

Abstract
The many protein processing reactions of the ATP-hydrolyzing Hsp70s are regulated by J cochaperones, which contain J domains that stimulate Hsp70 ATPase activity and accessory domains that present protein substrates to Hsp70s. We report the structure of a J domain complexed with a J responsive portion of a mammalian Hsp70. The J domain activates ATPase activity by directing the linker that connects the Hsp70 nucleotide binding domain (NBD) and substrate binding domain (SBD) towards a hydrophobic patch on the NBD surface. Binding of the J domain to Hsp70 displaces the SBD from the NBD, which may allow the SBD flexibility to capture diverse substrates. Unlike prokaryotic Hsp70, the SBD and NBD of the mammalian chaperone interact in the ADP state. Thus, while both nucleotides and J cochaperones modulate Hsp70 NBD:linker and NBD:SBD interactions, the intrinsic persistence of those interactions differs in different Hsp70s and this may optimize their activities for different cellular roles.
Keywords: Hsp70, Hsc70, J Cochaperone, Molecular Chaperone, X-ray crystallography, DnaJ, DnaK, Protein:Protein Interactions
Introduction
ATP hydrolysis in the Hsp70 NBD drives cycles of conformational changes in the SBD, leading alternately to binding and release of protein substrates (Mayer et al., 2001). Cycles of substrate binding and release by these motors are harnessed by cellular machineries to refold or remodel proteins and protein complexes or to move proteins between subcellular compartments (Sousa and Lafer, 2006). Hsp70s alone have broad substrate specificity (Rudiger et al., 1997), low ATPase rates and little chaperone activity. Specification, activation, and coordination of Hsp70 activity is achieved by J cochaperones (James et al., 1997; Misselwitz et al., 1998). These proteins display a J domain that binds Hsp70s and stimulates their ATPase activity, and one or more accessory domains that bind and present substrates to the chaperone (Walsh et al., 2004).
Some Hsp70s will interact with multiple J cochaperones, while others are specific for a single J protein (Walsh et al., 2004). The basis for such specificity is not well understood, but is, at least in part, believed to reside in sequence variations within the J domain of the cochaperone (Hennessy et al., 2005). An obstacle to structural studies of these interactions is their transience and ATP dependence: upon ATP hydrolysis the J cochaperone:Hsp70 complex dissociates (Bukau and Horwich, 1998; Misselwitz et al., 1999). The NBD is believed to make the primary interactions with the J domain of the cochaperone (Garimella et al., 2006; Suh et al., 1998) and is also the recipient of its ATPase activating signal, but isolated NBDs are not stimulated by J cochaperones (Laufen et al., 1999). It is not clear if this reflects a requirement for direct SBD:J cochaperone contacts or a requirement for the presence of the SBD to allow the Hsp70 to undergo the ATP-driven conformational change that then allows it to bind the cochaperone.
One of the better-characterized Hsp70:J cochaperone pairs is the constitutively expressed mammalian Hsc70 and the neuronal specific J cochaperone auxilin. Auxilin contains a J domain and a clathrin binding domain (Holstein et al., 1996; Ungewickell, 1985; Ungewickell et al., 1997). Together, auxilin and Hsc70 remove clathrin coats from clathrin coated vesicles during synaptic vesicle recycling (Chappell et al., 1986; Morgan et al., 2001; Ungewickell et al., 1995). Structures of the auxilin J domain are known (Gruschus et al., 2004; Jiang et al., 2003), as is the structure of a bovine Hsc70 that contains both the NBD and SBD but is missing the 10 kD C-terminal oligomerization domain (denoted the 2-domain Hsc70 or Hsc70ΔC) (Jiang et al., 2005). Here we describe the structure of auxilin J domain complexed with bovine Hsc70 NBD and with an Hsc70 that lacks the SBD but contains both the NBD and the interdomain linker central to the allosteric mechanism of DnaK and mammalian Hsc70. The linker confers a high basal ATPase rate and J domain responsiveness on the NBD, and J domain binding alters linker conformation, directing it towards a hydrophobic patch on the NBD that transmits the J signal from the linker to the nucleotide in the active site. J domain binding to the NBD requires displacement of the SBD:NBD interaction. This could allow the SBD a greater range of motion so as to accept diverse substrates presented by different J cochaperones.
Unlike what has been reported (Swain et al., 2007) for E. coli Hsp70 (DnaK), we find that bovine Hsc70 NBD and SBD interact in the ADP state of the chaperone. The picture of Hsp70 regulation that emerges is one in which both the nucleotide and J domain modulate SBD:NBD and linker:NBD interactions to control Hsp70 kinetics, and in which the persistence of such interactions may differ in different Hsp70s to tune different Hsp70s for their different cellular roles.
Results
The interdomain linker confers a constitutively high ATPase rate and J cochaperone sensitivity on the NBD
Isolated Hsp70 NBDs and substrate free Hsp70s exhibit low basal ATPase rates (Szabo et al., 1994). Binding of protein substrate to the SBD stimulates this constitutively low ATPase rate several-fold. A fragment of DnaK that lacks the SBD but contains the NBD (aa 1-382) and the interdomain linker (aa 383-392) exhibits a constitutively high ATPase rate (Swain et al., 2007; Vogel et al., 2006). To determine if the corresponding fragment of a mammalian Hsc70 would also exhibit a high basal ATPase rate we measured single-turnover ATPase rates for a bovine Hsc70 construct that encompasses the NBD (aa 1-383) and interdomain linker (aa 384-394). ATPase rates for intact Hsc70, for a functional Hsc70 in which the C-terminal 10 kD oligomerization domain is deleted (Hsc70ΔC), for the Hsc70 NBD (aa 1-386) alone, and for a fragment that encompasses the NBD and interdomain linker (NBD_Linker: aa 1-394) are presented in fig. 1A. The ATPase rate of Hsc70 is stimulated by auxilin J domain (‘+J’: aa 810-910) and by 1 μM Hsc70 substrate peptide (‘+Pep’, seq: ALLLSAPRRGAGKKC). The basal ATPase rate of Hsc70ΔC is higher than that of Hsc70 because it is auto stimulated by binding of its unfolded C-terminus in its substrate-binding site and it is not further stimulated by a substrate peptide (Jiang et al., 2005), but it is stimulated by the J domain. The low ATPase rate of the isolated NBD is not stimulated by J domain, indicating that either the SBD or linker, or both, are required for J cochaperone stimulation. The basal ATPase rate of the NBD_Linker is ~10-fold higher than that of Hsc70, and is further stimulated by the J domain. When we compare the fold stimulation in ATPase rates of the Hsc70ΔC and NBD_Linker as a function of J domain concentration we find that both enzymes are maximally stimulated 5–6 fold by J domain, but the J domain concentration required for half-maximal stimulation of Hsc70ΔC is 1.5 μM, while half-maximal stimulation with the NBD_Linker requires a 10-fold higher J domain concentration (fig. S1). We conclude that: (1) The presence of the interdomain linker confers a high basal ATPase rate on the NBD of mammalian Hsc70, just as is seen with bacterial DnaK; (2) The linker also confers J domain sensitivity on the NBD; (3) Since the maximal stimulation of the ATPase rate is similar for Hsc70 and the NBD_Linker, but requires a higher J domain concentration with the latter, the J domain may make interactions with the SBD that are important for J domain:Hsc70 affinity, but not for stimulation of ATP hydrolysis.
Figure 1. Stimulation of Hsc70 ATPase by free and disulfide-linked J domains.
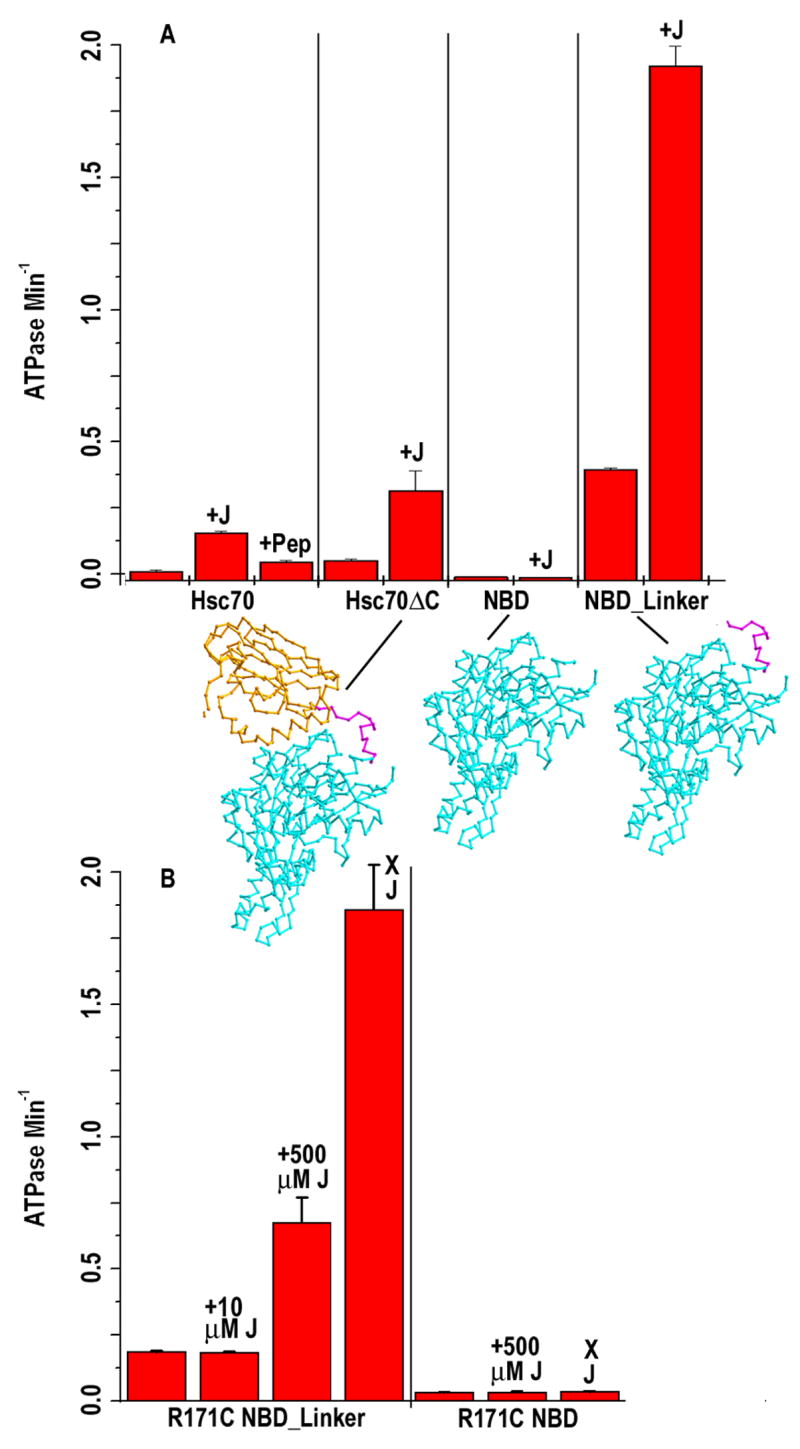
A: Single-turnover ATPase rates for 1 μM Hsc70, Hsc70ΔC (aa 1-554), NBD (aa 1-386) or NBD_Linker (aa 1-394) alone or with 10 μM added J domain (“+ J”). Error bars are ±s.e. for n=3–6. B: As in A, but with R171C substituted NBD or NBD_Linker with D876C J domain either added in solution (at 10 or 500 μM, as indicated) or cross-linked to the Hsc70 (“X J”).
Preparation and crystallization of functional disulfide-linked J domain:Hsc70 complexes
Efforts to crystallize NBD or NBD_Linker complexed with J domain yielded crystals of the individual proteins, but never of the complex, irrespective of the nucleotides or analogs present during crystallization. This may be a consequence of the low affinity and ATP requirement of the Hsc70:auxilin interaction. Though mutant Hsc70s that eliminate ATP hydrolysis have been identified, these mutations also abolish the conformational change in Hsc70 that allows it to bind the J cochaperone(Johnson and McKay, 1999). Nor could we identify non-hydrolysable analogs that stabilized Hsc70 or the NBD_Linker in the J-binding competent conformation. We therefore sought a different route to form a stable J domain:Hsc70 complex for crystallization. Cysteines were substituted for D876 and R171 of auxilin J domain and Hsc70, respectively. These correspond to residues in DnaJ and DnaK that, by genetic co-suppression, are suggested to interact in the DnaJ:DnaK complex (Suh et al., 1998). Air oxidation of D876C J domain with R171C Hsc70 NBD or NBD_Linker led to formation of disulfide-linked complexes, while control experiments with D847C or D849C J domains exhibited little complex formation (fig. S2). The presence of ATP enhanced formation of the complex with the NBD_Linker, but not with the NBD alone.
To determine whether the J domain was coupled to the Hsc70 in a functionally relevant manner, we compared ATPase rate stimulation by J domain in solution to that in the disulfide-linked complex. The basal ATPase rate of the R171C NBD_Linker is not stimulated by 10 μM D876C J domain (fig. 1B). This may reflect reduced affinity of the D876C J domain for R171C NBD_Linker, consistent with results that mutation of the corresponding residues in DnaK or DnaJ reduces DnaJ:DnaK affinity (Jiang et al., 2003; Suh et al., 1998). That this is true is indicated by the observation that a high J domain concentration overcomes this reduced affinity and leads to stimulation of R171C NBD_Linker ATPase by D876C J domain (fig. 1B: ‘+500 μM J’). Thus, the cysteines introduced into the J domain or NBD_Linker reduce the affinity of this interaction but do not abrogate ATPase stimulation. The ATPase rate of the disulfide cross-linked R171C NBD_Linker:D876C J domain complex (fig. 1B: ‘X J’) is 10–11 fold greater than of R171C NBD_Linker alone. The linked J domain is therefore interacting with the NBD_Linker in a manner that stimulates the latter’s ATPase rate. In fact, the fold stimulation in the ATPase rate by the cross-linked J domain is ~2-fold greater than seen with the wt Hsc70 and J domain in solution (figs. 1A, S1), possibly as a consequence of the covalent linkage holding the J domain on the NBD_Linker more persistently in a configuration that leads to ATPase stimulation. In agreement with data obtained with the wt NBD, the R171C NBD is not stimulated by J domain even when the latter is added at very high concentrations (fig 1B: R171C NBD, ‘+500 μM J’). Furthermore, even when the R171C NBD is cross-linked to the J domain (fig. 1B: ‘X J’), its ATPase rate is not enhanced. This reveals that the inability of the NBD to be stimulated by the J domain is not due to the latter failing to bind or binding inappropriately to the NBD, since, when identically cross-linked to the NBD and NBD_Linker the J domain stimulates the ATPase rate of the latter, but not the former. Instead, these observations show that the linker is both necessary and sufficient to confer J domain sensitivity on the NBD.
Since these assays revealed that the J domain is cross-linked to the Hsc70 in a functional (ATPase stimulating) configuration, we screened both NBD:J domain and NBD_Linker:J domain complexes for crystallization and obtained crystals of ADP*Pi and AMPPNP forms. Structures of the auxilin J domain and Hsc70 NBD in isolation had been previously determined, but to allow for comparison of the structures of all of these proteins in their isolated vs. J domain complexed forms we also determined crystal structures of the isolated NBD_Linker in different nucleotide states. Statistics on these 7 structures are in Table s1.
The intermolecular interface in the complex identifies residues critical for the J cochaperone:Hsc70 interaction
The J-domain is bound in the cleft between lobes IA and IIA of the Hsc70 NBD, with the Hsc70 interdomain linker and the linker proximal portion of this cleft forming the Hsc70 side of the intermolecular interface (fig. 2A, B). Though the cross-linked complex in solution is functional, as shown by effects on NBD ATPase rates, the 2 partners in this complex can rotate about the disulfide linkage, so it is possible that the crystal has captured a non-functional orientation of the J domain. If this structure does reveal the functional complex then it should corroborate previous analyses of J cochaperone:Hsp70 interactions and should predict previously unidentified interactions. The intermolecular interactions in the crystal bury 1028 Å of protein surface and involve most of J domain helix 3 and the helix 2 C-terminus (including the signature ‘HPD’ motif; fig. 2A–C). These are the elements identified by NMR and mutational analysis in polyomavirus T antigen J domain (PyJ) as binding mammalian Hsc70 (Fewell et al., 2002; Garimella et al., 2006; Whalen et al., 2005).
Figure 2. The auxilin J domain:NBD_Linker complex.
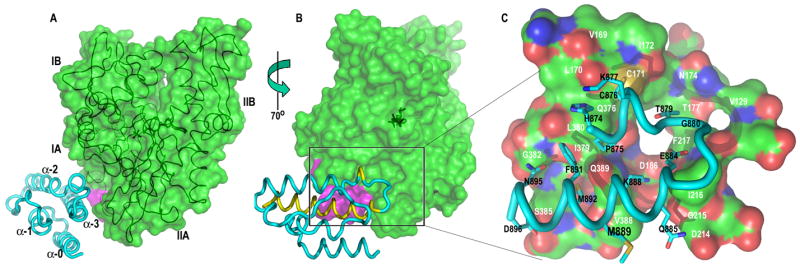
A: Auxilin J Domain:NBD_Linker complex with J domain (cyan) in ribbon representation and NBD_Linker rendered as a transparent surface (green; with aa 383–390 in magenta) with the path of the polypeptide chain shown as a coil and the bound nucleotide in stick representation. B: Model from A rotated as indicated. In yellow on the J domain are regions corresponding to those mapped by NMR (in the polyoma virus T-antigen) to be involved in interaction with Hsc70 (Garimella et al., 2006). C: The region indicated by the box in B expanded to identify residues important for the J domain:Hsc70 interaction. These are labeled with white lettering on the surface of the Hsc70, which is colored green, red, and blue for carbon, oxygen, and nitrogen atoms, respectively, and with black lettering on the J domain with stick representations of the side-chains of relevant J domain residues colored cyan, red, and blue for carbon, oxygen, and nitrogen atoms, respectively.
The intermolecular interaction involves H-bonds between the J domain H784 imidazole ring and the Hsc70 L170 carbonyl (fig. S2), and between the D896 and S385 side-chains (fig. 2C). L380 and I179 make hydrophobic interactions with F891 and HP874,875. The V388 side- and main-chains, respectively, pack on M892 and M889. The C-terminal portion of the turn between J domain helix 2 and 3 (aa 878-885) and aliphatic elements of the E884 and Q885 side-chains are packed against the I216 side-chain. In addition to these direct interactions there are several water-mediated H-bonds contributed by the main and side-chain atoms of residues identified in fig. 2C. Many of these residues have been previously shown to be important for Hsp70 interaction with the J cochaperone. Mutations in the Hsc70 or DnaK interdomain linker reduce or eliminate J domain stimulation of ATPase activity (Jiang et al., 2005; Laufen et al., 1999), and mutations of V388 and L393 reduce binding to auxilin, as have mutations of Hsc70 I216 (Jiang et al., 2005). Significantly, I216 mutants were identified as exceptional amongst several of Hsc70 mutants, in that they exhibited the most severe effects on auxilin binding and auxilin dependent clathrin coat disassembly, but had less severe effects on ATP dependent Hsc70 conformational changes, leading to the speculation that I216 mutations affect conformational mechanisms in Hsc70 that were not detected in the assays used (Jiang et al., 2005). This speculation now appears unwarranted, as the effects of the I216 mutants are most simply explained as due to direct effects on auxilin binding. Finally, Gross and colleagues previously identified not only DnaK R167 (corresponding to Hsc70 R171), but also N170 and T173 (corresponding to N174 and T177) as important for the DnaK:DnaJ interaction (Suh et al., 1998). Both of these residues form part of the intermolecular interface in the complex (fig. 2C) and part of a network of water-mediated H-bonds that includes J domain residues T879 and E884.
To test the ability of the complex structure to predict new interactions we constructed an Hsc70 L380G mutant. This mutant was unresponsive to the J domain (fig. 3), consistent with an important role for this residue in the J domain interaction. We also characterized the ability of the J domain to stimulate the ATPase activity of a previously described set of Hsc70 mutants (Jiang et al., 2005). Most of these mutants were similar to WT in their response to the J domain (not shown), though some actually exhibited increased stimulation (fig. 6). As expected, an I216T mutant was not stimulated by the J domain (fig. 3), and at low J domain concentrations V388C and L393C mutants were also unresponsive (not shown). Unexpectedly, at high J domain concentrations the ATPase rates of the linker mutants were inhibited ~3-fold by J (fig. 3). This result highlights the central role of the interdomain linker in mediating the response to J domain: sufficiently high concentrations of J can overcome the lower affinity of the linker mutants, but the resulting complex is catalytically inhibited.
Figure 3. Effects of J domain on the ATPase rates of WT and mutant Hsc70ΔC enzymes.
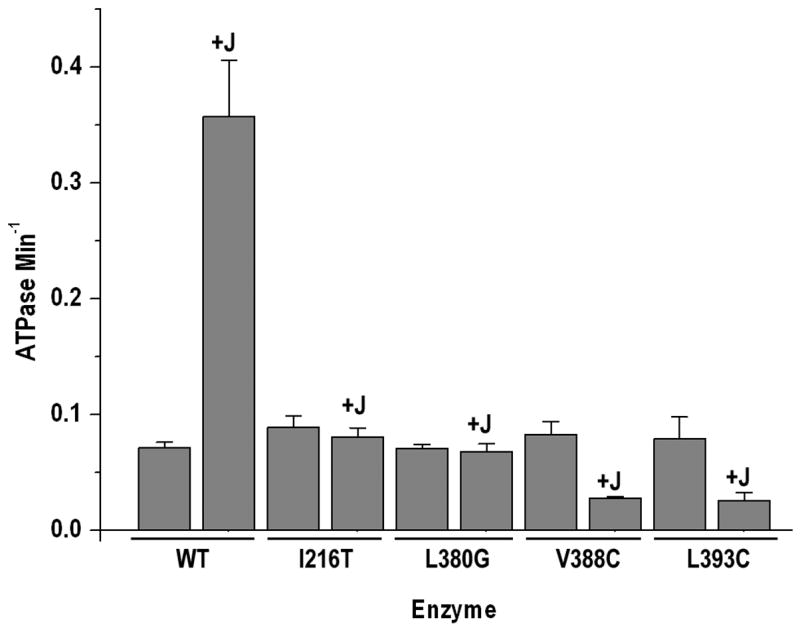
Experimental conditions as in fig. 1A, but with J domain (+J) added at 20 μM. Error bars are ±s.e. for n=3.
Figure 6. J domain stimulation of Hsc70 ATPase activity requires displacement of the SBD from the NBD.
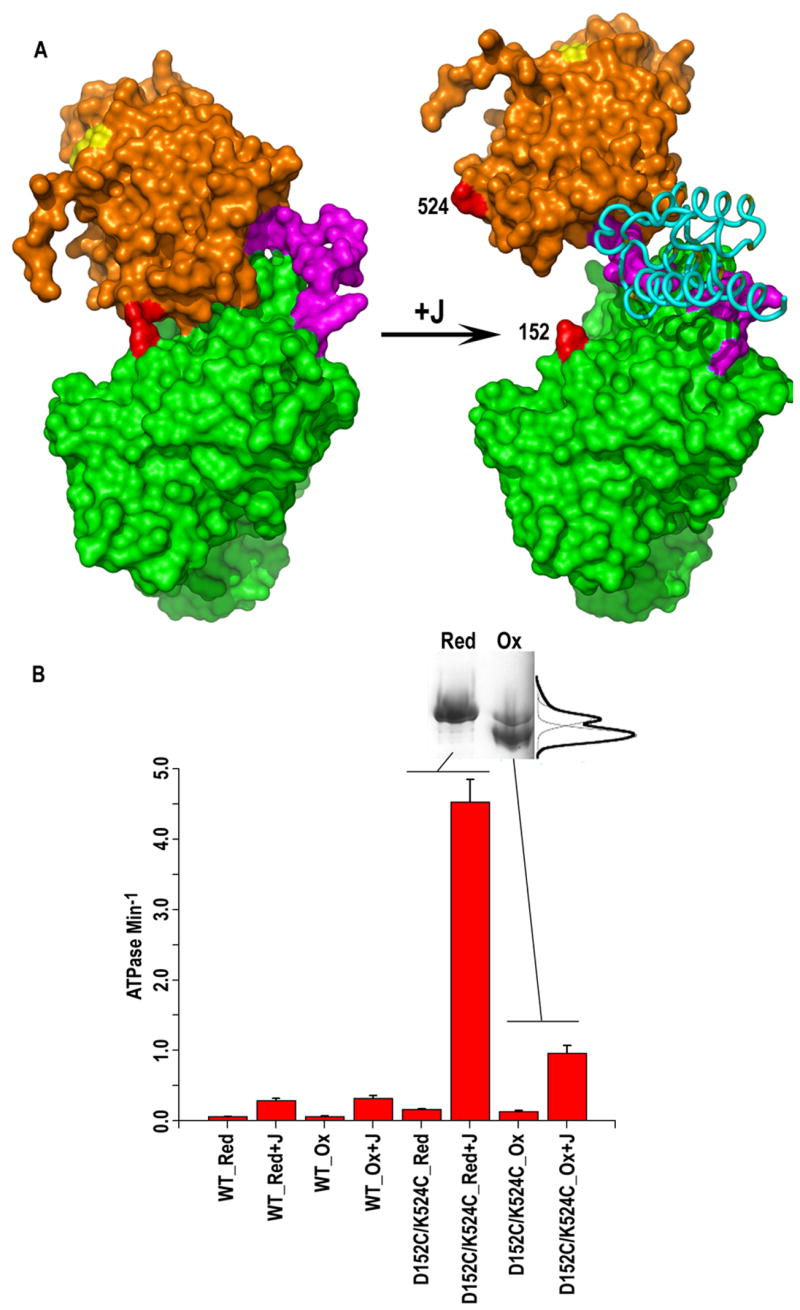
A. Left: Structure of the 2-domain Hsc70 (Hsc70ΔC: pdb 1YUW(Jiang et al., 2005)) with the SBD (aa 397-554) in orange, the NBD (aa 1-382) in green, the interdomain linker (aa 383-396) in magenta, and residues K524 and D152 in red. Right: Structure of the NBD_Linker:J domain complex with the J domain in cyan and NBD and linker (extending to aa 390) colored as on the left. The SBD and linker (aa 392-554) from the left-hand structure are placed to suggest how the SBD would have to move away from the NBD to allow the J domain to bind and to allow the linker to interact with the NBD. B: Rates of ATP hydrolysis for WT or D152C/K524C 2-domain (aa 1-554) Hsc70 under oxidizing (“Ox”) or reducing (“Red”=2 mM DTT) conditions in the absence or presence of J domain. The inset gel of D152C/K524C resolved on SDS PAGE under reducing or oxidizing conditions shows that ~80% of the enzyme forms a faster migrating, disulfide bonded species in the oxidizing environment.
J domain binding modulates linker conformation
While the structure of the auxilin J-domain (pdb 1NZ6) has been previously determined (Jiang et al., 2003), structures of an Hsp70 NBD_Linker fragment have not been previously described. We therefore determined structures of the NBD_Linker (aa 1-394) in nucleotide free, ADP, and ADP*Vi states (Table S1) to allow comparison of the isolated domains to their conformations in the complex. The conformations of the J-domain and aa 1-382 of the NBD in the free and complexed states were similar with mean RMS differences of 0.3–0.4 Å, but the linker conformation in the complex differed from that seen in the isolated NBD_Linker or 2-domain Hsc70 (pdb 1YUW). Structures of the isolated NBD_Linker in nucleotide free, ADP, and ADP*Vi states revealed electron density only to aa 384, as did crystals of isolated NBDs (aa 1-386) in ADP*Vi or nucleotide free conformations. Therefore, even though the presence of the linker alters the biochemical properties of the NBD (fig. 1), it is disordered in the structure of the isolated NBD_Linker. This suggests either that all 3 NBD_Linker structures have captured a state that is not reflective of the linker conformation in solution, or that the ATPase stimulating effect of the linker is mediated by transient interactions with the NBD that may be stabilized by J domain binding to stimulate ATPase activity.
Consistent with the latter idea, in the crystal of the J domain:NBD_Linker complex, clear electron density for the linker extends to residue 386, and weaker electron density consistent with 2 alternate linker conformations extends to residue 390 (fig. 4; The absence of electron density for the complete linker does not reflect proteolysis as mass spec analysis of dissolved crystals revealed that in over 90% of the sample the linker extends to residue 394 with the remaining fraction corresponding to a fragment that extends to aa 393). In one of these conformations (fig. 4B: “Linker in”) the linker approaches Hsc70 residues I181 and Y371. These correspond, respectively, to DnaK L177 and I373, both identified by NMR chemical shift analysis as residues likely to contact the linker in an a DnaK NBD_Linker fragment (Swain et al., 2007). To determine if these residues are, in fact, involved in mediating the ATPase stimulating effect of the interdomain linker and the J cochaperone, we constructed I181A and Y371A mutants and measured their ATPase rates in the absence and presence of the J domain (fig. 4C). The unstimulated ATPase activity of I181A is modestly higher than that of the WT enzyme, but addition of J domain inhibits the ATPase activity of I181A ~10-fold, an effect that is qualitatively similar, but quantitatively greater, than that seen with the V388C and L393C linker mutants. Y371A, on the other hand, exhibits a 7-fold increase in its unstimulated ATPase rate, relative to WT, but this higher rate is stimulated only ~1.4 fold by the J cochaperone. Y371A therefore appears to be constitutively J-stimulated and unresponsive to further J-stimulation, while the I181A phenotype implies that inappropriate NBD:linker interactions in the J cochaperone complex can strongly catalytically inhibit the NBD. These phenotypes, together with our x-ray and previous NMR results (Swain et al., 2007), suggest a direct role for Y371 and I181 in mediating the ATPase stimulating effect of the J cochaperone through interactions with the interdomain linker.
Figure 4. J domain induced changes in linker conformation may activate ATPase through interactions with Y371 and I181.
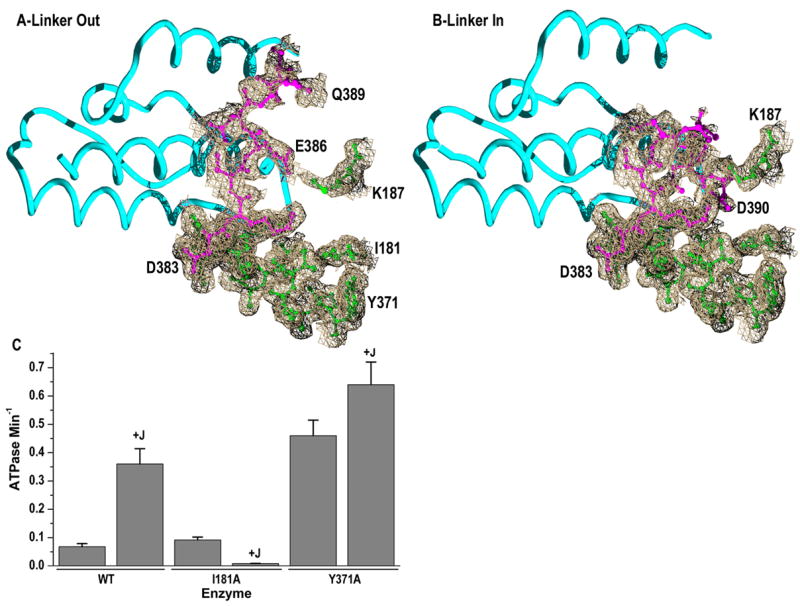
A: Structures of the J domain (cyan) and Hsc70 residues 371–389, 181, and 187 with the linker in the ‘Out’ conformation. Hsc70 linker residues 383–389 and 371–382+181+187 are in magenta and green, respectively. The ED around the illustrated Hsc70 residues is contoured at 0.5 σ. B: As in A, but with the linker in the ‘In’ conformation and extending to residue 390; average B-factors for linker residues 383–389 (“Out”) or 383–390 (“In”) are 55 and 56, respectively, while the average B-factor for residues 3–382 of the NBD is 28. C: Effects of J domain on the ATPase rates of WT and mutant Hsc70ΔC enzymes. Experimental conditions as in fig. 1, but with Hsc70ΔC and J domain (+J) at 10 and 25 μM, respectively. Error bars are ±s.e. for n=3.
Elements of transmission of the J signal to the NBD active site
The nucleotide and active site structures of free and J domain complexed NBDs with bound ADP*Pi are similar and do not reveal how J stimulates ATPase activity. Crystals of J domain:NBD_Linker complexed with the slowly hydrolyzed analog AMPPNP were therefore prepared. These crystals grew under identical conditions as the ADP*Pi crystals and in the same space group and unit cell (Table s1) but data from these crystals was consistently more mosaic than from the ADP*Pi crystals, with poorer electron density and higher B-factors in the refined structures (Table s1), implying a higher degree of disorder or flexibility in the AMPPNP vs. ADP*Pi crystals. In the active site, electron density for the base, ribose, and α- and β-phosphates of the AMPPNP was identical to that seen in the ADP*Pi crystals, but the electron density suggested 2 locations for the γ-phosphate, one corresponding to the Pi seen in the product (ADP*Pi) complex, and the other corresponding to a γ-phosphate in intact AMPPNP, suggesting that the AMPPNP crystal is composed of a mixture of intact and hydrolyzed forms of nucleotide.
The AMPPNP*J domain:NBD_Linker structure was compared to that of an isolated AMPPNP*NBD (Flaherty et al., 1994). If these structures are superimposed by optimizing the alignment of the ribose, α- and β-phosphates of their respective nucleotides, then the γ-phosphate in the J:NBD_Linker complex is seen to shifted by 3.5 Å towards E175 and residues 172–181 are shifted by 0.6–1.3 Å between the two structures (fig. 5A; these shifts are larger than the overall coordinate errors, estimated at 0.1–0.15 Å for the 1.7 Å ADP*Pi structures and 0.2–0.3 Å for the 2.2 Å AMPPNP structure; Table S1). The latter changes involve elements that interact with the J domain (aa 172-177) or that mediate transduction of the J signal to the NBD through interactions with the linker (aa 181). It is unclear if these small, localized changes can explain how the J domain stimulates Hsc70 ATPase activity. It is possible that the activated conformation of the ATPase is not revealed in this structure, either because that conformation is transient, or because AMPPNP does not stabilize that conformation or crystal packing interactions inhibit its formation. However, catalysis at the Hsc70 ATPase site is sensitive to small changes in the position of catalytic groups (Wilbanks and McKay, 1998), and the 5–6 fold stimulation in ATPase rates conferred by the J could domain could be induced, at least in part, through relatively small local changes in the structure of Hsc70 elements that interact with both the ATP and the J domain or linker. Inspection of fig. 5A suggests that E175, which has been previously shown to catalytically important (Johnson and McKay, 1999) and is within a stretch of residues that interact with the J domain and that transmit the J signal from the linker to the NBD, could be an active site residue that transmits the J signal directly to the nucleotide. This would predict that mutation of E175 would affect the ability of the Hsc70 to be stimulated by the J domain. To test this we, constructed and characterized an E175S NBD_Linker mutant. Ultrafiltration confirmed that this mutant associates with the J domain in an ATP dependent manner. The basal ATPase rate of E175S is 100-fold lower than that of the wt NBD_Linker (fig. 5B). This low rate is not stimulated by the J domain but is, in fact, reduced 30–40% by J (fig. 5B: ‘+J’). Failure to be stimulated by the J domain is not simply an attribute of any catalytically compromised mutant, as we tested an active site T204A mutant that had an 8-fold lower basal ATPase rate and found that it was still responsive to J domain stimulation (not shown). Instead, these results are consistent with involvement of E175 in transmission of the J signal to the nucleotide.
Figure 5. Elements of transmission of the J signal to the NBD.
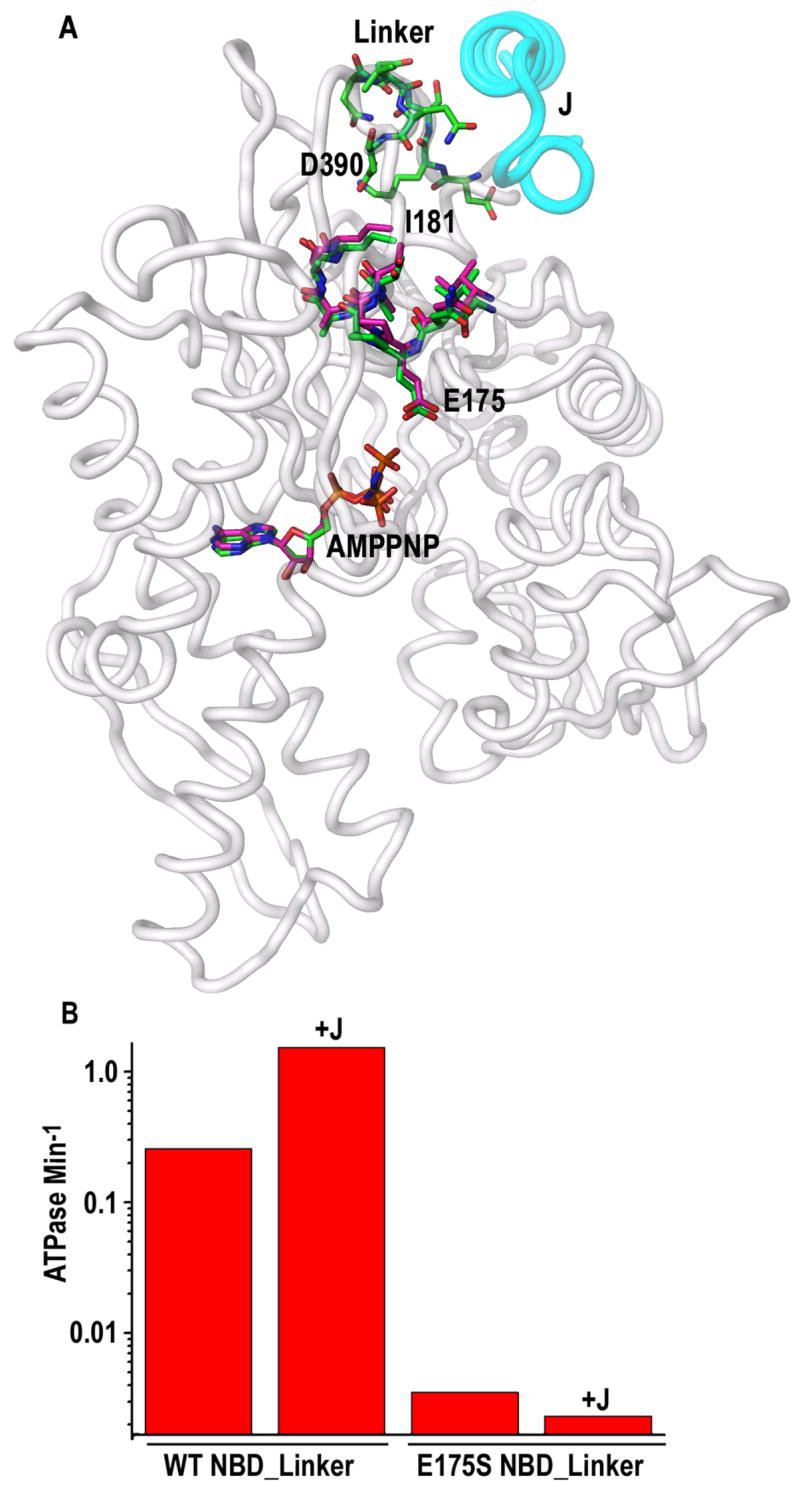
A: Stick representations of the AMPPNP, Hsc70 residues 172–181, 383–390 (“Linker”) from the J Domain:NBD_Linker structure are shown superimposed on the AMPPNP and residues 172–181 from an isolated NBD complexed with AMPPNP (pdb 1NGJ (Flaherty et al., 1994)). Alignment of the ribose, α- and β-phosphates of the nucleotides from each structure was optimized. Nitrogens, oxygens, and phosphates are colored blue, red, and orange, respectively. The AMPPNP γ-phosphate in J-domain complexed structure has swung towards E175. Carbons from the J-domain complexed or uncomplexed NBD are colored, respectively, green and magenta. Ribbon representaions of J-domain residues 874–895 and NBD residues 3–386 are shown in cyan and light grey, respectively. B. ATPase rates of WT or E715S NBD_Linker in the absence or presence (‘+J’) of 10 μM J domain.
Binding of the J cochaperone displaces the SBD:NBD interaction
Superposition of the J domain:NBD_Linker complex on the structure of the 2-domain Hsc70 generates steric clashes between the SBD and J domain, implying that the SBD must be displaced from the NBD for J domain to bind (fig. 6A). To test this we prepared a 2-domain Hsc70 in which the SBD and NBD are locked by a disulfide between cysteines substituted for D152 and K524 in the NBD and SBD, respectively. Though the distance between these 2 residues’ α-carbons (8.5 Å) is not optimal for a disulfide, air oxidation of D152C/K524C leads to formation of a disulfide bonded species in ~80% of the molecules as assessed by migration on non-reducing vs. reducing SDS PAGE (inset in fig. 6B; the remaining 20% cannot be induced to form a disulfide, cannot be removed with thiol-sepharose and may represent mixed sulfoxides). Single (D152C or K542C) mutants and WT enzymes showed no shifts in migration upon similar treatment (not shown). ATPase rates for the WT enzyme measured under oxidizing or reducing conditions were similar, and were both increased ~5-fold by addition of saturating levels of J domain (fig. 6B; oxidation/reduction also did not affect the basal or stimulated ATPase rates of the single mutants). The unstimulated ATPase rates of the oxidized or reduced D152C/K524C mutant were also similar but were 2–3-fold greater than the unstimulated rate of the WT enzyme (fig. 6B). The increased basal ATPase rate is a consequence of mutation of D152 and has been noted previously in Hsc70 (Jiang et al., 2005) and for mutants of the corresponding D148 in DnaK (Gassler et al., 1998). The ATPase activity of reduced D152C/K524C enzyme is strongly stimulated (~20-fold) by J domain, but the degree of stimulation of the oxidized enzyme is only ~20% of that seen with the reduced form (fig. 6B). This residual stimulation can be entirely accounted for by the ~20% that does not form a disulfide bond upon air oxidation. Since oxidation/reduction does not affect basal or stimulated ATPase rates of the WT or singly mutated enzymes, this indicates that it is the interdomain linkage that inhibits J domain stimulation of ATPase activity and that such stimulation requires displacement of the SBD:NBD interaction.
The mammalian Hsc70 NBD and SBD interact in the ADP State
The observation that a cross-link between the SBD and NBD abrogates Hsc70 response to the J domain implies that J binding modulates SBD:NBD interactions (and vice versa). Nucleotide binding is also believed to regulate interdomain interactions. Since we are able to induce formation of a disulfide bond between the NBD and SBD under nucleotide free conditions this suggests that the NBD and SBD interact, at least transiently, under such conditions. The crystal structure of a functional 2-domain Hsc70 in a nucleotide free state reveals extensive interdomain interactions (Jiang et al., 2005). However, a recent NMR study of E. coli DnaK concluded that the NBD and SBD interact only when ATP binds, and that in the ADP and nucleotide free states (which are believed to be conformationally similar) the 2-domains do not interact and behave like ‘beads-on-a-string’ (Swain et al., 2007). The latter conclusion would be at odds with an analysis of deuterium exchange in DnaK that concluded that, even in the nucleotide free state, the NBD and SBD in DnaK mutually stabilize each others’ structures as revealed by reductions in deuteron exchange rates in the intact enzyme vs. the isolated domains (Rist et al., 2006). However, it’s possible that transient interdomain interactions in the 2-domain DnaK are sufficient to induce mutual structural stabilization. The conclusion that the NBD and SBD in DnaK do not interact in the ADP or nucleotide free states was based on the observation that the peaks for an isolated NBD (or SBD) in an HSQC spectrum were identical to the corresponding peaks in a 2-domain DnaK unless ATP was present (Swain et al., 2007). To see if a similar result would obtain with bovine Hsc70 we collected HSQC spectra for the ADP states of an isolated NBD and a 2-domain Hsc70 (fig. 7). Superposition of these spectra reveals many peaks from the NBD spectrum (red and purple outlines in fig. 7) that are either missing or chemically shifted in the spectrum of the 2-domain protein (black and gray peaks). This indicates changes in the chemical environment of many residues in the NBD vs. 2-domain enzyme and well as peak broadening in the 2-domain Hsc70, due either to its larger size and/or exchange between multiple conformations. An effective understanding of these conformational mechanisms will require a more extensive NMR analysis, but inspection of the spectra in fig. 7 reveals that, whatever the case for DnaK, bovine Hsc70 in the ADP state cannot be described as 2 noninteracting domains.
Figure 7.
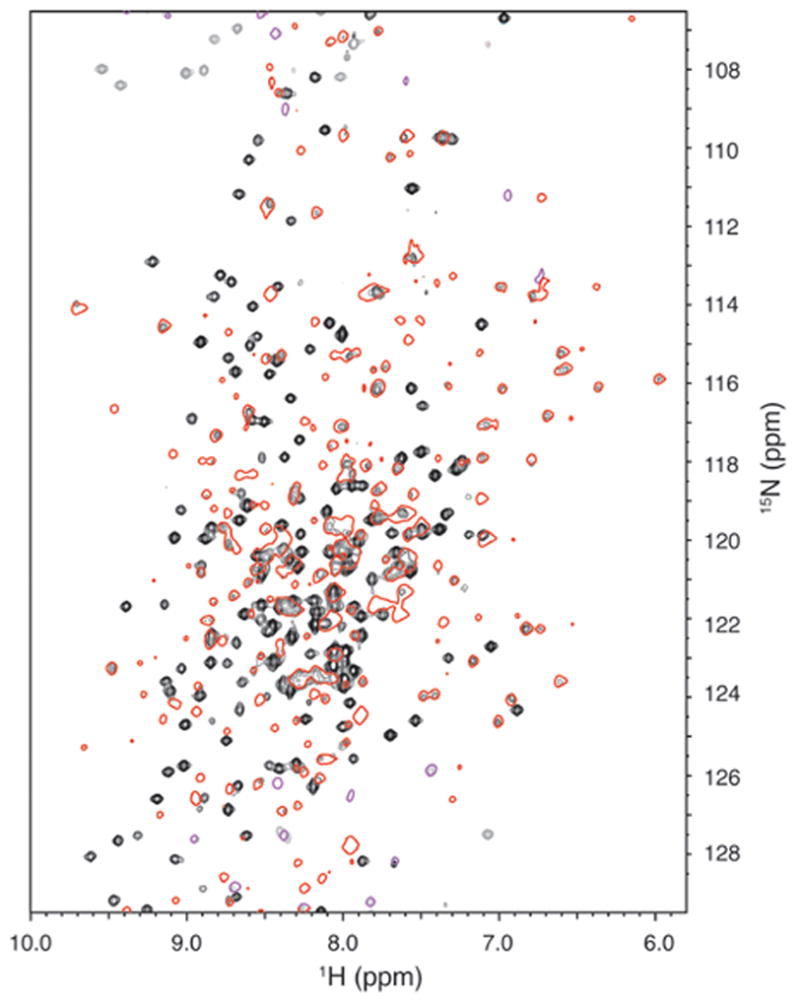
NMR spectra reveal interactions between the Hsc70 NBD and SBD in the ADP state. HSQC spectrum of 2-domain (aa 1-554) Hsc70 (black with folded peaks in gray) superimposed on that of the NBD (aa 1-386; red outlines with folded peaks in purple) in 1 mM ADP.
Discussion
Approaches analogous to that described here, in which a disulfide bond is used to stabilize a protein:protein complex for crystallization, were previously used to cross-link HIV-1 RT and DNAP I to DNA to restrict the lateral mobility of these proteins on their templates and facilitate crystallization (Beese et al., 1993; Huang et al., 1998). The crystal structure of a protein:protein complex cross-linked by engineered cysteines has also been described (Guo et al., 2004) but, in that case, corresponded to a stable complex whose structure had previously been determined without cross-links. Here we have used cross-linking to crystallize a transient and previously uncrystallized protein:protein complex. Such an approach can be generally applied to study transiently interacting proteins but biochemical data must confirm the functional relevance of such linked complexes. In our case, not only is the complex structure able to account for and predict intermolecular interactions identified by mutational and NMR analyses (fig. 2), we also demonstrate its functionality in its higher ATPase activity relative to the isolated Hsc70 (fig. 1B). This allows us to unambiguously conclude that the interdomain linker is required to transmit the J signal to Hsc70 and not merely for J cochaperone binding since we observe ATPase stimulation in the NBD_Linker:J domain complex but not in the NBD:J complex (even though the structures reveal J domain bound in identical orientations in both complexes).
A role for the linker in Hsp70 allostery was first suggested by observations of alternate conformations of a partial linker in a DnaK SBD fragment (Zhu et al., 1996). A central role for the linker in Hsp70 mechanism was explicitly proposed based on a characterization of 23 Hsc70 mutants that revealed that direct or allosteric disruption of an ATP-driven reduction in linker accessibility to protease resulted in loss of chaperone function (Jiang et al., 2005). It was proposed that this ATP-driven change in linker conformation involved insertion of the exposed linker into a hydrophobic patch in the Hsc70 interdomain interface. Analysis of deuterium exchange rates in DnaK confirmed an ATP-driven exposed to buried transition in the linker (Rist et al., 2006). In the structures described here, the J domain induces ordering of most of the linker, which is otherwise invisible in electron density maps of the NBD_Linker alone. In one of the 2 observed conformations the linker is directed towards a hydrophobic patch on the NBD surface (fig. 4b). This patch includes amino acids Y371 and I181, whose DnaK homologues (I373 and L177) were identified by NMR as residues with which the linker may interact to activate the ATPase (Swain et al., 2007). The phenotypes of Y731A and I181A support their identification as key residues through which the J cochaperone stimulates Hsc70 ATPase activity (fig. 4c). These data indicate that transmission of the J signal follows the pathway: J→Linker→(Y371, I181).
Subsequent steps in the transmission of this signal, which must reach into the active site, are less certain since it is unclear if the small differences in nucleotide and active site structure between an AMPPNP*NBD structure and an AMPNP*J*NBD_Linker structure explain J stimulation of Hsc70 ATPase activity. However, the DnaK active site residue E171 was previously identified as important in DnaK allostery (Buchberger et al., 1994) and possibly in regulation by DnaJ (Fan et al., 2003). The corresponding residue in Hsc70 is E175 and--given its 100-fold contribution to rate enhancement and proximity to residues that interact with the J domain and mediate ATPase stimulation--a role for this residue in transmission of the J signal to the nucleotide is plausible and is supported by the observation that its mutation abolishes J stimulation (figs. 5A,B).
An important question is whether unstimulated and J stimulated ATP hydrolysis proceed through the same conformational state. It would be simplest to explain J stimulation as occurring by stabilization of linker:NBD interactions that form even in the absence of the J cochaperone. This is especially so since, even in the absence of the J cochaperone, ATP binding leads to a reduction in linker accessibility that could most easily be explained if the linker is interacting identically with the same hydrophobic patch with which it interacts during J stimulated ATP hydrolysis. However, our mutational analyses may rule out this simple explanation. Mutation of linker residues V388 or L393 or of NBD I181 causes the Hsc70 ATPase rate to be inhibited, rather than stimulated, by the J cochaperone (fig. 3, 4), with the I181A mutant exhibiting a dramatic 10-fold inhibition of its ATPase rate by the J cochaperone (corresponding to 50–60 fold inhibition when compared to the J-stimulated rate of WT Hsc70). However, these mutations have no effect on the unstimulated ATPase rate. If unstimulated and J-stimulated ATP hydrolysis depend on identical linker:I181 interactions that are simply stabilized by the cochaperone, then the I181 and linker mutations should affect both stimulated and unstimulated ATPase rates. Instead, these observations suggest that linker:NBD interactions differ during J-stimulated and unstimulated ATP hydrolysis.
However, the ATP driven conformational change in the linker may be coupled to partial disruption of the NBD:SBD interaction (Jiang et al., 2005) and is required for J cochaperone binding to Hsc70 (Misselwitz et al., 1999; Misselwitz et al., 1998). In fact, we find that displacement of the SBD from the NBD is required to allow J domain to bind as well as to allow linker access to the hydrophobic patch on the NBD that is otherwise buried by the SBD (fig. 6A). Interestingly, the ATPase rate of the D152C/K524C mutant is stimulated ~4-fold more than the WT Hsc70 by the J domain (fig. 6B). The singly substituted enzymes show an identical quantitative increase in their response to the J domain (not shown) and were also shown to display enhanced binding to auxilin (Jiang et al., 2005). We do not fully understand such effects but these mutations may weaken NBD:SBD interactions to facilitate SBD displacement and productive J domain binding. This displacement may have other functions. Substrates presented by J proteins to Hsp70s are heterogeneous (i.e., denatured proteins or protein aggregates) and many Hsp70s work with multiple J cochaperones, each of which will have a distinct substrate presentation domain. It seems unlikely that these different J cochaperones will all present their distinct and heterogeneous substrates to the Hsp70 with identical geometry. Displacement of the SBD from the NBD could allow the SBD to sample a wider range of positions so as to increase its possibility of capturing substrate.
These results reveal a picture of Hsp70 regulation in which not only the nucleotide, but also the J cochaperone, modulate NBD:SBD and NBD:linker interactions, but our conclusion that the SBD and NBD of mammalian Hsc70 interact in the ADP state (figs. 5, 6) differs from that reached in a recent study of a 2-domain DnaK (Swain et al., 2007). Other studies have also suggested that the equilibria between different conformational states may be set differently in different Hsp70s. While NMR analyses of a 2-domain E. coli DnaK indicate that these domains interact only in the ATP state (Swain et al., 2007), similar studies of a 2-domain T. thermophilus DnaK concluded that ‘the nucleotide binding domain and substrate binding domain are closely associated in all ligand states’, with the interdomain interface being tighter in the ADP vs. ATP state (Revington et al., 2005). These differences might be attributed to differences in the nature of the mutations and truncations introduced into these 2-domain proteins to facilitate their characterization (Swain et al., 2007), rather than to intrinsic species differences in conformational equilibria. However, even comparison of wt NBDs from different Hsp70s reveals variations in intrinsic conformational dynamics. Thus, NMR studies indicate that the T. thermophilus DnaK NBD in the ADP state is in slow exchange between 2 conformations that collapse into a single conformation when ATP binds (Revington et al., 2004). The E. coli DnaK NBD apparently exhibits the opposite behavior and is conformationally more heterogeneous in the ATP vs. ADP state (Swain et al., 2007), while the bovine Hsc70 NBD exhibits exchange between multiple conformations in both ATP and ADP states (Zhang and Zuiderweg, 2004). It is possible that all Hsp70s share the same fundamental mechanism but the equilibria between different conformational states may be set differently in different Hsp70s to tune them for different cellular roles.
Methods
Preparation of WT and mutant proteins and disulfide linked complexes
Full-length bov taurus Hsc70 (aa 1-650), Hsc70ΔC or 2-domain Hsc70 (aa 1-554), NBD_Linker (aa 1-394), and NBD (aa 1-386) were expressed without fusion tags and purified as described (Jiang et al., 2005). Bov taurus auxilin J-domain (aa 810-910) was prepared as an N-terminal GST fusion protein and purified by glutathione affinity chromatography (Jiang et al., 2003). 15N-labeled proteins were prepared by growth in minimal media supplemented with 15N NH4Cl. To form disulfide-linked complexes D876C GST-auxilin J domain was mixed with either R171C Hsc70 NBD or NBD_Linker at J domain:Hsc70 molar ratios of 1:5 in 10 mM Tris pH 8.0, 50% glycerol, 1mM DTT, 5 mM MgCl2, 50 mM KCl, 2.5 mM ATP and dialyzed overnight into 2×100-fold excess of the same buffer without DTT. This led to near complete conversion of the J-domain to disulfide-linked complex. The complex was purified by glutathione affinity chromatography using on-column thrombin digestion to remove the complex from the resin, followed by ion-exchange chromatography on Q- and SP-sepharose. Complexes were concentrated to 40 mg/ml and stored in 10 mM Tris pH 8.0, 1mM EDTA, 50% glycerol, and 1 mM oxidized glutathione.
Crystallization and structure determination
Complexes were screened for crystallization directly from storage buffer after being brought to 5 mM MgCl2, 50 mM KCl, and 2 mM ATP or AMPPNP as described (Jiang et al., 2006). Crystals appeared in a variety of conditions but the best grew as long prisms in 0.2M Ammonium Acetate, 0.1M Tris pH 8.5, 25% PEG3350 (for NBD:J domain) or 0.2 M Calcium Acetate, 20% PEG3350 (NBD_Linker:J domain). Crystals were frozen directly from the mother liquor. Data were collected at the Advanced Synchrotron Light Source beamline 5.0.1 and on a Rigaku Cu rotating anode X-ray source with imaging plate detector and processed with DENZO and SCALEPACK(Otwinowski et al., 2003). Molecular replacement and refinement were done with Phaser (Storoni et al., 2004), MolRep(Vagin and Teplyakov, 2000), CNS(Brunger et al., 1990), and RefMac (Winn et al., 2003), and model building with O (Jones et al., 1991).
ATPase assays
ATPase rates were measured as described (Jiang et al., 2005).
NMR data collection and analysis
Trosy-HSQC spectra (Pervushin et al., 1997; Rance et al., 1999) were collected at 27 °C with 15N labeled samples at concentrations of 30 mg mL−1 in 20 mM KH2PO4 pH 7.5, 5 mM MgCl2, 1 mM ADP, 5% 2H2O using a Bruker Avance 700 spectrometer equipped with a 1H-(13C,15N) cryoprobe. Spectra were recorded with 96 scans, a recycle time of 2 sec, and acquisition times of 0.110 and 0.0935 s in the F1 and F2 dimensions, respectively.
Supplementary Material
Acknowledgments
Supported by GM-52522, AQ-1486, and 0755057Y from the NIH, Welch Foundation, and AHA, respectively (to R.S.) and NS29051 from NINDS (to E.M.L.). Support for the X-ray Crystallography Core, the Mass Spectroscopy Core, the NMR Core and the UTHSCSA Center for Macromolecular Interactions from the UTHSCSA Executive Research Committee and San Antonio Cancer Institute is acknowledged. Coordinates and structure factors for the following have been deposited at the PDB: NBD-linker/apo (PDB ID: 2QW9), NBD-linker/ADP (PDB ID: 2QWL), NBD-linker/ADP*Vi (PDB ID: 2QWM), NBD:J/ADP*Pi (PDB ID: 2QWN), NBD-linker:J/ADP*Pi (#1) (PDB ID: 2QWO), NBD-linker:J/ADP*Pi (#2) (PDB ID: 2QWP), NBD-linker:J/AMPPNP hydrolyzed form (PDB ID: 2QWQ), NBD-linker:J/AMPPNP intact form (PDB ID: 2QWR)
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Beese LS, Derbyshire V, Steitz TA. Structure of DNA polymerase I Klenow fragment bound to duplex DNA. Science. 1993;260:352–355. doi: 10.1126/science.8469987. [DOI] [PubMed] [Google Scholar]
- Brunger AT, Krukowski A, Erickson JW. Slow-cooling protocols for crystallographic refinement by simulated annealing. Acta Crystallogr A . 1990;46(Pt 7):585–593. doi: 10.1107/s0108767390002355. [DOI] [PubMed] [Google Scholar]
- Buchberger A, Valencia A, McMacken R, Sander C, Bukau B. The chaperone function of DnaK requires the coupling of ATPase activity with substrate binding through residue E171. Embo J. 1994;13:1687–1695. doi: 10.1002/j.1460-2075.1994.tb06433.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bukau B, Horwich AL. The Hsp70 and Hsp60 chaperone machines. Cell. 1998;92:351–366. doi: 10.1016/s0092-8674(00)80928-9. [DOI] [PubMed] [Google Scholar]
- Chappell TG, Welch WJ, Schlossman DM, Palter KB, Schlesinger MJ, Rothman JE. Uncoating ATPase is a member of the 70 kilodalton family of stress proteins. Cell. 1986;45:3–13. doi: 10.1016/0092-8674(86)90532-5. [DOI] [PubMed] [Google Scholar]
- Fan CY, Lee S, Cyr DM. Mechanisms for regulation of Hsp70 function by Hsp40. Cell Stress Chaperones. 2003;8:309–316. doi: 10.1379/1466-1268(2003)008<0309:mfrohf>2.0.co;2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fewell SW, Pipas JM, Brodsky JL. Mutagenesis of a functional chimeric gene in yeast identifies mutations in the simian virus 40 large T antigen J domain. Proc Natl Acad Sci U S A. 2002;99:2002–2007. doi: 10.1073/pnas.042670999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Flaherty KM, Wilbanks SM, DeLuca-Flaherty C, McKay DB. Structural basis of the 70-kilodalton heat shock cognate protein ATP hydrolytic activity. II. Structure of the active site with ADP or ATP bound to wild type and mutant ATPase fragment. J Biol Chem. 1994;269:12899–12907. [PubMed] [Google Scholar]
- Garimella R, Liu X, Qiao W, Liang X, Zuiderweg ER, Riley MI, Van Doren SR. Hsc70 contacts helix III of the J domain from polyomavirus T antigens: addressing a dilemma in the chaperone hypothesis of how they release E2F from pRb. Biochemistry. 2006;45:6917–6929. doi: 10.1021/bi060411d. [DOI] [PubMed] [Google Scholar]
- Gassler CS, Buchberger A, Laufen T, Mayer MP, Schroder H, Valencia A, Bukau B. Mutations in the DnaK chaperone affecting interaction with the DnaJ cochaperone. Proc Natl Acad Sci U S A. 1998;95:15229–15234. doi: 10.1073/pnas.95.26.15229. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gruschus JM, Han CJ, Greener T, Ferretti JA, Greene LE, Eisenberg E. Structure of the functional fragment of auxilin required for catalytic uncoating of clathrin-coated vesicles. Biochemistry. 2004;43:3111–3119. doi: 10.1021/bi0354740. [DOI] [PubMed] [Google Scholar]
- Guo M, Bhaskar B, Li H, Barrows TP, Poulos TL. Crystal structure and characterization of a cytochrome c peroxidase-cytochrome c site-specific cross-link. Proc Natl Acad Sci U S A. 2004;101:5940–5945. doi: 10.1073/pnas.0306708101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hennessy F, Nicoll WS, Zimmermann R, Cheetham ME, Blatch GL. Not all J domains are created equal: implications for the specificity of Hsp40-Hsp70 interactions. Protein Sci. 2005;14:1697–1709. doi: 10.1110/ps.051406805. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Holstein SE, Ungewickell H, Ungewickell E. Mechanism of clathrin basket dissociation: separate functions of protein domains of the DnaJ homologue auxilin. J Cell Biol. 1996;135:925–937. doi: 10.1083/jcb.135.4.925. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang H, Chopra R, Verdine GL, Harrison SC. Structure of a covalently trapped catalytic complex of HIV-1 reverse transcriptase: implications for drug resistance. Science. 1998;282:1669–1675. doi: 10.1126/science.282.5394.1669. [DOI] [PubMed] [Google Scholar]
- James P, Pfund C, Craig EA. Functional specificity among Hsp70 molecular chaperones. Science. 1997;275:387–389. doi: 10.1126/science.275.5298.387. [DOI] [PubMed] [Google Scholar]
- Jiang J, Lafer EM, Sousa R. Crystallization of a Functionally Intact Bovine Hsc70. Acta Crystallogr F. 2006;62 doi: 10.1107/S1744309105040303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jiang J, Prasad K, Lafer EM, Sousa R. Structural Basis of Interdomain Communication in the Hsc70 Chaperone. Molecular Cell. 2005;20:513–524. doi: 10.1016/j.molcel.2005.09.028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jiang J, Taylor AB, Prasad K, Ishikawa-Brush Y, Hart PJ, Lafer EM, Sousa R. Structure-function analysis of the auxilin J-domain reveals an extended Hsc70 interaction interface. Biochemistry. 2003;42:5748–5753. doi: 10.1021/bi034270g. [DOI] [PubMed] [Google Scholar]
- Johnson ER, McKay DB. Mapping the role of active site residues for transducing an ATP-induced conformational change in the bovine 70-kDa heat shock cognate protein. Biochemistry. 1999;38:10823–10830. doi: 10.1021/bi990816g. [DOI] [PubMed] [Google Scholar]
- Jones TA, Zou JY, Cowan SW, Kjeldgaard M. Improved methods for building protein models in electron density maps and the location of errors in these models. Acta Crystallogr A . 1991;47(Pt 2):110–119. doi: 10.1107/s0108767390010224. [DOI] [PubMed] [Google Scholar]
- Laufen T, Mayer MP, Beisel C, Klostermeier D, Mogk A, Reinstein J, Bukau B. Mechanism of regulation of hsp70 chaperones by DnaJ cochaperones. Proc Natl Acad Sci U S A. 1999;96:5452–5457. doi: 10.1073/pnas.96.10.5452. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mayer MP, Brehmer D, Gassler CS, Bukau B. Hsp70 chaperone machines. Adv Protein Chem. 2001;59:1–44. doi: 10.1016/s0065-3233(01)59001-4. [DOI] [PubMed] [Google Scholar]
- Misselwitz B, Staeck O, Matlack KE, Rapoport TA. Interaction of BiP with the J-domain of the Sec63p component of the endoplasmic reticulum protein translocation complex. J Biol Chem. 1999;274:20110–20115. doi: 10.1074/jbc.274.29.20110. [DOI] [PubMed] [Google Scholar]
- Misselwitz B, Staeck O, Rapoport TA. J proteins catalytically activate Hsp70 molecules to trap a wide range of peptide sequences. Mol Cell. 1998;2:593–603. doi: 10.1016/s1097-2765(00)80158-6. [DOI] [PubMed] [Google Scholar]
- Morgan JR, Prasad K, Jin S, Augustine GJ, Lafer EM. Uncoating of clathrin-coated vesicles in presynaptic terminals: roles for Hsc70 and auxilin. Neuron. 2001;32:289–300. doi: 10.1016/s0896-6273(01)00467-6. [DOI] [PubMed] [Google Scholar]
- Otwinowski Z, Borek D, Majewski W, Minor W. Multiparametric scaling of diffraction intensities. Acta Crystallogr A. 2003;59:228–234. doi: 10.1107/s0108767303005488. [DOI] [PubMed] [Google Scholar]
- Pervushin K, Riek R, Wider G, Wuthrich K. Attenuated T2 relaxation by mutual cancellation of dipole-dipole coupling and chemical shift anisotropy indicates an avenue to NMR structures of very large biological macromolecules in solution. Proc Natl Acad Sci U S A. 1997;94:12366–12371. doi: 10.1073/pnas.94.23.12366. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rance M, Loria JP, Palmer AGr. Sensitivity improvement of transverse relaxation-optimized spectroscopy. J Magn Reson. 1999;136:92–101. doi: 10.1006/jmre.1998.1626. [DOI] [PubMed] [Google Scholar]
- Revington M, Holder TM, Zuiderweg ER. NMR study of nucleotide-induced changes in the nucleotide binding domain of Thermus thermophilus Hsp70 chaperone DnaK: implications for the allosteric mechanism. J Biol Chem. 2004;279:33958–33967. doi: 10.1074/jbc.M313967200. [DOI] [PubMed] [Google Scholar]
- Revington M, Zhang Y, Yip GN, Kurochkin AV, Zuiderweg ER. NMR investigations of allosteric processes in a two-domain Thermus thermophilus Hsp70 molecular chaperone. J Mol Biol. 2005;349:163–183. doi: 10.1016/j.jmb.2005.03.033. [DOI] [PubMed] [Google Scholar]
- Rist W, Graf C, Bukau B, Mayer MP. Amide hydrogen exchange reveals conformational changes in hsp70 chaperones important for allosteric regulation. J Biol Chem. 2006;281:16493–16501. doi: 10.1074/jbc.M600847200. [DOI] [PubMed] [Google Scholar]
- Rudiger S, Germeroth L, Schneider-Mergener J, Bukau B. Substrate specificity of the DnaK chaperone determined by screening cellulose-bound peptide libraries. Embo J. 1997;16:1501–1507. doi: 10.1093/emboj/16.7.1501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sousa R, Lafer EM. Keep the traffic moving: mechanism of the Hsp70 motor. Traffic. 2006;7:1596–1603. doi: 10.1111/j.1600-0854.2006.00497.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Storoni LC, McCoy AJ, Read RJ. Likelihood-enhanced fast rotation functions. Acta Crystallogr D Biol Crystallogr. 2004;60:432–438. doi: 10.1107/S0907444903028956. [DOI] [PubMed] [Google Scholar]
- Suh WC, Burkholder WF, Lu CZ, Zhao X, Gottesman ME, Gross CA. Interaction of the Hsp70 molecular chaperone, DnaK, with its cochaperone DnaJ. Proc Natl Acad Sci U S A. 1998;95:15223–15228. doi: 10.1073/pnas.95.26.15223. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Swain J, Dinler G, Sivendran R, Montgomery D, Stotz M, Gierasch L. Hsp70 Chaperone Ligands Control Domain Association via an Allosteric Mechanism Mediated by the Interdomain Linker. Mol Cell. 2007;26:27–39. doi: 10.1016/j.molcel.2007.02.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Szabo A, Langer T, Schroder H, Flanagan J, Bukau B, Hartl FU. The ATP hydrolysis-dependent reaction cycle of the Escherichia coli Hsp70 system DnaK, DnaJ, and GrpE. Proc Natl Acad Sci U S A. 1994;91:10345–10349. doi: 10.1073/pnas.91.22.10345. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ungewickell E. The 70-kd mammalian heat shock proteins are structurally and functionally related to the uncoating protein that releases clathrin triskelia from coated vesicles. Embo J. 1985;4:3385–3391. doi: 10.1002/j.1460-2075.1985.tb04094.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ungewickell E, Ungewickell H, Holstein SE. Functional interaction of the auxilin J domain with the nucleotide- and substrate-binding modules of Hsc70. J Biol Chem. 1997;272:19594–19600. doi: 10.1074/jbc.272.31.19594. [DOI] [PubMed] [Google Scholar]
- Ungewickell E, Ungewickell H, Holstein SE, Lindner R, Prasad K, Barouch W, Martin B, Greene LE, Eisenberg E. Role of auxilin in uncoating clathrin-coated vesicles. Nature. 1995;378:632–635. doi: 10.1038/378632a0. [DOI] [PubMed] [Google Scholar]
- Vagin A, Teplyakov A. An approach to multi-copy search in molecular replacement. Acta Crystallogr D Biol Crystallogr. 2000;56:1622–1624. doi: 10.1107/s0907444900013780. [DOI] [PubMed] [Google Scholar]
- Vogel M, Mayer MP, Bukau B. Allosteric regulation of hsp70 chaperones involves a conserved interdomain linker. J Biol Chem. 2006;281:38705–38711. doi: 10.1074/jbc.M609020200. [DOI] [PubMed] [Google Scholar]
- Walsh P, Bursac D, Law YC, Cyr D, Lithgow T. The J-protein family: modulating protein assembly, disassembly and translocation. EMBO Rep. 2004;5:567–571. doi: 10.1038/sj.embor.7400172. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Whalen KA, de Jesus R, Kean JA, Schaffhausen BS. Genetic analysis of the polyomavirus DnaJ domain. J Virol. 2005;79:9982–9990. doi: 10.1128/JVI.79.15.9982-9990.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wilbanks SM, McKay DB. Structural replacement of active site monovalent cations by the epsilon-amino group of lysine in the ATPase fragment of bovine Hsc70. Biochemistry. 1998;37:7456–7462. doi: 10.1021/bi973046m. [DOI] [PubMed] [Google Scholar]
- Winn MD, Murshudov GN, Papiz MZ. Macromolecular TLS refinement in REFMAC at moderate resolutions. Methods Enzymol. 2003;374:300–321. doi: 10.1016/S0076-6879(03)74014-2. [DOI] [PubMed] [Google Scholar]
- Zhang Y, Zuiderweg ER. The 70-kDa heat shock protein chaperone nucleotide-binding domain in solution unveiled as a molecular machine that can reorient its functional subdomains. Proc Natl Acad Sci U S A. 2004;101:10272–10277. doi: 10.1073/pnas.0401313101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhu X, Zhao X, Burkholder WF, Gragerov A, Ogata CM, Gottesman ME, Hendrickson WA. Structural analysis of substrate binding by the molecular chaperone DnaK. Science. 1996;272:1606–1614. doi: 10.1126/science.272.5268.1606. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.