


Abstract
Precise, controllable single-molecule force spectroscopy studies of RNA and RNA-dependent processes have recently shed new light on the dynamics and pathways of RNA folding and RNA-enzyme interactions. A crucial component of this research is the design and assembly of an appropriate RNA construct. Such a construct is typically subject to several criteria. First, single-molecule force spectroscopy techniques often require an RNA construct that is longer than the RNA molecules used for bulk biochemical studies. Next, the incorporation of modified nucleotides into the RNA construct is required for its surface immobilization. In addition, RNA constructs for single-molecule studies are commonly assembled from different single-stranded RNA molecules, demanding good control of hybridization or ligation. Finally, precautions to prevent RNase- and divalent cation-dependent RNA digestion must be taken. The rather limited selection of molecular biology tools adapted to the manipulation of RNA molecules, as well as the sensitivity of RNA to degradation, make RNA construct preparation a challenging task. We briefly illustrate the types of single-molecule force spectroscopy experiments that can be performed on RNA, and then present an overview of the toolkit of molecular biology techniques at one's disposal for the assembly of such RNA constructs. Within this context, we evaluate the molecular biology protocols in terms of their effectiveness in producing long and stable RNA constructs.
INTRODUCTION
RNA molecules can be divided into several classes that are responsible for a diverse set of metabolic and regulatory functions in vivo. As early as the 1940s, it was shown that RNA molecules are the carriers of the genetic information in tobacco mosaic virus (1). Since then, many other viruses with RNA genomes have been reported in the literature (2–4). In 1956, ‘DNA-like RNA’ was discovered (5), which serves as an intermediate between DNA and proteins and was later renamed messenger RNA. Further identification and characterization showed that RNA molecules can perform a diverse set of biological reactions such as peptidyl transferase reaction, degradation and ligation of nucleic acids and recognition of metabolites (6–17). Furthermore, a diverse class of short RNA molecules involved in the regulation of gene expression has recently emerged (18–22).
A host of bulk biophysical techniques have been successfully applied to elucidate the thermodynamics and kinetics of these RNA-dependent processes. More recently, the biological importance of RNA has stimulated a wealth of single-molecule research. The single-molecule approach offers several advantages over ensemble studies. For example, single-molecule techniques enable sensitive micromanipulation of individual biomolecules (e.g. extension by force, supercoiling) (23–25), which can be used to lay bare the states a molecule can adopt, or to precisely control its initial state. Also, by observing a single molecule or a single event, it is possible to measure individual fluctuations in thermodynamic or kinetic parameters, thus eliminating the averaging inherent in ensemble studies (26). Furthermore, the amounts of sample required for single-molecule studies are usually far smaller than for ensemble studies (27,28). Finally, single-molecule detection can be used to investigate biological systems in vitro as well as in the living cell (29–31). In this manner, single-molecule studies of RNA have provided new insights into the polymer physics of RNA (32,33), RNA folding (34–43) and enzyme–RNA interactions (44–49). For example, single-molecule force spectroscopy has been performed to measure the persistence lengths of both homopolymeric single-stranded RNA (ssRNA) (33) and random sequence of double-stranded RNA (dsRNA) (Figure 1A) (32). Elegant experiments based on optical tweezers have been carried out by Liphardt and coworkers (38) to measure the folding thermodynamics and kinetics of RNA hairpins (Figure 1B and C). Similar experimental schemes have been used to investigate step-wise unwinding of an RNA hairpin by an RNA helicase (Figure 1D) (45).
Figure 1.

Examples of single-molecule studies of RNA and RNA-dependent processes. (A) Measurement of the persistence length of double-stranded RNA (dsRNA) with magnetic tweezers (32). A force-extension curve for dsRNA is obtained on an 8.3 kb dsRNA. The experimental points were fitted to the worm-like chain model (smooth line). The fit yielded a persistence length of dsRNA of 63.0 ± 2.5 nm. (Reprinted with the permission of the Biophysical Journal and the original authors.) The inset: The experimental configuration used to measure the persistence length of dsRNA. The dsRNA is suspended between the magnetic bead and glass surface. At a given applied magnetic force, the distance between the glass surface and the magnetic bead is measured, yielding the force-extension curve. (B) An experimental configuration for single-molecule study of RNA hairpin unfolding by force or through the activity of an RNA helicase (38,45). The RNA hairpin is suspended between two beads. One bead is optically trapped while the other bead is held by a micropipette that can be moved with a nanometer precision to apply a force. (C) An example of a force-extension curve obtained during the RNA hairpin unfolding by externally applied force (38). The force on the RNA construct is increased by moving the micropipette in the direction indicated. The RNA extension is obtained from measurement of distance between the centers of the two beads, corrected for the bead diameter. The RNA hairpin unfolding occurs at the point indicated by the red arrow. (Reprinted with the permission of Science Magazine and the original authors.) (D) An increase in RNA extension at constant force in the presence of RNA helicases (45). The RNA construct extension is the result of RNA helicase binding to the bottom of the RNA hairpin (position 1) and subsequent unwinding of the RNA hairpin to produce a completely denatured RNA molecule (position 2). Between the binding of RNA helicase and complete denaturation of the RNA hairpin, several plateaus in the construct extension can be observed, suggesting pauses in helicase activity. (Reprinted with the permission of Nature Publishing Group and the original authors.)
In order to study RNA and RNA-dependent processes with single-molecule force spectroscopic techniques, specialized RNA constructs have to be designed that differ from bulk RNA substrates in several respects: (i) single-molecule force-spectroscopy studies can require long RNAs of lengths greater than ∼1 kb (32,33,38,39,50); (ii) the incorporation of modified nucleotides is often necessary at one or both ends of the RNA construct to immobilize the RNA to a surface or to suspend it between two surfaces, respectively; and (iii) the RNA construct is often composed of different sections that need to be assembled by hybridization or ligation. These specific requirements can present several difficulties for the assembly of an RNA construct. For example, the maximum length of RNA that can be chemically synthesized is often limited to ∼80 nt. The low yield of longer RNA oligomers renders non-enzymatic RNA synthesis unfeasible for such applications. Enzymatic synthesis of RNA by viral polymerases provides an alternative, but the presence of termination signals and the limited processivity of RNA polymerase nonetheless substantially reduce the yield of RNAs longer than 10 kb. In addition, the yield of many enzymatic reactions necessary for the assembly of RNA constructs (e.g. RNA ligation) decreases considerably as the RNA length increases beyond ∼1 kb (51). A controlled joining of different sections of RNA construct presents additional challenges due to lack of RNA restriction enzymes (50). Finally, due to the chemical nature of RNA and the prevalence of RNA-degrading enzymes special precautions need to be taken when working with RNA. High pH (>9), RNase contamination and/or the presence of divalent cations can decrease the yield of active RNA constructs and thus decrease the probability to observe the biophysical processes of interest. While RNases can be inhibited through the use of RNase inhibitors, the presence of divalent cations often cannot be avoided, since their presence is necessary for a variety of RNA-dependent processes (e.g. RNA folding, RNA polymerization). The limited selection of molecular biology tools for RNA construct synthesis and RNA sensitivity to degradation by RNases and divalent cations make the design of suitable RNA constructs a challenging task.
Here, we provide an overview of the toolkit available for RNA construct assembly, in particular for single-molecule force spectroscopy techniques that require RNA molecules longer than those usually handled in bulk studies or in other single-molecule techniques such as FRET [the assembly protocols for other single-molecule techniques have been reviewed elsewhere (52,53)]. We focus on RNA construct design for magnetic and optical tweezers experiments, and illustrate techniques by examples from both the literature and our own results. This article is divided into several chapters. The first section gives an introduction to the basic design of the RNA construct for single-molecule studies, and defines the basic steps of the RNA construct assembly. Examples of RNA constructs from the literature are given to illustrate the type of experiments that can be performed. This is followed by sections that discuss RNA synthesis, labeling and joining of different sections of a ssRNA construct, which we have evaluated in our laboratory. A separate section is devoted to the assembly of dsRNA constructs, which includes a demonstration of the synthesis of dsRNA molecules longer than 15 kb and an evaluation of the ligation requirements for dsRNA molecules. To complete the overview of protocols, we discuss the methods for RNA construct isolation and purification. We conclude by describing the persisting challenges in the field of RNA construct assembly.
Throughout this article, we refer to the RNA under consideration either as an ‘RNA molecule’, or an ‘RNA construct’. This is to distinguish building blocks (i.e. ‘the RNA molecules’) from the final product used in the experimental setup (i.e. ‘the RNA construct’). Also, as mentioned above, RNA length is an important factor in most of the steps of RNA construct assembly, as it usually affects the yield of the enzymatic reactions. If an exact length of a specific RNA is not given, it is described with qualitative terms ‘long’ or ‘short’. This is to indicate when the RNA length starts to affect the reaction yield of a particular step in the RNA construct assembly. The approximate length separating the ‘long’ and ‘short’ RNA is discussed for each step of the assembly process.
MATERIALS AND METHODS
3′-labeling of DNA and RNA oligomers with Terminal deoxynucleotidyl transferase
The dry pellets of DNA oligomer with a sequence 5′-GTACCGGCTGTCTGGTATGTATGAG-3′, and RNA oligomer with a sequence 5′-UCCGAUGGCUGGCUGCG-3′ (Biolegio, The Netherlands) were re-suspended in 1× TE to a final concentration of 500 mM. For every 3′-end labeling reaction, 10 μl reaction mixtures were prepared that contained 100 pmol of either DNA or RNA oligomer, 10 U of Tdt (Fermentas, Canada), 1× Tdt reaction buffer (Fermentas, Canada) (200 mM potassiumcacodylate, 25 mM Tris, 1 mM CoCl2 and 0.01% (v/v) TritonX-100, pH 7.2), and varying concentrations of dUTP (Roche, The Netherlands) or digoxigenin-labeled dUTP (Roche, The Netherlands). The reaction mixtures were incubated at 37°C for 1 h, and subsequently denatured by the addition of an equal volume of 9 M urea. The denatured samples were loaded on a urea-denaturing PAGE (20% polyacrylamide, 8.3 M urea, 1× TBE). The 3′-labeling efficiency was evaluated by staining the gel with SYBR® Gold (Invitrogen Molecular Probes, USA) and imaging the bands under the UV light.
Synthesis and hybridization of complementary ssRNA molecules
Two 4.1 kb and a 0.5 kb PCR fragments were generated using pBADb10 (a recombinant pBAD vector) encompassing the sequences from base pair 230 to base pair 4373, and base pair 230 to base pair 681, respectively. The PCR primers (Biolegio, The Netherlands) were designed to incorporate a T7 RNA polymerase promoter sequence at one end of each PCR fragment (Figure S1 and Table S1). In the 4.1 kb PCR fragments, the T7 RNA polymerase promoter was inserted at position 230 or 4373 for the synthesis of the sense or antisense 4.1 kb ssRNA (4.1 kb ssRNA), respectively (Figure S1). The T7 RNA promoter sequence was inserted at the position 681 in the 0.5 kb PCR fragment to yield a 0.5 kb ssRNA complementary to the 3′-end of the sense 4.1 kb ssRNA (Figure S1). (The positions of the promoter sequence insertion refer to the base pair in the original pBADb10 vector.) Each 50 μl PCR reaction contained 0.01 U Phusion™ High-Fidelity DNA Polymerase (Finnzymes, Finland), 1× Phusion™ HF (Finnzymes, Finland), 0.2 μM of each primer, 200 μM of each dNTP (Promega, USA), 3% DMSO and 5 ng of the pBADb10 vector. The PCR temperature cycling was programmed as suggested by the manufacturer with an annealing temperature of 55°C. PCR fragments were purified with a NucleoSpin® Extract II clean-up kit (Macherey-Nagel, Germany). The lengths of the PCR fragments were confirmed on a 0.75% agarose gel.
In vitro run-off transcription of the three ssRNA molecules was performed with T7 RiboMAX™ Large Scale RNA Production System (Promega, USA) according to the manufacturer's protocol. The in vitro run-off transcription reactions were purified with RNeasy MiniElute Cleanup kit (Qiagen, USA). The aliquots of purified ssRNA were denatured with RNA Sample Loading Buffer (Sigma–Aldrich, USA) and loaded on a 0.75% agarose gel to confirm the length of the in vitro run-off transcripts. The concentrations of ssRNA transcripts were determined spectroscopically by measuring the absorbance at 260 nm using an extinction coefficient of 25 l cm−1g−1.
To hybridize the complementary 4 kb ssRNA molecules, the sense and the antisense 4.1 kb ssRNA transcripts were mixed in a 1:1 molar ratio at a total ssRNA concentration of 2 μg per hybridization reaction. The same quantity of the sense 4.1 kb ssRNA was hybridized with 0.5 kb ssRNA at 1:1 and 1:4 molar ratios. The final ssRNA concentrations depended on the hybridization volume, which is noted below for each of the hybridization conditions. Hybridization reactions under aqueous conditions were carried out in 0.5×SSC (Promega, USA) supplemented with 1× TE (Sigma–Aldrich, USA) (SSCTE buffer) with a final volume of 150 μl. Hybridization of the sense 4.1 kb ssRNA and 0.5 kb ssRNA was also performed in 150 μl 1× TE. Hybridization reactions were also performed in a formamide-based buffer used by Liphardt and coworkers (38). Briefly, 1 μg of sense 4.1kb ssRNA and 1 μg of antisense 4.1 kb ssRNA were re-suspended in 17.5 μl 1× TE and mixed with 80 μl of the formamide buffer [prepared by mixing 100% formamide, 1 M EDTA (pH = 8.0), 1 M Pipes (pH = 6.3), and 5 M NaCl in a 400:1:20:40 volume ratio, respectively] (38). Two different temperature programs were tested for hybridization. First, a ‘gradual-cool’ temperature program was adopted from Dekker and coworkers (50) (denaturation for 1 h at 65°C followed by a hybridization step with 1.5°C/5 min cooling rate with the final temperature of 25°C). Second, a ‘step-cool’ temperature program was adopted from Liphardt and coworkers (38) (denaturation at 85°C for 10 min, followed by 1 h incubation at 62°C, 1.5 h incubation at 52°C and finished with a cooling to 10°C within 10 min.) Hybridization reactions were purified with the RNeasy MiniElute Cleanup kit (Qiagen, USA), and analyzed on a 0.75% agarose gel.
Synthesis of dsRNA constructs
Long random sequences of ssRNA were synthesized using polynucleotide phosphorylase (Pnpase) from Bacillus stearothermophilus (Sigma–Aldrich, USA). Four microgram of Pnpase was incubated in 100 mM Tris–HCl buffer (pH = 9), supplemented with 5 mM MgCl2, 0.4 mM EDTA and 0.04% w/v BSA with nucleotide diphosphates. The total nucleotide diphosphates (NDP) concentration was 20 mM. CDP, UDP, ADP and GDP at 9, 9, 1 and 1 mM, concentrations, respectively (Dr R. Tuma, personal communication). The total reaction volume of 200 μl was incubated at 37°C for 15 min. The reaction mixture was purified with RNeasy MiniElute Cleanup kit (Qiagen, USA), and the ssRNA eluted with 20 μl 1× TE. To obtain dsRNA, ssRNA was subsequently incubated with 5 μg of Ф6 P2 RNA-dependent RNA polymerase (Finnzymes, Finland) in the presence of 50 mM Tris–HCl (pH = 8.9), 80 mM NH4OAc, 2 mM DTT, 0.1 mM EDTA, 5 mM MnCl2 and 2.5 mM of each NTP. The final reaction volume was 200 μl. The reaction was incubated at room temperature for 1 h and subsequently purified with RNeasy MiniElute Cleanup kit (Qiagen, USA) and loaded on a 0.75% agarose gel. 15–20 kb dsRNA was then gel-extracted using a TAKARA Recochip (Takara Bio, Japan). AFM images of the gel-extracted dsRNA on a Ni-coated mica surface were taken as previously described (46).
Intramolecular ligation of 5′-overhangs in dsRNA molecules
The sense and antisense 4.1 kb ssRNA were synthesized as described above. In addition, an antisense 4.1 kb ssRNA with an eight-base extension at its 5′-end (5′-GGGCUACC-3′) was transcribed from a PCR fragment as described previously by Dekker and coworkers (50). Briefly, a forward primer used for the PCR fragment of the antisense 4.1 kb ssRNA was modified to include a 5′-GCTACC-3′ sequence between the T7 promoter sequence and the sequence complementary to the pBADb10 vector (Table S1). The reverse primer was the same as the one used to obtain the PCR fragment for the antisense 4.1 kb ssRNA. The 5′-extended antisense 4.1 kb ssRNA was alternatively transcribed in vitro in the absence or in the presence of GMP as described by Doudna and coworkers (54) to obtain 5′-triphosphate and 5′-monophosphate transcripts of 4.1 kb ssRNA, respectively. The sense 4.1 kb ssRNA was hybridized to the antisense 4.1 kb ssRNA, and to 5′-extended antisense 4.1 kb ssRNA (alternatively synthesized with or without GMP) in three separate hybridization reactions. The SSCTE buffer and the ‘gradual-cool’ temperature program were used. The hybridization reactions were purified with RNeasy MiniElute Cleanup kit (Qiagen, USA), and analyzed on a 0.75% agarose gel. The intramolecular ligation of the hybrids was carried out in a 50 μl volume containing 2 μg of hybridized dsRNA, 4 U of RNA Ligase 2 (RNL2) (New England BioLabs, MA, USA), 1× RNL2 New England BioLabs buffer and 20 U Superase•In™ (Ambion, TX, USA). The ligation reactions were incubated at 37°C for 1 h and purified with RNeasy MiniElute Cleanup kit (Qiagen, USA). An aliquot of each reaction was denatured with RNA Sample Loading Buffer (Sigma–Aldrich, USA) and loaded on 0.75% agarose gel.
RNA CONSTRUCT ASSEMBLY FOR SINGLE-MOLECULE FORCE SPECTROSCOPY STUDIES
As illustrated by the examples in Figure 1 single-molecule force spectroscopy can be used to sensitively monitor and manipulate RNA molecules and RNA-dependent processes. We now describe the general principles underlying the assembly of suitable RNA constructs. For force spectroscopy, molecules are typically tethered on both ends to allow the application of force. Therefore, an RNA construct can be divided into a central section and two terminal sections (Figure 2A). The central section contains the RNA sequence that is to be investigated experimentally in terms of its mechanical properties or its interactions with other biomolecules such as proteins or small ligands. The two terminal sections, commonly referred to as ‘handles’, are necessary to suspend the RNA construct between the two attachment points [i.e. a glass surface and a magnetic bead in the magnetic tweezers, a glass surface and a small gold particle for the analysis of tethered particle motion (TPM), or two beads in the optical tweezers] (Figure 2B). The attachment of the RNA construct to the glass surface or beads is facilitated by formation of bonds between molecules immobilized on the surface and their ligands incorporated in the terminal sections. Each terminal section has to incorporate a unique ligand to allow for controlled anchoring of the RNA construct to the two surfaces (Figure 2B). The terminal section can be labeled with a single or multiple surface-binding molecules (Figure 2C and D) (38,50). Multiple surface-binding molecules are often needed to increase the stability of anchoring of the RNA construct at high forces. In special cases, single-molecule applications do not require labeled terminal sections on both ends of the central section. For example, force spectroscopy measurements in which forces are applied via the intermediaries of fluid flow or electric fields, as in optical tweezers measurements combined with flow or with nanopores, can be performed with a labeled terminal section attached to only one end of the central section (55,56). A second example is the case in which the second extremity of the nucleic acid is tethered via the intermediary of surface-immobilized molecular motors (e.g. polymerases, helicases, transferases), in which case a labeled terminal section at only one end of the construct suffices (44,57).
Figure 2.

The basic design of the RNA construct for single-molecule experiments. (A) The RNA construct is divided into three sections. The central section denoted in black contains the RNA sequence or structure to be studied. The two labeled terminal sections of the RNA construct contain one or multiple nucleotides modified with ligands that can bind to coated surfaces. This enables a controlled mode of RNA construct binding between two surfaces. The scheme shows a single nucleotide modification per terminal section. Modification ligands are shown as red forks and green crescents at the ends of the respective terminal sections. (B) The RNA construct is suspended between surfaces in the single-molecule experiment. The nucleotide modifications in the terminal sections serve as ligands that bind to surface-immobilized molecules. The surfaces are shown as gray spheres. The surface-immobilized molecules are shown either as red squares or green circles, which specifically bind to the red forks and green crescents, respectively. (C) A schematic of the RNA construct by Liphardt and co-workers (38) that was successfully used to study the kinetics of RNA folding. The central section contains an RNA hairpin. The terminal sections consist of a DNA:RNA hybrid with single modified nucleotides at the terminal ends of DNA strands. (D) Schematic of an RNA construct used to measure the persistence length of a double-stranded RNA (dsRNA) (32). The central section contains dsRNA. The terminal sections have been ligated with RNA Ligase 2 to the central section, and contain multiple modified nucleotides.
Different protocols have been reported in the literature to assemble RNA constructs (32,33,38,39,45). These protocols can be divided into several steps that are the focus of this review: (i) Synthesis of the central section of the RNA construct; (ii) synthesis of labeled terminal sections of the RNA construct; (iii) joining of the central and terminal sections of the RNA construct and (iv) isolation and purification of the final RNA construct. While the above four steps describing the assembly of RNA constructs may seem straightforward, many single-molecule studies impose a complication, namely the RNA construct length. Here, we present each step in RNA construct assembly and point out limitations arising from this particular constraint. For simplicity, we first discuss the complete assembly of RNA constructs for studies of ssRNA, and subsequently generalize this to include the assembly of dsRNA constructs for single-molecule experiments.
THE CENTRAL SECTION SYNTHESIS FOR ssRNA CONSTRUCT
The central section of the ssRNA construct may be several kilobases (kb) in length, in part, because the central section is usually extended at both ends to provide for subsequent attachment of the labeled terminal sections (Figure 2C). Two main experimental approaches have been adopted for the synthesis of such long central sections; in vitro run-off transcription with RNA polymerase (32,38,39), and polymerization of NDP using Pnpase (33). In vitro run-off transcription has been used in the synthesis of the central sections shorter than 10 kb, and in the synthesis of central sections that contain specific sequences (e.g. naturally occurring RNA sequences, RNA structural motifs) (38,39,45). In contrast, Pnpase has been used to synthesize central sections longer than 10 kb, but could be adopted only for random or homopolymeric RNA sequences (33) (Dr R. Tuma, personal communication). Alternatively, the central section that contains only the naturally occurring RNA sequence and does not require any sequence extensions for subsequent attachment of the labeled terminal sections can be isolated directly from its biological source (34).
DNA templates for in vitro run-off transcription of the central section of the ssRNA construct
In vitro run-off transcription is most frequently used to synthesize the central section of the ssRNA construct. In this case, the first step is the design of a DNA fragment that serves as a template for in vitro run-off transcription. The optimal strategy for obtaining this DNA template depends on the type of RNA sequence desired. Cloning of a DNA sequence into a plasmid is commonly used to study short, naturally occurring RNA sequences with a specific biological function (e.g. ribozyme, t-RNA) or to produce a central section of a ssRNA construct that folds into a specific RNA structural motif (e.g. a hairpin) (38,39,45). In contrast, a transcription termination signal-free section of a plasmid sequence is usually sufficient to generate kilobase-long central sections that can be used to study polymer properties of RNA (e.g. persistence length) (32). These two basic approaches require different protocols that we now discuss.
Most of the single-molecule studies of naturally occurring RNA sequences and structural motifs take advantage of the protocol developed by Liphardt and coworkers (38) to obtain DNA templates for in vitro run-off transcription. First, two synthetic DNA oligomers are hybridized to form a primary DNA fragment containing the sequence of interest in the center and two different restriction overhangs at the respective ends. The primary DNA fragment is then cloned into a plasmid that has been doubly digested to yield overhangs complementary to the overhangs of the primary DNA fragment. Finally, an RNA-polymerase promoter sequence for in vitro run-off transcription (Table 1) is inserted upstream of the cloned sequence by PCR to yield a secondary DNA fragment. The choice of primers in this PCR step determines the extent to which the final RNA sequence extends up- and downstream of the cloned primary DNA fragment. These extensions at the ends of the central section enable subsequent joining of the labeled terminal sections of the ssRNA construct (please, see below for the discussion on labeling of ssRNA constructs) (38,39). This approach has been limited to relatively short, naturally occurring sequences and RNA structural motifs (< 120 bp) due to the length limitations of the chemical synthesis of DNA oligomers. To circumvent this problem, Mangeol and coworkers (39) have chosen to clone cDNA sequences obtained from the RNA to be studied. Once the cDNA sequence was cloned into a plasmid, the secondary DNA fragment was obtained as in the protocol described above. This approach was successfully applied to study the unfolding and refolding of 173 and 735 nucleotide long fragments of 23S Escherichia coli ribosomal RNA (39).
Table 1.
Promoter sequences of commonly used viral RNA polymerases
| Type of RNA polymerase | Promoter sequence |
|---|---|
| SP6 RNA polymerase | 5′-ATTTAGGTGACACTATA AAGNG-3′ AAGNG-3′ |
| T3 RNA polymerase | 5′-AATTAACCCTCACTAAAGGAGA-3′ |
| T7 RNA polymerase | 5′-TAATACGACTCACTATAG-3′ |
The bolded base indicates the first nucleotide to be transcribed by a given viral RNA polymerase. The bases downstream from the TATA box are often not reported as part of the promoter sequence. However, these bases often influence the promoter strength. The reported bases are known to enhance the promoter strength.
A simplified DNA template synthesis scheme can be utilized for single-molecule studies that do not require a specific naturally occurring RNA sequence or structural motif. In order to produce these central sections by in vitro run-off transcription, it is common to amplify a section of a plasmid with PCR using a pair of primers, one of which contains an RNA-polymerase promoter sequence at its 5′-end (32,39,50). The choice of plasmid for this type of DNA template synthesis is crucial if the length of RNA required for single-molecule studies exceeds kilobases (32,33,39), since the probability of encountering transcription termination signals that cause abortive RNA transcripts starts to increase significantly. Prediction of the number of transcription termination signals is challenging, since the signals themselves can be quite diverse and indeed vary for different RNA polymerases. For example, it has been shown that certain sequences and structural motifs on the DNA template and in the nascent RNA can affect the activity of bacteriophage T7 RNA polymerase (58–62). Due to the complexity of transcription termination signals, it is nearly impossible to predict termination sites for even the little-regulated viral RNA polymerases commonly used for in vitro run-off transcription. The problem of termination signals is further compounded when designing DNA fragments for dsRNA constructs (please, see the section below on dsRNA constructs), since in this case both DNA strands have to be free of termination signal. Hence, rather than searching for the termination signals within a given DNA sequence, it is typically more efficient to use plasmids that have a demonstrated lack of termination signals for a given RNA polymerase (32,38,39,50).
In vitro run-off transcription of the central section of the ssRNA construct
In vitro run-off transcription of RNA from the DNA template is routinely carried out using viral RNA polymerases (e.g. T7 or SP6 RNA polymerase). Viral RNA polymerases have a number of advantages: (i) the ability to transcribe in the absence of transcription factors; (ii) selective transcription initiation from well-defined promoter sequences and (iii) high polymerization rates (63). However, there are also several known issues that need to be considered when in vitro run-off transcription of ssRNA molecules is performed. For instance, viral RNA polymerases tend to initiate transcription from within the promoter sequence (Table 1). This results in an extension of the RNA transcript at its 5′-end by an RNA sequence of a defined length and base composition (64,65). Furthermore, viral RNA polymerases are known to add a random number of nucleotides to the 3′-end of the RNA transcripts (65–68). The exact sequence composition and length of the 3′-end extension are not known, although the relative base composition of the 3′-extension can be influenced by the relative concentrations of the free nucleotides during the in vitro run-off transcription (data not shown). Several protocols have been developed in bulk studies to overcome this limitation and obtain well-defined 3′-ends. First, the random extension at the 3′-end has been corrected with site-specific RNase H cleavage, stimulated by the hybridization of a short oligomer to the cleavage site (69–74). Also, cis- or trans-acting ribozyme sequences have been attached to the 3′-end of the RNA transcripts to obtain homogenous 3′-ends after in vitro run-off transcription (75–77). However, these site-specific RNase H cleavage and ribozyme sequences have only been used successfully with high specificity for editing the 3′-ends of RNA transcripts shorter than 1 kb (69,75–77). In the case of RNase H cleavage, the presence of RNA secondary structure can prevent specific binding of short oligomers to the cleavage site (69). In addition, the folding of the ribozyme sequence into its active structure may be less efficient when attached to an RNA sequence longer than 1 kb (77). Therefore, to prevent the random 3′-end extension of RNA transcripts longer than 1 kb, a more successful strategy may be the use of a DNA template obtained with a reverse PCR primer that contains 2′-methoxyl modifications at its last two 5′ nucleotides. In this case, random nucleotide addition by T7 RNA polymerase should be prohibited (78).
In addition to the terminal extensions of the in vitro run-off transcripts, abortive transcripts are a common problem associated with in vitro run-off transcription. In the absence of the transcription termination sites on DNA template and nascent RNA, most of the commercially available viral RNA polymerases readily transcribe DNA templates of up to 10 kb in length. The abortive transcripts are produced when transcription termination sites are present on the DNA template and/or nascent RNA. Furthermore, limited RNA polymerase processivity can also cause the occurrence of the abortive transcripts. Thus, the onset of abortive transcription depends on the type of RNA polymerase used to transcribe RNA as well as on the particular plasmid from which the DNA template is derived.
Polymerization of long random and homopolymeric central sections of ssRNA constructs
An alternative synthesis scheme can be used to obtain ssRNA molecules of up to 20 kb in length, provided a known RNA sequence is not required. This scheme relies on the use of the Pnpase enzyme for the synthesis of the central section of the ssRNA construct (33). Although Pnpase functions in vivo as an RNA exonuclease (79,80), it has been used in vitro to polymerize NDP into long RNA polymers in a primer-independent fashion (33,81,82). When the Pnpase is provided with only one type of NDP, the enzyme will generate a homopolymer. Alternatively, an incubation of the enzyme with all four NDPs will generate random RNA sequences. While the advantage of Pnpase over RNA polymerases is its ability to synthesize RNA molecules longer than 10 kb, Pnpase-assisted RNA synthesis results in a wide distribution of RNA molecule lengths. The resulting absence of well-defined lengths can present difficulties in characterizing RNA molecules in the single-molecule experiment (33)
LABELING OF THE TERMINAL SECTIONS OF THE SSRNA CONSTRUCT
Having described the techniques that can be used to synthesize the central section of the ssRNA construct for use in single-molecule force spectroscopy experiments, we next address the introduction of labeled terminal sections for tethering of such RNA molecules to the glass or bead surfaces (Figure 2A and B). The anchoring of each terminal section of the ssRNA construct is achieved by the formation of a strong bond between the surface-immobilized molecule and its ligand in the terminal section. The most commonly used bonds are those between digoxigenin and its monoclonal antibody (i.e. antidigoxigenin) or between biotin and streptavidin. The surface is usually coated with either antidigoxigenin or streptavidin, whereas the corresponding terminal sections of the ssRNA constructs are labeled with digoxigenin or biotin, respectively. As an alternative, fluorescein-labeled RNA molecules have been recently used for immobilization on an antifluorescein-coated surface (34). Even though Lambert and coworkers (34) applied the fluorescein-labeled RNA in the context of a TPM experiment in the absence of applied force, it has been used in DNA single-molecule force spectroscopy experiments (83,84).
Labels in the terminal sections of the ssRNA construct are introduced by incorporating digoxigenin- or biotin-modified nucleotides into its terminal sections. The terminal section of ssRNA construct are principally obtained in three ways: (i) chemical modifications of the 3′- and 5′-end of the central section can be utilized for the attachment of a variety of labels (e.g. biotin) (34,85–87), (ii) by direct enzymatic extension of the 3′- and 5′-ends of the ssRNA construct central section itself with modified nucleotides or (iii) by the hybridization of a labeled RNA or DNA complementary to the ends of the central sections (Figure 2C).
Chemical labeling of the central sections of ssRNA construct
A selective 3′- or 5′-end chemical labeling of the ssRNA central section has been the least utilized way of RNA constructs labeling in single-molecule force spectroscopy experiments. However, this method holds several advantages over the more common enzymatic labeling and hybridization of complementary labeled strands discussed below, namely high efficiency even at ssRNA lengths longer than 1 kb, and insensitivity to the sequence at the end of the central section.
Chemical labeling consists of two steps (34,85–87). First, the 3′- or 5′-end of the ssRNA central section is chemically activated. In a subsequent step, a chemically activated label is attached to the activated end of the ssRNA central section. The most common chemical activation of 3′-end of ssRNA is periodate oxidation, resulting in a cis-diol ribose modification at the 3′-end (85,86). The cis-diol ribose modified 3′-end then readily reacts with hydrazide-activated labels such as biotinamidocaproyl hydrazide (85,86). The 5′-end labeling of the ssRNA construct can be carried out with initial chemical activation of the 5′-end in the presence of carbohydrazide and EDC (1-ethyl 3-(3-diethylaminopropyl)carbodiimide), followed by a reaction with isothiocyanate derivative of the label of interest (34,87). These chemical labeling protocols have been successfully applied to 3′- and 5′- labeling of 1.6 kb long 16S rRNA from E. coli and synthetic polyU ssRNA molecules of up to 5.5 kb (34).
Enzymatic labeling of the ends of the central section of the ssRNA construct
Enzymatic extension of the central section of the RNA construct can be carried out by several different enzymes [e.g. Poly(A) polymerase (Pap), Terminal deoxynucleotidyl transferase (Tdt) and T4 RNA Ligase 1 (RNL1)]. Most of the labeling efficiencies cited in the literature have been measured for RNA oligomers (88–90). It is expected that the yield of labeling ssRNA for a typical single-molecule force spectroscopy experiment is lower than for an RNA oligomer. However, even a relatively low efficiency of enzymatic labeling of the 3′- and 5′-ends of ssRNA molecules does not necessarily pose an insurmountable problem in single-molecule research, provided the unlabeled ssRNA constructs do not anchor and thus do not interfere with single-molecule experiments.
Direct labeling is possible for both the 3′- and 5′-ends of RNA molecules. The 3′-end of the central section can be extended with a single modified nucleotide by using Pap in the presence of a 3′-deoxy version of the modified nucleotide which, following its incorporation, prevents any further addition of modified nucleotides at the new 3′-end (89). This reaction typically has an efficiency of ∼50% for RNA oligomers (data not shown) (89). Similarly, the enzyme Tdt has also been reported to be able to attach labeled deoxynucleotides at the 3′-end of RNA oligomers with at least 50% efficiency (Figure 3A) (90). Our experiments indicate however, that the Tdt labeling efficiency of RNA oligomers is consistently below 10% (Figure 3B). While Tdt efficiently adds multiple modified or unmodified deoxynucleotides at the 3′-end of DNA oligomers (Lanes 2 and 3 in Figure 3B, respectively), it exhibits poor modification yield for RNA oligomers even at higher concentrations of deoxynucleotides (Lanes 4–8 in Figure 3B). It should be noted that in the case of RNA oligomers an intriguing higher modification yield is observed with modified compared to unmodified deoxynucleotides (Figure 3B). The 5′-end of ssRNA molecule can be labeled using T4 RNA ligase 1 (88). The labeling efficiency of this method is ∼50% for RNA oligomers (88).
Figure 3.

3′-end labeling of DNA and RNA oligomers using Terminal deoxynucleotidyl transferase (Tdt). (A) DNA or RNA oligomers are incubated with Tdt in the presence of labeled deoxynucleotides, which are added to the 3′-end of the DNA or RNA oligomers by the Tdt. Usually, more than one labeled deoxynucleotide is added to the DNA oligomers. Previously, the addition of multiple labeled deoxynucleotide was reported for the RNA oligomers (75). (B) An agarose gel of Tdt labeling reactions in the presence of DNA or RNA oligomers and varying concentrations of dTTP, dUTP or digoxigenin-labeled dUTP (dig-dUTP). DNA or RNA oligomers were incubated with Tdt and respective deoxynucleotide in the 1× Tdt reaction buffer for 1 h at 37°C (Materials and Methods section). The reactions were stopped with the addition of EDTA and denatured with urea prior to loading them on a denaturing 20% PAGE. Lanes: M, single-stranded DNA marker. The 20 and 30 nt long oligomers are indicated; 1, DNA oligomer incubated in the absence of deoxynucleotides; 2, DNA oligomer labeled with 50 μM dig-dUTP; 3, DNA oligomer extended at its 3′-end with 50 μM dTTP; 4, RNA oligomer incubated in the absence of deoxynucleotides; 5, RNA oligomer labeled with 50 μM dig-dUTP; 6, RNA oligomer labeled with 250 μM dig-dUTP; 7, RNA oligomer incubated with 50 μM of dTTP; 8, RNA oligomer incubated with 10 mM dTTP.
Hybridization of labeled complementary ssRNA and ssDNA to the ends of the central section of ssRNA construct
An alternative to the enzymatic labeling of the central section of the RNA construct is the hybridization of labeled complementary ssRNA or ssDNA to the ends of the central section (Figure 2C). While the enzymatic labeling of the central section is the faster alternative, it can suffer from low yield due to the length of the central section. Such low yield can be problematic since an incomplete enzymatic modification of ssRNA construct termini may result in a low yield of tethered ssRNA constructs in TPM or tweezers measurements, where one end of the construct binds to the bead and the opposite end is left unlabeled and free to attach to a surface-immobilized protein. In this type of an experimental setup, the yield of tethered constructs would be decreased by unlabeled construct competition for the binding to the surface-immobilized motors (44).
Labeling via hybridization consists of two steps: (i) synthesis of complementary ssRNA or ssDNA with modified nucleotides or deoxynucleotides, respectively, and (ii) hybridization of the complementary strands to the ends of the central section of the RNA construct. The labeled ssRNA or ssDNA molecules must be shorter than the central section itself (Figure 2C). The labeled ssRNA and ssDNA experimentally used are typically 500–2000 bp in length. This provides the stability required in tweezers experiments in the presence of applied forces and/or low loading rates (39,91). Labeling via hybridization offers a greater degree of flexibility since the labeled molecules can contain single or multiple modified nucleotides. Synthesis of the complementary ssRNA transcript with a single modified nucleotide is carried out enzymatically as described above for the central section of the ssRNA construct. The complementary ssRNA labeled with multiple modified nucleotides is obtained by in vitro run-off transcription in the presence of modified nucleotides (32,50). Similarly to the complementary ssRNA, ssDNA can be labeled with a single or multiple modified deoxynucleotides. The labeling of the ssDNA with a single modified nucleotide at the 3′-end can be obtained by labeling of the corresponding dsDNA with Tdt prior to denaturation (92). Alternatively, 3′-end labeling of ssDNA can be achieved by performing a replacement reaction on a corresponding dsDNA using T4 DNA polymerase (38). Labeling of ssDNA with a single modified deoxynucleotide at the 5′-end can be obtained by labeling of the corresponding dsDNA with T4 RNA ligase 1 (RNL1) prior to denaturation (88,90). More commonly however, the 5′-labeled PCR primers can be used to obtain a 5′-labeled dsDNA, which is denatured in the subsequent step to obtain a 5′-labeled complementary ssDNA (38). To obtain ssDNA with multiple modified nucleotides, a PCR is performed in the presence of modified deoxynucleotides to obtain dsDNA (93). The ssDNA required is produced by denaturing the dsDNA during the subsequent hybridization step (see subsequently).
Hybridization of the labeled complementary ssRNA or ssDNA to the ends of the central section of the RNA construct is subsequently performed. The basic protocol for the hybridization of complementary ssRNA or ssDNA to the central section of the RNA construct starts with a denaturation step followed by the actual hybridization. The denaturation of labeled ssRNA and dsDNA requires temperatures above 60°C and can be promoted by the presence of denaturing agents such as formamide (94). The subsequent hybridization is usually achieved by slow cooling from the denaturing temperature to the ambient temperature, which promotes the formation of the thermodynamically stable state of RNA:RNA or DNA:RNA hybrids between the ends of the central section and labeled complementary single-stranded nucleic acids. The higher thermodynamic stability of DNA:RNA compared to DNA:DNA allows for the formation of DNA:RNA hybrid when labeled dsDNA is used in formamide-based buffers (94–97). At the high temperatures required for the denaturation, care must be taken to avoid degradation of RNA. The presence of chelating agents (e.g. EDTA, sodium citrate) is essential to prevent RNA hydrolysis by trace amounts of divalent ions (98–101). RNA degradation by RNases is typically avoided through the use of commercially available RNase inhibitors, but this does not apply in the case of hybridization, as most of the commercially available RNase inhibitors are inactivated at temperatures above 60°C. In this case, care must be taken to use only RNase-free materials.
Several different RNA hybridization protocols have been applied in single-molecule research (32,38,39,50). They differ in the composition of the denaturing buffer as well as in the cooling rate used for the hybridization step. A comparison of the denaturing buffer and the cooling rate effects on hybridization of complementary 4.1 kb long ssRNA molecules is shown in Figure 4A. It can be concluded that the complementary RNA molecules hybridize equally well in aqueous-based and formamide-based buffers, as judged by the identical electrophoretic mobilities of hybridization products (Figure 4A). The two temperature programs reported in the literature (38,50) also did not notably affect the final result of hybridization as judged by agarose gel electrophoresis (Lane 1 versus Lane 2, and Lane 3 versus Lane 4 in Figure 4A). Conversely, the molar ratio of complementary RNA strands did influence the efficiency of the reaction. Figure 4B shows that the efficiency of hybridization reaction of a 0.5 kb ssRNA to a section of 4.1 kb ssRNA is higher when the short strand is in a molar excess regardless of the denaturation buffer used in the reaction.
Figure 4.

Comparison of hybridization of RNA molecules under various sets of experimental conditions. (A) Hybridization of the sense and antisense 4.1 kb single-stranded RNA molecules (4.1 kb ssRNA) in aqueous or formamide denaturing buffer at a 1:1 molar ratio. Lanes: M, double-stranded DNA marker; 1, hybridization of the sense and antisense 4.1 kb ssRNA performed in SSCTE buffer. Hybridization reaction was performed using ‘gradual-cool’ temperature program; 2, hybridization performed in SSCTE buffer. Hybridization reaction carried out according to ‘step-cool’ temperature protocol; 3, hybridization performed in FEPS buffer. Hybridization reaction was done using the ‘gradual-cool’ temperature program; 4, RNA hybridization in FEPS buffer using the ‘step-cool’ temperature protocol. (B) Hybridization of a 0.5 kb ssRNAto the 5′-end of the sense 4.1 kb ssRNA. All the hybridizations were performed using the ‘gradual-cool’ temperature program. The sense 4.1 kb ssRNA and the hybrid of 0.5 kb ssRNA and the sense 4.1 kb ssRNA are indicated. Lanes: 1–3, hybridization with a 1:1 strand molar ratio in TE buffer (Lane 1), SSCTE buffer (Lane 2) or FEPS buffer (Lane 3); 4–6, hybridization with a 4:1 strand molar ratio with an excess of the 0.5 kb ssRNA in TE buffer (Lane 4), SSCTE buffer (Lane 5) or FEPS buffer (Lane 6).
An ssRNA construct obtained with the hybridization protocol discussed above has been used successfully in the force-extension experiments employing terminal sections with a single modified nucleotide each (38,39). This is not surprising as the high rapture forces of streptavidin–biotin, digoxigenin–monoclonal antibody pairs enable the ssRNA construct with a single modified nucleotide to resist forces up to 100 pN (102–106).
JOINING CENTRAL AND LABELED TERMINAL SECTIONS OF THE SSRNA CONSTRUCT BY LIGATION
Ligation of the single-stranded central and labeled terminal sections with RNA Ligase 1 (RNL1) can represent an alternative to labeling of the ends of the central section, which often exhibit low efficiency for long ssRNA molecules. In addition, ligation can substitute for hybridization of complementary ssRNA or ssDNA to the ends of the central section, if completely ssRNA construct is required (e.g. measurement of the persistence length of ssRNA). An overview of possible ligation reactions performed by RNL1, which catalyze the formation of a phosphodiester bond between the 3′-OH and 5′-monophosphate termini of nucleic acids, is given in Table 2. The 3′- and 5′-ends of two ssRNA molecules can be ligated by an intermolecular reaction using RNL1 (107,108). The efficiency of this reaction decreases with an increase in ssRNA length and hence often requires high concentrations of the substrate molecules (107,108). Fortunately, the rate of ligation of two ssRNA molecules can be increased, if a DNA splint is used to bring the 3′- and 5′-end of the two ssRNA molecules into close proximity prior for ligation with RNL1 to form an RNA loop structure (51,107,109–112). An alternative way to increase the rate of ligation is Y-ligation (Table 2) (107). Briefly, the 3′- and 5′-end of two ssRNA molecules are hybridized to obtain a short stem structure with free 3′- and 5′-ends. In the Y-ligation step, the free 3′- and 5′-ends are joined by RNL1 to form an RNA loop structure (113). The length of the resulting RNA loop should not exceed 15 nt (113).
Table 2.
An overview of RNA ligation schemes using RNA ligase 1 (RNL1) or RNA ligase 2 (RNL2)
| Ligation type | Ligation structure | Ligation conditions |
|---|---|---|
| Single-stranded RNAs ligation with RNL1 |  |
Molecule I: RNA Molecule II: RNA or DNA |
| Splint ligation with RNL1 | 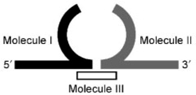 |
Molecule I: RNA or DNA Molecule II: RNA Molecule III: DNA Resulting hairpin length <8 nt |
| Y-ligation with RNL1 | 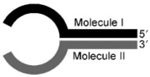 |
Molecule I: RNA Molecule II: RNA or DNA Resulting hairpin length <15 nt |
| Nick ligation with RNL2 |  |
Molecule I: RNA Molecule II: RNA or DNA Molecule III: RNA or DNA |
The directionality of the ligated nucleic acid strands are denoted by the non-ligated end of each strand. ‘Ligation type’ column includes a common name for a particular ligation reaction and RNA ligase type to be used for a particular ligation. RNA Ligase 1, and RNA Ligase 2 are abbreviated with RNL1 and RNL2, respectively. The ‘Ligation structure’ column provides a schematic representation of the RNA structure to be ligated. The ‘Ligation conditions’ column lists the combinations of nucleic acids resulting in an efficient ligation (107,109).
When ligating RNA molecules synthesized by in vitro run-off transcription, it should be kept in mind that while the viral RNA polymerases create a 3′-OH at the end of RNA transcripts, they yield a triphosphate at the 5′-end of the RNA transcript. This 5′-triphosphate can be removed after in vitro run-off transcription by use of calf intestinal phosphatase (CIP) and then the desired monophosphate moiety is added by a kinase (32,50). Alternatively, the in vitro run-off transcription reaction can be supplemented with an excess of nucleotide monophosphate (NMP) corresponding to the first transcribed nucleotide (54). For example, GMP was added in a 20-time molar excess compared to GTP in a transcription reaction with T7 RNA polymerase (45). The 20:1 molar excess of GMP to GTP ensures a 95% yield of transcripts with a monophosphate at their 5′-end, assuming equal binding affinity and reaction kinetics for the two nucleotides (54).
DOUBLE-STRANDED RNA CONSTRUCTS
So far, the assembly of ssRNA constructs has been discussed due to its importance in single-molecule studies of naturally occurring ssRNA sequences and RNA structural motifs. However, the interest in dsRNA has steadily grown in recent years, prompted by a growing appreciation of dsRNA as an important molecule in the cellular regulatory processes (18–22). As in the case of ssRNA constructs several different protocols have been applied to obtain dsRNA constructs for single-molecule studies. Dekker and coworkers (32,50) have obtained ∼8-kb long dsRNA by in vitro run-off transcribing the antisense and sense RNA strands from the same DNA fragment with a subsequent hybridization to form a dsRNA under the solution conditions discussed above. The length of these dsRNA molecules is limited by the same factors affecting ssRNA in vitro run-off transcription. To circumvent the limits of in vitro run-off transcription, we have recently used RNA-dependent RNA polymerase (RdRP) to obtain a dsRNA molecule longer than 8 kb by using ssRNA molecule obtained with Pnpase as a template (Figure 5A). Using this scheme, the products of the Pnpase polymerization reaction were used to obtain dsRNA molecules (Lane 2 in Figure 5B). The dsRNA molecules between 15 and 20 kb in length were then gel-extracted (Lane 3 in Figure 5B) and imaged with AFM to verify their lengths (Figure 5C). The combined use of Pnpase and RdRP for the synthesis of dsRNA constructs longer than 10 kb (Figure 5A) does not enable a control over the final sequence of dsRNA since it can be used only to synthesize homopolymeric or random dsRNA sequences. This limits the use of these dsRNA construct to studies that require either a homopolymeric RNA sequence or permit the use of an unknown random RNA sequence.
Figure 5.

Synthesis of dsRNA with a combination of Polynucleotide phosphorylase (Pnpase) and RNA-dependent RNA polymerase (RdRP) yields longer dsRNA molecules than standard in vitro run-off transcription. (A) A reaction scheme for dsRNA synthesis. ssRNA molecules are first synthesized enzymatically with Pnpase in the presence of four nucleotide diphosphates (rADP, rGDP, rUDP and rCDP). In the subsequent step, ssRNA molecules are used as template strands for RdRP replication reaction. (B) An agarose gel of the products of dsRNA synthesis with PnPase and RdRp. Lanes: 1, double-stranded DNA marker obtained with digestion of 49 kb λDNA. The 21 kb double-stranded DNA band is indicated; 2, dsRNA products of the RdRp replication reaction. The length of synthesized dsRNA varies from several hundred base pairs to molecules longer than 15 kb. This is due to de novo initiation of Pnpase polymerization that yields a broad length distribution of ssRNA molecules in the first step of the reaction scheme in (A) 3, the gel-extracted dsRNA. The range of RNA lengths extracted is indicated by the white rectangle in Lane 2. (C) A representative AFM image of the gel-extracted dsRNAfrom Lane 3 in (B). The gel-extracted dsRNA was adsorbed on a Ni-functionalized mica surface and imaged as described in the Materials and Methods section. We measured an average dsRNA length of 5 μm, which corresponds to the length of ∼15 kb dsRNA. The scale bar represents 1 μm.
Terminal sections of the dsRNA constructs
By analogy with the methods used to assemble DNA constructs for single-molecule manipulation (114), the addition of terminal sections to a dsRNA construct has been achieved by ligation of one double-stranded terminal section to each end of the double-stranded central section of the RNA construct. This is most effectively accomplished using RNA Ligase 2 (RNL2) (Figure 2D) (50), which has a higher affinity than RNL1 for nicked dsRNA substrates (115). The double-stranded terminal sections were labeled with multiple modified nucleotides on both strands (Figure 2D) (50). One end in each terminal section was engineered to contain an overhang that was complementary to the one of the engineered overhangs on the double-stranded central section (Figure 2D) (50). After hybridization of the complementary overhangs, the junctions were sealed with RNL2 (Figure 2D). Figure 6A shows an agarose gel of unligated control and products of a ligation of a double-stranded central section and two double-stranded terminal sections through complementary overhangs using RNL2. The additional two bands with lower electrophoretic mobility in Lane 3 represent the two products of the ligation reaction, namely the 4.1 kb dsRNA with 0.5 kb dsRNA ligated to one or both of its ends (Figure 6A). Comparison of Lane 2 with Lane 3 in Figure 6A suggests that the ligation reaction did not go to completion, as unligated product was still observed. There are several factors that can decrease the yield of ligation of the terminal and central sections of the dsRNA construct, including: (i) low rate of ligation due to a low concentration of the overhangs; (ii) a folding of the complementary overhangs back onto themselves into hairpins, thus producing an intramolecular nick which can be sealed by RNL2; (iii) the presence of random extensions at the 3′-end of the terminal and central sections as a result of imperfect viral RNA polymerase transcription. These extensions would yield an imperfect junctions, that have been shown to be poor substrates for RNL2 (112) and (iv) partial digestion of the complementary overhangs during the ligation reaction would result in an unstable junction or in a gapped junction, that has been shown to be a poor substrate for RNL2 (112). In this particular case, the incomplete ligation reaction is not due to the low concentration of overhangs, as the yield of the ligation reaction did not increase with longer reaction times (data not shown). Furthermore, the intramolecular hairpin ligation did not affect the yield of intermolecular ligation during the ligation time as the yield of intramolecular ligation of a 4.1 kb dsRNA with a 5′-overhang at one end was negligible. This was suggested by the low intensity of 8.2 kb long ssRNA (Lane 3 in Figure 6B) that would result from such an intramolecular ligation. It is thus possible that the random 3′-end extensions and/or overhang digestion decreased the yield of the dsRNA ligation reaction. If the digestion of the overhangs results in a low yield of the intermolecular ligation reaction, an increase in the RNL2 reaction rate may prove effective. This can be accomplished by using RNA:DNA hybrids as the double-stranded terminal sections, as these have been shown to present a more efficient substrate for RNL2 activity than RNA:RNA junctions (Table 2) (115).
Figure 6.

Ligation of double-stranded RNA molecules with complementary 5′-overhangs using RNA Ligase 2 (RNL2). (A) Intermolecular ligation of a 0.5 kb dsRNA molecule to each end of a 4.1 kb dsRNA with RNL2 through complementary 5′-overhangs (Figure 2D) (gel image from (50), and used with the permission of NAR). Lanes: 1, double-stranded DNA marker. The corresponding dsDNA lengths are noted next to each band; 2, 4.1 kb dsRNA with 5′-overhang at both ends incubated in the RNL2 ligation buffer; 3, ligation of 0.5 kb dsRNA to the 3′- and 5′-ends of 4.1 kb dsRNA with RNL2. (B) The efficiency of intramolecular ligation with RNL2 was tested with a 4.1 kb dsRNA with a monophosphate at its 5′-overhang (activated dsRNA). Two control molecules 4.1 kb dsRNA with a triphosphate at its 5′-overhang (deactivated dsRNA), and the 4.1 kb dsRNA with a blunt end (blunt dsRNA) were also incubated with RNL2. Lanes: 1, 4.1 kb ssRNA; 2, activated dsRNA; 3, activated dsRNA ligated with RNL2; 4, deactivated dsRNA; 5, deactivated dsRNA ligated with RNL2; 6, blunt dsRNA; 7, blunt dsRNA ligated with RNL2. The unligated dsRNA molecules run as 4.1 kb ssRNA after denaturation whereas intramolecularly ligated dsRNA molecules run as 8.2 kb ssRNA after dentauration. The 4.1 and 8.2 kb ssRNA mobilities are indicated in the gel image.
ISOLATION AND PURIFICATION OF RNA MOLECULES AND CONSTRUCTS
As we have shown above, the assembly of RNA constructs for single-molecule measurements is a process that consists of several steps, typically requiring purification after completion of each step. In addition, it can be advantageous to isolate the final RNA construct from a complex mixture of side products of RNA construct assembly. The isolation and purification protocols for RNA molecules and constructs reported in the literature are almost exclusively based on agarose gel-extraction and ethanol precipitation, respectively. (116). The stability of RNA molecules and constructs during all these purification and isolation procedures should always be given consideration. Usually, the addition of RNase inhibitors for inactivation of RNases (e.g. RNasin) and the addition of EDTA for chelation of the divalent cations is sufficient to prevent appreciable RNA degradation.
Agarose gel extraction is the most common way of RNA construct isolation. Once a gel slice containing the desired RNA molecule or construct has been selectively cut out of the gel, the RNA can be extracted by centrifuging a crushed gel slice at high speed, which sediments the gel particles while RNA remains in the supernatant (117). Alternatively, the polymerized agarose can be enzymatically digested to release the RNA construct into the solution (118). Several precautions must be noted with gel separation and extraction. First, the yield of agarose gel-extraction strongly depends on the length of the RNA construct. In addition to the length of the RNA molecule, we find that the isolation of dsRNA exhibits higher yields and lower degrees of RNA degradation compared to the ssRNA molecules, presumably due to higher stability of dsRNA. Certain homopolymeric sequences of ssRNA cannot be detected on agarose gels due to their poor staining with standard nucleic acid dyes. Agarose gel electrophoresis results in poor length separation for solutions with wide and continuous RNA length distributions as in the case of Pnpase polymerization products. In cases where gel extraction results in a low yield, HPLC has been shown to be a plausible alternative (Dr R, Tuma, personal communication).
CONCLUSIONS
RNA is involved in many of the most important processes in biology. The ability to make stable and active RNA constructs for single-molecule experiments enables one to complement RNA bulk studies by taking the advantage of the unique property of single-molecule techniques to measure the properties of the biological processes in the absence of ensemble averaging. We have both discussed and analyzed a number of protocols and molecular biology tools for the assembly of RNA construct that have been successfully used to study RNA and RNA-related processes on a single-molecule level. Many relevant RNA properties and RNA-dependent processes have thus been investigated on a single-molecule level (32,33,35–49), however many still remain relatively unexplored at the single-molecule level (e.g. protein translation). Currently, the scope of many RNA single-molecule studies is limited by the inability to synthesize ss- or dsRNA molecules of known sequence that are longer than 10 kb. The progress in the molecular biology of RNA should thus focus on further increasing the maximum length of the attainable RNA of a known sequence. Recently a subsequent use of in vitro run-off transcription and replication with RdRP has been used by Bamford and coworkers to synthesize long dsRNAs. This protocol successfully reduces the effect of termination signals on the maximum length of attainable dsRNA by requiring only one of the two RNA strands to be transcribed in vitro. Consequently, ssRNA and dsRNA of known sequences have an identical length limit, determined by the presence of the termination signals on the transcribed strand and DNA fragment used for transcription. Nevertheless, some single-molecule experiments may require even longer RNA molecules of known sequence (e.g. optical tweezers combined with nanopores). Furthermore, a higher efficiency of labeling with the modified nucleotides and higher RNA stability under RNA construct assembly conditions (i.e. hybridization temperatures above 60°C) should prove beneficial for high-throughput single-molecule studies. Several commercial RNase inhibitors have been optimized to provide for RNA stability at temperatures above 60°C (e.g. RNasin), however they are guaranteed their efficiency only for a limited reaction time. In addition to RNases and divalent cations, RNA construct stability can also be affected by the interaction with glass and bead surfaces, which has not been systematically investigated.
SUPPLEMENTARY DATA
Supplementary Data are available at NAR Online.
ACKNOWLEDGEMENTS
We thank Jeroen Abels, Michiel Bruinink, and Peter Veenhuizen for developing preliminary experiments on RNA in our laboratory, and Roman Tuma and Dennis Bamford for a critical reading of the manuscript. We would also like to thank the reviewers for their insightful comments and suggestions to improve the manuscript. The research was supported by NanoNed, FOM and NWO. Funding to pay the Open Access publication charges for this article was provided by NanoNed, FOM, NWO.
Conflict of interest statement. None declared.
REFERENCES
- 1.Pennazio S, Roggero P. Tobacco mosaic virus RNA as genetic determinant: Genesis of a discovery. Riv. Biol. Forum. 2000;93:431–455. [PubMed] [Google Scholar]
- 2.Domingo E, Escarmis C, Sevilla N, Moya A, Elena SF, Quer J, Novella IS, Holland JJ. Basic concepts in RNA virus evolution. FASEB J. 1996;10:859–864. doi: 10.1096/fasebj.10.8.8666162. [DOI] [PubMed] [Google Scholar]
- 3.Domingo E, Holland JJ. RNA virus mutations and fitness for survival. Annu. Rev. Microbiol. 1997;51:151–178. doi: 10.1146/annurev.micro.51.1.151. [DOI] [PubMed] [Google Scholar]
- 4.Holland JJ, Delatorre JC, Steinhauer DA. RNA virus populations as quasi-species. Curr. Top. Microbiol. Immunol. 1992;176:1–20. doi: 10.1007/978-3-642-77011-1_1. [DOI] [PubMed] [Google Scholar]
- 5.Volkin E, Astrachan L. Phosphorus incorporation in Escherichia coli ribonucleic acid after infection with bacteriophage-T2. Virology. 1956;2:149–161. doi: 10.1016/0042-6822(56)90016-2. [DOI] [PubMed] [Google Scholar]
- 6.Stahley MR, Strobel SA. RNA splicing: group I intron crystal structures reveal the basis of splice site selection and metal ion catalysis. Curr. Opin. Struct. Biol. 2006;16:319–326. doi: 10.1016/j.sbi.2006.04.005. [DOI] [PubMed] [Google Scholar]
- 7.Weinger JS, Parnell KM, Dorner S, Green R, Strobel SA. Substrate-assisted catalysis of peptide bond formation by the ribosome. Nat. Struct. Mol. Biol. 2004;11:1101–1106. doi: 10.1038/nsmb841. [DOI] [PubMed] [Google Scholar]
- 8.Moore MJ, Sharp PA. Evidence for 2 active-sites in the spliceosome provided by stereochemistry of premessenger RNA splicing. Nature. 1993;365:364–368. doi: 10.1038/365364a0. [DOI] [PubMed] [Google Scholar]
- 9.Kazantsev AV, Pace NR. Bacterial RNase P: a new view of an ancient enzyme. Nat. Rev. Microbiol. 2006;4:729–740. doi: 10.1038/nrmicro1491. [DOI] [PubMed] [Google Scholar]
- 10.Beebe JA, Fierke CA. A kinetic mechanism for cleavage of precursor tRNA(asp) catalyzed by the RNA component of Bacillus-subtilis ribonuclease-P. Biochemistry. 1994;33:10294–10304. doi: 10.1021/bi00200a009. [DOI] [PubMed] [Google Scholar]
- 11.Steitz TA, Steitz JA. A general 2-metal-ion mechanism for catalytic RNA. Proc. Natl Acad. Sci. USA. 1993;90:6498–6502. doi: 10.1073/pnas.90.14.6498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Smith D, Pace NR. Multiple magnesium-ions in the ribonuclease-P reaction-mechanism. Biochemistry. 1993;32:5273–5281. doi: 10.1021/bi00071a001. [DOI] [PubMed] [Google Scholar]
- 13.Bergman NH, Johnston WK, Bartel DP. Kinetic framework for ligation by an efficient RNA ligase ribozyme. Biochemistry. 2000;39:3115–3123. doi: 10.1021/bi992654u. [DOI] [PubMed] [Google Scholar]
- 14.Birikh KR, Heaton PA, Eckstein F. The structure, function and application of the hammerhead ribozyme. Eur. J. Biochem. 1997;245:1–16. doi: 10.1111/j.1432-1033.1997.t01-3-00001.x. [DOI] [PubMed] [Google Scholar]
- 15.Brimacombe R, Stiege W. Structure and function of ribosomal-RNA. Biochem. J. 1985;229:1–17. doi: 10.1042/bj2290001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Serganov A, Polonskaia A, Phan ATN, Breaker RR, Patel DJ. Structural basis for gene regulation by a thiamine pyrophosphate-sensing riboswitch. Nature. 2006;441:1167. doi: 10.1038/nature04740. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Winkler WC, Nahvi A, Roth A, Collins JA, Breaker RR. Control of gene expression by a natural metabolite-responsive ribozyme. Nature. 2004;428:281. doi: 10.1038/nature02362. [DOI] [PubMed] [Google Scholar]
- 18.Ahlquist P. RNA-dependent RNA polymerases, viruses, and RNA silencing. Science. 2002;296:1270–1273. doi: 10.1126/science.1069132. [DOI] [PubMed] [Google Scholar]
- 19.Fire A, Xu S, Montgomery MK, Kostas SA, Driver SE, Mello CC. Potent and specific genetic interference by double-stranded RNA in Caenorhabditis elegans. Nature. 1998;391:806. doi: 10.1038/35888. [DOI] [PubMed] [Google Scholar]
- 20.Haley B, Zamore PD. Kinetic analysis of the RNAi enzyme complex. Nat. Struct. Mol. Biol. 2004;11:599–606. doi: 10.1038/nsmb780. [DOI] [PubMed] [Google Scholar]
- 21.Hannon J. RNA interference. Nature. 2002;418:244–251. doi: 10.1038/418244a. [DOI] [PubMed] [Google Scholar]
- 22.McManus MT, Sharp PA. Gene silencing in mammals by small interfering RNAs. Nat. Rev. Genet. 2002;3:737–747. doi: 10.1038/nrg908. [DOI] [PubMed] [Google Scholar]
- 23.Charvin G, Strick TR, Bensimon D, Croquette V. Tracking topoisomerase activity at the single-molecule level. Annu. Rev. Biophys. Biomol. Struct. 2005;34:201–219. doi: 10.1146/annurev.biophys.34.040204.144433. [DOI] [PubMed] [Google Scholar]
- 24.Williams MC, Rouzina I, Bloomfield VA. Thermodynamics of DNA interactions from single molecule stretching experiments. Acc. Chem. Res. 2002;35:159–166. doi: 10.1021/ar010045k. [DOI] [PubMed] [Google Scholar]
- 25.Williams MC, Wenner JR, Rouzina I, Bloomfield VA. Entropy and heat capacity of DNA melting from temperature dependence of single molecule stretching. Biophys. J. 2001;80:1932–1939. doi: 10.1016/S0006-3495(01)76163-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Tamarat P, Maali A, Lounis B, Orrit M. Ten years of single-molecule spectroscopy. J. Phys. Chem. A. 2000;104:1–16. doi: 10.1021/jp992505l. [DOI] [PubMed] [Google Scholar]
- 27.Hong F, Root DD. Downscaling functional bioassays by single-molecule techniques. Drug Discov. Today. 2006;11:640–645. doi: 10.1016/j.drudis.2006.05.003. [DOI] [PubMed] [Google Scholar]
- 28.Skinner GM, Visscher K. Single-molecule techniques for drug discovery. Assay Drug Dev. Technol. 2004;2:397–405. doi: 10.1089/adt.2004.2.397. [DOI] [PubMed] [Google Scholar]
- 29.Mashanov GI, Molloy JE. Automatic detection of single fluorophores in live cells. Biophys. J. 2007;92:2199–2211. doi: 10.1529/biophysj.106.081117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Sako Y, Uyemura T. Total internal reflection fluorescence microscopy for single-molecule imaging in living cells. Cell Struct. Funct. 2002;27:357–365. doi: 10.1247/csf.27.357. [DOI] [PubMed] [Google Scholar]
- 31.Sako Y, Yanagida T. Single-molecule visualization in cell biology. Nat. Cell. Biol. 2003:SS1–SS5. [PubMed] [Google Scholar]
- 32.Abels JA, Moreno-Herrero F, van der Heijden T, Veenhuizen PTM, Bruinink MM, Dekker C, Dekker NH. Single-molecule measurements of the persistence length of double-stranded RNA. Biophys. J. 2005;88:2737–2744. doi: 10.1529/biophysj.104.052811. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Seol Y, Skinner GM, Visscher K. Elastic properties of a single-stranded charged homopolymeric ribonucleotide. Phys. Rev. Lett. 2004;93 doi: 10.1103/PhysRevLett.93.118102. Art. No. 118102. [DOI] [PubMed] [Google Scholar]
- 34.Lambert MN, Vocker E, Blumberg S, Redemann S, Gajraj A, Meiners JC, Walter NG. Mg2+-induced compaction of single RNA molecules monitored by tethered particle microscopy. Biophys. J. 2006;90:3672–3685. doi: 10.1529/biophysj.105.067793. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Bokinsky G, Rueda D, Misra VK, Rhodes MM, Gordus A, Babcock HP, Walter NG, Zhuang XW. Single-molecule transition-state analysis of RNA folding. Proc. Natl Acad. Sci. USA. 2003;100:9302–9307. doi: 10.1073/pnas.1133280100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Bokinsky G, Zhuang XW. Single-molecule RNA folding. Acc. Chem. Res. 2005;38:566–573. doi: 10.1021/ar040142o. [DOI] [PubMed] [Google Scholar]
- 37.Li PTX, Bustamante C, Tinoco I. Unusual mechanical stability of a minimal RNA kissing complex. Proc. Natl Acad. Sci. USA. 2006;103:15847–15852. doi: 10.1073/pnas.0607202103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Liphardt J, Onoa B, Smith SB, Tinoco I, Bustamante C. Reversible unfolding of single RNA molecules by mechanical force. Science. 2001;292:733–737. doi: 10.1126/science.1058498. [DOI] [PubMed] [Google Scholar]
- 39.Mangeol P, Cote D, Bizebard T, Legrand O, Bockelmann U. Probing DNA and RNA single molecules with a double optical tweezer. Eur. Phys. J. E. 2006;19:311–317. doi: 10.1140/epje/i2005-10060-4. [DOI] [PubMed] [Google Scholar]
- 40.Onoa B, Tinoco I. RNA folding and unfolding. Curr. Opin. Struct. Biol. 2004;14:374–379. doi: 10.1016/j.sbi.2004.04.001. [DOI] [PubMed] [Google Scholar]
- 41.Pan T, Sosnick T. RNA folding during transcription. Annu. Rev. Biophys. Biomol. Struct. 2006;35:161–175. doi: 10.1146/annurev.biophys.35.040405.102053. [DOI] [PubMed] [Google Scholar]
- 42.Tinoco I, Li PTX, Bustamante C. Determination of thermodynamics and kinetics of RNA reactions by force. Q. Rev. Biophys. 2006;39:325–360. doi: 10.1017/S0033583506004446. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Zhuang XW, Rief M. Single-molecule folding. Curr. Opin. Struct. Biol. 2003;13:88–97. doi: 10.1016/s0959-440x(03)00011-3. [DOI] [PubMed] [Google Scholar]
- 44.Davenport RJ, Wuite GJL, Landick R, Bustamante C. Single-molecule study of transcriptional pausing and arrest by E. coli RNA polymerase. Science. 2000;287:2497–2500. doi: 10.1126/science.287.5462.2497. [DOI] [PubMed] [Google Scholar]
- 45.Dumont S, Cheng W, Serebrov V, Beran RK, Tinoco I, Pyle AM, Bustamante C. RNA translocation and unwinding mechanism of HCVNS3 helicase and its coordination by ATP. Nature. 2006;439:105–108. doi: 10.1038/nature04331. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Henn A, Medalia O, Shi HP, Steinberg M, Franceschi F, Sagi I. Visualization of unwinding activity of duplex RNA by DbpA, a DEAD box helicase, at single-molecule resolution by atomic force microscopy. Proc. Natl Acad. Sci. USA. 2001;98:5007–5012. doi: 10.1073/pnas.071372498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Rueda D, Bokinsky G, Rhodes MM, Rust MJ, Zhuang XW, Walter NG. Single-molecule enzymology of RNA: essential functional groups impact catalysis from a distance. Proc. Natl Acad. Sci. USA. 2004;101:10066–10071. doi: 10.1073/pnas.0403575101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Yin H, Wang MD, Svoboda K, Landick R, Block SM, Gelles J. Transcription against an applied force. Science. 1995;270:1653–1657. doi: 10.1126/science.270.5242.1653. [DOI] [PubMed] [Google Scholar]
- 49.Zhuang X, Bartley LE, Babcock HP, Russell R, Ha T, Herschlag D, Chu S. A single-molecule study of RNA catalysis and folding. Science. 2000;288:2048–2051. doi: 10.1126/science.288.5473.2048. [DOI] [PubMed] [Google Scholar]
- 50.Dekker NH, Abels JA, Veenhuizen PTM, Bruinink MM, Dekker C. Joining of long double-stranded RNA molecules through controlled overhangs. Nucleic Acids Res. 2004;32 doi: 10.1093/nar/gnh138. Art. No. e140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Stark MR, Pleiss JA, Deras M, Scaringe SA, Rader SD. An RNA ligase-mediated method for the efficient creation of large, synthetic RNAs. RNA-Publ. RNA Soc. 2006;12:2014–2019. doi: 10.1261/rna.93506. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Walter NG. Structural dynamics of catalytic RNA highlighted by fluorescence resonance energy transfer. Methods. 2001;25:19–30. doi: 10.1006/meth.2001.1212. [DOI] [PubMed] [Google Scholar]
- 53.Qin PZF, Pyle AM. Site-specific labeling of RNA with fluorophores and other structural probes. Methods. 1999;18:60–70. doi: 10.1006/meth.1999.0757. [DOI] [PubMed] [Google Scholar]
- 54.Ke A, Doudna JA. Crystallization of RNA and RNA-protein complexes. Methods. 2004;34:408–414. doi: 10.1016/j.ymeth.2004.03.027. [DOI] [PubMed] [Google Scholar]
- 55.Amitani I, Baskin RJ, Kowalczykowski SC. Visualization of Rad54, a chromatin remodeling protein, translocating on single DNA molecules. Mol. Cell. 2006;23:143–148. doi: 10.1016/j.molcel.2006.05.009. [DOI] [PubMed] [Google Scholar]
- 56.Keyser UF, Koeleman BN, Van Dorp S, Krapf D, Smeets RMM, Lemay SG, Dekker NH, Dekker C. Direct force measurements on DNA in a solid-state nanopore. Nat. Phys. 2006;2:473–477. [Google Scholar]
- 57.Yin H, Landick R, Gelles J. Tethered particle motion method for studying transcript elongation by a single RNA-polymerase molecule. Biophys. J. 1994;67:2468–2478. doi: 10.1016/S0006-3495(94)80735-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Hartvig L, Christiansen J. Intrinsic termination of T7 RNA polymerase mediated by either RNA or DNA. EMBO J. 1996;15:4767–4774. [PMC free article] [PubMed] [Google Scholar]
- 59.He B, Kukarin A, Temiakov D, Chin-Bow ST, Lyakhov DL, Rong MQ, Durbin RK, McAllister WT. Characterization of an unusual, sequence-specific termination signal for T7 RNA polymerase. J. Biol. Chem. 1998;273:18802–18811. doi: 10.1074/jbc.273.30.18802. [DOI] [PubMed] [Google Scholar]
- 60.Jeng ST, Gardner JF, Gumport RI. Transcription termination by Bacteriophage T7 RNA polymerase at Rho-independent terminators. J. Biol. Chem. 1990;265:3823–3830. [PubMed] [Google Scholar]
- 61.Lyakhov DL, He B, Zhang X, Studier FW, Dunn JJ, McAllister WT. Pausing and termination by bacteriophage T7 RNA polymerase. J. Mol. Biol. 1998;280:201–213. doi: 10.1006/jmbi.1998.1854. [DOI] [PubMed] [Google Scholar]
- 62.Macdonald LE, Durbin RK, Dunn JJ, McAllister WT. Characterization of 2 types of termination signal for bacteriophage T7 RNA polymerase. J. Mol. Biol. 1994;238:145–158. doi: 10.1006/jmbi.1994.1277. [DOI] [PubMed] [Google Scholar]
- 63.Logel J, Dill D, Leonard S. Synthesis of cRNA probes from PCR-generated DNA. Biotechniques. 1992;13:604–610. [PubMed] [Google Scholar]
- 64.Melton DA, Krieg PA, Rebagliati MR, Maniatis T, Zinn K, Green MR. Efficient in vitro synthesis of biologically active RNA and RNA hybridization probes from plasmids containing a bacteriophage-SP6 promoter. Nucleic Acids Res. 1984;12:7035–7056. doi: 10.1093/nar/12.18.7035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Milligan JF, Groebe DR, Witherell GW, Uhlenbeck OC. Oligoribonucleotide synthesis using T7 RNA polymerase and synthetic DNA templates. Nucleic Acids Res. 1987;15:8783–8798. doi: 10.1093/nar/15.21.8783. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Draper DE, White SA, Kean JM. Preparation of specific ribosomal-RNA fragments. Method Enzymol. 1988;164:221–237. doi: 10.1016/s0076-6879(88)64045-6. [DOI] [PubMed] [Google Scholar]
- 67.Kholod N, Vassilenko K, Shlyapnikov M, Ksenzenko V, Kisselev L. Preparation of active tRNA gene transcripts devoid of 3′-extended products and dimers. Nucleic Acids Res. 1998;26:2500–2501. doi: 10.1093/nar/26.10.2500. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Milligan JF, Uhlenbeck OC. Determination of RNA-protein contacts using thiophosphate substitutions. Biochemistry. 1989;28:2849–2855. doi: 10.1021/bi00433a016. [DOI] [PubMed] [Google Scholar]
- 69.Donis-Keller H. Site specific enzymatic cleavage of RNA. Nucleic Acids Res. 1979;7:179–192. doi: 10.1093/nar/7.1.179. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Lapham J, Crothers DM. RNase H cleavage for processing of in vitro transcribed RNA for NMR studies and RNA ligation. RNA. 1996;2:289–296. [PMC free article] [PubMed] [Google Scholar]
- 71.Li J, Wartell RM. RNase H1 can catalyze RNA/DNA hybrid formation and cleavage with stable hairpin or duplex DNA oligomers. Biochemistry. 1998;37:5154–5161. doi: 10.1021/bi9730801. [DOI] [PubMed] [Google Scholar]
- 72.Lima WF, Crooke ST. Cleavage of single strand RNA adjacent to RNA-DNA duplex regions by Escherichia coli RNase H1. J. Biol. Chem. 1997;272:27513–27516. doi: 10.1074/jbc.272.44.27513. [DOI] [PubMed] [Google Scholar]
- 73.Yu YT, Steitz JA. A new strategy for introducing photoactivatable 4-thiouridine ((4S)U) into specific positions in a long RNA molecule. RNA. 1997;3:807–810. [PMC free article] [PubMed] [Google Scholar]
- 74.Zamaratski E, Pradeepkumar PI, Chattopadhyaya J. A critical survey of the structure-function of the antisense oligo/RNA heteroduplex as substrate for RNase H. J. Biochem. Biophys. Methods. 2001;48:189–208. doi: 10.1016/s0165-022x(01)00149-x. [DOI] [PubMed] [Google Scholar]
- 75.FerreDamare AR, Doudna JA. Use of cis- and trans-ribozymes to remove 5′ and 3′ heterogeneities from milligrams of in vitro transcribed RNA. Nucleic Acids Res. 1996;24:977–978. doi: 10.1093/nar/24.5.977. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Walker SC, Avis JM, Conn GL. General plasmids for producing RNA in vitro transcripts with homogeneous ends. Nucleic Acids Res. 2003;31 doi: 10.1093/nar/gng082. Art. No. e82. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Wichlacz A, Legiewicz M, Ciesiolka J. Generating in vitro transcripts with homogenous 3′ ends using trans-acting antigenomic delta ribozyme. Nucleic Acids Res. 2004;32 doi: 10.1093/nar/gnh037. Art. No. e39. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Kao C, Zheng M, Rudisser S. A simple and efficient method to reduce nontemplated nucleotide addition at the 3′ terminus of RNAs transcribed by T7 RNA polymerase. RNA-Publ. RNA Soc. 1999;5:1268–1272. doi: 10.1017/s1355838299991033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Grunbergmanago M, Ochoa S. Enzymatic synthesis and breakdown of polynucleotides - Polynucleotide phosphorylase. J. Am. Chem. Soc. 1955;77:3165–3166. [Google Scholar]
- 80.Grunbergmanago M, Ortiz PJ, Ochoa S. Enzymatic synthesis of nucleic acid-like polynucleotides. Science. 1955;122:907–910. doi: 10.1126/science.122.3176.907. [DOI] [PubMed] [Google Scholar]
- 81.Adami RC, Rice KG. Metabolic stability of glutaraldehyde cross-linked peptide DNA condensates. J. Pharm. Sci. 1999;88:739–746. doi: 10.1021/js990042p. [DOI] [PubMed] [Google Scholar]
- 82.Vanzi F, Vladimirov S, Knudsen CR, Goldman YE, Cooperman BS. Protein synthesis by single ribosomes. RNA-Publ. RNA Soc. 2003;9:1174–1179. doi: 10.1261/rna.5800303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Zlatanova J, Lindsay SM, Leuba SH. Single molecule force spectroscopy in biology using the atomic force microscopy. Prog. Biophys. Mol. Biol. 2000;74:37–61. doi: 10.1016/s0079-6107(00)00014-6. [DOI] [PubMed] [Google Scholar]
- 84.Gore J, Bryant Z, Nollmann M, Le MU, Cozzarelli NR, Bustamante C. DNA overwinds when stretched. Nature. 2006;442:836–839. doi: 10.1038/nature04974. [DOI] [PubMed] [Google Scholar]
- 85.Hansske F, Cramer F. Modification of the 3′ terminus of tRNA by periodate oxidation and subsequent reaction with hydrazides. Meth. Enzymol. 1979;59:172–181. doi: 10.1016/0076-6879(79)59079-x. [DOI] [PubMed] [Google Scholar]
- 86.Willkomm DK, Hartmann RK. In: Handbook of RNA Biochemistry. Hartmann RK, Bindereif A, Schön A, Westhof E, editors. Vol. 1. Weinheim: Wiley-VCH GmbH & Co. KGaA; 2005. pp. 86–94. [Google Scholar]
- 87.Bakin AV, Borisova OF, Shatsky IN, Bogdanov AA. Spatial-organization of template polynucleotides on the ribosome determined by fluorescence methods. J. Mol. Biol. 1991;221:441–453. doi: 10.1016/0022-2836(91)80065-3. [DOI] [PubMed] [Google Scholar]
- 88.Kinoshita Y, Nishigaki K, Husimi Y. Fluorescence-, isotope- or biotin-labeling of the 5′-end of single-stranded DNA/RNA using T4 RNA ligase. Nucleic Acids Res. 1997;25:3747–3748. doi: 10.1093/nar/25.18.3747. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Martin G, Keller W. Tailing and 3′-end labeling of RNA with yeast poly(A) polymerase and various nucleotides. RNA-Publ. RNA Soc. 1998;4:226–230. [PMC free article] [PubMed] [Google Scholar]
- 90.Rosemeyer V, Laubrock A, Seibl R. Nonradioactive 3′-end labeling of RNA molecules of different lengths by terminal deoxynucleotidyltransferase. Anal. Biochem. 1995;224:446–449. doi: 10.1006/abio.1995.1068. [DOI] [PubMed] [Google Scholar]
- 91.Green NH, Williams PM, Wahab O, Davies MC, Roberts CJ, Tendler SJB, Allen S. Single-molecule investigations of RNA dissociation. Biophys. J. 2004;86:3811–3821. doi: 10.1529/biophysj.103.026070. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Itakura K, Rossi JJ, Wallace RB. Synthesis and use of synthetic oligonucleotides. Annu. Rev. Biochem. 1984;53:323–356. doi: 10.1146/annurev.bi.53.070184.001543. [DOI] [PubMed] [Google Scholar]
- 93.Koster DA, Croquette V, Dekker C, Shuman S, Dekker NH. Friction and torque govern the relaxation of DNA supercoils by eukaryotic topoisomerase IB. Nature. 2005;434:671–674. doi: 10.1038/nature03395. [DOI] [PubMed] [Google Scholar]
- 94.Casey J, Davidson N. Rates of formation and thermal stabilities of RNA-DNA and DNA-DNA duplexes at high concentrations of formamide. Nucleic Acids Res. 1977;4:1539–1552. doi: 10.1093/nar/4.5.1539. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Bodkin DK, Knudson DL. Assessment of sequence relatedness of double-stranded-RNA genes by RNA-RNA blot hybridization. J. Virol. Methods. 1985;10:45–52. doi: 10.1016/0166-0934(85)90087-4. [DOI] [PubMed] [Google Scholar]
- 96.Bolton ET, McCarthy BJ. A general method for isolation of RNA complementary to DNA. Proc. Natl Acad. Sci. USA. 1962;48:1390–1397. doi: 10.1073/pnas.48.8.1390. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.McConaug BL, Laird CD, McCarthy BJ. Nucleic acid reassociation in formamide. Biochemistry. 1969;8:3289–3295. doi: 10.1021/bi00836a024. [DOI] [PubMed] [Google Scholar]
- 98.Butzow JJ, Eichhorn GL. Different susceptibility of DNA and RNA to cleavage by metal ions. Nature. 1975;254:358–359. doi: 10.1038/254358a0. [DOI] [PubMed] [Google Scholar]
- 99.Eichhorn GL, Clark P, Tarian E. Interaction of metal ions with polynucleotides and related compounds.13. Effect of metal ions on enzymatic degradation of ribonucleic acid by bovine pancreatic ribonuclease and of deoxyribonucleic acid by bovine pancreatic deoxyribonuclease I. J. Biol. Chem. 1969;244:937–942. [PubMed] [Google Scholar]
- 100.Kaga E, Nakagomi O, Uesugi S. Thermal degradation of RNA-RNA hybrids during hybridization in solution. Mol. Cell. Probes. 1992;6:261–264. doi: 10.1016/0890-8508(92)90026-t. [DOI] [PubMed] [Google Scholar]
- 101.Tenhunen J. Hydrolysis of single-stranded RNA in aqueous solutions - Effect on quantitative hybridizations. Mol. Cell. Probes. 1989;3:391–396. doi: 10.1016/0890-8508(89)90018-2. [DOI] [PubMed] [Google Scholar]
- 102.Dammer U, Hegner M, Anselmetti D, Wagner P, Dreier M, Huber W, Guntherodt HJ. Specific antigen/antibody interactions measured by force microscopy. Biophys. J. 1996;70:2437–2441. doi: 10.1016/S0006-3495(96)79814-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Lee GU, Kidwell DA, Colton RJ. Sensing discrete streptavidin biotin interactions with atomic force microscopy. Langmuir. 1994;10:354–357. [Google Scholar]
- 104.Moy VT, Florin EL, Gaub HE. Intermolecular forces and energies between ligands and receptors. Science. 1994;266:257–259. doi: 10.1126/science.7939660. [DOI] [PubMed] [Google Scholar]
- 105.Pincet F, Husson J. The solution to the streptavidin-biotin paradox: the influence of history on the strength of single molecular bonds. Biophys. J. 2005;89:4374–4381. doi: 10.1529/biophysj.105.067769. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Merkel R, Nassoy P, Leung A, Ritchie K, Evans E. Energy landscapes of receptor-ligand bonds explored with dynamic force spectroscopy. Nature. 1999;397:50–53. doi: 10.1038/16219. [DOI] [PubMed] [Google Scholar]
- 107.Persson T, Willkomm DK, Hartmann RK. In: Handbook of RNA Biochemistry. Hartmann RK, Bindereif A, Schön A, Westhof E, editors. Vol. 1. Weinheim: Wiley-VCH GmbH & Co. KGaA; 2005. pp. 53–74. [Google Scholar]
- 108.Sugino A, Goodman HM, Heyneker HL, Shine J, Boyer HW, Cozzarelli NR. Interaction of Bacteriophage T4 RNA and DNA ligases in joining of duplex DNA at base-paired ends. J. Biol. Chem. 1977;252:3987–3994. [PubMed] [Google Scholar]
- 109.Bain JD, Switzer C. Regioselective ligation of oligoribonucleotides using DNA splints. Nucleic Acids Res. 1992;20:4372. doi: 10.1093/nar/20.16.4372. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Kurschat WC, Muller J, Wombacher R, Helm M. Optimizing splinted ligation of highly structured small RNAs. RNA-Publ. RNA Soc. 2005;11:1909–1914. doi: 10.1261/rna.2170705. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Moore MJ, Query CC. Joining of RNAs by splinted ligation. Methods in Enzymology. 2000;317:109–123. doi: 10.1016/s0076-6879(00)17009-0. [DOI] [PubMed] [Google Scholar]
- 112.Nandakumar J, Ho CK, Lima CD, Shuman S. RNA substrate specificity and structure-guided mutational analysis of bacteriophage T4 RNA ligase 2. J. Biol. Chem. 2004;279:31337–31347. doi: 10.1074/jbc.M402394200. [DOI] [PubMed] [Google Scholar]
- 113.Nishigaki K, Taguchi K, Kinoshita Y, Aita T, Husimi Y. Y-ligation: an efficient method for ligating single-stranded DNAs and RNAs with T4 RNA ligase. Mol. Divers. 1998;4:187–190. doi: 10.1023/a:1009644028931. [DOI] [PubMed] [Google Scholar]
- 114.Strick TR, Allemand JF, Bensimon D, Bensimon A, Croquette V. The elasticity of a single supercoiled DNA molecule. Science. 1996;271:1835–1837. doi: 10.1126/science.271.5257.1835. [DOI] [PubMed] [Google Scholar]
- 115.Bullard DR, Bowater RP. Direct comparison of nick-joining activity of the nucleic acid ligases from bacteriophage T4. Biochem. J. 2006;398:135–144. doi: 10.1042/BJ20060313. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Shapiro DJ. Quantitative ethanol precipitation of nanogram quantities of DNA and RNA. Anal. Biochem. 1981;110:229–231. doi: 10.1016/0003-2697(81)90139-1. [DOI] [PubMed] [Google Scholar]
- 117.Wu W, Welsh MJ. A method for purification of DNA species by high-speed centrifugation of agarose gel slices. Anal. Biochem. 1995;229:350–352. doi: 10.1006/abio.1995.1425. [DOI] [PubMed] [Google Scholar]
- 118.Yaphe W. The use of agarase from Pseudomonas atlantica in the identification of agar in marine algae (Rhodophyceae) Can. J. Microbiol. 1957;3:987–993. doi: 10.1139/m57-109. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.