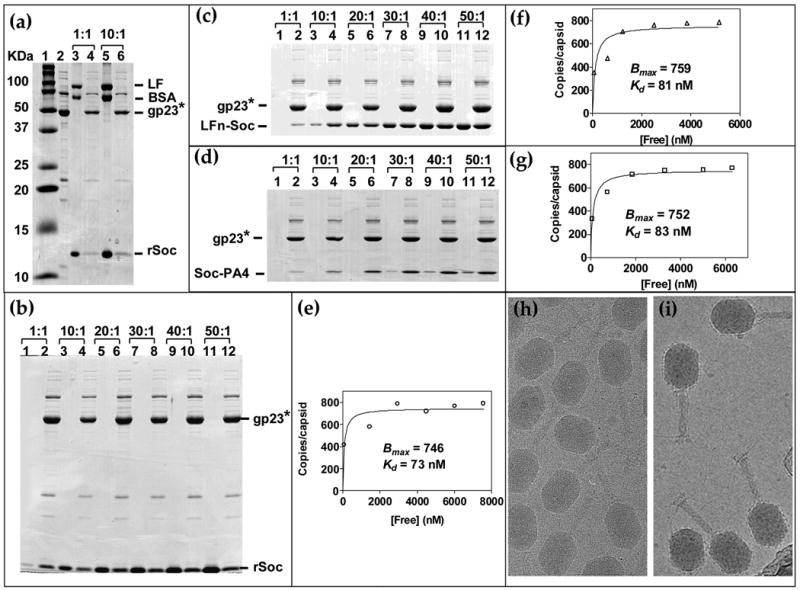
In vitro binding of purified proteins to hoc−soc− phage was performed according to the procedure described in Materials and Methods. (a): Specificity of rSoc binding. A mixture of purified rSoc, BSA, and LF were incubated with hoc−soc− phage and the supernatant (unbound) and pellet (phage-bound) fractions were analyze by SDS-PAGE. Lanes: 1, mol. wt. standards; 2, control hoc−soc− phage; 3 and 5, unbound fraction; 4 and 6, phage-bound fraction. The ratios at the top represent the ratio of number of molecules of each protein (rSoc, BSA, and LF) to the number of Soc binding sites on phage (~2 × 1010 hoc−soc− particles) in the reaction mixture. (b, c and d): Saturation binding of rSoc, LFn-Soc, and Soc-PA4 respectively, on phage T4. About 2 × 1010 hoc−soc− phage particles were incubated with increasing ratio of rSoc, LFn-Soc, or Soc-PA4 molecules to Soc binding sites (1:1 to 50:1, shown at the top). Lanes: 1, 3, 5, 7, 9, and 11, unbound protein; Lanes 2, 4, 6, 8, 10, and 12, phage-bound protein. The density volumes of the bound and unbound proteins and gp23* in the respective lane were determined by laser densitometry, normalized to the copy number of gp23* (930 copies per capsid) and the copy number of each fusion protein per phage particle was determined. The data were plotted as a saturation binding curve (e, f and g) from which the apparent Kd (association constant) and Bmax (maximum copy number per phage particle) were calculated using the GraphPad PRISM-4 software. (h, i): Cryo electron micrographs of wild-type and LFn-Soc-T4 phage, respectively (micrographs kindly provided by Drs. Andrei Fokine and Michael Rossmann, Purdue University).
To test whether fusion to N- and C-termini of Soc affected binding, the ratio of LFn-Soc (40 kDa) or Soc-PA4 (29 kDa) to capsid binding sites was varied at a fixed concentration of hoc− soc− phage. Binding of both LFn-Soc and Soc-PA4 reached saturation at a ratio of 30:1, the same as native Soc (Figure 3(c, d)). The Bmax was 759 and 752, and the apparent Kd for binding, 81 nM and 83 nM respectively, for LFn-Soc and Soc-PA4 (Figure 3(f, g)). Thus the basic parameters of Soc binding are not altered by fusion to either terminus of Soc. Cryo-EM of the LFn-Soc-T4 phage showed decoration of the capsid surface with a layer of the displayed antigen mass (Figure 3(i); also see Ref. 18).
Bulky protein fusions to Soc assemble on phage T4
Considering that Soc is a small protein (80 aa, 10 kDa) and tightly meshed with the capsid lattice,1 attachment of bulky mass could disrupt binding, as well as cause steric problems. Data with three independent clones, PA-Soc, Soc-PA, and LF-Soc, (fused mass up to 93 kDa) showed good binding, reaching saturation at a fusion protein to binding site ratio of 30:1 (Figure 4(a–c)). However, the apparent Kd for LF-Soc, Soc-PA, and PA-Soc binding has increased by about 10-fold to 745 nM, 761 nM, and 784 nM, respectively and the Bmax decreased by about 2-fold to 358, 352, and 365, respectively (Figure 4(d–f)).
Figure 4. Assembly of bulky full-length anthrax toxins on phage T4.
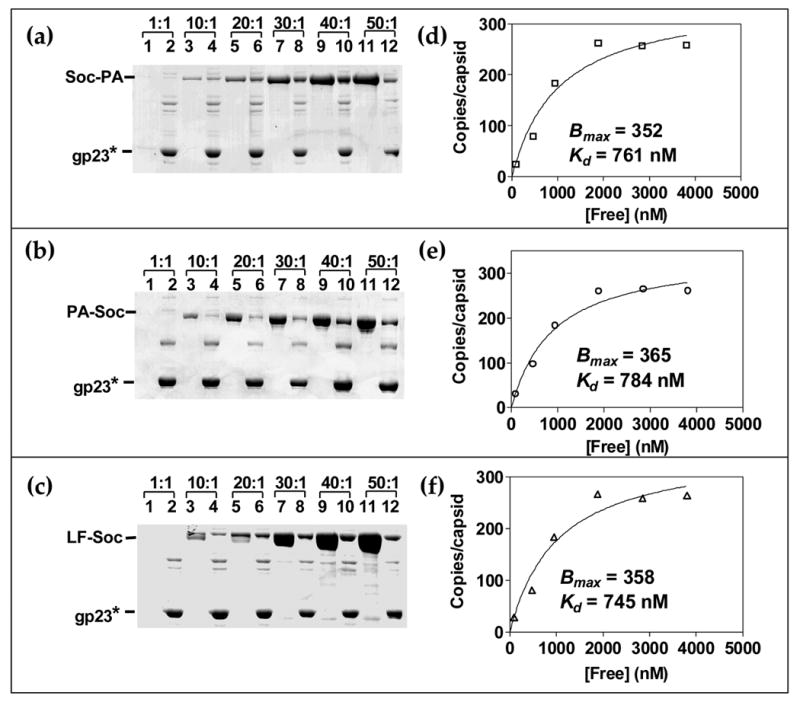
(a, b and c): Saturation binding of Soc-PA, PA-Soc, and LF-Soc respectively, on phage T4. The in vitro assembly and quantitative analyses were performed as described in the legend to Figure 3 and Materials and Methods. About 2 × 1010 hoc−soc− phage particles were incubated with increasing ratio of the respective protein to Soc binding sites, as shown at the top. Lanes: 1, 3, 5, 7, 9, and 11, unbound protein; 2, 4, 6, 8, 10, and 12, phage-bound protein. (d, e and f): Saturation binding curves showing the apparent Kd and Bmax for each protein.
Domains fused to both N- and C-termini of Soc efficiently assemble on phage T4
Can both the N- and C-termini of Soc be simultaneously fused and displayed? Soc was fused to LFn at the N-terminus, and to PA4 at the C-terminus, and its binding behavior was analyzed. Surprisingly, the LFn-Soc-PA4 bound to hoc−soc− T4 phage as efficiently as the individual fusions (Figure 5(a)). Binding reached saturation at a relatively low LFn-Soc-PA4 to capsid binding site ratio of about 20:1 with an apparent Kd of 93 nM and Bmax of 769 (Figure 5(b)). The double domain fusion in essence increased the display capacity to 1538 sites, about twice the number of Soc binding sites.
Figure 5. Binding of the double domain fusion, LFn-Soc-PA4, on phage T4.
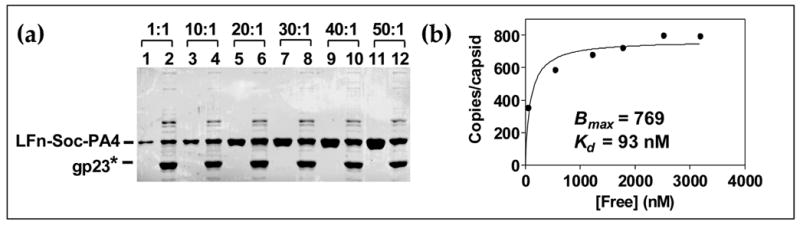
The protocol for in vitro binding of LFn-Soc-PA4 and quantitative analyses are the same as described in the legend to Figure 3 and Materials and Methods. (a): Lanes: 1, 3, 5, 7, 9, and 11, unbound protein; 2, 4, 6, 8, 10, and 12, phage-bound protein. (b): Saturation binding curve showing the apparent Kd and Bmax for LFn-Soc-PA4 binding to phage.
Multicomponent display
LFn-Soc and Soc-PA4 were added to the binding reaction to simultaneously display two proteins on the same capsid. Both the proteins bound to the capsid and the copy number was proportional to the ratio of the proteins in the binding reaction (Figure 6(a, c)). Adding a third protein, PA-Soc or Soc-PA, to the binding reaction resulted in the assembly of all three proteins on the capsid (Figure 6(b)).
Figure 6. Multicomponent display of anthrax toxin antigens on phage T4.
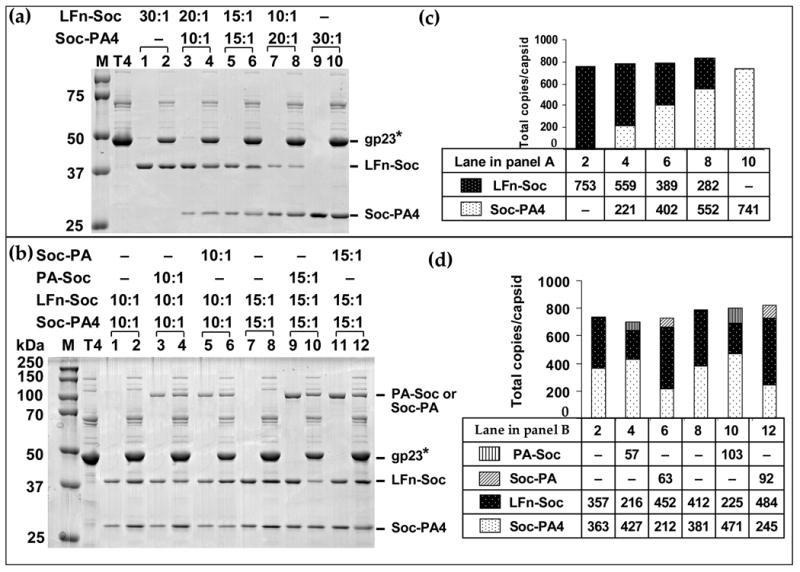
(a, b): About 2 × 1010 hoc−soc− phage particles were incubated with a mixture of LFn-Soc, Soc-PA4, PA-Soc, or Soc-PA and assembly was carried out as described in Materials and Methods and legend to Figure 3. The ratios of anthrax-Soc fusion proteins to the number of Soc binding sites are shown at the top. Lanes: M, mol. wt. standards; T4, hoc−soc− phage control; 1, 3, 5, 7, 9, and 11, unbound proteins; lanes 2, 4, 6, 8, 10, and 12, phage-bound proteins. (c, d): Histograms depicting the copy number of the fusion proteins at different ratios, as calculated from the respective lanes in panels (a) and (b).
Copy number control and orientation of Soc on capsid
When two fusion proteins of approximately the same size were added to the binding reaction, binding was competitive showing no particular preference for the N-terminal vs C-terminal fusion. For instance, at a ratio of 15:1 each, the total copy number of bound Soc was divided equally between the two proteins (Figure 6(a), C, lane 6). However when the bulkier 89 kDa fusion was added, it selectively inhibited the assembly of the smaller protein that was also fused to the same terminus of Soc, not the other. In the presence of PA-Soc (N-terminal fusion), Soc-PA4 binding was normal whereas LFn-Soc binding was strongly inhibited (Figure 6(b); compare lanes 4 and 10 to lanes 2 and 8, respectively). When Soc-PA was used, inhibition shifted to Soc-PA4 whereas LFn-Soc binding was normal (compare lanes 6 and 12 to lanes 2 and 8, respectively). In fact, the copy number of the protein fused to the opposite terminus was somewhat enhanced in the presence of the bulky fusion. These data suggest that binding of bulky Soc-PA or PA-Soc at a given site excludes the binding of another Soc fused to the same terminus because the same termini are facing each other at the dimeric and trimeric junctions (two-fold and three-fold symmetry axes); there is not enough room to accommodate both the bulky 87 kDa his-tagged PA and the PA4 or LFn at the interfaces. Instead, binding of Soc fused to the other terminus is favored. Since the cryo-EM reconstruction of LFn-Soc-T4 showed the LFn mass at the dimeric junctions, it appears that the N-termini are present at the dimeric junctions and the C-termini at the trimeric junctions (see schematic in Figure 10; A. Fokine, V. Bowman, A. Battisti, Q. Li, P. Chipman, V. Rao and M. Rossmann, submitted).
Figure 10. Schematic of the disposition of Soc subunits in various anthrax antigen-phage T4 recombinants.
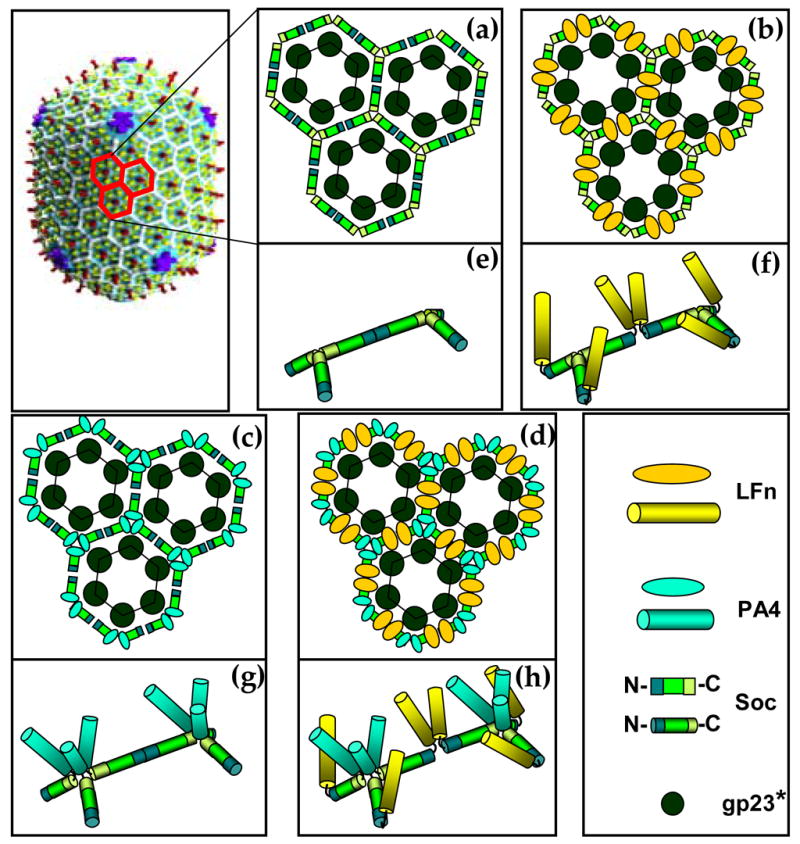
The Cryo-EM reconstruction of phage T4 is shown in the top left corner.1 The gp23* subunits are shown in green, gp24* in blue, Hoc in red, and Soc in white. The Soc subunits of three adjacent gp23* hexons are painted in red and enlarged in panels (a) to (d) (Hoc is not shown in these panels). Soc subunits bind between two adjacent gp23* subunits, forming trimers where three hexons meet (local three-fold symmetry) and dimers at the local two-fold symmetry axes. Soc is shown as a rod with three rectangular compartments; the one in the middle represents the capsid binding site, and the flanking ones, the N- and C-termini. The C-termini face each other at the trimeric junctions and the N-termini, at the dimeric junctions. (a): wild-type Soc; (b): LFn-Soc (N-terminal fusion); (c): Soc-PA4 (C-terminal fusion); (d): LFn-Soc-PA4 (both N- and C-terminal fusion). The panels (e)-(h) are close-up representations of two adjacent Soc trimers corresponding to (a)-(d), respectively. The shapes and colors used are shown in the right lower corner. Note that the representation of inter-Soc subunit contacts in the wild-type (e), and opening up of these contacts in the respective fusion proteins (f, g, and h), is based on the results from this study.
Inter-Soc subunit interactions are critical for maintaining phage infectivity
Cryo-EM reconstructions show evidence of interactions between Soc subunits at the trimeric junctions. Indeed, Soc is visualized as a “propeller-like” structure with the blades emanating from a central contact point.6 Interaction at the dimeric junctions is however ambiguous. Although Soc binding to capsid is not lost by fusion, the inter-Soc subunit interactions would likely be disrupted by fusion of large domains or proteins to the termini. If so, the connected scaffold-like Soc network around the capsid would be interrupted. Would this affect capsid integrity and phage infectivity?
First, the efficiency of plating of LFn-Soc-T4, Soc-PA4-T4, LFn-Soc-PA4-T4, rSoc-T4, and hoc−soc− phages was determined. The particle number was obtained by quantification of the gp23* band following SDS-PAGE and Coomassie blue staining of each phage. No significant differences were observed suggesting that display of Soc-fused antigens did not affect the plaque forming efficiency (pfu per particle) of phage T4.
The LFn-Soc-T4 (dimer links would be interrupted), Soc-PA4-T4 (trimer links would be interrupted), and LFn-Soc-PA4-T4 (both dimer and trimer links would be interrupted) phages were treated with pH 10.4 – 10.6 buffers at 37°C for 1 hr, neutralized to pH 7, and the infectious virus titer was determined (Figure 7(a)). Consistent with the previous report,5 the wild-type, or the hoc−soc− phage saturated with rSoc (equivalent to hoc− phage), retained 75–85% of the titer after the treatment (the titer gradually decreased with increasing pH), whereas the hoc−soc− phage lost >98% of the titer (the titer of each phage type in control pH7 buffer was taken as 100% (Table, column 1), and the titer after treatment was normalized to the respective control). The LFn-Soc-PA4-T4 phage, expected to have lost both dimer and trimer links, showed a loss of 91–97% of the titer. However, this titer is about 2–5 fold greater than that of hoc−soc− phage. The Soc-PA4-T4 phage, which would have lost the trimer links, showed 91–96% loss of the phage titer. This loss was significantly greater than that of LFn-Soc-T4 phage (86–92% loss), which would have lost the dimer links. SDS-gel analysis of the treated phage showed that the observed loss of the recombinant phage titers was not due to dissociation of the capsid subunits, nor were they due to dissociation of Soc from the capsid (Figure 7(b)). In fact, the Soc-capsid interactions are remarkably stable at pH 10.4 since the amounts of capsid protein and Soc retained after treatment at pH 10.4 were exactly the same as that at pH 7 (compare lanes 4, 6, 8, and 10 with 3, 5, 7, and 9). Not surprisingly, much of the hoc−soc− phage was dissociated at pH 10.4 (compare lane 2 to lane 1).
Figure 7. Inter-Soc subunit interactions are critical for capsid integrity.

(a): Infectious phage titers after treatment with high pH. Equivalent numbers of phage particles, as determined by plaque assay and SDS-PAGE, were used. Sucrose-gradient purified wild-type and hoc−soc− phages were directly used as positive and negative controls, respectively. For the rest, in vitro assembly was first carried out using the purified phage and the recombinant protein(s) as described in Materials and Methods, and a small aliquot of the binding reaction mixture was used. Phage were diluted into high pH glycine-NaOH buffer to give rise to a pH between 10.4 to 10.6, as shown in the Table, or the pH 7 assembly buffer. The samples were incubated at 37°C for 1 hr and neutralized to pH 7 by adding excess assembly buffer. The infectious phage titer was determined in duplicates by the plaque assay and the average values are shown. The titer of each phage type in control pH 7 buffer is taken as 100% (column 1 in Table) and the titers of pH 10 treated samples were normalized to the control. (b): Capsid integrity after high pH treatment. Phage were treated with pH 10.4 as above, and sedimented by centrifugation at 16,000 × g for 40 min. The phage pellet was washed twice with pH 10.4 buffer to remove any dissociated phage, and washed once with pH 7 assembly buffer. The final pellet was resuspended in 10 μl of SDS-buffer and subjected to SDS-PAGE. Lanes: M, mol. wt. standards; 1, 3, 5, 7, and 9, phage treated with pH 7; 2, 4, 6, 8, and 10, phage treated with pH 10.4.
Bipartite display using both Hoc and Soc
Since the apparent Kd for Hoc and Soc display are similar (~70 nM),18,21 a cocktail of Hoc and Soc fusion proteins, LFn-Soc, Soc-PA4, and EF-Hoc (EFg, edema factor; 89 kDa), were added to the binding reaction to analyze bipartite Hoc plus Soc display.h The ratio of EF-Hoc molecules to Hoc binding sites was varied from 1:1 to 30:1 for individual (EF-Hoc) display (Figure 8(a, b)) or bipartite display (Figure 8(c, d)). The data showed that, not only all three proteins assemble on the capsid, the EF-Hoc assembly exhibited no interference on LFn-Hoc/Soc-PA4 assembly. The apparent Kd for EF-Hoc binding was 81 nM for bipartite display and 74 nM for individual display, demonstrating that Hoc and Soc binding sites (total: 965) are filled independently. The same binding behavior was observed using the double domain Soc fusion, LFn-Soc-PA4, plus EF-Hoc in the same reaction mixture (Figure 9(a)). In this case, the total copy number of displayed anthrax antigens reached 1662, the highest reported for any phage display system. The disposition of the displayed antigens on the capsid surface according to scale indicates that this strategy allows engineering of essentially all the available surface on the T4 capsid (Figure 9(b)).
Figure 8. Bipartite display using both Soc and Hoc.
In vitro binding and analyses were performed as described in Materials and Methods and legend to Figure 3. About 2 × 1010 hoc−soc− T4 phage were incubated with EF-Hoc (a) or a mixture of EF-Hoc, LFn-Soc, and Soc-PA4, at the ratios indicated (b) (a): display of EF-Hoc. (b): bipartite display of EF-Hoc, LFn-Soc, and Soc-PA4. Lanes: M, mol. wt. standards; T4: hoc− soc− phage control; 1, 3, 5, 7, and 9: unbound proteins; lanes 2, 4, 6, 8, and 10: phage-bound proteins. (c) and (d): Histograms depicting the copy number of displayed proteins at different ratios, as calculated from the respective lanes in panels (a) and (b).
Figure 9. Maximizing phage T4 display.
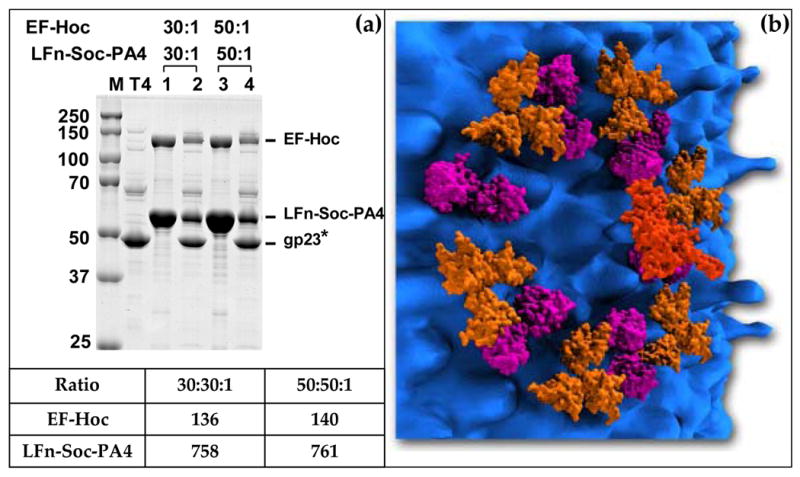
Combining fusions at both ends of Soc and Hoc display, up to 1775 antigens can be displayed. (a): The in vitro assembly was performed with a mixture of LFn-Soc-PA4 and EF-Hoc as described in Materials and Methods and legend to Figure 3. Lanes: M, mol. wt. standards; T4, hoc−soc− phage control; 1 and 3, unbound proteins; lanes 2 and 4, phage-bound proteins. The table shows the copy number of the displayed proteins at the respective ratios. (b): Depiction of the T4 capsid surface following display as in (a). The densities of the displayed antigens, LFn, PA4, and EF, are attached to the predicted positions of Soc and Hoc on T4 capsid surface. LFn (aa 1–262, shown in Purple; PDB ID# 1J7N) is attached to the N-terminus of Soc at the dimeric interface; PA-4 (aa 596–735, shown in Gold; PDB ID# 1ACC) is attached to the C-terminus of Soc at the trimeric interface; EF (aa 301–800; the N-terminal 299 aa are truncated in the x-ray structure, shown in Brick-Red; PDB ID# 1K8T) is attached to the exposed tip of Hoc monomer. Only a single gp23* hexamer with the associated Soc and Hoc subunits are shown. The sizes of T4 capsid proteins as well as the displayed antigens are shown according to scale. The figure is designed with the expert assistance of Mr. Steven McQuinn, Utah Science Center, Salt Lake City, UT.
Discussion
It is becoming increasingly clear that many icosahedral viruses encode triplex proteins, which assemble as trimers on the outer capsid surface.9–14 Here, using a recombinant approach, we show that the phage T4 triplex protein, Soc, exhibits unusual plasticity in its interactions with the capsid, allowing the production of arrays of particulate recombinant anthrax antigens.
In a defined in vitro assembly system consisting of purified soc− phage and Soc, or Soc-anthrax fusion proteins, Soc binding is specific, of high affinity (apparent Kd: 73 nM at 8°C and 54 nM at room temperature), and saturable at low protein concentration (4.6 μM). Multiple Soc-fusion proteins can be simultaneously assembled, and the copy number of bound proteins can be controlled by the relative ratios of proteins in the binding reaction (eg., Figure 6). The LFn-Soc-PA4 binding is particularly notable as in this construct both the Soc termini are simultaneously modified without altering binding parameters, Kd and Bmax (Figure 5). This effectively doubled the capacity of Soc display to 1620 antigens per T4 capsid, up to 95% of which can be filled by in vitro assembly.
It is remarkable that the 10 kDa, 80aa-long Soc can stably anchor a protein mass of up to 93 kDa attached to either end of the molecule (Figure 4). However, unlike the 30 kDa fusions, the Kd for PA/LF-Soc increased by 10-fold and the Bmax decreased by 2-fold. This is not unexpected since, as visualized in the cryo-EM reconstruction,1 the Soc subunits are tightly meshed with the capsid lattice, leaving limited space to accommodate bulky proteins. Moreover, the same termini facing trimeric and dimeric interfaces essentially exclude binding of bulky fusion at half of the available sites (see below). Even so, the copy number of the in vitro assembled PA or LF is 3.5 times greater than the best achieved by in vivo display using much smaller proteins.16,25,26
Both Hoc and Soc fusions can be simultaneously assembled onto the capsid (Figure 8). The Kd and Bmax for EF-Hoc binding are unaffected by the presence of LFn-Soc and Soc-PA, indicating that these proteins assemble independently. This is consistent with the fact that Hoc is ~60Å away from the nearest Soc, as well as raised 60Å above the capsid surface.1 Using Hoc plus Soc (both termini) display, the possible copy number of displayed antigens is raised to 1775 per capsid. About 94% of this maximum, 761 each of LFn and PA4 domains, and 140 molecules of EF (total 1662 antigens), could be reached by in vitro assembly (Figure 9), the highest reported to date in any display system. This approach also demonstrates that all important components of the tripartite anthrax toxin can be incorporated into the phage structure in a single binding reaction.
Soc is a rod shaped molecule.1 Our data suggest that the Soc termini are surface-exposed and present at the edges, whereas the capsid binding site is located in the middle of the structure (see Figure 10). Otherwise, fusion of bulky PA or LF to the termini, or simultaneous fusion of both LFn and PA4 to the termini, should have disrupted, or at least significantly compromised capsid binding, which was not the case. Even pH 10.4 treatment did not dissociate the capsid-bound fusion proteins (Figure 7; see below). Cryo-EM reconstructions show that Soc binds to all quasi three-fold symmetry axes except those at the five-fold vertices where gp23* and gp24* subunits join.6 Here, only one Soc is present between the gp23* subunits and none between the two gp23*-gp24* junctions (Figure 1). Cryo-EM data further show that the Soc density contacts two flanking capsomers, essentially clamping the adjacent gp23* subunits.6 Alignments of seven Soc sequences, two of which having significantly diverged from T4 Soc (50% similarity), revealed two strictly conserved regions having a β-strand structure (Figure 11). Considering that gp23* of these phages has not diverged significantly (>94% similarity), these regions may correspond to the two gp23* binding sites that form an intermolecular clamp (experiments are underway to define the binding site residues).
Figure 11. Multiple sequence alignment and secondary structure predictions of Soc.
Seven T4 Soc-like sequences were aligned by ClustalW (www.ebi.ac.uk/clustalw/) and JPRED (www.compbio.dundee.ac.uk/~www-jpred/) to generate multiple sequence alignment and secondary structure predictions, respectively (accession numbers: T4_Soc: P03715; RB32_Soc: YP_802970; T2_Soc: AAK66983; RB69_Soc: NP_861717. Soc sequences of RB3, T6, and RB14 were from Tulane T4-like genome website, http://phage.bioc.tulane.edu/). The numbers on the left and right of sequence correspond to the number of aa in the respective Soc sequence. Identical aa are highlighted in black, and conserved aa, in pink. For secondary structure predictions, each Soc sequence was submitted in FASTA format to ClustalW to generate a multiple sequence alignment. The output file was submitted to the Jpred server to produce a secondary structure prediction (ssp) based on the consensus sequence. The secondary structure prediction of T4 Soc sequence was generated using PSIPRED (http://bioinf.cs.ucl.ac.uk/psipred/); E = β strand (green); dashes indicate no predicted structure.
Same Soc termini appear to face at the two-fold and three-fold interfaces (see Figure 10). Addition of PA-Soc to the binding reaction consisting of equimolar concentrations of LFn-Soc and Soc-PA4 inhibits LFn-Soc binding, but not Soc-PA4 binding (Figure 6). Conversely, addition of Soc-PA inhibits the binding of Soc-PA4 but not LFn-Soc. Thus the binding of Soc-PA or PA-Soc at a given site excludes the binding of another Soc fused to the same terminus at the neighboring site, suggesting that there is steric interference to accommodate both the bulky 90 kDa PA and the PA4 or LFn domain at the same interface. This is consistent with the recent cryo-EM reconstruction of LFn-Soc-T4, which showed additional density emanating only from the dimer interfaces, and the conclusion that the N-termini of Soc are oriented towards the dimeric interfaces and the C-termini, towards the trimeric interfaces (A. Fokine, V. Bowman, A. Battisti, Q. Li, P. Chipman, V. Rao and M. Rossmann, submitted).
Soc-capsid interactions are not only unaltered by fusion to a large protein but also exhibit unusually high resilience to high pH. It is striking that no dissociation of Soc-anthrax fusions occurred when the recombinant particles were exposed to pH 10.4 (Figure 7(b)). Although these interactions stabilized the capsid, these alone are not sufficient. In the recombinant phage particles in which the inter-Soc subunit interactions are compromised, >90% of infectious titer was lost upon exposure to pH 10.4 to 10.6, whereas the wild-type phage or rSoc-supplemented hoc−soc− phage were stable (Figure 7(a)). Thus, reinforcement through inter-Soc subunit interactions at the dimeric and trimeric interfaces is essential for full capsid stabilization. The data further show that the interactions at the trimeric interfaces impart greater reinforcement (8% survived at pH 10.6) when compared to those at the dimeric interfaces (4.2% survived at pH 10.6). This is consistent with the near complete merging of Soc subunit densities at the trimeric interfaces, whereas the same at the dimeric interfaces is not entirely continuous, and in fact looks separated in some areas.1,6,7 Unlike other triplex proteins, which form trimers at the local three-fold axes, Soc covers both the three-folds and the two-folds, apparently cementing the gp23* subunits for maximum stabilization.
Soc-Soc interactions per se must be weak since rSoc or any of the anthrax-Soc fusion proteins do not oligomerize in solution and behave entirely as a monomer by gel filtration (data not shown). Trimerization and dimerization must therefore occur following Soc binding to the capsid. Since the binding curves are not sigmoidal, it is unlikely that oligomerization and binding are cooperative. The termini may be precisely positioned after Soc binding, possibly following a conformational change,19 facilitating inter-subunit interactions. This is analogous to phage HK97 where rigid body movements in the capsid protein domains during expansion precisely position certain residues for covalent crosslinking, resulting in capsid stabilization.27–29
The theme that emerges from this and other studies is that the capsid of large icosahedral viruses must be reinforced by clamping, gluing, or crosslinking of the inter-capsid protein subunit contacts in order to contain the enormous pressure the capsid is under by the densely packed DNA, further exacerbated by the physical and chemical challenges imposed by extracellular environments.6,9,11,28 In T4, as well as other phages, expansion maturation first causes a major rearrangement of the capsid subunits, stabilizing the capsid.19 Additionally, capsid expansion in many phages including T4 and λ creates sites for binding of triplex proteins such as Soc or gpD respectively, which further stabilize the capsid. These occur at the same time the internal capsid pressure is building due to DNA packaging by the powerful translocase motor.30,31 Our data show that, although Soc binding is efficient and practically irreversible, inter-Soc subunit links at the local three-fold and two-fold axes, generating a nearly continuous cage-like scaffold enclosing the icosahedral capsid, hold the key to stabilize the pressurized capsid and preserve infectivity.
Finally, this study describes the basic aspects of phage T4 Soc in vitro display, the most flexible and the highest density platform reported to date. In many respects this system is more versatile than, as well as complements, the recently described Hoc and complex display systems.18,21, 32 Soc in particular allows customized engineering of T4 particle surface to display multiple, large, functionally well-characterized proteins at high density. Such recombinant particles with unique surface properties offer novel applications in basic molecular biology and biotechnology, including i) in proteomics to “fish-out” interacting partner(s) from cell extracts; ii) in molecular diagnostics to detect bacterial pathogens; iii) in structural biology to generate cryo-EM structures of domains and proteins that are hard to crystallize; iv) in nanotechnology as polyvalent inhibitors; and v) in gene therapy for targeted delivery of genes and proteins. Considering that the phage T4 displayed antigens are highly immunogenic without adjuvant,32,33 the high density Soc-anthrax recombinant particles are prime candidates for developing the next generation multicomponent anthrax vaccine. Indeed, formulations of Soc-T4 displayed anthrax antigens generated strong lethal toxin neutralizing antibodies in mice and rabbits (Rao et al., in preparation). The same could be envisioned to develop vaccines against an array of pathogenic diseases. This study provides the basic foundation to investigate the feasibility of the above approaches, some of which are currently underway.
Materials and Methods
Bacteria, phage, and plasmids
E. coli P301 (sup−) was used to prepare hoc−soc− (hoc.Q21am-soc.del) phage stocks. E. coli XL-10 Gold cells were used for initial transformation and maintenance of recombinant constructs. The clones were then transferred into the expression strain, E. coli BL21 (DE3) RIPL (Stratagene), to allow IPTG induced over-expression of recombinant fusion proteins.34 The T7 expression plasmid pET15b (Ampr) and pET28b (Kanr) (EMD Biosciences, Inc) were used as the cloning vectors. Plasmid pPA26 was used as a template for amplification of PA gene (pagA) and PA4 gene (pagA4); pLF7 (E687C), for amplification of LFE687C gene (lef) and LFn gene (lfn); pSE42, for amplification of EFK346R gene (cya), and phage T4 genomic DNA, for amplification of Hoc (hoc) gene and Soc gene (soc), respectively.
Gene fusions
Gene fusions of were constructed using the SOE strategy as previously described.21, 35, 36 As shown in Figure 1, these include N-terminal Soc fusions (LF-Soc, PA-Soc, and LFn-Soc), C-terminal fusions (Soc-PA and Soc-PA4), and the double-domain fusion to both N- and C-termini (LFn-Soc-PA4). A flexible linker sequence, SASA, was incorporated between the anthrax toxin and Soc genes. Each fusion had a 25 aa hexa-histidine sequence at the N-terminus. The recombinant gene fusions following amplification and purification by gel electrophoresis were inserted in-frame into either the BamH1 site of pET-15b or Nhe I-Bam HI sites of pET-28b. The 5′-end primers were designed so that the insertion resulted in in-frame fusion of the hexa-histidine sequence from the vector with the recombinant sequence.
Purification of the rSoc and anthrax-Soc fusion proteins
Liquid cultures were induced with 1 mM IPTG at 30°C for 2 hrs and harvested by centrifugation at 6,000 × g for 15 min. Some proteins, e.g., rSoc and Soc-PA4, were completely insoluble. The rest of the fusion proteins were partially soluble; the solubility varied between 20–50% of the expressed protein. LFn-Soc, PA-Soc, and Soc-PA were purified both from insoluble as well as soluble fractions, and both the preparations behaved the same way in the functional assays and assembly experiments. For the data reported here, all the experiments were performed using the proteins purified from the soluble fraction.
The cells were lysed by French press and lysates centrifuged at 27,000 × g for 30 min. The supernatants containing the soluble proteins were first purified by HisTrap column chromatography (AKTA-prime, GE Healthcare). For purification of proteins from the insoluble fraction (rSoc and Soc-PA4), the French press lysate was centrifuged at 8,000 × g for 10 min. The pellet was washed twice with the HisTrap bind buffer (50 mM Tris-HCl, pH 8, 20 mM imidazole and 300 mM NaCl), and the protein was solubilized with the denaturing buffer (8 M Urea, 50 mM Tris-Cl, pH 8, 20 mM imidazole and 300 mM NaCl) and loaded onto HisTrap column. The protein was then renatured by a decreasing urea gradient (8-0 M) and eluted with 20–500 mM linear imidazole gradient. The peak fractions containing the fusion protein were desalted by passing through the Hiprep10/26 column and concentrated by Amicon Ultra-4 centrifugal filtration (5 kDa cut-off; Millipore). The protein (from either the soluble or the insoluble fraction) was then purified by gel filtration on Hi-Load 16/60 Superdex 200 column (prep-grade) (AKTA-FPLC, GE Healthcare). The purified protein fractions were concentrated and stored frozen at −70°C.
Functional analysis of the anthrax toxin-Soc fusions
About 50 μg of PA, PA-Soc, and Soc-PA were nicked with 20 ng of trypsin (Sigma) at 37°C for 45 min in 20 μl of cleavage buffer (25 mM HEPES pH 7.3, 10 mM CaCl2, and 5 mM EDTA).23 The reaction was terminated by adding 20 μg of trypsin inhibitor (SBTI, Sigma) and the mixture was further incubated at room temperature for 30 min. The nicked PA preparations (nPA; nPA-Soc and Soc-nPA) were incubated with LF, LFn-Soc, LF-Soc, or LFn-Soc-PA4, at 37°C for 45 min in the binding buffer (50 mM Tris-Cl, pH 8.8, 2 mg/ml CHAPS). The samples were centrifuged at 16,000 × g for 10 min (4°C) to remove any aggregates. The supernatants were electrophoresed on a 4–12% gradient native polyacrylamide gel (PAG) (Invitrogen) and stained with Coomassie blue.
In vitro assembly of rSoc and anthrax-Soc fusion proteins on phage T4 particles
The hoc−soc− phage was purified by 25–50% linear sucrose gradient centrifugation (Biocomp Inc. NB, Canada, sucrose gradient maker). About 2 × 1010 T4 hoc−soc− phage particles were centrifuged at 16,000 × g for 40 min using low-bind Eppendorf tubes. The pellets were resuspended in assembly buffer (50 mM sodium phosphate buffer, pH 7.0, 75 mM NaCl and 1 mM MgSO4) and various recombinant Soc fusion proteins were added in a total reaction volume of 100 μl. The samples were incubated at 8°C for 30 min and the phages were sedimented by centrifugation as above. The phage pellet was washed twice with excess assembly buffer and the final pellet was resuspended in 10 μl of assembly buffer and transferred to a new Eppendorf tube for analysis by SDS-PAGE (10–13% polyacrylamide). After Coomassie blue staining and destaining, the gels were scanned by a laser densitometer (PDSI, GE Healthcare) and the density volumes of the displayed Soc fusion protein bands and the internal control band, T4 gp23*, were quantified as described previously21. Each lane was individually quantified so that the precise copy number of the displayed antigen was obtained in comparison with the gp23* internal control for which the copy number was established to be 930 per particle. Saturation binding curves and Scatchard plots were generated from the data and the Bmax and Kd values for each binding experiment were determined by non-linear regression analysis using GraphPad PRISM-4 software (San Diego, CA). The apparent binding constant, Kd, is a measure of binding affinity, not a thermodynamic quantity. It is defined as the molar concentration of Hoc or Soc fusion protein (ligand) at which half the available capsid binding sites are occupied with the ligand. The Bmax is defined as the maximum number of binding sites occupied by the displayed antigen per capsid particle as determined from the saturation binding curve.
Soc-Hoc bipartite display
About 2 × 1010 hoc−soc− phage particles were incubated with EF-Hoc individually or together with Soc fusion proteins, LFn-Soc, Soc-PA4, and LFn-Soc-PA at room temperature for 10 min. The binding reaction and data quantification were carried out as described above.
Acknowledgments
It is a pleasure to thank Drs. Andrei Fokine and Michael Rossmann for thoughtful discussions, the cryo-EM micrographs shown in Figure 3, sharing unpublished reconstruction data, and reviewing the manuscript. The expert assistance of Mr. Steven McQuinn, Utah Science Center, Salt Lake City, UT, to design Figure 9(b) is gratefully acknowledged. Our collaborative research on anthrax vaccine development and discussions with Drs. Carl Alving, Mangala Rao, Gary Matyas, and Kristina Peachman at the Division of Retrovirology, Walter Reed Army Institute of Research, Washington, DC, and Dr. Stephen Leppla, NIAID, NIH, are instrumental to conduct the studies reported here. This work was supported by the grant AI056443 from the National Institute of Allergy and Infectious Diseases (NIAID).
The abbreviations used are
- aa
amino acid(s)
- EF
edema factor
- gp
gene product(s)
- LF
lethal factor
- LFn
N-terminal domain of lethal factor
- LTx
lethal toxin
- nt
nucleotide(s)
- PA
protective antigen
- PA4
PA domain-4
- PAGE
polyacrylamide gel electrophoresis
- rSoc
recombinant Soc
- SDS
sodium dodecyl sulfate
- SOE
splicing by overlap extension
Footnotes
“*” represents the cleaved capsid protein following T4 capsid maturation cleavages by the prohead protease.
The terms “assembly”, “binding”, and “display” are interchangeably used to refer to the binding of rSoc or anthrax-Soc antigens on phage T4.
null mutants of LF and EF, LFE687C and EFK346R, which exhibit no MAPKK protease and adenyl cyclase activities respectively, were used to eliminate the toxicity of anthrax toxin complexes.22 The mutants retain binding functions and immunogenicity.
All Soc binding experiments except the bipartite display using both Soc and Hoc were performed at 8°C because Soc tends to precipitate at room temperature, generating some background. No significant Soc precipitation was observed at 8°C. In addition, each Soc (or Hoc) fusion protein was centrifuged at high speed (17,000 × g) prior to addition to the binding reaction in order to remove any aggregates or precipitates present in the purified protein sample.
The apparent binding constant, Kd, is defined as the molar concentration of Hoc or Soc fusion protein (ligand) at which half the available capsid binding sites are occupied with the ligand. The Bmax is defined as the maximum number of binding sites occupied by the displayed antigen per capsid particle, as determined from the saturation binding curve (see Materials and Methods). Note that the Kd for rSoc binding at room temperature is 54 nM as opposed to 73 nM at 8°C.
Edema factor, a calmodulin-activated adenyl cyclase, is a component of the tripartite anthrax toxin, causing accumulation of fluids within and between cells.22
The bipartite display experiments were performed at room temperature since Hoc binding at 8°C is less than optimal. The Soc- and Hoc-fusion proteins were centrifuged at high speed (17,000 × g) prior to addition to the binding reaction in order to remove any aggregates or precipitates present in the purified protein sample. Any Soc precipitation during incubation was subtracted by using a control reaction containing no phage.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Fokine A, Chipman PR, Leiman PG, Mesyanzhinov VV, Rao VB, Rossmann MG. Molecular architecture of the prolate head of bacteriophage T4. Proc Natl Acad Sci USA. 2004;101:6003–6008. doi: 10.1073/pnas.0400444101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Leiman PG, Kanamaru S, Mesyanzhinov VV, Arisaka F, Rossmann MG. Structure and morphogenesis of bacteriophage T4. Cell Mol Life Sci. 2003;60:2356–2370. doi: 10.1007/s00018-003-3072-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Black LW, Showe MK, Steven AC. Morphogenesis of the T4 Head. In: Karam JD, editor. Molecular Biology of Bacteriophage T4. ASM press; Washington, D. C.: 1994. pp. 218–258. [Google Scholar]
- 4.Ishii T, Yanagida M. The two dispensable structural proteins (soc and hoc) of the T4 phage capsid; their purification and properties, isolation and characterization of the defective mutants, and their binding with the defective heads in vitro. J Mol Biol. 1977;109:487–514. doi: 10.1016/s0022-2836(77)80088-0. [DOI] [PubMed] [Google Scholar]
- 5.Ishii T, Yamaguchi Y, Yanagida M. Binding of the structural protein soc to the head shell of bacteriophage T4. J Mol Biol. 1978;120:533–544. doi: 10.1016/0022-2836(78)90352-2. [DOI] [PubMed] [Google Scholar]
- 6.Iwasaki K, Trus BL, Wingfield PT, Cheng N, Campusano G, Rao VB, Steven AC. Molecular architecture of bacteriophage T4 capsid: vertex structure and bimodal binding of the stabilizing accessory protein, Soc. Virology. 2000;271:321–333. doi: 10.1006/viro.2000.0321. [DOI] [PubMed] [Google Scholar]
- 7.Olson NH, Gingery M, Eiserling FA, Baker TS. The structure of isometric capsids of bacteriophage T4. Virology. 2001;279:385–391. doi: 10.1006/viro.2000.0735. [DOI] [PubMed] [Google Scholar]
- 8.Sternberg N, Weisberg R. Packaging of coliphage lambda DNA. II The role of the gene D protein. J Mol Biol. 1977;117:733–759. doi: 10.1016/0022-2836(77)90067-5. [DOI] [PubMed] [Google Scholar]
- 9.Yang F, Forrer P, Dauter Z, Conway JF, Cheng N, Cerritelli ME, Steven AC, Pluckthun A, Wlodawer A. Novel fold and capsid-binding properties of the lambda-phage display platform protein gpD. Nat Struct Biol. 2000;7:230–237. doi: 10.1038/73347. [DOI] [PubMed] [Google Scholar]
- 10.Gilcrease EB, Winn-Stapley DA, Hewitt FC, Joss L, Casjens SR. Nucleotide sequence of the head assembly gene cluster of bacteriophage L and decoration protein characterization. J Bacteriol. 2005;187:2050–2057. doi: 10.1128/JB.187.6.2050-2057.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Tang L, Gilcrease EB, Casjens SR, Johnson JE. Highly discriminatory binding of capsid-cementing proteins in bacteriophage L. Structure. 2006;14:837–845. doi: 10.1016/j.str.2006.03.010. [DOI] [PubMed] [Google Scholar]
- 12.van Oostrum J, Burnett RM. Molecular composition of the adenovirus type 2 virion. J Virol. 1985;56:439–448. doi: 10.1128/jvi.56.2.439-448.1985. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Parks RJ. Adenovirus protein IX: a new look at an old protein. Mol Ther. 2005;11:19–25. doi: 10.1016/j.ymthe.2004.09.018. [DOI] [PubMed] [Google Scholar]
- 14.Saad A, Zhou ZH, Jakana J, Chiu W, Rixon FJ. Roles of triplex and scaffolding proteins in herpes simplex virus type 1 capsid formation suggested by structures of recombinant particles. J Virol. 1999;73:6821–6830. doi: 10.1128/jvi.73.8.6821-6830.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Sternberg N, Hoess RH. Display of peptides and proteins on the surface of bacteriophage lambda. Proc Natl Acad Sci USA. 1995;92:1609–1613. doi: 10.1073/pnas.92.5.1609. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Ren Z, Lewis GK, Wingfield PT, Locke EG, Steven AC, Black LW. Phage display of intact domains at high copy number: a system based on SOC, the small outer capsid protein of bacteriophage T4. Protein Sci. 1996;5:1833–1843. doi: 10.1002/pro.5560050909. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Jiang J, Abu-Shilbayeh L, Rao VB. Display of a PorA peptide from Neisseria meningitidis on the bacteriophage T4 capsid surface. Infect Immun. 1997;65:4770–4777. doi: 10.1128/iai.65.11.4770-4777.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Li Q, Shivachandra SB, Leppla SH, Rao VB. Bacteriophage T4 capsid: a unique platform for efficient surface assembly of macromolecular complexes. J Mol Biol. 2006;363:577–588. doi: 10.1016/j.jmb.2006.08.049. [DOI] [PubMed] [Google Scholar]
- 19.Steven AC, Heymann JB, Cheng N, Trus BL, Conway JF. Virus maturation: dynamics and mechanism of a stabilizing structural transition that leads to infectivity. Curr Opin Struct Biol. 2005;15:227–236. doi: 10.1016/j.sbi.2005.03.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Rao VB, Black LW. DNA packaging of bacteriophage T4 proheads in vitro. Evidence that prohead expansion is not coupled to DNA packaging. J Mol Biol. 1985;185:565–578. doi: 10.1016/0022-2836(85)90072-5. [DOI] [PubMed] [Google Scholar]
- 21.Shivachandra SB, Rao M, Janosi L, Sathaliyawala T, Matyas GR, Alving CR, Leppla SH, Rao VB. In vitro binding of anthrax protective antigen on bacteriophage T4 capsid surface through Hoc-capsid interactions: a strategy for efficient display of large full-length proteins. Virology. 2006;345:190–198. doi: 10.1016/j.virol.2005.10.037. [DOI] [PubMed] [Google Scholar]
- 22.Leppla SH. Bacillus anthracis toxins. In: Alouf JE, Popoff MR, editors. The Comprehensive Sourcebook of acterial Protein Toxins. Academic Press; Burlington, MA: 2006. pp. 323–347. [Google Scholar]
- 23.Singh Y, Klimpel KR, Goel S, Swain PK, Leppla SH. Oligomerization of anthrax toxin protective antigen and binding of lethal factor during endocytic uptake into mammalian cells. Infect Immun. 1999;67:1853–1859. doi: 10.1128/iai.67.4.1853-1859.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Mogridge J, Cunningham K, Lacy DB, Mourez M, Collier RJ. The lethal and edema factors of anthrax toxin bind only to oligomeric forms of the protective antigen. Proc Natl Acad Sci USA. 2002;99:7045–7048. doi: 10.1073/pnas.052160199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Ren Z, Black LW. Phage T4 SOC and HOC display of biologically active, full-length proteins on the viral capsid. Gene. 1998;215:439–444. doi: 10.1016/s0378-1119(98)00298-4. [DOI] [PubMed] [Google Scholar]
- 26.Wu J, Tu C, Yu X, Zhang M, Zhang N, Zhao M, Nie W, Ren Z. Bacteriophage T4 nanoparticle capsid surface SOC and HOC bipartite display with enhanced classical swine fever virus immunogenicity: A powerful immunological approach. J Virol Methods. 2007;139:50–60. doi: 10.1016/j.jviromet.2006.09.017. [DOI] [PubMed] [Google Scholar]
- 27.Wikoff WR, Liljas L, Duda RL, Tsuruta H, Hendrix RW, Johnson JE. Topologically linked protein rings in the bacteriophage HK97 capsid. Science. 2000;289:2129–2133. doi: 10.1126/science.289.5487.2129. [DOI] [PubMed] [Google Scholar]
- 28.Ross PD, Cheng N, Conway JF, Firek BA, Hendrix RW, Duda RL, Steven AC. Crosslinking renders bacteriophage HK97 capsid maturation irreversible and effects an essential stabilization. EMBO J. 2005;24:1352–1363. doi: 10.1038/sj.emboj.7600613. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Hendrix RW. Bacteriophage HK97: assembly of the capsid and evolutionary connections. Adv Virus Res. 2005;64:1–14. doi: 10.1016/S0065-3527(05)64001-8. [DOI] [PubMed] [Google Scholar]
- 30.Smith DE, Tans SJ, Smith SB, Grimes S, Anderson DL, Bustamante C. The bacteriophage straight phi29 portal motor can package DNA against a large internal force. Nature. 2001;413:748–752. doi: 10.1038/35099581. [DOI] [PubMed] [Google Scholar]
- 31.Rao VB, Black LW. DNA packaging in bacteriophage T4. In: Catalano C, editor. Viral Packaging Machines: Genetics, Structure, and Mechanism. Landes Bioscience; Georgetwo, TX: 2005. pp. 40–58. [Google Scholar]
- 32.Shivachandra SB, Li Q, Peachman KK, Matyas GR, Leppla SH, Alving CR, Rao M, Rao VB. Multicomponent anthrax toxin display and delivery using bacteriophage T4. Vaccine. 2007;25:1225–1235. doi: 10.1016/j.vaccine.2006.10.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Sathaliyawala T, Rao M, Maclean DM, Birx DL, Alving CR, Rao VB. Assembly of human immunodeficiency virus (HIV) antigens on bacteriophage T4: a novel in vitro approach to construct multicomponent HIV vaccines. J Virol. 2006;80:7688–7698. doi: 10.1128/JVI.00235-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Studier FW, Rosenberg AH, Dunn JJ, Dubendorff JW. Use of T7 RNA polymerase to direct expression of cloned genes. Methods Enzymol. 1990;185:60–89. doi: 10.1016/0076-6879(90)85008-c. [DOI] [PubMed] [Google Scholar]
- 35.Horton RM, Hunt HD, Ho SN, Pullen JK, Pease LR. Engineering hybrid genes without the use of restriction enzymes: gene splicing by overlap extension. Gene. 1989;77:61–68. doi: 10.1016/0378-1119(89)90359-4. [DOI] [PubMed] [Google Scholar]
- 36.Kuebler D, Rao VB. Functional analysis of the DNA-packaging/terminase protein gp17 from bacteriophage T4. J Mol Biol. 1998;281:803–814. doi: 10.1006/jmbi.1998.1952. [DOI] [PubMed] [Google Scholar]