

Abstract
Peroxiredoxins (Prx) are widely distributed and abundant proteins, which control peroxide concentrations and related signaling mechanisms. Prx1 is found in the cytoplasm and nucleus, but little is known about compartmentalized Prx1 function during redox signaling and oxidative stress. We targeted expression vectors to increase Prx1 in nuclei (NLS-Prx1) and cytoplasm (NES-Prx1) in HeLa cells. Results showed that NES-Prx1 inhibited NF-κB activation and nuclear translocation. In contrast, increased NLS-Prx1 did not affect NF-κB nuclear translocation but increased activity of a NF-κB reporter. Both NLS-Prx1 and NES-Prx1 inhibited NF-κB p50 oxidation, suggesting that oxidation of the redox-sensitive cysteine in p50′s DNA binding domain is regulated via peroxide metabolism in both compartments. Interestingly, following treatment with H2O2, nuclear thioredoxin-1 (Trx1) redox status was protected by NLS-Prx1, and cytoplasmic Trx1 was protected by NES-Prx1. Compartmental differences from increasing Prx1 show that the redox poise of cytoplasmic and nuclear thiol systems can be dynamically controlled through peroxide elimination. Such spatial resolution and protein-specific redox differences imply that the balance of peroxide generation/metabolism in microcompartments provides an important specific component of redox signaling.
Introduction
Reactive oxygen species (ROS) can be generated through multiple types of stimuli: physiologic (cytokine or growth factors), xenobiotics and toxicants, etc. Overproduction of ROS causes oxidative stress and can lead to macromolecule damage and eventual cellular toxicity. However, at lower, non-toxic concentrations, ROS can stimulate or inhibit certain elements of redox-sensitive signal transduction pathways to yield a specific response, implicating ROS as effective second messengers. The production and duration of an ROS-mediated response can be regulated by antioxidants, such as glutathione and α-tocopherol, and antioxidant enzyme systems, such as superoxide dismutase, catalase and thioredoxin/peroxiredoxin.
Peroxiredoxins (Prx) are antioxidant enzymes that have peroxidase functions and are found in various subcellular compartments. Peroxiredoxin-1, -2 and -5 are found in both the cytoplasm and nucleus and work with thioredoxin-1 to effectively detoxify hydrogen peroxide (H2O2). Typical 2-cysteine Prxs, such as Prx1 and Prx2, reduce H2O2 to yield water and form a covalent Prx dimer through the formation of an intermolecular disulfide bond. Oxidized typical 2-cysteine Prxs can be reduced by reduced thioredoxin (Trx). Oxidized Trx is then reduced by thioredoxin reductase using NADPH as an electron donor [1, 2]. The presence of multiple peroxiredoxins distributed among subcellular compartments suggests that the peroxiredoxins could play important and perhaps distinct roles at different sites within cells.
This possibility is supported by recent studies which show that embryonic fibroblasts from Prx1 -/- mice have a preferential accumulation of ROS within the nucleus, while Prx1 +/+ show a preferential accumulation of ROS within the cytoplasm [3]. These studies suggest that Prx1 is a critical element for the regulation of ROS specifically in the nucleus. Nuclear Prx5 targeted to the nucleus confers resistance to oxidant-induced cell death as well as to DNA damage [4].
During redox signaling, some Prxs have also been implicated in the regulation of NF-κB through the initial activation in the cytoplasm by controlling the components affecting I-κB phosphorylation and subsequent dissociation [5]. In principle, Prxs could have a different function in the nucleus because NF-κB interactions with DNA are governed by a redox-sensitive cysteine (Cys62) on the p50 subunit of the NF-κB dimer [6]. Oxidation of Cys62 inhibits NF-κB binding and decreases the effectiveness of NF-κB signaling [7]. Past studies have shown that targeting Trx1 to the nucleus enhances NF-κB and other transcription factor activities [8, 9]. The effect of nuclear Trx1 is believed to be a result of the reduction of redox-sensitive cysteines in the DNA binding domain in these transcription factors. Thus, nuclear Prxs could contribute to control of nuclear NF-κB activity by altering the concentration of oxidant which drives the oxidative inactivation of the transcription factor.
Because DNA binding is a nuclear event, it is feasible that nuclear components are primarily responsible for the regulation of this process. Indeed, recent research shows that nuclear Trx1 is more reduced than cytoplasmic Trx1 and preferentially protected against oxidation during metabolic energy limitation induced by glucose- and glutamine-free media [10]. Here, we utilize nuclear- and cytoplasmic-targeted Prx1 to investigate compartment-specific redox events during oxidative stress and redox signaling. Nuclear content is increased by expressing a fusion protein of Prx1 containing 3 nuclear localization signals (NLS-Prx1), and cytoplasmic content is increased by expressing a fusion protein containing a nuclear export signal (NES-Prx1). Nuclear translocation of NF-κB p50 is used as a reporter of cytoplasmic activation, redox state of p50 is used as an indicator of the balance of redox-sensitive oxidation/reduction of the critical DNA-binding component, and an NF-κB reporter is used to measure overall activity of the NF-κB system.
Experimental Procedures
Construction of NES-Prx1 and NLS-Prx1 expression vectors
Human peroxiredoxin-1 expressed in pENTR(tm)221 was obtained from Invitrogen Life Technologies (Carlsbad, CA) and cloned into pCMV/myc/nuc (Invitrogen, Carlsbad, CA) between the NcoI and XhoI restriction sites in the multiple cloning site. This vector contains a nuclear localization sequence (NLS) from the SV40 large T antigen and a c-myc tag. The MAPKK nuclear export signal (NES), ALQKKLEELELDE [11], was synthesized and cloned into c-myc-tagged pCMV/myc/cyto expression vector (Invitrogen, Carlsbad, CA) between the NcoI and XhoI sites in the multiple cloning region. Human peroxiredoxin-1 in pENTR(tm)221 (Invitrogen, Carlsbad, CA) was cloned into pCMV/myc/cyto/NES between the XhoI and NotI restriction sites in the multiple cloning region. C-myc tags and additional localization sequences were placed on the c-terminus of the human Prx1and increased Prx1s size by approximately 3-5 kDa.
Localization of targeted Prx1
Subcellular localization was confirmed by nuclear/cytoplasmic fractionation using the NE-PER kit (Pierce, Rockland, IL) followed by standard Western blotting. Briefly, HeLa cells (10 cm dish) were transfected using Fugene 6 (Roche Applied Science, Indianapolis, IN) per manufacturers’ instructions. 24 h after transfection, cells were trypsinized and lysed following the NE-PER protocol for cellular fractionation. Fractionated lysates were stored at -80°C until Western blot analysis. Protein samples (50 μg) were separated by SDS/12% PAGE, transferred onto a PVDF membrane and probed for myc-tag (Cell Signaling Technologies, Boston, MA, USA) and Prx1 (LabFrontiers, S. Korea). Bands were visualized using the Odyssey scanner (LI-COR, Lincoln, NE). These results also show significant quantities of Prx1 in both compartments under normal, untreated conditions.
MTT assay
HeLa cells in a 96-well plate were transfected with either empty vector, NES-Prx1 or NLS-Prx1 for 24 h prior to exposure to H2O2. Transfected cells were dosed with 0-2000 μM H2O2 for 16 h after which toxicity was evaluated by MTT conversion to a formazan as described by Denizot and Lang [12]. Briefly, after H2O2 exposure, cells were loaded with 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide (MTT) (5 mg/ml final concentration) for 2 h. Medium was then removed and DMSO was added. Absorbance was read on a M5 microplate reader (Molecular Devices, Sunnydale, CA) at 550 nm and correlates to the amount of live cells.
NF-κB activity
HeLa cells were co-transfected with NF-κB luciferase reporter constructs (Invitrogen) and with either empty vector, NES-Prx1 or NLS-Prx1 for 24 h. Transfected cells were dosed with 0-2000 μM H2O2 for 4 h and then collected in reporter lysis buffer (Promega, Madison, WI). Luciferase assays (Promega) were performed by measuring luminescence on a M5 microplate reader as suggested by the manufacturer. Cells were transfected with a β-galactosidase vector which was used to normalize luciferase activity and evaluate transfection efficiency.
Transfected cells were treated with 0-2000 μM H2O2 for 1 h after which they were fractionated for the collection of nuclear and cytoplasmic proteins. Collection was achieved with the Nu-Per nuclear/cytoplasmic protein kit (Pierce, Rockford, IL) per the manufacturer’s instructions. Fractions were normalized to protein content and then separated by SDS-PAGE, electroblotted to a nitrocellulose membrane and probed with a mouse antibody for p50 (Santa Cruz Biotechnology, Santa Cruz, CA). The secondary antibody, anti-mouse AlexaFluor 680, was purchased from Invitrogen. Membranes were scanned using the Odyssey detection system (LI-COR, Lincoln, NE).
Evaluation of p50 redox state
The redox state of p50 was determined by methods analogous to those described by Kim et al. [13] for the determination of thioredoxin reductase redox states. HeLa cells were transfected with either empty vector, NES-Prx1 or NLS-Prx1 for 24 h and then treated with H2O2 for 20 min. Cells were collected in an oxygen-free lysis buffer (50 mM Bis-Tris-HCl (pH 6.5), 0.5% Triton X-100, 0.5% deoxycholate, 0.1% SDS, 150 mM NaCl, 1 mM EDTA and protease inhibitors) containing 20 μM biotinylated iodoacetamide (BIAM). Samples were incubated at 37°C for 10 min in the dark after which the reaction was stopped by the addition of 5 mM unbiotinylated iodoacetamide (IAM). p50 was immunoprecipitated by anti-p50 rabbit polyclonal antibodies using the Protein G Immunoprecipitation Kit (Sigma) per the manufacturers instructions. Isolates were separated by SDS-PAGE electrophoresis and electroblotted to nitrocellulose membranes. Membranes were probed with a streptavidin labeled with Alexa Fluor 680 (Invitrogen). Membranes were scanned using the Odyssey detection system (LI-COR, Lincoln, NE). Intensity of the bands directly corresponds to the amount of reduced p50.
Nuclear and cytoplasmic Trx1 redox state
HeLa cells were transfected with either empty, NES-Prx1 or NLS-Prx1 expression vectors for 24 h as described above. Transfected cells were then treated with 1 mM H2O2 for up to 120 min. Cultures were collected at 0, 20, 60 and 120 min of treatment. Nuclear and cytoplasmic fractionation techniques for Trx1 redox states were performed as previously described (Watson, 2003). Oxidized and reduced fractions were separated by non-reducing electrophoresis and then electroblotted to nitrocellulose membranes. Membranes were probed for Trx1 with an antibody to human Trx1 (American Diagnostica, Greenwich, CT). An alexafluor 680 anti goat antibody was used for secondary detection. Membranes were scanned using the Odyssey detection system (LI-COR, Lincoln, NE). Band intensities were then used for determination of the Trx1 re dox potential (Eh) by the Nernst equation [14, 15].
Results
NLS-Prx1, but not NES-Prx1, confers resistance to oxidant-induced apoptosis
Successful targeting of Prx1 to the nucleus and cytoplasm of HeLa cells was verified by the immunoblotting of nuclear and cytoplasmic fractionation lysates from transiently transfected cells and probing with a Prx1 antibody (Figure 1).
Figure 1.

Immunoblot of nuclear and cytoplasmic targeted Prx1. HeLa cells were transfected with empty vector, NLS-Prx1 and NES-Prx1 and fractionated for nuclear and cytosolic lysates. Fractions were separated by SDS-PAGE, transferred to nitrocellulose and probed with a Prx1 antibody. Results show successful targeting of Prx1 to both the nucleus and cytoplasm.
Transfected cells were dosed with 0-2000 μM H2O2 for 16 h. MTT assay of treated cells shows a dose-dependent increase in toxicity in empty vector transfected cells (Figure 2). Interestingly, no significant differences were observed in cells transfected with NES-Prx1, showing nearly the exact same sensitivity to H2O2. NLS-Prx1 transfection conferred significantly more protection from H2O2 at 500 μM and higher concentrations. This difference could be due to enhanced protection by increased activation of the NF-κB system (see below) or a result of increased expression of the NLS-Prx1 vs. NES-Prx1 (see Figure 1).
Figure 2.
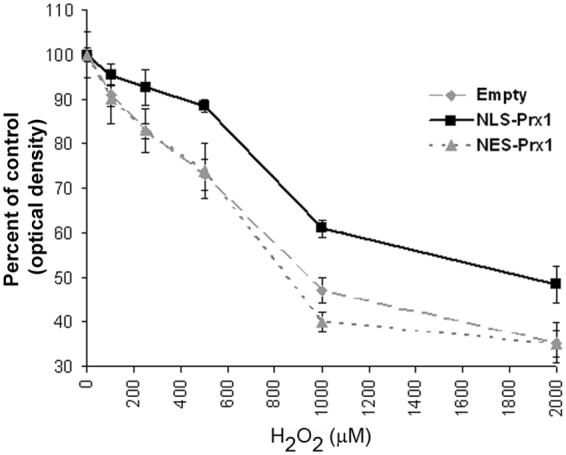
MTT assay for H2O2-induced cell death with empty vector, NES-Prx1 and NLS-Prx1 expression. Data are expressed as percent changes from respective, untreated controls and show that oxidant-induced toxicity was significantly decreased in cells overexpressing NLS-Prx1 at concentrations in excess of 500 μM H2O2. Cells transfected with NES-Prx1 showed no significant differences from empty vector controls. Asterisks (*) denote a statistically significant difference (p<0.05).
NES-Prx1 alters NF-κB activation
To examine effects of increased nuclear and cytoplasmic Prx1 on NF-κB activation in the cytoplasm, contents of the p50 subunit in the respective fractions were measured at 1 h after treatment with an activating pulse of H2O2. In the cytoplasmic fraction of cells treated with an empty vector control, cells showed a decrease in p50 levels with increasing concentrations of H2O2 (Figure 3A). Cells transfected with NLS-Prx1 showed a similar pattern of cytoplasmic p50 concentrations, consistent with the interpretation that nuclear Prx1 does not affect cytoplasmic activation of NF-κB. In contrast, cells treated with the NES-Prx1 construct showed only a minor decrease in cytoplasmic p50 concentrations at the highest (2000 μM H2O2) concentration.
Figure 3.
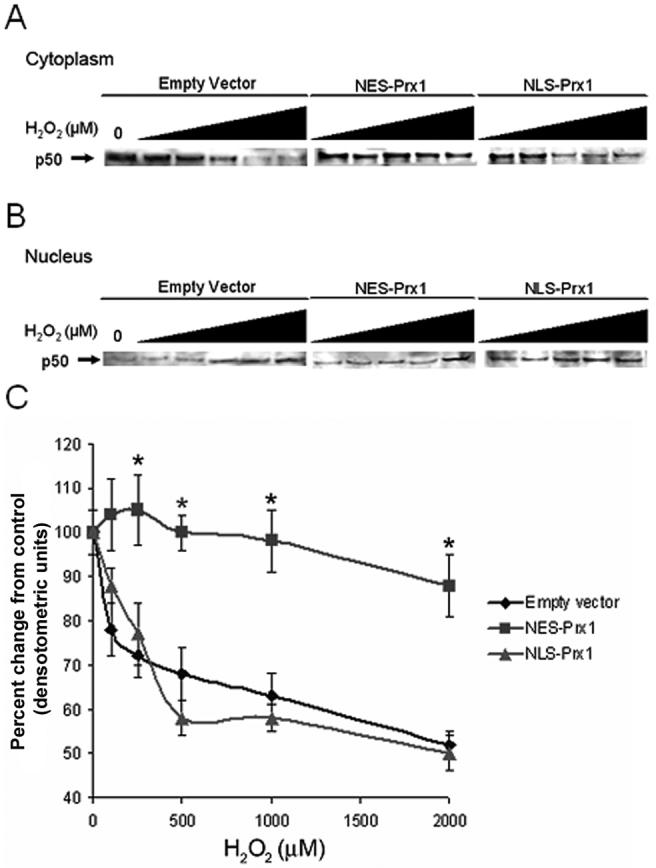
p50 subcellular localization is regulated by compartmented Prx1. Cytoplasmic (A) and nuclear (B) fractions were isolated in HeLa cells transfected with empty vector, NES-Prx1 or NLS-Prx1 following treatment with 0-2000 μM H2O2 for 1 h. Changes in p50 compartmentalization was determined by densitometry of immunoblotted bands (C). H2O2 treatment caused a redistribution of p50 from the cytoplasm to the nucleus in empty vector controls and NLS-Prx1 transfected cells, but higher concentrations of H2O2 were needed to cause the translocation of p50 to the nucleus in NES-Prx1 transfected cells. Asterisks (*) denote statistically significant (p<0.05) difference from empty vector controls. Averages are representative of experiments performed at least twice.
Analysis of nuclear fractions showed that the loss of cytoplasmic p50 in control and NLS-Prx1 cells was due to translocation into the nucleus. In both empty vector controls and cells transfected with NLS-Prx1, nuclear p50 content increased in a dose-dependent manner with H2O2 (Figure 3B). Translocation was inhibited by NES-Prx1, showing that cytoplasmic activation is prevented by increased cytoplasmic Prx1. Densitometric measurements show quantification of p50 loss from the cytoplasm (Figure 3C). These results show that increased nuclear Prx1 has no effect on preventing H2O2-dependent cytoplasmic activation while cytoplasmic Prx1 prevents this process. Consequently, the data imply that the peroxide tone in the subcellular compartments, at least under conditions of exogenously added peroxide, are differentially controlled at the level of the microcompartment.
NLS-Prx1 enhances NF-κB reporter activity
Cells were co-transfected with an NF-κB reporter to determine whether nuclear translocation of p50 was directly related to increased transcriptional activity in the presence of NES-Prx1 and NLS-Prx1. HeLa cells transfected with empty vector and stimulated with H2O2 (0-2000 μM) showed an increase in NF-κB reporter activity at 250 μM H2O2. However, reporter activity returned to baseline at 500 μM H2O2 and decreased at higher concentrations (1000-2000 μM) (Figure 4). Cells transfected with NES-Prx1 showed a decreased NF-κB reporter activity with 100-500 μM H2O2 concentrations, which is likely to be a result of failure for NF-κB to be activated. At higher concentrations of H2O2 (1000-2000 μM) that may have been sufficient to overcome excessive cytoplasmic Prx1, nuclear translocation was seen with NES-Prx1 and NF-κB reporter activity was also observed.
Figure 4.
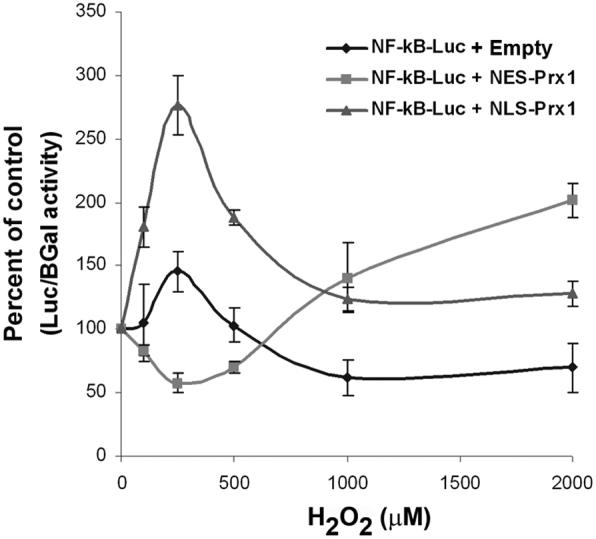
Prx1 affects oxidant-induced NF-κB reporter activity. Cells were transfected with empty vector, NES-Prx1 and NLS-Prx1 and NF-κB-luciferase reporter vectors. In empty vector controls at lower hydrogen peroxide concentrations, NF-κB activation is stimulated, but at higher concentrations, activity is inhibited. NES-Prx1 expression inhibited NF-κB activity at lower concentrations but activity increased with higher concentrations of H2O2. NLS-Prx1 expression increased NF-κB activity at lower concentrations from control levels of NF-κB activity and decreased with increasing hydrogen peroxide concentrations.
In contrast to the NES-Prx1 data, cells transfected with NLS-Prx1 showed a robust increase in NF-κB activity with low concentrations of H2O2 (100-500 μM). NF-κB activity returned closer to baseline at higher concentrations (1000-2000 μM) with a pattern similar to the vector control, but at a higher level of activity. The difference in pattern of responses with NES-Prx1 and NLS-Prx1 clearly shows that the Prx1 has different effects on the NF-κB system in the two compartments.
Prx1 overexpression in either compartment maintains p50 in a reduced state
To measure redox state of p50, biotinylated iodoacetamide (BIAM) was used to label reactive thiols [13] so that reduced fractions could be detected by immunoprecipitation, gel electrophoresis and probing with streptavidin. Addition of H2O2 to HeLa cells transfected with empty vector caused a dose-dependent decrease in concentrations of reduced p50 (Figure 5) as demonstrated by a decrease in band intensity. Oxidation of p50 was significantly different from control at H2O2 concentrations greater than 100 μM. These H2O2-induced changes in band intensity show a decrease in total amount of reduced p50 compared to untreated cells. Interestingly, HeLa cells transfected with either NES-Prx1 or NLS-Prx1 prevented a significant loss in reduced p50 by H2O2. Even with treatment with 2000 μM H2O2, NES-Prx1 and NLS-Prx1 transfected cells showed relatively little decrease in band intensity, suggesting that most of the p50 remained reduced. In combination with the above data, the results show that Prx1 in either compartment protected against p50 oxidation, and hence preserved nuclear DNA binding activity, while only the cytoplasmic Prx1 altered the cytoplasmic activation and translocation into the nucleus.
Figure 5.

Prx1 maintains p50 in a reduced state. HeLa cells were transfected with either empty vector, NLS-Prx1 or NES-Prx1 expression vector for 24 h and then dosed with 0-2000 μM H2O2 for 20 min. BIAM (biotinylated iodoacetamide) labeling was used as a determinant of reduced proteins (see methods). p50 was immunoprecipitated from cell lysates, separated by SDS-PAGE and transferred to nitrocellulose. Membranes were probed with streptavidin-conjugated fluorophore. A decrease in band intensity in empty vector cells shows an oxidation of p50 in a dose-dependent manner. NES-Prx1 and NLS-Prx1 expression inhibit oxidation of p50 at all concentrations of H2O2. Data is representative of experiments, which were performed independently three times.
Nuclear and cytoplasmic Prx1 regulate Trx1 redox state
Measurement of Trx1 redox states in nuclei and cytoplasm provide an alternate means to assess redox effects of targeted expression of Prx1. Using this approach, empty vector control cultures showed a significant (p<0.05) oxidation of cytosolic Trx1 after 60 min (-274 mV) compared to 0 min (-282 mV), and Trx1 became increasingly oxidized at 120 min (-270 mV) (Figure 6A, C). Cultures transfected with NES-Prx1 showed an initial, non-significant oxidation of cytosolic Trx1 at 20 min (-276 mV) compared to control (-281 mV) and this reverted to a normal redox potential at 60 min (-279 mV) and 120 min (-285 mV). NLS-Prx1 transfected culture showed significant oxidation at 20 min (-277 mV) and was still oxidized even after 120 min (-274 mV). Thus, the results showed that cytosolic Trx1 oxidation was protected by NES-Prx1 but not by NLS-Prx1.
Figure 6.
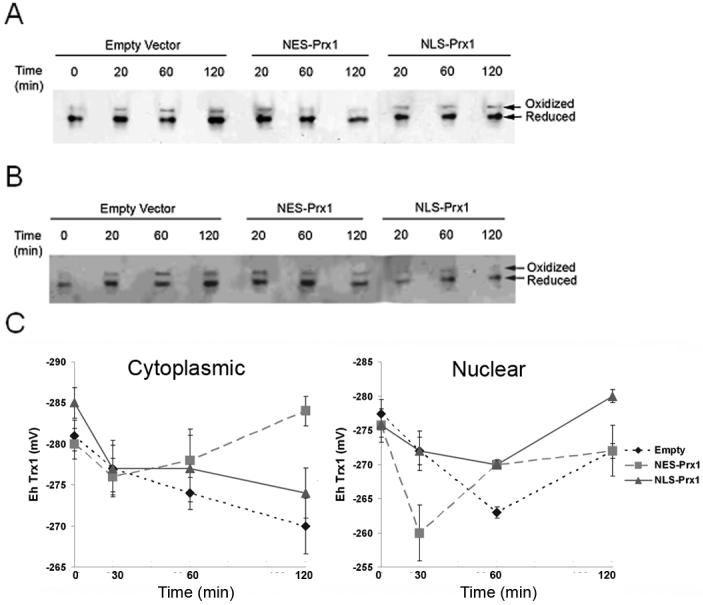
Nuclear and cytoplasmic Trx1 redox state is regulated by compartmented Prx1. Oxidized and reduced Trx1 in the cytoplasm and nucleus were determined via redox Western techniques. (A) Cytoplasmic Trx1 is protected from oxidation by overexpression of NES-Prx1. (B) Nuclear Trx1 is protected from oxidation by overexpression of NLS-Prx1. (C) Cytoplasmic and nuclear Trx1 redox potentials were determined as described in the methods section using the Nernst equation and are quantified. Each experiment was performed independently at least three times. Astersiks (*) denote a statistically significant difference (p<0.05) from empty vector controls.
Nuclear Trx1 redox in empty vector control cultures showed a more extensive oxidation than cytosolic Trx1 at 60 min (-262 mV) but rebounded to -271 mV by 120 min (Figure 6B, C). With NES-Prx1, nuclear Trx1 was oxidized more rapidly and recovered more rapidly than the vector control. In contrast, nuclear Trx1 did not become significantly at any time point with NLS-Prx1 (Figure 6B, C). These data show that steady-state redox status of nuclear Trx1 in response to exogenously added H2O2 is better protected by nuclear Prx1 than by cytoplasmic Prx1.
Discussion
Previous studies show that subcellular compartments have different thiol/disulfide redox environments, with mitochondria being most reduced, endoplasmic reticulum being most oxidized, and nuclei and cytoplasm being intermediate [14, 16, 17]. Distinction between nuclear and cytoplasmic thiol/disulfide redox systems has, however, been limited by methodologic issues. Earlier research on GSH being present at a higher concentration in nuclei than in cytoplasm has been controversial [18-20], but convincing evidence for distinct redox regulation came from the demonstration that Trx1 is translocated into nuclei in response to oxidative stress [9]. Recent research using redox western blot analysis of Trx1 redox state in nuclei and cytosol shows that Trx1 is more reduced in nuclei during deprivation of metabolic energy supply [10]. Moreover, the ratio of S-glutathionylation of protein to protein thiol content (PrSSG/PrSH) was lower in nuclei than cytosol and resistant to increase under these energy-limiting conditions [10]. Thus, available data show that the thiol/disulfide systems in nuclei and cytoplasm are at least semiautonomously controlled, with the nuclear compartment being more reduced and more resistant to oxidation [10].
These compartmental differences in redox are consistent with functional studies which show that key transcription factors, including AP-1, NF-κB and Nrf2, are activated by oxidative signals in the cytoplasm and inactivated by excessive oxidative stress in the nuclei [21]. For instance, NF-κB activation is a critical survival response to different physiologic stimuli, such as TNF-α, upregulating the expression of anti-apoptotic proteins such as SOD, Bcl-2, Bcl-xL and γ-glutamylcysteinyl ligase (Gcl) [22]. Unactivated NF-κB is sequestered in the cytosol by I-κB, and oxidant-induced activation causes the dissociation of the NF-κB/I-κB complex, freeing NF-κB to enter the nucleus [23]. In the nucleus, DNA binding requires a conserved, redox-sensitive cysteine residue to be in the reduced form [9, 24]. Thus, relatively low levels of oxidants in the cytoplasm can activate the system while higher concentrations of oxidants in the nucleus inhibit the system.
Trx1 appears to have an important role in regulating the redox sensitivity of NF-κB in both the cytoplasm and nucleus. Increased Trx1 inhibits cytoplasmic activation and nuclear translocation of NF-κB and also protects against inactivation of p50 in the nucleus. The present data show that NES-Prx1 inhibits nuclear translocation of p50. Because there is no evidence that Prxs directly act as a general reductant to oxidized proteins, these data suggest that Trx1 inhibits cytoplasmic NF-κB activation by peroxiredoxin-dependent decrease in peroxide concentration, thereby preventing IκB oxidation. Protection against such macromolecular oxidation by peroxide removal is mechanistically distinct from the well characterized protein-repair function of Trx1 which is thought to protect against p50 inactivation. Previous studies have shown that following stimulation with phorbol esters, nuclear p50 is much more reduced than p50 in the cytoplasm [25]. The redox pathway for this reduction includes Trx1, which reduces Ref1, a catalyst to “repair” oxidized p50 and allow DNA binding. The distinction in mechanism between prevention of oxidation in cytoplasm and repair of oxidized protein in nuclei appears to be especially important in the context of the present data, which show that nuclear protection can also occur by the Trx1-dependent peroxidase, Prx1.
Subcellular targeting of Prxs has previously been shown to be protective in the mitochondria, nucleus and cytoplasm [4, 26]. In our study, nuclear targeting of Prx1 showed a protection against H2O2-induced toxicity while cytoplasmic targeting conferred no resistance. These data could mean that inhibition of cytoplasmic oxidative activation interferes with the protective NF-κB system while inhibition of nuclear oxidation protects against injury regardless of whether NF-κB is activated. Such an interpretation suggests that under the conditions of these experiments, the Ref1-dependent repair function supported by Trx1 is secondary to the function of Trx1 in peroxiredoxin-dependent peroxide metabolism. This suggests that a primary function of the Trx1/Prx1 system could be to control the peroxide tone, a function which may be needed to maintain dynamic H2O2-dependent redox signaling.
Peroxiredoxins have been implicated as key regulatory factors in redox signaling [27]. Active metabolism of H2O2 by Prx can quench the available second messenger H2O2 and inhibit signal transduction. Overoxidation of Prx from a cysteine-sulfenic acid (Cys-SOH) to a cysteine-sulfinic acid (Cys-SO2H) stops H2O2 metabolism, allowing ample second messenger H2O2 concentration and propagation of the signal. Reduction of Cys-SO2H to Cys-SOH is achieved through the actions of sulfiredoxin, which restores Prx-mediated H2O2 metabolism [28, 29]. Similar mechanisms may also function during stimulation by cytokines, like tumor necrosis factor-α (TNF-α). Overexpression of mitochondrial Trx2 blocked TNF-α-induced ROS generation and NF-κB activation [30]. Moreover, overexpression of cytosolic and mitochondrial Prxs in HeLa cells eliminated H2O2 and blocked NF-κB activation by TNF-α [31, 32]. Other antioxidant systems have shown similar patterns of compartmental regulation. Together with earlier data showing that hydrogen peroxide generated within either peroxisomes or cytoplasm was metabolized within each respective compartment [33], these data show that the regulation of H2O2 as a signal occurs within specific compartments and is managed by specific antioxidant systems within each respective compartment.
In summary, the present data add to the understanding of cytoplasmic and nuclear regulation of redox signaling by providing evidence that compartment-specific over-expression of Prx1 regulates compartment-specific NF-κB activation, thioredoxin-1 redox state and sensitivity of cells to oxidant-induced cell death. The data can be most readily interpreted to mean that Prx1 controls the compartment-specific H2O2 concentration, which serves to establish the peroxide tone for local redox signaling events. Cytoplasmic Prx1 regulates H2O2-dependent NF-κB activation and nuclear translocation, and nuclear Prx1 regulates NF-κB/DNA binding through elimination of H2O2 as a p50 subunit oxidant. The redox state of Trx1 recovered more rapidly following an oxidative challenge in the compartment in which Prx1 was overexpressed, further indicating that the redox states of individual proteins are dynamically controlled by the local peroxide concentration. The data showed that, unlike cytoplasmic Prx1 which inhibited oxidant-induced activation of NF-κB and did not protect against oxidant-induced cell death, nuclear Prx1 was protective. The results indicate that an endogenous peroxide tone functions in opposition to Trx1-dependent reduction to maintain a balance of oxidant and reductant potential, thereby supporting sensitive and rapidly responsive redox signaling mechanisms. Together, these findings suggest that nuclear and cytoplasmic Prx1 pools have distinct regulatory roles during oxidative stress and redox signaling and support the need for further study to understand mechanisms of compartmentalized antioxidant function.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Rhee SG, Chae HZ, Kim K. Peroxiredoxins: a historical overview and speculative preview of novel mechanisms and emerging concepts in cell signaling. Free Radic Biol Med. 2005;38:1543–1552. doi: 10.1016/j.freeradbiomed.2005.02.026. [DOI] [PubMed] [Google Scholar]
- 2.Rhee SG, Chang TS, Bae YS, Lee SR, Kang SW. Cellular regulation by hydrogen peroxide. J Am Soc Nephrol. 2003;14:S211–215. doi: 10.1097/01.asn.0000077404.45564.7e. [DOI] [PubMed] [Google Scholar]
- 3.Egler RA, Fernandes E, Rothermund K, Sereika S, de Souza-Pinto N, Jaruga P, Dizdaroglu M, Prochownik EV. Regulation of reactive oxygen species, DNA damage, and c-Myc function by peroxiredoxin 1. Oncogene. 2005;24:8038–8050. doi: 10.1038/sj.onc.1208821. [DOI] [PubMed] [Google Scholar]
- 4.Banmeyer I, Marchand C, Verhaeghe C, Vucic B, Rees JF, Knoops B. Overexpression of human peroxiredoxin 5 in subcellular compartments of Chinese hamster ovary cells: effects on cytotoxicity and DNA damage caused by peroxides. Free Radic Biol Med. 2004;36:65–77. doi: 10.1016/j.freeradbiomed.2003.10.019. [DOI] [PubMed] [Google Scholar]
- 5.Jin DY, Chae HZ, Rhee SG, Jeang KT. Regulatory role for a novel human thioredoxin peroxidase in NF-kappaB activation. J Biol Chem. 1997;272:30952–30961. doi: 10.1074/jbc.272.49.30952. [DOI] [PubMed] [Google Scholar]
- 6.Matthews JR, Wakasugi N, Virelizier JL, Yodoi J, Hay RT. Thioredoxin regulates the DNA binding activity of NF-kappa B by reduction of a disulphide bond involving cysteine 62. Nucleic Acids Res. 1992;20:3821–3830. doi: 10.1093/nar/20.15.3821. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Toledano MB, Leonard WJ. Modulation of transcription factor NF-kappa B binding activity by oxidation-reduction in vitro. Proc Natl Acad Sci U S A. 1991;88:4328–4332. doi: 10.1073/pnas.88.10.4328. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Hansen JM, Watson WH, Jones DP. Compartmentation of Nrf-2 redox control: regulation of cytoplasmic activation by glutathione and DNA binding by thioredoxin-1. Toxicol Sci. 2004;82:308–317. doi: 10.1093/toxsci/kfh231. [DOI] [PubMed] [Google Scholar]
- 9.Hirota K, Murata M, Sachi Y, Nakamura H, Takeuchi J, Mori K, Yodoi J. Distinct roles of thioredoxin in the cytoplasm and in the nucleus. A two-step mechanism of redox regulation of transcription factor NF-kappaB. J Biol Chem. 1999;274:27891–27897. doi: 10.1074/jbc.274.39.27891. [DOI] [PubMed] [Google Scholar]
- 10.Go YM, Ziegler T, Johnson J, Gu L, Hansen JM, Jones DP. Selective protection of nuclear thioredoxin-1 and glutathione redox systems against oxidation during glucose and glutamine deficiency in human colonic epithelial cells. Free Radic Biol Med. 2007 doi: 10.1016/j.freeradbiomed.2006.11.005. in press. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Fukuda M, Gotoh I, Adachi M, Gotoh Y, Nishida E. A novel regulatory mechanism in the mitogen-activated protein (MAP) kinase cascade. Role of nuclear export signal of MAP kinase kinase. J Biol Chem. 1997;272:32642–32648. doi: 10.1074/jbc.272.51.32642. [DOI] [PubMed] [Google Scholar]
- 12.Denizot F, Lang R. Rapid colorimetric assay for cell growth and survival. Modifications to the tetrazolium dye procedure giving improved sensitivity and reliability. J Immunol Methods. 1986;89:271–277. doi: 10.1016/0022-1759(86)90368-6. [DOI] [PubMed] [Google Scholar]
- 13.Kim JR, Lee SM, Cho SH, Kim JH, Kim BH, Kwon J, Choi CY, Kim YD, Lee SR. Oxidation of thioredoxin reductase in HeLa cells stimulated with tumor necrosis factor-alpha. FEBS Lett. 2004;567:189–196. doi: 10.1016/j.febslet.2004.04.055. [DOI] [PubMed] [Google Scholar]
- 14.Watson WH, Jones DP. Oxidation of nuclear thioredoxin during oxidative stress. FEBS Lett. 2003;543:144–147. doi: 10.1016/s0014-5793(03)00430-7. [DOI] [PubMed] [Google Scholar]
- 15.Watson WH, Pohl J, Montfort WR, Stuchlik O, Reed MS, Powis G, Jones DP. Redox potential of human thioredoxin 1 and identification of a second dithiol/disulfide motif. J Biol Chem. 2003;278:33408–33415. doi: 10.1074/jbc.M211107200. [DOI] [PubMed] [Google Scholar]
- 16.Hansen JM, Go YM, Jones DP. Nuclear and mitochondrial compartmentation of oxidative stress and redox signaling. Annu Rev Pharmacol Toxicol. 2006;46:215–234. doi: 10.1146/annurev.pharmtox.46.120604.141122. [DOI] [PubMed] [Google Scholar]
- 17.Hwang C, Sinskey AJ, Lodish HF. Oxidized redox state of glutathione in the endoplasmic reticulum. Science. 1992;257:1496–1502. doi: 10.1126/science.1523409. [DOI] [PubMed] [Google Scholar]
- 18.Bellomo G, Vairetti M, Stivala L, Mirabelli F, Richelmi P, Orrenius S. Demonstration of nuclear compartmentalization of glutathione in hepatocytes. Proc Natl Acad Sci U S A. 1992;89:4412–4416. doi: 10.1073/pnas.89.10.4412. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Cotgreave IA. Analytical developments in the assay of intra- and extracellular GSH homeostasis: specific protein S-glutathionylation, cellular GSH and mixed disulphide compartmentalisation and interstitial GSH redox balance. Biofactors. 2003;17:269–277. doi: 10.1002/biof.5520170126. [DOI] [PubMed] [Google Scholar]
- 20.Voehringer DW, McConkey DJ, McDonnell TJ, Brisbay S, Meyn RE. Bcl-2 expression causes redistribution of glutathione to the nucleus. Proc Natl Acad Sci U S A. 1998;95:2956–2960. doi: 10.1073/pnas.95.6.2956. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Arrigo AP. Gene expression and the thiol redox state. Free Radic Biol Med. 1999;27:936–944. doi: 10.1016/s0891-5849(99)00175-6. [DOI] [PubMed] [Google Scholar]
- 22.Garg AK, Aggarwal BB. Reactive oxygen intermediates in TNF signaling. Mol Immunol. 2002;39:509–517. doi: 10.1016/s0161-5890(02)00207-9. [DOI] [PubMed] [Google Scholar]
- 23.Baeuerle PA, Baltimore D. I kappa B: a specific inhibitor of the NF-kappa B transcription factor. Science. 1988;242:540–546. doi: 10.1126/science.3140380. [DOI] [PubMed] [Google Scholar]
- 24.Kabe Y, Ando K, Hirao S, Yoshida M, Handa H. Redox regulation of NF-kappaB activation: distinct redox regulation between the cytoplasm and the nucleus. Antioxid Redox Signal. 2005;7:395–403. doi: 10.1089/ars.2005.7.395. [DOI] [PubMed] [Google Scholar]
- 25.Nishi T, Shimizu N, Hiramoto M, Sato I, Yamaguchi Y, Hasegawa M, Aizawa S, Tanaka H, Kataoka K, Watanabe H, Handa H. Spatial redox regulation of a critical cysteine residue of NF-kappa B in vivo. J Biol Chem. 2002;277:44548–44556. doi: 10.1074/jbc.M202970200. [DOI] [PubMed] [Google Scholar]
- 26.Banmeyer I, Marchand C, Clippe A, Knoops B. Human mitochondrial peroxiredoxin 5 protects from mitochondrial DNA damages induced by hydrogen peroxide. FEBS Lett. 2005;579:2327–2333. doi: 10.1016/j.febslet.2005.03.027. [DOI] [PubMed] [Google Scholar]
- 27.Woo HA, Chae HZ, Hwang SC, Yang KS, Kang SW, Kim K, Rhee SG. Reversing the inactivation of peroxiredoxins caused by cysteine sulfinic acid formation. Science. 2003;300:653–656. doi: 10.1126/science.1080273. [DOI] [PubMed] [Google Scholar]
- 28.Chang TS, Jeong W, Woo HA, Lee SM, Park S, Rhee SG. Characterization of mammalian sulfiredoxin and its reactivation of hyperoxidized peroxiredoxin through reduction of cysteine sulfinic acid in the active site to cysteine. J Biol Chem. 2004;279:50994–51001. doi: 10.1074/jbc.M409482200. [DOI] [PubMed] [Google Scholar]
- 29.Rhee SG, Kang SW, Jeong W, Chang TS, Yang KS, Woo HA. Intracellular messenger function of hydrogen peroxide and its regulation by peroxiredoxins. Curr Opin Cell Biol. 2005;17:183–189. doi: 10.1016/j.ceb.2005.02.004. [DOI] [PubMed] [Google Scholar]
- 30.Hansen JM, Zhang H, Jones DP. Mitochondrial thioredoxin-2 has a key role in determining tumor necrosis factor-alpha-induced reactive oxygen species generation, NF-kappaB activation, and apoptosis. Toxicol Sci. 2006;91:643–650. doi: 10.1093/toxsci/kfj175. [DOI] [PubMed] [Google Scholar]
- 31.Kang SW, Chae HZ, Seo MS, Kim K, Baines IC, Rhee SG. Mammalian peroxiredoxin isoforms can reduce hydrogen peroxide generated in response to growth factors and tumor necrosis factor-alpha. J Biol Chem. 1998;273:6297–6302. doi: 10.1074/jbc.273.11.6297. [DOI] [PubMed] [Google Scholar]
- 32.Kang SW, Chang TS, Lee TH, Kim ES, Yu DY, Rhee SG. Cytosolic peroxiredoxin attenuates the activation of Jnk and p38 but potentiates that of Erk in Hela cells stimulated with tumor necrosis factor-alpha. J Biol Chem. 2004;279:2535–2543. doi: 10.1074/jbc.M307698200. [DOI] [PubMed] [Google Scholar]
- 33.Jones DP, Eklow L, Thor H, Orrenius S. Metabolism of hydrogen peroxide in isolated hepatocytes: relative contributions of catalase and glutathione peroxidase in decomposition of endogenously generated H2O2. Arch Biochem Biophys. 1981;210:505–516. doi: 10.1016/0003-9861(81)90215-0. [DOI] [PubMed] [Google Scholar]