

Abstract
Bullous pemphigoid (BP) is an inflammatory subepidermal blistering disease associated with an IgG autoimmune response to the hemidesmosomal protein BP180. Passive transfer of antibodies to the murine BP180 (mBP180) ectodomain triggers a blistering skin disease in mice that depends on complement activation and neutrophil infiltration and closely mimics human BP. In the present study, we show that mast cells (MCs) play a crucial role in experimental BP. Wild-type mice injected intradermally with pathogenic anti-mBP180 IgG exhibited extensive MC degranulation in skin, which preceded neutrophil infiltration and subsequent subepidermal blistering. In contrast, mice genetically deficient in MCs or MC-sufficient mice pretreated with an inhibitor of MC degranulation failed to develop BP. Further, MC-deficient mice reconstituted in skin with MCs became susceptible to experimental BP. Despite the activation of complement to yield C3a and C5a, in the absence of MCs, accumulation of neutrophils at the injection site was blunted. The lack of response due to MC deficiency was overcome by intradermal administration of a neutrophil chemoattractant, IL-8, or by reconstitution of the injection sites with neutrophils. These findings provide the first direct evidence to our knowledge that MCs play an essential role in neutrophil recruitment during subepidermal blister formation in experimental BP.
Introduction
Bullous pemphigoid (BP) is an acquired autoimmune skin disease characterized by autoantibodies against two hemidesmosomal antigens, BP230 (BPAG1) and BP180 (BPAG2), and subepidermal blisters (1). These anti-hemidesmosomal autoantibodies can be detected, along with complement proteins, bound to the dermal-epidermal junction (DEJ) of perilesional skin. In the skin lesions of these patients, basal keratinocytes detach from the underlying dermis at the level of the lamina lucida, leading to subepidermal blistering (1, 2). Eosinophils (3, 4), neutrophils (5), lymphocytes (6), monocyte/macrophages (7, 8), and mast cells (MCs; 7, 9) are present in the upper dermis of lesional areas in patients with BP. Interestingly, MC degranulation is a feature of BP (7, 9). Chemoattractants from MCs, including eosinophilic/neutrophilic chemotactic factors and histamine, are present at high concentrations in BP blister fluids (10, 11). Similar skin lesions are observed in the pregnancy-associated nonviral disorder herpes gestationis (12). These observations imply that MCs may play a role in blister formation.
We have used an experimental model of BP that involves the passive transfer of anti-mBP180 antibodies into neonatal BALB/c mice to reproduce the key immunopathological features of this human autoimmune disease: IgG and complement deposition at the DEJ, inflammatory infiltration of the upper dermis, and subepidermal blistering (13). The pathogenicity of anti-mBP180 antibodies depends on complement activation (14) and polymorphonuclear leukocyte (PMN) recruitment (15). In the present study, we have investigated the role of MCs in experimental BP using mice genetically deficient in MCs.
Methods
Laboratory animals.
Breeding pairs of MC-deficient WCB6F1-MgfSl/MgfSl-d (referred to as MgfSl/MgfSl-d), MC-deficient WBB6F1-Kitw/Kitw-v (referred to as Kitw/Kitw-v), and the congenic MC-sufficient (MC+/+) littermates (16) and C5-deficient B10-D2-OSN (C5–) and their matched normal control B10-D2-NSN (C5+) mice were purchased from The Jackson Laboratory (Bar Harbor, Maine, USA) and maintained at the Medical College of Wisconsin Animal Resource Center and the University of North Carolina at Chapel Hill Animal facility. The MC deficiency in MgfSl/MgfSl-d and Kitw/Kitw-v mice are caused by distinct mutations in MC growth factor and c-Kit, respectively (16). Neonatal mice (24–36 hours old, 1.4–1.6 g) were used for passive transfer experiments.
Preparation of pathogenic rabbit anti-mouse IgG.
The preparation of recombinant mBP180 and the immunization of rabbits were performed as described previously (13). The titers of rabbit anti-mBP180 antibodies in the rabbit sera and in the purified IgG fractions were assayed by indirect immunofluorescence (IF) using mouse skin cryosections as substrate (13). The pathogenicity of these IgG preparations were tested by passive transfer experiments, as described below.
Induction of experimental BP and animal evaluation.
A 50-μl dose of sterile IgG in PBS was administered to neonatal mice by intradermal injection (2.5 mg IgG per gram of body weight) as described previously (13, 15). The skin of neonatal mice from the test and control groups was examined 12 hours after the IgG injection. The extent of cutaneous disease was scored as follows: (–), no detectable skin disease; 1+, mild erythematous reaction with no evidence of the “epidermal detachment” sign (this sign was elicited by gentle friction of the mouse skin, which, when positive, produced fine, persistent wrinkling of the epidermis); 2+, intense erythema and epidermal detachment sign involving 10–50% of the epidermis in localized areas; and 3+, intense erythema with frank epidermal detachment sign involving more than 50% of the epidermis. The animals were then killed, and skin and serum specimens were obtained. Skin sections were taken for light microscopy (hematoxylin and eosin [H&E] staining) and for direct IF analysis to detect rabbit IgG and mouse C3 deposition at the basement membrane zone (BMZ). Sera of injected animals were used for indirect IF assay to determine the circulating titers of anti-mBP180 IgG. Direct and indirect IF analyses were performed as described previously (13). Monospecific FITC-conjugated goat anti-rabbit IgG was purchased from Kirkegaard & Perry Laboratories Inc. (Gaithersburg, Maryland, USA); monospecific goat anti-mouse C3 was purchased from Cappel Laboratories (Durham, North Carolina, USA).
Quantification of PMN accumulation at the skin site.
Myeloperoxidase (MPO) activity in skin sites of the injected animals was assayed as a measure of PMN infiltration as described elsewhere (17, 18). A standard reference curve was established using purified MPO (Athens Research and Technology Inc., Athens, Georgia, USA). The skin samples were extracted by homogenization in an extraction buffer containing 0.1 M Tris-Cl (pH 7.6), 0.15 M NaCl, and 0.5% hexadecyl trimethylammonium bromide. MPO activity in the supernatant fraction was measured by the change in optical density at 460 nm resulting from decomposition of H2O2 in the presence of o-dianisidine. MPO content was expressed as units of MPO activity per milligram of protein. Protein concentrations were determined by the Bio-RAD dye-binding assay (Bio-Rad Laboratories Inc., Hercules, California, USA) using BSA as a standard.
Quantification of MCs and MC degranulation.
MCs and MC degranulation in skin samples were quantified according to Wershil et al. (19) with modification. Briefly, lesional and nonlesional skin sections of IgG-injected mice were fixed in 10% formalin. Paraffin sections (5 μm thick) were prepared and stained with toluidine blue and H&E. The total number of MCs was counted and classified as degranulated (>10% of the granules exhibiting fusion or discharge) or normal in five fields under a light microscope as described previously. The results were expressed as percentage of MC degranulating.
Electron microscopic analysis of MC morphology.
The skin samples were dissected from the injected sites of the test and control animals and immersed in a fixative of 2% glutaraldehyde in 0.1 M cacodylate buffer (pH 7.2). The tissue blocks were post-fixed with OsO4 and then dehydrated with a series of graded ethanol. Thin sections were contrasted with uranyl acetate and lead citrate and examined with a Hitachi-600 transmission electron microscope (Hitachi Instruments Inc., San Jose, California, USA) at 75 KV.
In vivo inhibition of MC degranulation.
Neonatal wild-type mice were injected intradermally with pathogenic IgG (2.5 mg per gram of body weight) in the absence or presence of cromolyn sodium (Sigma Chemical Co., St. Louis, Missouri, USA) (10 μg per gram of body weight), an MC degranulation inhibitor (20, 21). The animals were examined for clinical blisters 12 hours after IgG injection, and the skin sections were analyzed by IF, H&E, and toluidine blue staining. Skin PMN infiltration was quantified by MPO assay.
PMN isolation.
Mouse PMNs were isolated from heparinized blood by dextran sedimentation followed by separation on a density gradient as described previously (22). Red blood cells were removed from the cell preparation by hypotonic lysis in 0.2% NaCl. PMNs were washed and resuspended in cold PBS/10 mM glucose, counted in a hemocytometer, and adjusted to a concentration of 1 × 107 cells/ml. PMN purity of the final cell preparation was consistently more than 96% as determined by cell-cytospin and LeukoStat staining (Fisher Diagnostics, Orangeburg, New York, USA). The viability of the neutrophils was >96% as determined by trypan blue exclusion.
Intradermal injection of PMNs.
PMNs were purified from MC-deficient and MC+/+ mice. MC-deficient mice were injected intradermally with pathogenic anti-mBP180 IgG (2.5 mg per gram of body weight per 50 μl PBS). Two hours later, these mice were injected with 5 × 105 PMNs intradermally (in 50 μl of PBS/10 mM glucose) at the same site (23). The animals were analyzed 12 hours after the IgG injections, as described above.
IL-8 pretreatment of MC-deficient mice.
Recombinant human IL-8 (hIL-8; R&D Systems Inc., Minneapolis, Minnesota, USA) was stored at 1 mg/ml in sterile PBS. A single intradermal injection of IL-8 (50 ng in 50 μl PBS) or an equivalent amount of BSA was given to neonatal MC-deficient mice, followed by intradermal injection of rabbit anti-mBP180 IgG (2.5 mg per gram of body weight in 50 μl) 1 hour later (15). Control animals received an equivalent amount of normal rabbit IgG, in place of the anti-mBP180 IgG. The animals were analyzed 12 hours after the IgG injections, as described above.
MC reconstitution.
Kitw/Kitw-v mice were repaired of their MC deficiency selectively and locally by the injection of growth factor–dependent bone marrow–derived cultured MCs into the skin (24, 25). Briefly, femoral bone marrow cells from congenic MC+/+ mice were maintained in vitro for 4 weeks in RPMI 1640 complete medium (Life Technologies Inc., Grand Island, New York, USA) supplemented with 20% WEHI-3–conditioned medium until MCs represented more than 95% of the total cells as determined by toluidine blue staining and flow cytometry analysis using antibodies specific for MC cell-surface markers FcεRI, c-Kit, and CD13 (25). Murine IgE and rat anti-mouse IgE were purchased from Southern Biotechnology Associates (Birmingham, Alabama, USA). FITC-labeled rat anti-mouse c-Kit and FITC-labeled rat anti-mouse CD13 were obtained from BD PharMingen (San Diego, California, USA). MCs (1 × 106 in 20 μl of medium) were injected intradermally into the right ears of MC-deficient mice. Media alone (20 μl) were injected intradermally into the left ears of the same mice as negative control. This procedure selectively and locally reconstitute dermal MC population without systemic effects (24). To confirm MC reconstitution, skin sections from MC-injected sites were stained by toluidin blue. Ten weeks after adoptive transfer of MCs, both ears of the mice were injected intradermally with pathogenic anti-BP180 IgG (2 mg/20 μl/site). Twenty-four hours later, ear skin biopsies were obtained and analyzed by H&E, toluidine blue staining, and MPO enzyme assay as described above.
Statistical analysis.
The data are expressed as mean ± SEM and were analyzed using the Student’s paired t test. A P value less than 0.05 was considered significant.
Results
MC degranulation precedes neutrophilic infiltration and subsequent dermal-epidermal separation.
We first determined whether and when MC infiltration and degranulation occur in experimental BP and how these events are related to PMN infiltration and subepidermal blistering over the 12-hour period after intradermal injection of pathogenic anti-mBP180 IgG. Five animals were used at each time point. Deposition of rabbit IgG and murine C3 at the cutaneous BMZ is first observed 1–2 hours after IgG injection (15). At 4 hours, an early stage of dermal-epidermal separation was seen histologically (Figure 1a). By 8 hours, relatively broad subepidermal blister vesicles could be seen, and complete detachment of the epidermis from the dermis occurred at 12 hours. Electron microscopy (Figure 1, b and c) revealed that MCs in the dermis of control mice (Figure 1b) had a large amount of cytoplasmic granules that present a variety of appearances. Large vacuoles, which were obviously resulted from degranulation of MCs, were easily detected in the dermis of the diseased mice 2 hours after IgG injection (Figure 1c). Although we saw no significant changes in MC abundance at any time points. MC degranulation was dramatic (Figure 1d). MC degranulation occurred within 1 hour after intradermal injection of anti-mBP180 IgG and reached a peak 1–2 hours before the first evidence of neutrophil recruitment to the skin site. PMN infiltration was first detected at 2–3 hours, then increased progressively over the next 5 hours, reaching a peak at 8 hours (Figure 1e).
Figure 1.

Time-course of MC degranulation in MC-sufficient mice injected intradermally with pathogenic rabbit anti-murine BP180 IgG and assessed 0–12 hours after injection. (a) Toluidine blue–stained sections showing progression of MC degranulation and separation of the epidermis from the dermis. d, dermis; e, epidermis; v, vesicle; arrow, site of antibody labeling. ×400. (b and c) Electron micrographs show degranulation of MCs in dermis of mice injected with pathogenic IgG. An MC in control animals (b) has a number of large cytoplasmic granules that appear in different density, whereas another MC from pathogenic IgG-injected animal (c) presents many empty vacuoles that apparently resulted from degranulation. Numerous microvilli are observed in the plasma membrane of degranulated MCs, indicating the activated cell status. (d) The MCs in the dermis were counted and classified as degranulated (>10% of the granules exhibiting fusion or discharge) or normal (see Methods). MC degranulation reached the peak level at 1–2 hours after pathogenic IgG injection. (e) Skin MPO activity (mean ± SEM) data reveal the onset of PMN infiltration at 2 hours after IgG injection (n = 5 for each group shown).
MC-deficient mice are resistant to experimental BP.
To determine whether MCs participate in experimental BP, we injected wild-type and MC-deficient mice with pathogenic IgG. The MC+/+ mice (n = 8) showed MC degranulation by toluidine blue staining and developed histological and clinical blisters 12 hours after IgG injection (Figure 2, a and d). In contrast, MC-deficient MgfSl/MgfSl-d mice (n = 8) injected with pathogenic IgG had no clinical or histological signs of blister formation (Figure 2, e and h), despite the fact that these animals showed as much deposition of IgG and C3 at the BMZ as did the wild-type mice (Figure 2, b, c, f, and g). Histological examination exhibited neither subepidermal blistering nor MC presence in the skin (Figure 2h). Similar phenotype was found in MC-deficient Kitw/Kitw-v mice when injected with pathogenic IgG (Table 1). Given that both strains of MC-deficient mice caused by distinct mechanisms are resistant to experimental BP, these results strongly suggest that MCs play an essential role in BP.
Figure 2.

Clinical and immunohistological analysis of neonatal MC-deficient and -sufficient mice injected with pathogenic anti-mBP180 IgG. The anti-mBP180 IgG (2.5 mg/g body weight) induced extensive blistering disease in MC-sufficient (+/+) mice (a). The skin of these animals showed linear deposition of rabbit IgG (b) and murine C3 (c) at the BMZ, as determined by direct IF. Toluidine blue staining revealed epidermal-dermal separation with MC degranulation (d). In contrast, MC-deficient (MgfSl/MgfSl-d) mice injected intradermally with pathogenic IgG showed no evidence of skin disease (e). Direct IF demonstrated BMZ deposition of rabbit IgG (f) and murine C3 (g). Toluidine blue staining showed no epidermal-dermal separation and absence of MCs (h). d, dermis; e, epidermis; v, vesicle; black arrow in a and e, site of clinical blister; white arrow, site of antibody labeling; black arrow in d and h, site of BMZ. ×400. Inset in d is a lower-magnification micrograph showing the edge of a subepidermal vesicle. ×100.
Table 1.
The role of MCs in BP blister formation

Reconstitution of MCs in skin restores BP in MC-deficient mice.
To directly demonstrate a requirement of MCs in subepidermal blistering in experimental BP, we injected pathogenic IgG into Kitw/Kitw-v mice (n = 6) locally repaired of their MC deficiency by adoptive transfer of cultured MCs derived from congenic MC+/+ mouse bone marrow cells. MCs, infiltrating neutrophils, and subepidermal blistering were found in the MC-reconstituted right ears but not in the MC-deficient left ears (Figure 3 and Table 1). The increase of neutrophils in the MC-reconstituted site was significantly higher than in the MC-deficient site (P < 0.01). These results clearly show that neutrophil infiltration and subsequent blister formation triggered by anti-BP180 IgG are indeed MC dependent. The MC reconstitution data also rule out the possibility that resistance of MC-deficient mice to experimental BP is due to other unidentified defects resulting from the c-Kit mutations in Kitw/Kitw-v mice, besides the MC deficiency of these animals.
Figure 3.

Effect of MC reconstitution on neutrophil infiltration and subsequent subepidermal blistering in pathogenic anti-BP180 IgG-injected MC-deficient mice. Neutrophil infiltration and blister formation were determined in the left (MC-deficient) (middle bar) and right (MC-reconstituted) (bottom bar) ears of Kitw/Kitw-v mice that underwent local reconstitution of the right ear with 1 × 106 bone marrow-derived MC+/+ MCs10 weeks before pathogenic IgG injection (2 mg/ear). MC+/+ littermates (top bar) were also injected with the same dose of IgG in the ear as positive control (see Methods for details). Significantly elevated levels of MPO activity (mean ± SEM) were seen in the right ear compared with the left ear of Kitw/Kitw-v mice. Like MC+/+ mice, the right but not the left ear skin sections exhibited dermal-epidermal separation as determined by H&E staining. MPO activity at 0 hours was 0.07 ± 0.01 for MC-deficient and 0.09 ± 0.02 (OD460nm/mg protein) for MC+/+ mice. (n = 6 for each group.) *P < 0.01, top vs. middle bar.
Pathogenic anti-mBP180 IgG induces subepidermal blistering in MC-deficient mice reconstituted with PMNs from MC-deficient mice or by intradermal injection of the neutrophil chemoattractant IL-8.
If the major function of MCs in experimental BP is to recruit PMNs from circulation to the IgG-injection site, then artificially recruiting them to the dermis of the MC-deficient mice should circumvent the requirement for MCs for disease phenotype. This is exactly what we observed when we injected MC-deficient mice with pathogenic IgG and reconstituted them with mouse PMNs. The MC-deficient mice reconstituted with 5 × 105 PMNs from wild-type mice (n = 5) developed subepidermal blisters 12 hours after IgG injection (Figure 4). The MC-deficient mice reconstituted with 5 × 105 PMNs from MC-deficient mice (n = 5) also became susceptible to pathogenic anti-mBP180 IgG, with a similar disease score indicating that PMNs are normal in MC deficiency (Table 1). In addition, MC-deficient mice (n = 5) given an intradermal injection of IL-8 before, or coincident with, the treatment with anti-mBP180 IgG, developed extensive blisters 12 hours later (Figure 5 and Table 1). The skin of these mice showed typical subepidermal separation and morphological and biochemical evidence of neutrophilic infiltration of the dermis (Figure 5). The mean MPO activity level in skin extracts of MC-deficient mice coinjected with anti-mBP180 IgG and IL-8 was 1.14 ± 0.18 OD460nm/mg protein (Figure 5), compared with 0.29 ± 0.02 (P < 0.01) for skin from the mice injected only with anti-mBP180 IgG. Taken together, these results clearly demonstrate that MCs mainly participate in PMN recruitment in experimental BP.
Figure 4.

Experimental BP in MC-deficient mice reconstituted with normal PMNs. Pathogenic rabbit anti-mBP180 IgG (intradermal injection, 2.5 mg/g body weight) induced skin blisters in MC-sufficient (+/+) mice (a) and toluidine blue staining showed the presence of MCs (b). Anti-mBP180 IgG failed to trigger the skin lesions in MC-deficient MgfSl/MgfSl-d mice (c), but R621 IgG triggered the skin disease in MgfSl/MgfSl-d mice reconstituted with PMNs from MC+/+ mice (e) or MC-deficient mice (data not shown). Toluidine blue staining confirmed the MC deficiency in these animals (d and f).
Figure 5.

In vivo reconstitution of PMNs at the tissue site by intradermal injection of IL-8 restores the pathogenic effect of anti-BP180 IgG in neonatal MC-deficient mice. MC-sufficient (+/+) (bar 1) or MC-deficient MgfSl/MgfSl-d mice (bars 2–6) were injected intradermally with either pathogenic anti-mBP 180 IgG alone (bars 1 and 4), control rabbit IgG alone (bar 2), hIL-8 alone (bar 3), or IgG plus hIL-8 (bars 5 and 6). MC-deficient mice coinjected with IL-8 and anti-mBP 180 IgG (bar 5), but not the control rabbit IgG (bar 6), developed subepidermal blisters. The IgG dose was 2.5 mg/g body weight. Doses for IL-8 was 50 ng/mouse. Tissue MPO activity (mean ± SEM) in skin at the injection site was determined 12 hours after IgG administration. Each group of mice (n = 5) without injection yielded an average of MPO activity of 0.08 ± 0.02 OD460nm/mg protein. *P < 0.01, Student’s t -test for paired samples (bar 4 versus bar 5). See Methods for details.
An inhibitor of MC degranulation blocks neutrophil recruitment and subsequent blistering.
Extensive MC degranulation takes place in the dermis of mice injected with pathogenic IgG. But is it required? Neonatal wild-type mice, which were pretreated with vehicle, followed by pathogenic anti-mBP180 IgG (n = 5) 90 minutes later, developed intense blisters by 12 hours (Table 1). In contrast, mice treated with the MC degranulation inhibitor cromolyn sodium, followed by pathogenic IgG (n = 5) did not develop skin lesions (Table 1). The skin of the cromolyn sodium–treated animals showed no evidence of subepidermal vesiculation and minimal MC degranulation, despite the presence of high levels of circulating rabbit anti-BP180 antibodies, in situ deposition of rabbit IgG and murine C3 at the BMZ (data not shown).
The cromolyn sodium–treated mice had significantly reduced PMN infiltration, compared with control mice. The relative mean MPO levels for mice treated with cromolyn sodium at 2, 4, and 12 hours were 0.23 ± 0.02, 0.26 ± 0.03, and 0.43 ± 0.05 OD460nm/mg protein, respectively, whereas the MPO levels for the control groups were 0.20 ± 0.03 (P > 0.05), 0.41 ± 0.05 (P < 0.05), and 1.13 ± 0.17 OD460nm/mg protein (P < 0.01), respectively. This is very similar to the phenotype seen in MC-deficient mice. We also ruled out the possibility that cromolyn may directly interfere with development of BP rather than blocking MC degranulation by pretreatment of MC-deficient mice with cromolyn followed by injection of pathogenic IgG plus IL-8. There was no difference in PMN influx and disease scores between MC-deficient mice coinjected with pathogenic IgG and IL-8 with or without cromolyn pretreatment (Table 1). Because initial PMN recruitment is impaired in both MC-deficient mice and in MC-sufficient mice treated with cromolyn sodium, MCs appear to play a key role in early steps of recruiting PMNs as a consequence of degranulation.
Complement is required for MC degranulation and PMN recruitment.
The requirement for neutrophil recruitment requires MCs. Complement is required for blister formation. Complement fragments can both induce PMN chemotaxis and MC degranulation (26). Because complement was still present at the BMZ in MC-deficient mice, we next determined whether complement activation was a key element for MC degranulation or PMN recruitment. As a genetic test of whether MC-mediated PMN infiltration depends on complement activation, C5+ and C5– mice were injected intradermally with pathogenic IgG. C5+ but not C5– mice developed subepidermal blisters with a neutrophil infiltrate 12 hours after IgG injection, as shown previously (14). Toluidine blue staining revealed extensive MC degranulation in the dermis at 2 hours (Figure 6a) and 12 hours (Figure 6b). At 2 hours, 68% of MC were degranulated and 17% at 12 hours, in C5+ mice. In contrast, C5– mice exhibited a minimal level of MC degranulation in the dermis at 2 hours and 12 hours (Figure 6, c and d). C5+ mice injected with F(ab′)2 fragments of the pathogenic IgG also lacked MC degranulation in the dermis (data not shown). We previously found that C5-sufficient mice injected with pathogenic IgG F(ab′)2 fragments, which do not fix complement, have F(ab′)2, but no C3 deposited at the BMZ and have only background levels of neutrophil infiltration and no signs of skin lesions (14). Thus, MC degranulation triggered by anti-BP 180 IgG depends on complement activation.
Figure 6.
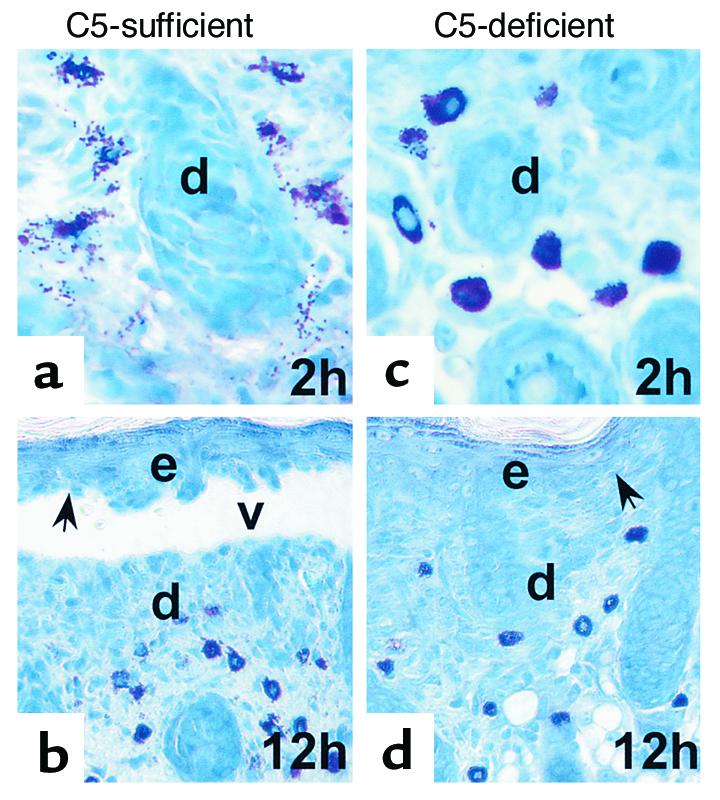
MC degranulation depends on complement activation. Neonatal C5-sufficient (a and b) and C5-deficient mice (c and d) were injected intradermally with pathogenic IgG R621 (2.5 mg/g body weight). C5+ (b) but not C5– mice (d) developed subepidermal blisters 12 hours after IgG injection. Toluidine blue staining analysis showed MC degranulation in the skin of C5+ mice at 2 hours (a) and 12 hours (b), whereas minimal levels of MC degranulation were seen in the dermis of C5– mice (b and d). d, dermis; e, epidermis; v, vesicle; arrow, site of basal keratinocyte. Original magnification ×400 (a and b), ×200 (c and d).
We next asked whether PMN infiltration occurred when complement was activated in the absence of MC. Intradermal PMN infiltration, quantified by MPO assay, was significantly lower in MC-deficient mice compared with wild-type controls both at 4 hours and 12 hours after IgG injection (Figure 7). Taken together, these results demonstrate a requirement of MCs for subepidermal blistering in experimental BP and suggest that the role of complement activation may be to activate MC degranulation.
Figure 7.

MPO activity of skin extracts from mice injected intradermally with pathogenic rabbit anti-mBP180 IgG. Neonatal MC-deficient MgfSl/MgfSl-d (bars 1 and 2), Kitw/Kitw-v (bars 3 and 4), and congenic normal (+/+) mice (bars 5 and 6) received 2.5 mg/g body weight pathogenic anti-mBP180 IgG. Tissue MPO activities (mean ± SEM) in the injection sites were determined 4 hours (bars 1, 3, and 5) and 12 hours (bars 2, 4, and 6) after the IgG injection (n = 5 for each group). *P < 0.01; **P < 0.005. Student’s t test for paired samples (bar 2 or 4 versus bar 6). The MPO values shown were corrected for control IgG controls. Each group of mice injected with control IgG yielded an average MPO activity of ∼0.13 OD460nm/mg protein.
Discussion
The aim of the present study was to assess the role of MCs in the pathogenesis of subepidermal blister formation in a mouse model of BP. The presence and degranulation of MCs at the BP lesional sites were first reported by Wintroub et al. in 1978 (7) and subsequently confirmed by other investigators (9, 27, 28). However, until our study, the functional significance of MCs in BP remained obscure. In this report, we find that the major role for MCs is as mediators in PMN recruitment and, thus, in subepidermal blister formation triggered by anti-mBP180 IgG. Pathogenic IgG failed to induce disease phenotype in MC-deficient mice. Not only did MC degranulate extensively in experimental BP (81% at the peak), but this degranulation was necessary for blistering. We have previously demonstrated that subepidermal blistering depends on complement activation (14) and neutrophil infiltration into the dermis (15). We now found that complement activating is required for MC degranulation. Injection of cromolyn sodium, which inhibits MC degranulation, prevented PMN recruitment and, subsequently, subepidermal blistering.
In these studies, we used two different strains of MC-deficient mice caused by distinct mechanisms: MgfSl/MgfSl-d mice have mutations in the c-Kit ligand MC growth factor (stem cell factor) and Kitw/Kitw-v mice have mutations in c-Kit. Owing to the nature of the mutations, transplantation of congenic MC+/+ MCs repairs the MC deficiency of Kitw/Kitw-v, but not MgfSl/MgfSl-d mice (16, 29). We found that both strains were resistant to the pathogenic activity of anti-BP180 IgG, independently implicating a critical role in subepidermal blistering. To demonstrate directly that MCs are absolutely required for the disease, we showed that MC-deficient Kitw/Kitw-v mice, which had been locally and selectively reconstituted with MCs from the congenic MC+/+ mice, like their normal control MC+/+ littermates, developed BP when injected with pathogenic antibodies.
MC degranulation precedes PMN infiltration and subsequent DEJ separation. Our results show a significant reduction in PMN infiltration both in the early and late stages of the disease process in MC-deficient mice. Blocking MC degranulation significantly reduced PMN recruitment into the skin and completely abolished experimental BP. Furthermore, when a potent PMN chemoattractant, such as IL-8, was coadministered with the anti-mBP180 antibodies, the MC-deficient mice showed both clinical and histological evidence of neutrophilic infiltration of the dermis and subepidermal blistering. Taken together, these findings provide the first in vivo evidence that subepidermal blistering induced by pathogenic anti-mBP180 antibodies depends on MCs, which, by degranulation step, play an essential role in recruiting neutrophils to the target tissue.
C3a and C5a have been widely used to stimulate activation and chemotaxis of PMNs (26). However, C5a also can induce MC degranulation (26). In human BP studies, complement components are fixed on the DEJ and are present in the blister fluid (1). Indeed, mice that are genetically deficient in C5 are resistant to experimental BP and show a significant reduction in PMN infiltration into the dermis relative to C5-sufficient controls (14). In the present study, we found that C5 deficiency completely abolished MC degranulation. These data suggest that a key function of complement system is MC activation triggered by pathogenic anti-BP 180 IgG. However, C5a also can induce PMN chemotaxis (26). From our data showing anti-mBP180 IgG-injected MC-deficient mice had 31% of MPO activity, and MC-sufficient mice treated with cromolyn sodium had 38% of MPO activity of diseased mice, we can conclude that MC degradation–dependent mechanism accounts for approximately two thirds of the PMN infiltration into the skin. Thus anti-mBP180 IgG triggers PMN infiltration by both an MC-dependent and an MC-independent pathway. But how do MCs regulate PMN infiltration? MCs can produce a variety of mediators such as leukotrienes, platelet-activating factor, and cytokines that contribute directly or indirectly to PMN recruitment (30, 31). In fact, high levels of histamine, leukotriene B4, IL-1, -2, -5, and -6, and TNF-α are present in BP blister fluids (11, 32–37). The MC-independent pathway probably involves direct activation of PMNs by the complement fragments activated by interaction between anti-mBP180 and its target antigen.
Support for our conclusion that the major role of MCs in experimental BP is as mediators of PMN infiltration comes from our findings that artificially recruiting PMNs to the dermis of MC-deficient mice circumvents the requirement for MCs for disease phenotype. These data are consistent with our previous observations that subepidermal blistering depends on PMNs, which are recruited into the skin site and, upon activation, release neutrophil elastase (38) and gelatinase B to damage DEJ (23). However, it is also possible that MCs contribute to other aspects of the pathogenic process. MCs could contribute to tissue damage directly by cleaving structural proteins in the DEJ or indirectly by activating gelatinase B in experimental BP. MCs contain numerous proteolytic enzymes (30). Activation and degranulation of MCs are associated with matrix degradation (39). The MC-specific serine protease MCP-4 (chymase) can activate gelatinase B, a protease required for BP (40).
It is known that our present studies, however, have not yet uncovered the mechanisms by which anti-BP180 IgG- and C′-dependent MC degranulation is linked to the neutrophils infiltration in experimental BP.
In summary, our present study provides direct evidence that subepidermal blistering triggered by anti-mBP180 antibodies is MC dependent. MCs participate in acute and chronic inflammatory responses (28, 41–44). Our current data extend their function to an autoimmune blistering disease, BP. MC products, such as TNF-α and MC tryptase, have been linked directly to neutrophils influx (45–47). Further investigations will be necessary to identify the MC-released molecule(s), which play a direct role in recruiting neutrophils downstream of anti-BP180 IgG- and complement-dependent MC degranulation, so we can better understand the immunopathogenesis of these complex reactions in experimental BP. Inhibition of MC degranulation could be a new therapeutic strategy for BP. These findings may have significant implications for other autoimmune subepidermal blistering disorders in which an anti-BP180 immune response and MCs have been implicated, such as cicatricial pemphigoid, linear IgA bullous dermatosis, lichen planus pemphigoid, epidermolysis bullosa acquisita, and lupus skin conditions (48–53).
Acknowledgments
This work was supported in part by US Public Health Service grants (R29 AI40768 to Z.Liu; R01 AR-32599 and R37-AR32081 to L.A. Diaz; and P60 AR-20684 and P01 CA-72006 to Z. Werb), grants from the NIH, and a VA Merit Review Grant (to L.A. Diaz).
References
- 1.Stanley, J.R. 1999. Bullous pemphigoid. In Fitzpatrick’s dermatology in general medicine. I.M. Freedberg et al., editors. McGraw-Hill. New York, New York, USA. 666–671.
- 2.Lever WF. Pemphigus. Medicine. 1953;32:1–123. doi: 10.1097/00005792-195302000-00001. [DOI] [PubMed] [Google Scholar]
- 3.Emmerson RW, Wilson-Jones E. Eosinophilic spongiosis in pemphigus. A report of an unusual histological change in pemphigus. Arch Dermatol. 1968;97:252–257. [PubMed] [Google Scholar]
- 4.Nishioka K, et al. Eosinophilic spongiosis in bullous pemphigoid. Arch Dermatol. 1984;120:1166–1168. [PubMed] [Google Scholar]
- 5.Natio K, Morioka S, Ikeda S, Ogawa H. Experimental bullous pemphigoid in guinea pigs: the role of pemphigoid antibodies, complement, and migrating cells. J Invest Dermatol. 1984;82:227–230. doi: 10.1111/1523-1747.ep12260057. [DOI] [PubMed] [Google Scholar]
- 6.Nestor MS, Cochran AJ, Ahmed AR. Mononuclear cell infiltrates in bullous disease. J Invest Dermatol. 1987;88:172–175. doi: 10.1111/1523-1747.ep12525315. [DOI] [PubMed] [Google Scholar]
- 7.Wintroub BU, Mihm MC, Jr, Goetzl EJ, Soter NA, Austen KF. Morphologic and functional evidence for release of mast-cell products in bullous pemphigoid. N Engl J Med. 1978;298:417–421. doi: 10.1056/NEJM197802232980803. [DOI] [PubMed] [Google Scholar]
- 8.Iwatsuki K, Tagami H, Yamada M. Induction of leukocyte adherence at the basement membrane zone with subsequent activation of their metabolic pathway by pemphigoid antibodies and complement. Acta Derm Venereol. 1983;63:495–500. [PubMed] [Google Scholar]
- 9.Dvorak AM, et al. Bullous pemphigoid, an ultrastructural study of the inflammatory response: eosinophil, basophil and mast granule changes in multiple biopsies from one patient. J Invest Dermatol. 1982;78:91–101. doi: 10.1111/1523-1747.ep12505711. [DOI] [PubMed] [Google Scholar]
- 10.Baba T, et al. An eosinophil chemotactic factor present in blister fluids of bullous pemphigoid patients. J Immunol. 1976;116:112–116. [PubMed] [Google Scholar]
- 11.Katayama I, Doi T, Nishioka K. High histamine level in the blister fluid of bullous pemphigoid. Arch Dermatol Res. 1984;276:126–127. doi: 10.1007/BF00511070. [DOI] [PubMed] [Google Scholar]
- 12.Shornick JK, Bangert JL, Freeman RG, Gilliam JN. Herpes gestationis: clinical and histological features of twenty-eight cases. J Am Acad Dermatol. 1983;8:214–224. doi: 10.1016/s0190-9622(83)70027-7. [DOI] [PubMed] [Google Scholar]
- 13.Liu Z, et al. A passive transfer model of the organ-specific autoimmune disease, bullous pemphigoid, using antibodies generated against the hemidesmosomal antigen, BP180. J Clin Invest. 1993;92:2480–2488. doi: 10.1172/JCI116856. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Liu Z, et al. The role of complement in experimental bullous pemphigoid. J Clin Invest. 1995;95:1539–1544. doi: 10.1172/JCI117826. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Liu Z, et al. A major role for neutrophils in experimental bullous pemphigoid. J Clin Invest. 1997;100:1256–1263. doi: 10.1172/JCI119639. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Galli SJ, Kitamura Y. Animal model of human disease. Genetically mast cell-deficient W/Wv and Sl/Sld mice: their value for the analysis of the roles of mast cells in biological responses in vivo. Am J Pathol. 1987;127:191–198. [PMC free article] [PubMed] [Google Scholar]
- 17.Bradley PP, Priebat DA, Christensen RD, Rothstein G. Measurement of cutaneous inflammation: estimation of neutrophil content with an enzyme marker. J Invest Dermatol. 1982;78:206–209. doi: 10.1111/1523-1747.ep12506462. [DOI] [PubMed] [Google Scholar]
- 18.Mulligan MS, et al. Inhibition of lung inflammatory reactions in rats by an anti-human IL-8 antibody. J Immunol. 1993;150:5585–5595. [PubMed] [Google Scholar]
- 19.Wershil BK, Wang Z, Jordon JR, Galli SJ. Recruitment of neutrophils during IgE-dependent cutaneous late phase responses in the mouse is mast cell dependent: partial inhibition of the reaction with antiserum against tumor necrosis factor-alpha. J Clin Invest. 1991;87:446–453. doi: 10.1172/JCI115016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Sheard P, Blair AM. Disodium cromoglycate activity in three in vitro models of the immediate hypersensitivity reaction in lung. Int Arch Allergy Appl Immunol. 1970;38:217–224. [PubMed] [Google Scholar]
- 21.Theoharides TC, Sieghart W, Greengard P, Douglas WW. Antiallergic drug cromolyn may inhibit histamine secretion by regulating phosphorylation of a mast cell protein. Science. 1980;207:80–82. doi: 10.1126/science.6153130. [DOI] [PubMed] [Google Scholar]
- 22.Metcalf, J.A., Gallin, J.I., Nauseef, W.M., and Root, R.K. 1985. Laboratory manual of neutrophil function. Raven Press. New York, New York, USA. 2–10.
- 23.Liu Z, et al. Gelatinase B-deficient mice are resistant to experimental bullous pemphigoid. J Exp Med. 1998;188:475–482. doi: 10.1084/jem.188.3.475. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Wershil BK, Mekori YA, Murakami T, Galli SJ. 125I-Fibrin deposition in IgE-dependent immediate hypersensitivity reactions in mouse skin: demonstration of the role of mast cells using genetically mast cell-deficient mice locally reconstituted with cultured mast cells. J Immunol. 1987;139:2605–2614. [PubMed] [Google Scholar]
- 25.Ryan JJ, et al. IL-4 inhibits mouse mast cell FcεRI expression through a STAT6-dependent mechanism. J Immunol. 1998;161:6915–6923. [PubMed] [Google Scholar]
- 26.Muller-Eberhard HJ. Molecular organization of the complement system. Annu Rev Biochem. 1988;57:321–347. doi: 10.1146/annurev.bi.57.070188.001541. [DOI] [PubMed] [Google Scholar]
- 27.Maynard B, Peters MS, Butterfield JH, Leiferman KM. Bullous pemphigoid: eosinophil, neutrophil and mast cell degranulation in lesional tissue. J Invest Dermatol. 1990;94:553A. [Google Scholar]
- 28.Borrego L, et al. Deposition of eosinophil granule proteins precedes blister formation in bullous pemphigoid. Comparison with neutrophil and mast cell granule proteins. Am J Pathol. 1996;148:897–909. [PMC free article] [PubMed] [Google Scholar]
- 29.Wershil, B.K., and Galli, S.J. 1994. The analysis of mast cell function in vivo using mast cell deficient-mice. In Immunobiology of proteins and peptides VII. M.Z. Atassi, editor. Plenum Press. New York, New York, USA. 39–54. [DOI] [PubMed]
- 30.Galli SJ, Gordon JR, Wershil BK. Cytokine production by mast cells and basophils. Curr Opin Immunol. 1991;3:865–873. doi: 10.1016/s0952-7915(05)80005-6. [DOI] [PubMed] [Google Scholar]
- 31.Galli SJ. New concepts about the mast cell. N Engl J Med. 1993;328:257–265. doi: 10.1056/NEJM199301283280408. [DOI] [PubMed] [Google Scholar]
- 32.Kawana S, Ueno A, Nishiyama S. Increased levels of immunoreactive leukotriene B4 in blister fluids of bullous pemphigoid patients and effects of a selective 5-lipoxygenase inhibitor on experimental skin lesions. Acta Derm Venereol. 1990;70:281–285. [PubMed] [Google Scholar]
- 33.Grando SA, et al. Mediators of inflammation in blister fluids from patients with pemphigus vulgaris and bullous pemphigoid. Arch Dermatol. 1989;125:925–930. [PubMed] [Google Scholar]
- 34.Takiguchi Y, et al. Cell-mediated immune reaction in the mechanism of blister formation in bullous pemphigoid. Dermatologica. 1989;179(Suppl. 1):137. doi: 10.1159/000248478. [DOI] [PubMed] [Google Scholar]
- 35.Endo H, Iwamoto I, Fujita M, Okamoto S, Yoshida S. Increased immunoreactive interleukin-5 levels in blister fluids of bullous pemphigoid. Arch Dermatol Res. 1992;284:312–314. doi: 10.1007/BF00372588. [DOI] [PubMed] [Google Scholar]
- 36.Tamaki K, So K, Furuya T, Furue M. Cytokine profile of patients with bullous pemphigoid. Br J Dermatol. 1994;130:128–129. doi: 10.1111/j.1365-2133.1994.tb06902.x. [DOI] [PubMed] [Google Scholar]
- 37.Zillikens D, et al. The interleukin-2 receptor in lesions and serum of bullous pemphigoid. Arch Dermatol Res. 1992;284:141–145. doi: 10.1007/BF00372706. [DOI] [PubMed] [Google Scholar]
- 38.Liu Z, et al. Neutrophil elastase plays a critical role in experimental bullous pemphigoid. J Clin Invest. 2000;105:113–123. doi: 10.1172/JCI3693. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Dabbous MK, et al. Mast cells and matrix degradation at sites of tumor invasion in rat mammary adenocarcinomas. Br J Cancer. 1986;54:459–465. doi: 10.1038/bjc.1986.198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Coussens LM, et al. Inflammatory mast cells up-regulate angiogenesis during squamous epithelial carcinogenesis. Genes Dev. 1999;13:1382–1397. doi: 10.1101/gad.13.11.1382. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Zhang Y, Ramos BF, Jakschik BA. Neutrophil recruitment by tumor necrosis factor from mast cells in immune complex peritonitis. Science. 1992;258:1957–1959. doi: 10.1126/science.1470922. [DOI] [PubMed] [Google Scholar]
- 42.Echtenacher B, Männel D, Hültner L. Critical protective role of mast cells in a model of acute septic peritonitis. Nature. 1996;381:75–77. doi: 10.1038/381075a0. [DOI] [PubMed] [Google Scholar]
- 43.Malaviya R, Ikeda T, Ross E, Abraham SN. Mast cell modulation of neutrophils influx and bacterial clearance at sites of infection through TNF-α. Nature. 1996;381:77–80. doi: 10.1038/381077a0. [DOI] [PubMed] [Google Scholar]
- 44.Prodeus A, Zhou X, Maurer M, Galli SJ, Carroll MC. Impaired mast cell-dependent natural immunity in complement C3-deficient mice. Nature. 1997;390:172–175. doi: 10.1038/36586. [DOI] [PubMed] [Google Scholar]
- 45.He S, Peng Q, Walls AF. Potent induction of a neutrophil and eosinophil-rich infiltrate in vivo by human mast cell tryptase: selective enhancement of eosinophil recruitment by histamine. J Immunol. 1997;159:6216–6225. [PubMed] [Google Scholar]
- 46.Huang C, et al. Induction of a selective and persistent extravasation of neutrophils into the peritoneal cavity by tryptase mouse mast cell protease 6. J Immunol. 1998;160:1910–1919. [PubMed] [Google Scholar]
- 47.Compton SJ, Cairns JA, Holgate SH, Walls AF. The role of mast cell tryptase in regulating endothelial cell proliferation, cytokine release, and adhesion molecule expression: tryptase induces expression of mRNA for IL-1β and IL-8 and stimulates the selective release of IL-8 from human umbilical vein endothelial cells. J Immunol. 1998;161:1939–1946. [PubMed] [Google Scholar]
- 48.Rothfield N, Rose HA, Minta JO, Lepow IH. Glomerular and dermal deposition of properdin in systemic lupus erythematosus. N Engl J Med. 1972;287:681–685. doi: 10.1056/NEJM197210052871402. [DOI] [PubMed] [Google Scholar]
- 49.Woodley DT, et al. Identification of the skin basement-membrane autoantigen in epidermolysis bullosa acquisita. N Engl J Med. 1984;310:1007–1013. doi: 10.1056/NEJM198404193101602. [DOI] [PubMed] [Google Scholar]
- 50.Bernard P, et al. The major cicatricial pemphigoid antigen is a 180-kD protein that shows immunologic cross-reactivities with the bullous pemphigoid antigen. J Invest Dermatol. 1992;99:174–179. doi: 10.1111/1523-1747.ep12616797. [DOI] [PubMed] [Google Scholar]
- 51.Chan LS, et al. Immune-mediated subepithelial blistering diseases of mucous membranes. Arch Dermatol. 1993;129:448–455. doi: 10.1001/archderm.129.4.448. [DOI] [PubMed] [Google Scholar]
- 52.Tamada Y, et al. Lichen planus pemphigoides: Identification of 180 kD hemidesmosome antigen. J Am Acad Dermatol. 1995;32:883–887. doi: 10.1016/0190-9622(95)91554-0. [DOI] [PubMed] [Google Scholar]
- 53.Zone JJ, Taylor TB, Meyer LJ, Peterson MJ. The 97 kDa linear IgA bullous disease antigen is identical to a portion of the extracellular domain of the 180 kDa bullous pemphigoid antigen, BPAg2. J Invest Dermatol. 1998;110:207–210. doi: 10.1046/j.1523-1747.1998.00129.x. [DOI] [PubMed] [Google Scholar]