

Abstract
Clostridium difficile is the etiological agent of antibiotic-associated diarrhea; the most common form of nosocomial infectious diarrhea. The basis for the shock-like systemic symptoms observed in severe cases of this infection are not known. It is hypothesized that the invasion of C difficile toxins A and/or B from the gut mucosa may contribute to these symptoms.
A polarized tissue culture model employing Caco-2 cells grown on transwell inserts was established to study the translocation of purified C difficile toxins A and B. C difficile toxins were 125I labelled and inoculated onto confluent polarized Caco-2 cell monolayers to study translocation dynamics. Electrical resistance measurements were used to monitor monolayer confluence and tight junction integrity. Samples were taken from the apical and basal sides of the insert, as well as the insert itself, and tested using the human foreskin fibroblasts cell cytotoxicity assay to monitor partitioning of the radiolabelled toxins. Toxin A produced a 50% reduction in electrical resistance in 3 h whereas the same concentration of toxin B required at least 7 h to achieve the same effect. Both toxins A and B were able to translocate across confluent monolayers of Caco-2 cells. The combination of toxin A and B together was synergistic with respect to promoting the translocation of toxin B. Although the addition of toxin A resulted in a 100% increase in the amount of toxin B able to translocate, no increases in toxin A translocation were observed. These findings suggest a model of pathogenesis in which C difficile toxin A facilitates the translocation of toxin B from the gut into submucosal areas where it may play a role in inflammatory damage.
Key Words: Clostridium difficile, Polarized-monolayers, Toxin A, Toxin B, Translocation
Clostridium difficile is the leading cause of nosocomial antibiotic-associated diarrhea and colitis. It is reported to be responsible for 10% to 25% of all cases of antibiotic-associated diarrhea, for 60% of antibiotic-associated colitis and for almost all cases of pseudomembranous colitis (1,2). C difficile is able to overgrow in the intestine of patients whose normal microflora has been disrupted by antimicrobial or antineoplastic drugs (3,4). Despite the proliferation in the bowel, studies have shown that the organism is noninvasive, and systemic spread of the organism has not been reported. In severe cases of infection, patients with pseudomembranous colitis may experience systemic complications resulting in death (5). Presently, the basis for these systemic symptoms are not known. Studies have shown that only strains capable of producing both toxins A and B have been associated with clinical disease (6). Toxin A is a 308 kDa enterotoxin responsible for promoting intestinal fluid secretion and inflammation. It is cytotoxic and can cause rounding of many mammalian cells (7-10). Toxin B is a 270 kDa protein. Although not enterotoxic, it has a 1000-fold greater cytotoxicity than toxin A (6,8,9,11). Despite extensive research on the two toxins using animal and biological models, the model of pathogenesis is still incomplete. Both toxins A and B are required for intestinal damage in animal studies, however, the exact role of toxin B in the disease progression is unclear. Some animal studies have shown that systemic injection of minute amounts of toxin B are fatal (12-14).
Despite having large molecular sizes, both toxins are internalized and act intracellularly (15,16). Once internalized, the toxins glycosylate small GTP-binding glycosylate proteins of the Rho subfamily at residue threonine 37 (17). The glycosylate process leads to an inactivation of the signalling molecules and results in a breakdown of the actin filament network, an integral network for maintaining the cytoskeletal structure of cells. The resultant depolymerization of F actin to G actin also leads to a compromise in the gastrointestinal tight junctions (18,19). Because gastrointestinal tight junctions play key roles in regulating cellular trafficking and maintenance of bowel integrity, disruption of their function could potentially lead to unregulated passage of foreign molecules into the submucosa from the gut lumen.
To our knowledge, there are no published data to demonstrate whether toxins A and/or B can cross the intestinal epithelium to gain access to the submucosa. It is plausible that systemic symptoms seen in severe cases of disease may be caused by the translocation of C difficile toxins from the gut into the submucosa. Once access is made into the submcosa, the toxins would be free to induce inflammatory mediated submucosal damage and/or to act systemically.
The aim of the present study was to use a polarized tissue culture model employing Caco-2 cells grown on transwell inserts to determine if either toxin A or B of C difficile can alone or in combination translocate across an intact monolayer.
MATERIALS AND METHODS
Cell culture
Caco-2 cells (ATCC HTB 37), a human colon adenocarcinoma cell line, was maintained in RPMI 1640 medium (ICN Biomedicals Inc, USA) supplemented with 20% fetal bovine serum (Gibco BRL, USA), 1mM sodium pyruvate (ICN Biomedicals Inc, USA) and 2 mM L-glutamine (ICN Biomedicals Inc, USA). Human foreskin fibroblasts (HFF), a nontransformed cell line derived from pooled foreskins from children, was maintained in RPMI 1640 medium supplemented with 10% fetal bovine serum, 1 mM sodium pyruvate and 2 mM L-glutamine. The tissue culture cells were incubated at 35°C in 5% (vol/vol) CO2 in a water-saturated incubator. Cells were passaged biweekly using a 0.05% trypsin solution in 0.53 mM EDTA-4Na (1x) to disperse the monolayer followed by a 1:2 split into fresh medium.
Polarized Caco-2 cell monolayers
Caco-2 cells were seeded onto 0.4 µm pore transwell inserts (Corning Costar, USA) by adding 0.5 mL of a cell suspension containing 1.0x105 cells/mL. Tissue culture medium consisting of RPMI 1640 supplemented with 20% fetal bovine serum, 1 mM sodium pyruvate and 2 mM L-glutamine was added (1.5 mL) to the basal side of the insert in each cluster plate. After 24 h, nonadherent cells were removed from the apical side of the insert and replaced with fresh tissue culture medium. Electrical resistance measurements were determined using MILLICELL-ERS probes (Millipore, USA). Experiments were carried out on confluent monolayers of Caco-22 cells as determined by incubating until high stable resistance readings (400 ohm cm2 or greater) were established. This usually required four to seven days of incubation postinoculation. Each test condition was assayed by taking triplicate readings at each time interval. Three transwell inserts were used for each test case giving a total of nine replicate readings.
Purified C difficile toxin A and toxin B
Highly purified C difficile toxin A (Lot: 0197005, Cat #T3001) and toxin B (Lot: 0197010, Cat #T3002) were generously provided by Dr David Lyerly (TechLab Inc, USA). Each toxin was purified to homogeneity at a concentration of 0.46 mg/mL for toxin A and 0.21 mg/mL for toxin B. The toxin preparations were stored at 4°C until used.
Cytotoxin assay
HFF cells (100 µL) were seeded onto 96 cell wells (Corning glass works, USA) at a concentration of 1.0x105 cells/mL and allowed to grow to confluence. Test samples consisting of tissue culture medium sampled (50 µL) from the basal side of the transwell at various time intervals was inoculated onto the HFF monolayers. The HFF monolayers were then incubated for 48 h and examined for cytopathic effect (CPE). Wells with 50% or more cells exhibiting CPE were scored as positive. Specificity of the CPE produced by the purified toxin preparations was confirmed by neutralizing the toxic effect using polyvalent anti-C difficile toxin B serum (Bartels Inc, USA). Negative and positive controls were included for each HFF CPE assay performed.
Radiolabelling
Purified test protein (50 µg) was radiolabelled with carrier free 125I in a reaction vessel containing 100 µL of phosphate buffered saline using IODO-BEADS (Pierce chemical co, USA) according to the manufacturers directions. The radiolabelling reaction was allowed to occur for 5 min at room temperature. Radiolabelled proteins were separated from free 125I using a 30 cm x 3.5 mm column packed with Sephadex G100 (Pharmacia, Sweden). Biological activity of the radiolabelled proteins was determined by using a HFF cytotoxin assay and by measuring electrical resistance changes in Caco-2 cells. The titre of biological activity of the radiolabelled versus nonradiolabelled proteins was essentially unchanged.
Translocation experiments
Medium from transwells showing high stable resistance measurements was removed and replaced with tissue culture medium containing unlabelled and/or radiolabelled proteins in various combinations. Resistance measurements were recorded at various intervals. The distribution of labelled proteins during the time course of the experiment was assessed by sampling tissue culture media from apical and basal sides of the insert, as well as, determining the counts per minute (cpm)/µL. The tissue culture monolayer was also assessed by washing the monolayer on the insert three times with fresh tissue culture medium and then cutting the rigid insert (with the monolayer attached) out of the transwell apparatus. All samples were counted for 1 min using a Gamma 5500 (Fullerton, USA). The efficiency of this gamma counter was 75%.
RESULTS
Transport of compounds across the mucosal barrier of the intestine is a difficult parameter to study because the organ exhibits considerable cellular heterogeneity and geometric structure. Compounding this problem is the lack of easy access to the serosal side of the epithelium due to the presence of subadjacent tissues (20). With the recent use of cultured intestinal cells, this problem has been alleviated. The human colonic carcinoma cell line, Caco-2, is known to show signs of spontaneous in vitro enterocytic differentiation characterized by tight junction and brush border microvilli formation (21). In the present study, the authors have employed a polarized tissue culture insert model using Caco-2 cells to study the translocation of C difficile toxins A and B. This model allows access to both apical (top) and basolateral (bottom) sides of epithelial cells, a feat possible using conventional tissue culture cells grown on plastic surfaces. The ability to assess the cell monolayers from both apical and basolateral domains allows for the study of the transport across the cell monolayer; permeability of the cell monolayer; and electrophysiology of the cell monolayers including transepithelial resistance.
Preliminary results from experiments which treated Caco-2 monolayers with C difficile toxins indicated that toxin A was much more efficient at eliciting tight-junction changes compared with the same concentration of toxin B (resistance dropped to less than 200 ohms 4 h postexposure for toxin A compared with 24 h for toxin B). Samples of tissue culture media from the basal side of these transwell inserts were inoculated onto confluent HFF cells to examine for translocation of a toxic component using a conventional HFF cell CPE assay. Translocation of a CPE-causing component was not detected when only toxin A was tested, but was detected when toxin B alone was tested. The time required for translocation of a CPE-causing component was shortened when both toxin A and toxin B were combined (24 h for toxin B alone compared with 8 h when both toxin A and B were combined). Although toxin B is 1000-fold more effective as a cytotoxin than toxin A, it was not possible to conclude that only toxin B had enhanced translocation when both toxins were inoculated together onto the apical side of the transwell insert. Attempts at using monoclonal and polyclonal antibodies targeted against toxin A and toxin B were unsuccessful in identifying which toxin(s) had translocated.
To investigate further, a more sensitive method involving 125I radiolabelling of toxin A and toxin B was employed. Radiolabelled bovine serum albumen (BSA) served as the control protein in translocation experiments. The monolayer confluence and tight junction integrity of Caco-2 cells grown on transwell inserts was tested by inoculating inserts with 125I-BSA. Confluent epithelial cell monolayers have defined tight junctions and regulated cellular trafficking. Under such conditions, cells will not allow unregulated passage of large molecular weight proteins. Treatment of inserts with 125I-BSA resulted in no change in transepithelial resistance at 10 h or 24 h (data not shown). There was also only a minimal change in the distribution of 125I-BSA (apical side inoculated with 1.37x104 cpm) in the transwell insert over a 24 h period (1.7% of radioactivity found on basal side after 10 h and 7.3% of radioactivity found on basal side after 24 h incubation). These results suggest that monolayer integrity was maintained and that over the 24 h test period, proteins such as BSA (66,430 molecular weight) are unable to translocate efficiently. (Figure 1) shows treatment of inserts with unlabelled toxins A and B in conjunction with 125I-BSA. In contrast to treatment with 125I-BSA alone, there was a significant drop in the transepithelial resistance measurements of the Caco-2 cells when toxin A and toxin B were included (Figure 1). The time required for a drop in transepithelial resistance to 50% of the original value (RD50) was only three hours. Analysis of the distribution of 125I-BSA in the insert model shows a gradual increase in the amount of radiolabelled BSA appearing on the basal side over the 10 h test period. The results indicate a compromise in the integrity of the Caco-2 cell monolayer elicited by toxin A and B, followed by nonspecific passage of labelled BSA from the apical to the basal side of the insert. In the next series of experiments, inserts were also treated with 125I-toxin A (Figure 2) and examined for translocation and/or binding of the radiolabelled toxin A to epithelial cells. Similar to unlabelled preparations, 125I-toxin A alone was sufficient to cause an RD50 after only 3 h. Analysis of the distribution of 125I-toxin A showed a marked increase in the amount of labelled toxin A appearing on the basal side of the insert at each time point tested when compared with treatment with 125I-BSA alone. The amount of 125I-toxin A appearing on the basal side of the insert was approximately 100% greater than that of 125I-BSA at each time point tested. The results suggest that the translocation of toxin A may occur by a specific mechanism, and that the process may be directly regulated by eukaryotic cells. There are at least two plausible mechanisms by which toxin A translocation across an intact Caco-2 monolayer may occur: when toxin A is internalized, transported through the cell, and then expelled from the basal side of the monolayer; or when toxin A is able to pass through tight junction openings of Caco-2 cells created by C difficile toxins. Binding of toxin A to receptors on Caco-2 cell monolayers grown on transwell inserts was also assessed by cutting the insert out and performing counts. Results indicated that the amount of toxin A bound to Caco-2 cell monolayers was at background levels. Finding no appreciable amounts of radiolabelled toxin A associated with Caco-2 cells was unexpected because previously published data (15) indicated that toxin A was internalized by eukaryotic cells.
Figure 1.
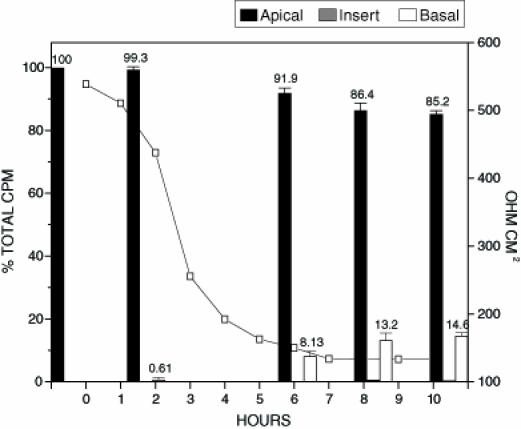
Translocation of 125I-bovine serum albumen (BSA) in the presence of toxin A and toxin B. Confluent Caco-2 monolayers on transwell inserts were inoculated with 250 µL of both toxin A and toxin B (both toxins at 0.250 µg/mL) and 100 µL 125I-BSA (0.250 µg/mL). Samples from apical, basal and insert were tested for per cent of the total input counts per minute (cpm). The figure represents the average of three separate experiments. The electrical resistance measurements indicate the level of tight junction integrity. The total amount of radioactivity inoculated onto the insert in the form of radiolabelled BSA was 2.8 x103 cpm
Figure 2.
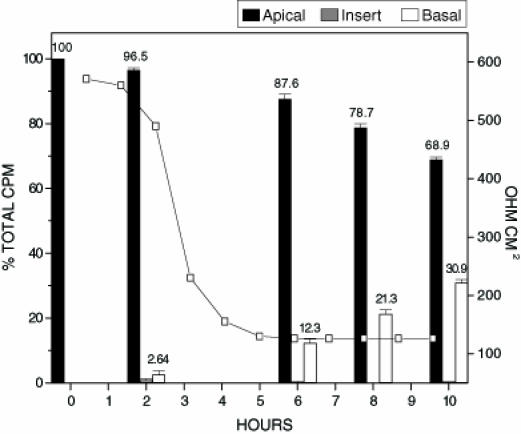
Translocation of 125I-toxin A. Confluent Caco-2 monolayers on transwell inserts were inoculated with 500 µL of 125I-toxin A (0.250 µg/mL). Samples from apical, basal, and insert were tested for per cent of the total input counts per minute (cpm). Each data point represents the average of three separate experiments. The electrical resistance measurements indicate the level of tight junction integrity. The total amount of radioactivity inoculated onto the insert in the form of 125I-toxin A was 9.2 x103 cpm
To determine if the addition of toxin B would have a synergistic effect on the translocation of toxin A, inserts were treated with unlabelled toxin B and 125I-toxin A (Figure 3). The addition of toxin B did not appear to increase the rate at which transepithelial resistance drops occurred. These findings suggest that toxin A plays the main role in the disruption of epithelial tight junctions, and that the limiting factor for tight junction modulation may be the amount of toxin A present. The distribution of radiolabelled toxin A in this test condition also paralleled that of treatment of inserts with toxin A alone. The addition of toxin B did not seem to influence or augment the amount of toxin A capable of translocating across an intact Caco-2 monolayer (Figure 3).
Figure 3.
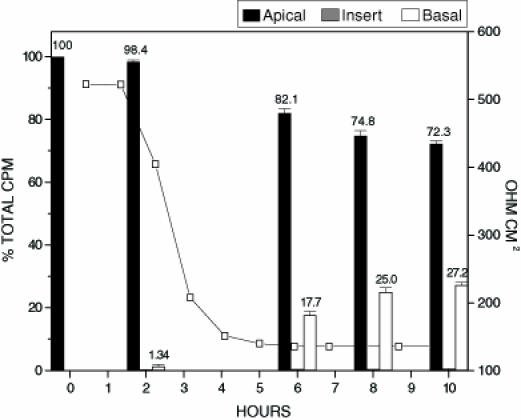
Radioactive count distribution for inserts treated with 125I labelled toxin A and unlabeled toxin B. Inserts were inoculated with 250 µL of 125I labelled toxin A (0.250 µg/mL) and 250 µL of unlabelled toxin B (0.250 µg/mL). Samples from apical, basal and insert were tested for percent of the total input counts per minute (cpm). The figure represents the average of three separate experiments. The electrical resistance measurements indicate the level of tight junction integrity. The total amount of radioactivity inoculated onto the insert in the form of radiolabelled toxin A was 4.6 x103 cpm
Toxin B is a potent cytotoxin capable of causing rearrangements in cytoskeletal structure. To examine whether toxin B is able to modulate tight junctions and translocate across an intact Caco-2 cell monolayer, inserts were inoculated with 125I-toxin B (Figure 4). In contrast to treatment with toxin A alone, the RD50 for toxin B was 7 h. This corresponded to a two-fold increase in the time required to elicit the same tight junction effects when compared with toxin A. When analyzing the distributions of 125I-toxin B and 125I-toxin A in experiments which tested the toxins individually, it was found that toxin B translocation was approximately 5% lower at each time frame. This may be attributed to the decreased ability of toxin B to affect tight junctions, thereby limiting the amount of toxin B capable of translocating through holes created in the tight junctions. To determine if the addition of toxin A would have a synergistic effect on the translocation of toxin B, inserts were treated with unlabelled toxin A and 125I-toxin B (Figure 5). The addition of toxin A had a dramatic impact on the ability of 125I-toxin B to translocate across intact Caco-2 cell monolayers. The amount of toxin B capable of translocating the monolayer was increased by approximately 100% at each of the time intervals tested. The addition of toxin A also reduced the RD50 of the monolayer to 3 h.
Figure 4.
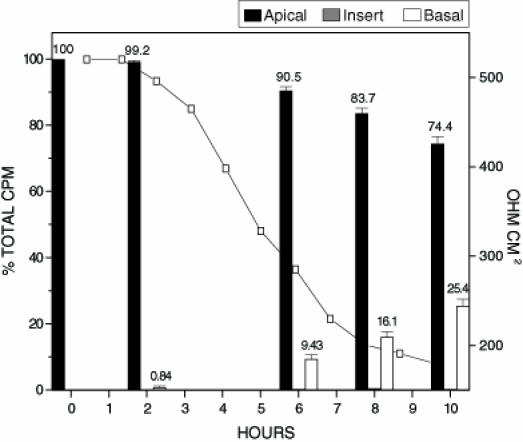
Radioactive count distribution for inserts treated with 125I labelled toxin B. Inserts were inoculated with 500 µL of 125I labelled toxin B (0.250 µg/mL). Samples from apical, basal and insert were tested for per cent of the total input counts per minute (cpm). The figure represents the average of three separate experiments. The electrical resistance measurements indicate the level of tight junction integrity. The total amount of radioactivity inoculated onto the insert in the form of radiolabelled toxin B was 7.0 x103 cpm
Figure 5.
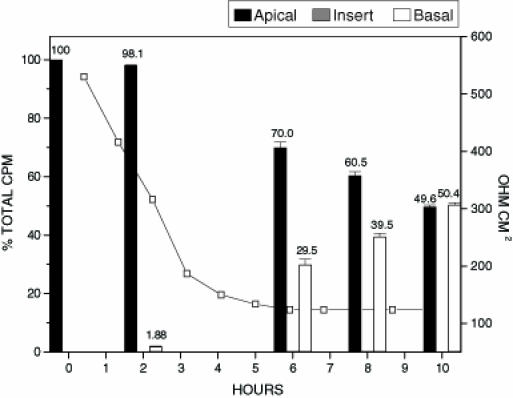
Radioactive count distribution for inserts treated with 125I labelled toxin B and unlabelled toxin A. Inserts were inoculated with 250 µL of 125I labelled toxin B (0.250 µg/mL) and 250 µL of unlabelled toxin A. Samples from apical, basal and insert were tested for per cent of the total input counts per minute (cpm). The figure represents the average of three separate experiments. The electrical resistance measurements indicate the level of tight junction integrity. The total amount of radioactivity inoculated onto the insert in the form of radiolabelled toxin B was 7.0 x103 cpm
Based on these data, which indicate that toxin B is preferentially translocated, a model of pathogenesis (Figure 6) which illustrates the synergistic roles of both C difficile toxins is proposed. In contrast to Sears and Kaper's (22) model for toxin A translocation, it is suggested that toxin B is the predominant toxin breaching the submucosal barrier to elicit the inflammatory responses.
Figure 6.
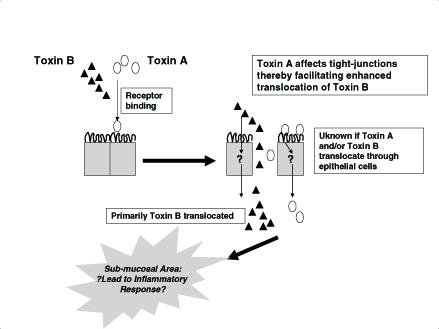
Proposed model for synergistic translocation of Clostridium difficile toxins A and B. Toxin A binds to epithelial cells and disrupts epithelial tight junctions. This allows both toxins to gain access to submucosal tissue to elicit an inflammatory response. Toxin B has enhanced ability to translocate compared with toxin A
DISCUSSION
C difficile has been referred to as "a pathogen of the nineties" (23). It is the most common cause of infectious nosocomial diarrhea in hospital and long term care facilities. The disease has significant implications for costs associated with hospitalization, as well as patient morbidity and mortality (23,24). Despite the significance of this infection, the model of pathogenesis is still incomplete. The relative contributions of toxin A and toxin B to human intestinal mucosal damage continue to be investigated. It was speculated that toxin A was the most important component in disease production because it elicited extensive tissue damage and fluid accumulation (6,25). Indeed, Sears and Kaper (22) hypothesized that translocation of toxin A into the submucosa may be responsible for the submucosal inflammatory response seen in histological sections of damaged bowel from patients with C difficile associated diarrhea. However, it is now clear that both toxins A and B are important for eliciting these disease symptoms. Two studies (13,26) support the shared role of toxin A and toxin B, reporting that it was necessary to vaccinate hamsters against both toxin A and toxin B in order to confer immunity.
Despite their large molecular sizes, both toxins A and B appear to have a more efficient mode of translocation across confluent Caco-2 monolayers compared with BSA. These findings suggest that the translocation process for the two toxins is not passive, and that an active transport mechanism may be involved. Several lines of evidence support this hypothesis. Although the cellular receptors for toxin B are unknown, researchers have already identified the putative receptor for toxin A: a 163 kDa glycoprotein found on the surface of brush border membranes (27). This receptor is believed to be involved in the attachment process which leads to the internalization of part or all of toxin A. Using biochemical and electron microscopy studies, investigators have also revealed endocytic pathways for both toxins A and B (15,28). Indeed, evidence supports an active transport model for C difficile toxin internalization and translocation. The results of our experiments are also in agreement with these findings becuase data indicated an increased amount of translocation of toxin A and toxin B in comparison with BSA. Furthermore, our data suggest that the intra- or transcellular trafficking of toxin A is likely different from that of toxin B, because our data show that the translocation of toxin B across Caco-2 monolayers is enhanced compared with toxin A.
When Caco-2 cell monolayers were exposed to toxins A and B, a synergistic effect was measured for the amount of toxin B that translocated. A molecular basis for the differential trafficking of toxin B compared with toxin A cannot be provided from the data presented; however, several explanations may be proposed. C difficile toxins A and B belong to the large clostridial cytotoxin family (29). These toxins have clostridial repetitive oligopeptide (CROP) sequences at their carboxyl termini which are believed to be involved in cellular attachment. Each CROP segment consists of either 20 amino acids or 50 amino acids repeated 14 to 30 times (7,30,31). Competition assays have shown the ability of CROPs to inhibit binding of toxin A to cell surfaces (32). There are 30 CROPs in toxin A and 19 CROPs in toxin B (16). Neutralization experiments using monoclonal antibodies have revealed a putative hexapeptide sequence (TIDGKK) believed to be involved in interaction between the toxin and its cellular receptor (32,33). This hexapeptide sequence was found in six copies in the CROP region of toxin A, however, no copies were found in toxin B. These findings suggest that cellular attachment specificity for the two toxins are different and may explain why toxin B does not bind to brush border membranes. Because toxin B cannot bind to cellular receptors, it would have an increased ability to pass through gaps created in the tight junctions, resulting in greater translocation efficiencies. The results of our experiments are in agreement with these findings because our data indicated an increase in translocation for 125I-toxin B when tested in conjunction with unlabelled toxin A. Research (16,17) has also shown that toxin A and toxin B have different sites of localization within endosomal compartments. Because precise cellular trafficking mechanisms for translocation are unknown, it is possible for the two toxins to use different mechanisms of translocation (15). In tests which used radiolabelled toxin A or toxin B alone, the amount of radiolabelled toxin translocating at each time point remained at similar levels. Because toxin A is more efficient at modulating tight junctions, we would have expected an increased amount of toxin A translocating as a result of passage through holes in tight junctions. However, this was not observed and may be attributable in part to the binding of toxin A to cellular receptors in a manner that does not allow it to translocate.
The results of our experiments suggest a synergistic role for C difficile toxins in promoting disease symptoms. If correct, our model would predict that C difficile isolates capable of producing both toxins would be more virulent because translocation of toxins would be more efficient. Indeed, most clinically significant infections have been caused by strains capable of producing both toxins. Strains deficient in toxin A production have been reported (34). One such strain, Serogroup F, lacks toxin A and is found only in neonates (35). Although cytotoxic activity has been detected in stool samples, the strain has not been associated with clinically significant disease and most carriers are asymptomatic. Futhermore, axenic mice inoculated with this strain do not develop diarrhea. Other Tox A-, Tox B+ strains have been described, that are associated with disease in humans. In these strains, it is not totally clear whether or not toxin A is totally deficient. Most of these Tox A-, Tox B+ strains have a truncated portion of the toxin A gene and some may produce a truncated version of toxin A which allows for enhanced translocation of toxin B.
To our knowledge, our data provides the first experimental evidence of the synergistic effect of toxins A and B on the translocation process. Furthermore, it is the first report to indicate that toxin B is more efficiently translocated across the bowel than toxin A. Our results would suggest that toxin B would be the primary component responsible for the submucosal bowel damage seen in vivo. Indeed, Lyerly et al (6) speculated that toxin A facilitated the exit of toxin B from the gut.
Acknowledgments
We would like to acknowledge the kind donation of the highly purified toxin A and toxin B preparations by Dr David Lyerly.
References
- 1.Struelens MJ, Maas A, Nonhoff C, et al. Control of nosocomial transmission of Clostridium difficile on sporadic case surveillance.Am J Med 1991;91:138S-44S. [DOI] [PubMed] [Google Scholar]
- 2.Bartlett J. Clostridium difficile: Clinical considerations.Rev Infect Dis 1990;12(Suppl 2):S243-51. [DOI] [PubMed] [Google Scholar]
- 3.Bartlett JG. Antibiotic-associated diarrhea.Clin Infect Dis 1992;15:573-81. [DOI] [PubMed] [Google Scholar]
- 4.Anand A, Glatt AE. Clostridium difficile infection associated with antineoplastic chemotherapy: A review.Clin Infect Dis 1993;17:109-13. [DOI] [PubMed] [Google Scholar]
- 5.Fekety R. Antibiotic-associated colitis. In Mandell GL, Douglas RG, Bennett JE, eds. Principles and practices of infectious disease, 4th edn. New York:Churchill Livingstone, 1995;:978 [Google Scholar]
- 6.Lyerly DM, Krivan HC, Wilkins TD. Clostridium difficile: Its disease and toxins.Clin Microbiol Rev 1998;1:1-18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Dove CH, Wang SZ, Proce SB, et al. Molecular characterization of the Clostridium difficile toxin A gene.Infect Immun 1990;58:480-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Lyerly DM, Lockwood DE, Richardson SH, Wilkins TD. Biological activities of toxins A and B of Clostridium difficile.Infect Immun 1982;35:1147-50 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Taylor NS, Thorne GM, Bartlett JG. Comparison of two toxins produced by Clostridium difficile.Infect Immun 1981;34:1036-43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Triadafilopoulus G, Pothoulakis C, O'Brien M, LaMont JT. Differential effects of Clostridium difficile toxins A and B on rabbit ileum.Gastroenterology 1987;93:273-9. [DOI] [PubMed] [Google Scholar]
- 11.Pothoulakis C, Barone LM, Ely R, et al. Purification and properties of Clostridium difficile cytotoxin B.J Biol Chem 1986;261:1316-21. [PubMed] [Google Scholar]
- 12.Arnon SS, Mills DC, Day PA, Henrickson RV, Sullivan NM, Wilkins TD. Rapid death of infant rhesus monkeys injected with Clostridium difficile toxins A and B: Physiologic and pathologic basis.J Pediatr 1984;104:34-40 [DOI] [PubMed] [Google Scholar]
- 13.Libby JM, Jortner BS, Wilkins TD. Effects of the two toxins of Clostridium difficile in antibiotic-associated cecitis in hamsters.Infect Immun 1982;36:822-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Lyerly DM, Saum KE, MacDonald DK, Wilkins TD. Effects of Clostridium difficile toxins given intragastrically to animals.Infect Immun 1985;47:349-52. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Henriques B, Florin I, Thelestam M. Cellular internalisation of Clostridium difficile toxin A.Microb Pathog 1987;2:455-63. [DOI] [PubMed] [Google Scholar]
- 16.von Eichel-Streiber C, Boquet P, Sauerborn M, Thelestam M. Large clostridial cytotoxins - a family of glycosyltransferases modifying small GTP-binding proteins. 1996;4:375-82. [DOI] [PubMed] [Google Scholar]
- 17.Just I, Selzer J, Wilm M, von Eichel-Streiber C, Mann M, Aktories K. Glucosylation of Rho proteins by Clostridium difficile toxin B.Nature 1995;375:500-3. [DOI] [PubMed] [Google Scholar]
- 18.Moore R, Pothoulakis C, LaMont JT, Carlson S, Madara JL. C difficile toxin A increases intestinal permeability and induces Cl-secretion.Am J Physiol 1990;259:G165-72. [DOI] [PubMed] [Google Scholar]
- 19.Nusrat A, Giry M, Turner JR, et al. Rho protein regulates tight junctions and perijunctional actin organization in polarized epithelia.Proc Natl Acad Sci USA 1995;92:10629-33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Grasset E, Pinto M, Dussaulx E, Zweibaum A, Desjeux J. Epithelial properties of human colonic carcinoma cell line Caco-2: Electrical parameters.Am J Physiol 1984;247:C260-7. [DOI] [PubMed] [Google Scholar]
- 21.Pinto M, Robine-Leon S, Appay MD, et al. Enterocyte differentiation and polarization of the human colon carcinoma cell line Caco-2 in culture.Biol Cell 1983;47:323-30. [Google Scholar]
- 22.Sears CL, Kaper JB. Enteric bacterial toxins: Mechanisms of action and linkage to intestinal secretion.Microbiol Rev 1996;60:167-215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Riley TV. Clostridium difficile: A pathogen of the nineties.Eur J Clin Microbiol Infect Dis 1998;17:137-41. [DOI] [PubMed] [Google Scholar]
- 24.Wilcox MH, Cunniffe JG, Trundle C, Redpath C. Financial burden of hospital-acquired Clostridium difficile infection.J Hosp Infect 1996;34:23-30. [DOI] [PubMed] [Google Scholar]
- 25.Borrielo SP, Barclay FE, Welch AR, et al. Host and microbial determinants of the spectrum of Clostridium difficile mediated gastrointestinal disorders.Microecol Ther 1985;15:231-6. [Google Scholar]
- 26.Fernie DS, Thomson RO, Batty I, Walker PD. Active and passive immunization to protect against antibiotic associated caecitis in hamsters.Dev Biol Stand 1983;53:325-32. [PubMed] [Google Scholar]
- 27.Rolf RD, Song W. Purification of a functional receptor for Clostridium difficile toxin A from intestinal brush border membranes of infant hamsters.Clin Infect Dis 1993;17(Suppl 4):S219-27. [DOI] [PubMed] [Google Scholar]
- 28.von Eichel-Streiber C, Warfolomeow I, Knautz D, Sauerborn M, Hadding U. Morphological changes in adherent cells induced by Clostridium difficile toxins.Biochem Soc Trans 1991;19:1154-60. [DOI] [PubMed] [Google Scholar]
- 29.Bette P, Oksche A, Mauler F, von Eichel-Streiber C, Popoff MR, Habermann E. A comparative biochemical, pharmacological and immunological study of Clostridium novyi alpha-toxin, C. difficile toxin B and C. sordellii lethal toxin.Toxicon 1991;29:877-87. [DOI] [PubMed] [Google Scholar]
- 30.von Eichel-Streiber C, Laufenberg-Feldman R, Sartingen S, Schulze J, Sauerborn M. Comparative sequence analysis of the Clostridium difficile toxins A and B.Mol Gen Genet 1992;233:260-8. [DOI] [PubMed] [Google Scholar]
- 31.Hofman F, Herrmann A, Habermann E, von Eichel-Streiber C. Sequencing and analysis of the gene encoding the alpha-toxin of Clostridium novyi proves its homology to toxins A and B of Clostridium difficile.Mol Gen Genet 1995;247:670-9. [DOI] [PubMed] [Google Scholar]
- 32.Sauerborn M, et al. Monoclonal antibodies discriminating between Clostridium difficle toxins A and B. Zentralbl.Bakteriol Suppl 1991;23:144-5. [Google Scholar]
- 33.Frey SM, Wilkins TD. Localization of two epitopes recognized by monoclonal antibody PCG-4 on Clostridium difficile toxin A.Infect Immun 1992;60:2488-92. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Lyerly DM, Barroso LA, Wilkins TD, Depitre C, Corthier G. Characterization of a toxin A-negative, toxin B-positive strain of Clostridium difficile.Infect Immun 1992;60:4633-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Depitre C, Delmee M, Avesani V, et al. Serogroup F strains of Clostridium difficile produce toxin B but not toxin A.J Med Microbiol 1993;38:434-41. [DOI] [PubMed] [Google Scholar]