

Abstract
A capillary electrophoresis (CE) method for the characterization of recombinant NTPDases 1, 2, and 3, and for assaying NTPDase inhibitors has been developed performing the enzymatic reaction within the capillary. After hydrodynamic injection of plugs of substrate solution with or without inhibitor in reaction buffer, followed by a suspension of an enzyme-containing membrane preparation, and subsequent injection of another plug of substrate solution with or without inhibitor, the reaction took place close to the capillary inlet. After 5 min, the electrophoretic separation of the reaction products was initiated by applying a constant current of μA. The method employing a polyacrylamide-coated capillary and reverse polarity mode provided baseline resolution of substrates and products within a short separation time of less than 7 min. A 50 mM phosphate buffer (pH 6.5) was used for the separations and the products were detected by their UV absorbance at 210 nm. The Michaelis–Menten constants (Km) for the recombinant rat NTPDases 1, 2, and 3 obtained with this method were consistent with previously reported data. The inhibition studies revealed pronounced differences in the potency of reactive blue 2, pyridoxalphosphate-6-azophenyl-2-4-disulfonic acid (PPADS), suramin, and N6-diethyl-β,γ-dibromomethylene-ATP (ARL67156) towards the NTPDase isoforms. Notably, ARL67156 does not inhibit all NTPDases, having only a minor inhibitory effect on NTPDase2. Dipyridamole is not an inhibitor of the NTPDase isoforms investigated. The new method is fast and accurate, it requires only tiny amounts of material (nanoliter scale), no sample pretreatment and can be fully automated; thus it is clearly superior to the current standard methods.
Key words: at-inlet CE reaction, capillary electrophoresis, inhibitors, in-capillary enzymatic reaction, kinetics, Michaelis–Menten analysis, NTPDase assay
Introduction
Extracellular nucleotides such as ATP, ADP, UTP, and UDP can act on a variety of nucleotide receptors (P2 receptors) [1]. The activation of P2 receptors is controlled by ecto-nucleotidases capable of hydrolyzing nucleoside tri- and diphosphates [2]. Inhibition of ecto-nucleotidases can result in a potentiation of purinergic signaling, supporting the notion that endogenous ecto-nucleotidases reduce the effective concentration of the released nucleotide [3–5]. Similarly, metabolically stable analogs of ATP are considerably more effective in causing a biological response than ATP itself (for references see [6]). Inhibitors of ecto-nucleotidases could thus represent valuable tools for amplifying the biological effects induced by extracellularly released nucleotides. In addition, inhibition of ectonucleotidases is mandatory for both, studies of nucleotide release and the analysis of the potency on P2 receptors of nucleotides or their hydrolyzable analogs.
Inhibitors of ecto-nucleotidases should have no effect on P2 receptors and should not be dephosphorylated by ectonucleotidase. Ideally they would also reveal selectivity for individual E-NTPDase isoforms. Many inhibitors of P2 receptors also act as inhibitors of ecto-nucleotidases. These include suramin, pyridoxalphosphate-6-azophenyl-2′,4′-disulfonic acid (PPADS) and reactive blue 2 (for references see [6]) (Figure 1). To date only the ATP analog ARL67156 (FPL67156, N6-diethyl-β,γ-dibromomethylene-ATP, Figure 1) [4, 7] and 8-thiobutyladenosine 5′-triphosphate (8-Bu-S-ATP) [8] reveal enzyme inhibitory potential without significantly affecting nucleotide receptors. However, these compounds have not been tested on defined NTPDase isoforms. The development of novel inhibitors of ecto-nucleotidases requires fast and precise methods for analyzing catalytic activity.
Figure 1.

Structures of investigated NTPDase inhibitors.
Capillary electrophoresis (CE) has recently emerged as a versatile technique for enzyme assays [9]. CE systems have been successfully applied for assaying enzyme activity [10–12], including the determination of Michaelis-Menten constants (Km values) [13], and inhibition constants (Ki values for enzyme inhibitors) [14], exhibiting a number of advantages over conventional methods. These include rapid separation of substrate and product, ultra-low sample volume requirements, and high throughput by automation. CE is particularly useful for investigating enzymatic reactions involving charged substrates or products, e.g., for the monitoring of phosphorylation or dephosphorylation reactions [15–17]. Electrophoretically mediated microanalysis (EMMA), first described by Bao and Regnier [18] has been successfully used for in-line enzyme assays [19]. In this technique, the capillary is used as a microbioreactor as well as for the separation of substrates and products. There are two major types of EMMA methods. In the first continuous format [18], the capillary is filled with an appropriate substrate solution and upon injection of a zone of enzyme, the product will continuously form during the electrophoretic mixing of enzyme and substrate. One drawback of this method is that the separation buffer has to allow the enzymatic reaction to proceed.
In the second plug-plug technique [20], substrate and enzyme are introduced into the capillary as distinct plugs. Upon application of an electric potential, these zones mix with each other due to differences in their electrophoretic mobilities. The reaction proceeds during the mixing process. The resultant product is transported to the detector separately from enzyme and educts under the influence of an applied voltage, where they are individually detected. Even double enzyme-catalyzed reactions were studied by the latter method by injecting plugs of substrate and two different enzymes separately in reaction buffer. This technique was used to study the enzymatic reactions of hexokinase and apyrase as well as lactate dehydrogenase and glucose-6-phosphate dehydrogenase [21, 22].
In the present study, a method for analyzing the enzymatic reactions of NTPDase1, 2 and 3, three surface-located members of the ecto-nucleoside triphosphate diphosphohydrolase family (EC 3.6.1.5) [2] has been developed, performing the enzymatic reaction inside the capillary at the capillary inlet followed by electrophoretic separation of the reaction products. In the at-capillary inlet reaction technique, the plugs of enzyme, substrate and inhibitor are introduced into the capillary, where they are allowed to react by simple diffusion and not by voltage as in EMMA according to a previously described procedure for angiotensin-converting enzyme assays [23, 24].
Materials and methods
Reagents and chemicals
Reactive blue 2, suramin, pyridoxalphosphate-6-azophenyl- 2′,4′-disulfonic acid (PPADS), dipyridamole and 4-(2-hydroxyethyl)piperazine-1-ethanesulfonic acid (HEPES) were obtained from Sigma, Steinheim, Germany. ARL-67156 was from Tocris Cookson, Bristol, UK. ATP, ADP, AMP, UMP, MgCl2·6H2O, and tris(hydroxymethyl)-aminomethane (Trizma Base), were from Sigma (Taufkirchen, Germany). Culture medium was obtained from Invitrogen (Karlsruhe, Germany). Penicillin and streptomycin were purchased from Sigma-Aldrich (Deisenhofen, Germany). Leupeptin, pepstatin A, chymostatin, and antipain were from Calbiochem (Schwalbach, Germany).
Cell transfection and preparation of membrane fractions containing NTP Dases
Chinese hamster ovary (CHO) cells were cultured in HAM's F-12 medium containing 10% fetal calf serum, 100 U/ml penicillin and 100 µg/ml streptomycin. They were transfected by electroporation with plasmid-DNA containing rat NTPDase1 (GenBank Accession number U81295) [25], NTPDase2 (Y11835) [25], and NTPDase3 (AJ437217) [26], all cloned into the pcDNA3 plasmid. Transfection with the empty plasmid pcDNA3 served as a control. Transiently transfected CHO cells were used for preparation of membrane fractions 48 h after electroporation.
After removal of culture medium, cells were washed twice with buffer A (in mM: 140 NaCl, 5 KCl, 0.5 EDTA, 20 Hepes, pH 7.4) and scraped off with 5 ml of ice-cold buffer B (in mM: 140 NaCl, 5 KCl, 20 Hepes, pH 7.4) containing protease inhibitors (in µg/ml: 2, chymostatin; 1, pepstatin A; 150 benzamidine; 2, antipain; 2, leupeptin) and iodoacetamide (2 mM). Cells were centrifuged at 300 gav for 10 min at 4 °C. The cell pellet was resuspended in buffer B, homogenized using a Potter-Elvehjem homogenizer and sonicated. The homogenate was centrifuged for 10 min at 300 gav at 4 °C and the resulting supernatant fraction was centrifuged at 100,000 gav for 1 h at 4 °C. The pellet fraction was resuspended in buffer C containing 50% (v/v) glycerol, 2 mM iodoacetamide, 20 mM Hepes (pH 7.4), and stored at −20 °C. ATPase activity of individual membrane fractions was determined by analysis of free phosphate formed according to Lanzetta et al. [27]. Protein was determined according to the method of Spector [28]. The membrane preparations contained 4–6 µg of protein/µl.
CE instrumentation
All experiments were carried out using a P/ACE MDQ capillary electrophoresis system (Beckman Instruments, Fullerton, CA, USA) equipped with a UV detection system coupled with a diode-array detector (DAD). Data collection and peak area analysis were performed by the P/ACE MDQ software 32 KARAT obtained from Beckman Coulter. The capillary temperature was kept constant at 37 °C. The temperature of the sample storing unit was adjusted to 10 °C. The electrophoretic separations were carried out using an eCAP polyacrylamide-coated fused-silica capillary [(30 cm (20 cm effective length) × 50 µm internal diameter (I.D.) × 360 µm outside diameter (O.D.), obtained from CS-Chromatographie (Langerwehe, Germany)]. The separation was performed using an applied current of −60µA and a data acquisition rate of 8 Hz. Analytes were detected using direct UV absorbance at 210 nm. The capillary was conditioned by rinsing with water for 2 min and subsequently with buffer (phosphate 50 mM, pH 6.5) for 1 min. Sample injections were made at the cathodic side of the capillary.
At-inlet reaction procedure and automation of analytical process
The CE running buffer consisted of dipotassium hydrogen phosphate 50 mM, pH 6.5. The reaction buffer contained 140 mM NaCl, 5 mM KCl, 1 mM MgCl2, 2 mM CaCl2, and 10 mM Hepes, pH 7.4. Before use, a new capillary was washed with deionized water for 10 min. The automation cycle consisted of (1) washing with water for 2 min (40 p.s.i; 1 p.s.i = 6,894.76 Pa), (2) equilibration with the CE running buffer for 1 min (40 p.s.i), (3) injection of a plug of reaction solution containing 320 µM ATP (substrate) in reaction buffer and various concentrations of inhibitor stock solutions in reaction buffer, (4) injection of a plug of suitably diluted enzyme, (5) injection of another plug of reaction solution as in (3), (6) and finally injection of a plug of water. The plugs were then allowed to react, while the capillary ends were dipped into water, for a predetermined waiting period of 5 min. Then, a current of −60 µA was applied and the reaction products moved towards the detector end of the capillary. After each analysis the capillary was rinsed with CE running buffer for 2 min followed by deionized water for 1 min.
Each electropherogram was recorded over 7 min. The diluted membrane fractions containing enzyme were placed in the sample storage unit whose temperature was kept constant at 10 °C. At this temperature, enzyme activity remained unaffected during the approximately 24 h of instrument run. Substrate and buffers used in the enzyme reaction were also kept at 10 °C in the autosampler reservoir.
Quantitative determination of AMP and ADP and method validation
AMP and ADP were dissolved in enzyme assay buffer (140 mM NaCl, 5 mM KCl, 1 mM MgCl2, 2 mM CaCl2, 10 mM Hepes, pH 7.4) to obtain 1 mM stock solutions. Standard calibration curves were obtained with final concentrations of 2, 5, 10, 20, 30, and 50 µM. For validating the method, 20 µM of UMP was used as an internal standard. A polyacrylamide-coated capillary was used for the separation and quantitation of AMP and ADP. The procedure was as described above using enzyme preparations inactivated by heating to 99 °C for 3 min using an Eppendorf Thermomixer Comfort. Determinations were performed in triplicate. The calibration curves were obtained by plotting the corrected peak area of AMP or ADP, respectively, against their concentrations.
Investigation of NTPDase inhibitors by in-capillary reaction
For the determination of the IC50 and Ki values of NTPDase inhibitors (Figure 1), 6–8 different concentrations of inhibitor spanning about three orders of magnitude were used (see Figure 5–7, while a fixed substrate concentration of 320 µM of ATP was employed for all three NTPDases. Under the applied conditions less than 10% of substrate was converted by the enzymes. Membrane preparations derived from transfected cells and containing NTPDase1, NTPDase2, or NTPDase3, were appropriately diluted with reaction buffer for the inhibition assays. Control experiments were performed using membrane preparations of cells transfected with the empty plasmid (pcDNA3). Substrate and inhibitors were dissolved in the reaction buffer containing 140 mM NaCl, 5 mM KCl, 1 mM MgCl2, 2 mM CaCl2, 10 mM Hepes, pH 7.4. The ChengYPrusoff equation was used to calculate the Ki values from the IC50 values, determined by the non-linear curve fitting program PRISM 3.0 (GraphPad, San Diego, California, USA).
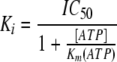 |
Figure 5.
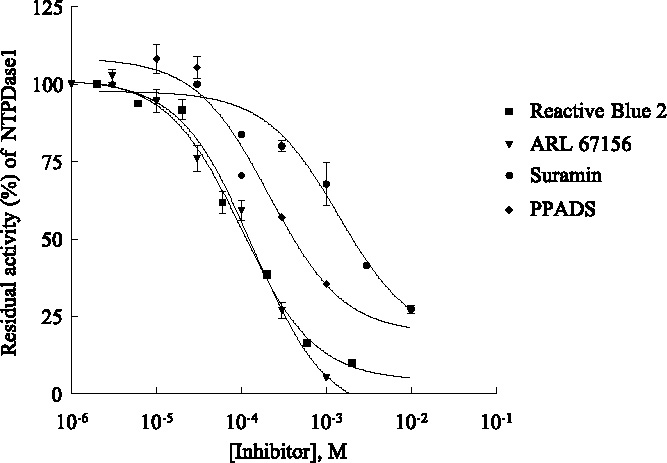
Concentration-dependent inhibition of NTPDase1 by reactive blue 2 (▪), ARL67156 (▾) suramin (•) and PPADS (dM) determined by capillary electrophoresis using in-capillary reaction at capillary inlet, using a substrate concentration of 320 µM ATP, a reaction buffer consisting of 140 mM NaCl, 5 mM KCl, 1 mM MgCl2, 2 mM CaCl2, and 10 mM Hepes, pH 7.4, and various concentrations of inhibitor. The separation conditions were 50 mM phosphate buffer, pH 6.5, neutral capillary, 30 cm length (20 cm to the detector), 50 µM I.D.; −60 µA, 7 kV; capillary cartridge temperature 37 °C; detection at 210 nm, pressure injection. Data points represent means T SD from three separate experiments, each run in duplicate.
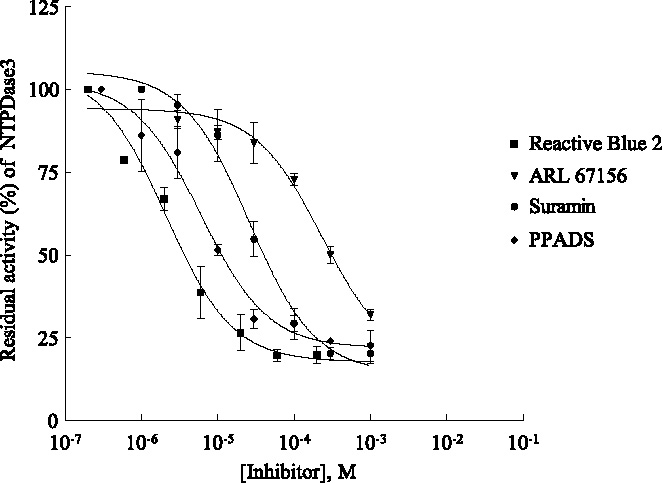
Figure 6.
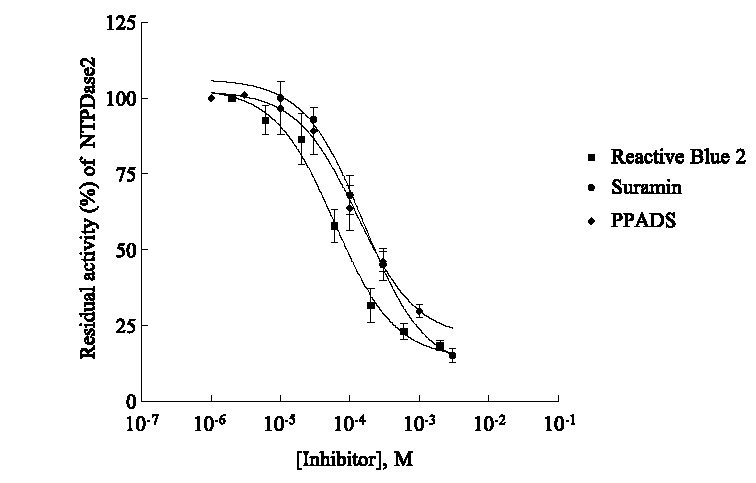
Concentration-dependent inhibition of NTPDase2 by reactive blue 2 (▪) suramin (•) and PPADS (dM) determined by capillary electrophoresis using in-capillary reaction at capillary inlet. For CE and assay conditions see Figure 5. Data points represent means T SD from three separate experiments, each run in duplicates. ARL67156 (not shown) exhibited only 50% inhibition at a concentration of 1 mM.
Michaelis-Menten constant (Km) and maximal velocity (Vmax ) determination
For the determination of the Michaelis-Menten constants (Km) and the maximum velocity (Vmax) eight different substrate concentrations of ATP were used, 10, 20, 30, 50, 100, 200, 250 and 1000 µM of ATP for NTPDase1 dissolved in reaction buffer, while the following ATP concentrations were used for NTPDase2 and 3: 25, 50, 100, 150, 200, 250, 300, 500 and 1000 µM. The capillary inlet reaction method was used as described above.
Results
Development of the on-capillary reaction technique
The on-capillary reaction technique has previously been successfully applied for inside capillary enzymatic reactions using electrophoretically mediated microanalysis (EMMA) [18, 19]. A related inside capillary enzymatic reaction methodology in which the enzymatic reaction was performed at the capillary inlet without electrophoretic mixing of enzyme and substrate had been applied for the determination of the Michaelis-Menten constant [23] and for inhibition studies of the angiotensin-converting enzyme [24]. In the present study we developed a CE method for the monitoring of reactions of NTPDase1, 2 and 3 by a modification of the described at-capillary inlet enzymatic reaction [23, 24]. Suitable conditions for the separation and quantitative determination of nucleotides and the monitoring of enzymatic nucleotide metabolism using CE had previously been developed in our group [16, 17]. However in those studies, the enzymatic reaction was performed in a vial outside the capillary and the samples were injected into the capillary and subjected to CE analysis only after the enzymatic reaction had been stopped [16]. A modification of the developed separation protocols was now used after performing the enzymatic reaction directly in the capillary close to the capillary inlet. Thus, a small aliquot of substrate-bearing reaction buffer was hydrodynamically injected into the capillary followed by enzyme and then again substrate, effectively sandwiching an aliquot of enzyme on either side by substrate. After the final plug of substrate a small plug of water was injected resulting in a stacking effect which improved the resolution of the peaks [16]. The sandwich mode was required because otherwise not enough product was formed. An inverse sandwich mode of two plugs of enzyme on either side of substrate as described by van Dyck et al. [24] for the reaction of angiotensin converting enzyme, proved to be unfavorable in our case since the rate of conversion of substrate to product was more than 10%. Figure 2 provides a schematic overview of the different steps of the capillary inlet reaction procedure. A reaction buffer containing 140 mM NaCl, 5 mM KCl, 1 mM MgCl2, 2 mM CaCl2 and 10 mM Hepes, pH 7.4, was found suitable for the enzymatic reaction, which was allowed to take place for 5 min. The separation of substrate and product(s) was then initiated by applying a constant current of −60 µA using a 50 mM phosphate buffer (pH 6.5) resulting in a voltage of 7 kV. The separation was completed within less than 6 min (see Figure 3).
Figure 2.

Schematic illustration of microscale reaction of NTPDases at capillary inlet. 1. Injection of a sample of 4 nl of 320 µM of ATP (substrate) in reaction buffer containing UMP (20 µM) as an internal standard in the absence or presence of test compound (potential inhibitors) (0.3 p.s.i., 5 s); 2. Injection of enzyme (0.3 p.s.i., 5 s); 3. Injection of 320 µM of ATP (substrate) in reaction buffer containing UMP (20 µM) as an internal standard in the absence or presence of test compound (0.3 p.s.i., 5 s); 4. Overlayed plugs are then allowed to stand during a predetermined period of 5 min; 5. Subsequently a −60 µA current is applied and the reaction products migrate to the detector. Electrophoresis conditions were as described in the experimental section.
Figure 3.

Overlay of five electropherograms after NTPDase3 on-line reaction at the capillary inlet with different concentrations of reactive blue 2 (inhibitor) added to the substrate plug. The concentration of NTPDase3 was 0.05 µg/µl of protein, ATP: 320 µM, UMP (internal standard): 40 µM. Waiting period (duration of enzymatic reaction): 5.0 min. CE conditions: Running buffer: 50 mM potassium phosphate, pH 6.5; constant current of −60 µA; detection at 210 nm, capillary cartridge temperature: 37 °C. Reactive blue 2 (RB2) concentrations: a) 20, b) 6, c) 2, d) 0.6 and e) 0.2 µM. The lowest concentration (0.2 µM) gave virtually the same electropherogram as the control without inhibitor.
A wavelength of 210 nm was chosen for the detection of the nucleotides due to the higher sensitivity that can be reached at this wavelength in comparison with higher wavelengths. The washing time and pressure had to be optimized for the enzyme assay, because inside the capillary there was a high concentration of salts, buffer ions, lipids and proteins. A period of 2 min of washing with buffer followed by 1 min with water at 40 p.s.i pressure was found to be sufficient for cleaning the capillary. In the method described for monitoring angiotensin converting enzyme [23, 24], van Dyck et al. had encountered some drawbacks, such as unpredictable migration time shifts and current breakdown. We believe that these problems were due to the adsorption of enzyme to the capillary wall because a fused silica capillary was used. In our study we used a polyacrylamide-coated capillary, which is not expected to show protein adsorption, thus we did observe neither current breakdown nor unpredicted migration time shifts. However, when we used a fused-silica capillary the method was not successful due to the high concentration of salts required in the reaction buffer and probably because of enzyme and lipid adsorption to the capillary wall resulting in peak broadening. Using a short, coated capillary, high precision of migration time and very good peak resolution was observed up to several hundreds of runs.
Quantitative analysis of AMP and ADP
The NTPDase activity and inhibition was determined by measuring the corrected peak area of AMP for NTPDase1, and ADP for NTPDase2 and 3, respectively. Validation of CE measurements of AMP and ADP were performed exactly the same way as for enzyme activity assays, i.e., (1) injection of plugs of different concentrations of AMP or ADP in reaction buffer containing UMP (20 µM) as an internal standard, (2) a suspension of a membrane preparation containing enzyme, followed by (3) another injection of AMP or ADP in reaction buffer containing UMP (20 µM) as an internal standard. An overview of the quantitative parameters of the method validation is provided in Table 1. A strictly linear correlation between AMP and ADP concentrations and the corrected peak area ratio was found: a correlation coefficient (R2) of 0.998 for AMP and 0.999 for ADP (n = 3) was calculated for a concentration range from 2.0 to 50.0 µM. The limit of quantification (LOQ) was found to be 2.95 µg/ml for AMP and 1.41 µg/ml for ADP. The limit of detection (LOD) of AMP was determined to be 0.80 µg/ml for AMP and 0.36 µg/ml for ADP. Standard deviations were generally low (Table 1).
Table 1.
Limits of detection, limits of quantification, migration times and linearity for AMP and ADP determination.
| Compound | AMP | ADP |
|---|---|---|
| Limit of detection T SD (μg/ml) | 0.80 ± 0.20 | 0.36 ± 0.10 |
| Limit of quantification ± SD (μg/ml) | 2.95 ± 0.21 | 1.41 ± 0.13 |
| Linearity of calibration curve; R2 | 0.998 | 0.999 |
| Mean value of migration time ±T SD (min) (n = 12) | 6.00 ± 0.03 | 4.77 ± 0.01 |
| % RSD of migration time (min) | 0.50 | 0.21 |
| Regression equation | y = 724.5x + 1.53, Sy,x = 775 | y = 432.2x + 0.70, Sy,x = 241 |
SD = standard deviation, RSD = relative standard deviation.
Determination of Michaelis-Menten constant (Km) and maximum velocity (Vmax)
The newly developed method was subsequently used to characterize the catalytic properties of defined members of the E-NTPDase family. Using the optimized conditions, Michaelis-Menten constants (Km) and maximal velocity (Vmax) for NTPDases were determined. The enzyme velocity was determined by measuring the peak areas of the products of the enzymatic reaction. Km values were obtained by using different concentrations of the substrate ATP. Each substrate concentration was analyzed in triplicate. The Michaelis-Menten plots are depicted in Figure 4. Estimated Km values of 76, 203, and 311 µM were obtained for NTPDase1, 2 and 3, respectively. The initial reaction velocities were calculated from the amounts of product formed, AMP in the case of NTPDase1 and ADP in the case of NTPDase2 and 3. Vmax values were 0.023, 0.021 and 0.010 µmol/min/mg of protein (membrane preparation) for NTPDase1, 2, and 3, respectively (Table 2).
Figure 4.
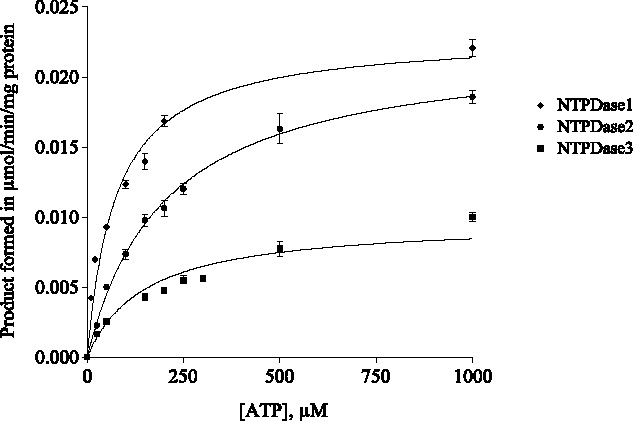
Michaelis-Menten plots for the enzymatic reaction of NTPDase1 (dM), NTPDase2 (•) and NTPDase3 (▪) of initial ATP concentrations with respect to the reaction velocity for the determination of Km and Vmax values by using in-capillary reaction at capillary inlet. For enzyme activity assay see Materials and methods and for CE conditions see Figure 3. Data points represent means T SD from three separate experiments each run in duplicate. For determined Km and Vmax values see Table 2.
Table 2.
Kinetic parameters of NTPDases.
| NTPDase1 | NTPDase2 | NTPDase3 | |
|---|---|---|---|
| Kμ ± SEM [μM] | 76 ± 12 | 203 ± 8 | 311 ± 4 |
| Vmax ± SEM [μmol/min/mg protein] | 0.023 ± 0.002 | 0.021 ± 0.002 | 0.010 ± 0.001 |
The results are means ± SEM of three separate experiments each run in duplicate.
Development of an enzyme inhibition assay
Four selected NTPDase inhibitors were investigated: The nucleotide analog ARL67156, the anthraquinone dye reactive blue 2, the pyridoxal phosphate derivative PPADS, and the symmetrical naphthalenesulfonic acid derivative suramin (Figure 1). While ARL67156 has been reported to be a weak but selective ectonucleotidase inhibitor without significant effects at P2 receptors, the other three compounds are also antagonists at certain P2 receptors [4, 29]. p ]Inhibition of NTPDases 1, 2 and 3 was determined by a range of concentrations of inhibitors spanning three orders of magnitude. The compounds entered the capillary inlet together with the substrate by hydrodynamic injection and the injected plugs of substrate/inhibitor and enzyme were then allowed to react for 5 min. The compounds reacted inside the capillary without applying any voltage, unlike in EMMA, where compounds are mixed electrophoretically by applying voltage before the reaction takes place. After the reaction, a constant current of −60 µA with reverse polarity was applied to separate the reaction products. As an example, an overlay of six electropherograms of the NTPDase3 enzymatic reaction is shown in Figure 3, in which the inhibitor concentration (reactive blue 2) was varied from 0.2 to 20 µM and the concentration of ATP as a substrate was fixed at 320 µM. By increasing the concentration of the inhibitor reactive blue 2, the peak height for ADP was decreased. Using NTPDase1, NTPDase2 and NTPDase3 and the newly developed CE method, a concentration-dependent inhibition by the standard inhibitors was observed for each of the enzymes (Figures 5–7). This allows a direct comparison of the effects of these inhibitors on a variety of identified NTPDases. The Ki values derived are summarized in Table 3.
Table 3.
Ki values for NTPDase1, 2 and 3 obtained for reactive blue 2, PPADS, suramin, and ARL67156, using the in-capillary electrophoresis method.
| Inhibitora | Ki ± SEM [μM] | ||
|---|---|---|---|
| NTPDase1 | NTPDase2 | NTPDase3 | |
| RB2 | 20.0 ± 0.003 | 24.2 ± 0.06 | 1.10 ± 0.03 |
| PPADS | 46.0 ± 0.01 | 44.2 ± 0.03 | 3.0 ± 0.001 |
| Suramin | 300 ± 0.1 | 65.4 ± 0.01 | 12.7 ± 0.03 |
| ARL 67156 | 27.0 ± 0.004 | Q 1000b | 112.1 ± 0.05 |
| Dipyridamole | > 1000c | > 1000c | > 1000c |
The results are means T SEM of three separate experiments each run in duplicate.
a For structures see Figure 1.
b50% inhibition at 1 mM concentration.
cNo inhibitory activity up to a concentration of 1 mM.
The Ki values clearly show that the various NTPDases are differentially susceptible to the individual inhibitors. The values for reactive blue 2 were similar for NTPDase1 and 2, but lower by a factor of 20 for NTPDase3. Similarly, Ki values for PPADS were similar for NTPDase1 and NTPDase2 but 15-fold lower for NTPDse3. ARL67156 revealed the highest inhibitory potency for NTPDase1, was considerably less effective on NTPDase3 and essentially ineffective on NTPDase2. In contrast, suramin revealed the lowest Ki values for NTPDase3 and was less effective on NTPDase2 and NTPDase1. Dipyridamole was not an inhibitor of the E-NTPDases investigated.
Discussion
The ecto-nucleoside triphosphate diphosphohydrolases (EC 3.6.1.5) represent a major and ubiquitous family of ecto-nucleotidases. They catalyze the sequential hydrolysis of the γ- and β-phosphate residues of nucleoside triand diphosphates, producing the corresponding nucleoside monophosphate derivatives [2]. To date four different cell surface-located isoforms of the enzyme family have been cloned and functionally characterized (NTPDase1, 2 and 3, and very recently NTPDase8) [30–32]. The four enzymes differ in substrate specificity and in the pattern of product formation. Whereas NTPDase1 hydrolyzes ATP and ADP about equally well, NTPDase2 has a high preference for the hydrolysis of ATP over ADP. NTPDase3 and NTPDase8 are functional intermediates. NTPDase1 hydrolyzes ATP directly to AMP, ADP is the preferential product of ATP hydrolysis by NTPDase2, and NTPDase3 and NTPDase8 hydrolyze ADP formed from ATP efficiently to AMP.
In previous studies, a variety of compounds has been tested regarding their potency for inhibiting ecto-nucleotidases, often in intact tissues or on cells with undefined enzyme species. Only few studies have used recombinant enzymes to clearly identify the isoform investigated [33, 34]. In addition, several methods have been used for the determination of Michaelis-Menten constants (Km values), and inhibition constants (Ki values for enzyme inhibitors) of NTPDases, including radioisotopic [29], HPLC [35, 36] and spectrophotometric assays [37]. All of these methods are time-consuming. Radiometric assays are very sensitive, but require tedious procedures and the use of radiolabeled substrates [29]. High performance liquid chromatography suffers from relatively high prices for columns, buffers and solvents; in addition, sample pretreatment to remove proteins and lipids is required. Spectrophotometric methods [8, 37] require large amounts of material and are prone to interference from other biological matrices. In addition, the analysis of the total of free phosphate formed confuses the additive contribution of the ATPase and ADPase activity of the identical enzyme, if the nucleoside triphosphate is applied. Our newly developed in-capillary electrophoresis method represents an easy, fast and convenient method for analyzing ecto-nucleotidase activity, including substrate analysis, enzyme kinetics, and the screening for novel inhibitors.
The validity of the method is underpinned by a comparison with previously obtained Km values. Reported Km values vary between species and investigators. Since in many cases the identity of the enzyme species was not determined [6], the values cannot easily be related to our data. However, the Km values we have obtained for the rat orthologs of the enzymes are in very good agreement with values obtained for the recombinant enzymes from other species. Reported Km values (ATP, µM) are 17 for human NTPDase1 [32] and 12 for mouse NTPDase1 [32]; 70, 210 and 394, respectively for human NTPDase2 [32, 38, 39] and 37 for mouse NTPDase2; 75 and 128, respectively for human NTPDase3 [32, 40] and 11 for mouse NTPDase3 [41]. Interestingly, the value obtained for rat NTPDase1 in our study (76 µM) is in excellent agreement with a previously reported value for recombinant rat NTPDase1 (75 µM, [42]). The Km values for NTPDase8, the most recently cloned member of the enzyme family are within the same range (13 µM for mouse and 46 µM for rat NTPDase8) [30, 32].
Using identified enzyme species we can clearly show that NTPDase1, 2 and 3 reveal a differential susceptibility to inhibitors. An inhibitory effect of suramin [3, 29, 43–49], of PPADS [29], of reactive blue 2 [29, 48–50], or of ARL67156 [4, 48, 51] on the hydrolysis of ATP by mammalian ecto-nucleotidases has previously been reported for various cellular systems. However, these studies did not identify the NTPDase isoform(s) expressed nor did they exclude a contribution to ATP hydrolysis by other types of ecto-nucleotidases.
In our study, reactive blue 2, PPADS and suramin inhibited NTPDase3 15- to 25-fold more effectively than NTPDase1. NTPDase2 was inhibited by reactive blue 2 and PPADS to a similar extent as NTPDase1 but the Ki value for suramin was five fold higher for NTPDase1. An analysis of detergent-solubilized NTPDase1 and NTPDase2 purified from porcine brain, revealed Ki values (ATP) for suramin of 1.8 mM and 2.1 mM, respectively [52]. These values are much higher than those obtained for the membrane-bound rat enzymes reported in this study. In contrast to the other compounds tested, ARL67156 revealed the highest inhibitory potency for NTPDase1. It was considerably less effective on NTPDase3 and essentially ineffective on NTPDase2. Since ARL67156 is a widely used inhibitor of ‘ecto-ATPase’ activity, this observation is a caveat for experiments in which this inhibitor is used without previous identification of the NTPDase involved. Dipyridamole was not an inhibitor of the NTPDases investigated but in a previous study it was found to be an effective inhibitor of ATP hydrolysis in rat superior cervical ganglionic cells at 10 µM concentration [53]. The compound has been found to be inactive in other systems [e.g., 16].
In conclusion, we have developed a new method for the characterization of E-NTPDases and the screening of potential substrates and inhibitors using capillary electrophoresis coupled with UV detection. In order to define the properties of individual isoforms, we determined the Km values of heterologously expressed NTPDase1, NTPDase2 and NTPDase3 and the Ki values for selected inhibitors. By using a neutral capillary, migration times of nucleotides were decreased in comparison with a fused silica capillary resulting in high reproducibility and precision of analysis. The developed in-capillary electrophoresis method is an easy, fast and convenient method for studying enzyme kinetics, and for searching for substrates and novel inhibitors.
The scale of the enzymatic reaction could be dramatically reduced to the nanoliter scale as compared to off-line analysis of the reaction carried out in a microcentrifuge tube. Moreover, since the capillary is used as a reaction vessel, all the assay steps (mixing, reaction, separation, and quantitation) are combined in a fully automated microscale activity assay. This process was carried out automatically using a temperature-controlled autosampler in order to eliminate routine handling and to speed-up the process. The newly developed fast, easy and versatile screening method will greatly facilitate the search for and development of potent and selective ectonucleotidase inhibitors which are urgently required for pharmacological studies.
Acknowledgements
N.B. and H.Z. were supported by a grant from the Deutsche Forschungsgemeinschaft (SFB 269, A4). C.E.M. is grateful for support by the Fonds der Chemischen Industrie.
Abbreviations
- ARL67156
N6-Diethyl-β,γ-dibromomethylene-ATP
- CE
capillary electrophoresis
- CHO
Chinese hamster ovary
- EMMA
electrophoretically mediated microanalysis
- (E)-NTPDase
(ecto)-nucleoside triphosphate diphosphohydrolase
- I.S.
internal standard
- PPADS
pyridoxalphosphate-6-azophenyl-2-4-disulfonic acid
- RB2
reactive blue 2
References
- 1.Ralevic V, Burnstock G. Receptors for purines and pyrimidines. Pharmacol Rev 1998; 50: 413-2. [PubMed]
- 2.Zimmermann H. Extracellular metabolism of ATP and other nucleotides. Naunyn–Schmiedeberg's Arch Pharmacol 2000; 362: 299–309. [DOI] [PubMed]
- 3.Crack BE, Beukers MW, McKechnie KC et al. Pharmacological analysis of ecto-ATPase inhibition: Evidence for combined enzyme inhibition and receptor antagonism in P2X-purinoceptor ligands. Br J Pharmacol 1994; 113: 1432-. [DOI] [PMC free article] [PubMed]
- 4.Crack BE, Pollard CE, Beukers MW et al. Pharmacological and biochemical analysis of FPL 67156, a novel, selective inhibitor of ecto-ATPase. Br J Pharmacol 1995; 114: 475-1. [DOI] [PMC free article] [PubMed]
- 5.Bultmann R, Driessen B, Goncalves J, Starke K. Functional consequences of inhibition of nucleotide breakdown in rat vas deferens: A study with Evans blue. Naunyn–Schmiedebergs Arch Pharmacol 1995; 351: 555-0. [DOI] [PubMed]
- 6.Zimmermann, H. Ecto-nucleotidases. Purinergic and Pyrimidergic Signalling. In Abbracchio MP, Williams M (eds): Handbook of Experimental Pharmacology. Heidelberg: Springer 2001; 209-0.
- 7.Kennedy C, Westfall TD, Sneddon P. Modulation of purinergic neurotransmission by ecto-ATPase. Semin Neurosci 1996; 8: 195-9. [DOI]
- 8.Gendron FP, Halbfinger E, Fischer B et al. Novel inhibitors of nucleoside triphosphate diphosphohydrolases: Chemical synthesis and biochemical and pharmacological characterizations. J Med Chem 2000; 43: 2239-7. [DOI] [PubMed]
- 9.Burns KL, May SW. Separation methods applicable to the evaluation of enzyme-inhibitor enzyme-substrate interactions. J Chromatogr, B Analyt Technol Biomed Life Sci 2003; 797: 175-0. [DOI] [PubMed]
- 10.Van Dyck S, Van Schepdael A, Hoogmartens J. Kinetic study of gamma-glutamyltransferase activity by electrophoretically mediated microanalysis combined with micellar electrokinetic capillary chromatography. Electrophoresis 2002; 23: 2854-. [DOI] [PubMed]
- 11.Viglio S, Zanaboni G, Luisetti M et al. Micellar electrokinetic chromatography: A convenient alternative to colorimetric and high performance liquid chromatographic detection to monitor protease activity. Electrophoresis 1998; 19: 2083-. [DOI] [PubMed]
- 12.Dai HJ, Parker CN, Bao JJ. Characterization and inhibition study of MurA enzyme by capillary electrophoresis. J Chromatogr, B Analyt Technol Biomed Life Sci 2002; 766: 123-2. [DOI] [PubMed]
- 13.Kanie Y, Kanie O. Electrophoretically mediated reaction of glycosidases at a nanoliter scale. Electrophoresis 2003; 24: 1111-. [DOI] [PubMed]
- 14.Whisnant AR, Johnston SE, Gilman SD. Capillary electrophoretic analysis of alkaline phosphatase inhibition by theophylline. Electrophoresis 2000; 21: 1341-. [DOI] [PubMed]
- 15.Meredith GD, Sims CE, Soughayer JS, Allbritton NL. Measurement of kinase activation in single mammalian cells. Nat Biotechnol 2000; 18: 309-2. [DOI] [PubMed]
- 16.Kaulich M, Qurishi R, Müller CE. Extracellular metabolism of nucleotides in neuroblastoma x glioma NG108-15 cells determined by capillary electrophoresis. Cell Mol Neurobiol 2003; 23: 349-4. [DOI] [PMC free article] [PubMed]
- 17.Qurishi R, Kaulich M, Müller CE. Fast, efficient capillary electrophoresis method for measuring nucleotide degradation and metabolism. J Chromatogr, A 2002; 952: 275-1. [DOI] [PubMed]
- 18.Bao J, Regnier FE. Ultramicro enzyme assays in a capillary electrophoretic system. J Chromatogr 1992; 608: 217-4. [DOI] [PubMed]
- 19.Telnarova M, Vytiskova S, Monincova M, Glatz Z. Electrophoretically mediated microanalysis with partial filling technique and indirect or direct detection as a tool for inhibition studies of enzymatic reaction. Electrophoresis 2004; 25: 1028-3. [DOI] [PubMed]
- 20.Telnarova M, Vytiskova S, Chaloupkova R, Glatz Z. Study of enzymatic reaction by electrophoretically mediated microanalysis in a partially filled capillary with indirect or direct detection. Electrophoresis 2004; 25: 290-. [DOI] [PubMed]
- 21.Zhang Y, Kaddis J, Silverio C et al. On-column enzyme-catalyzed microreactions using capillary electrophoresis: Quantitative studies. J Capillary Electrophor 2002; 7: 1–9. [PubMed]
- 22.Zhao DS, Gomez FA. Double enzyme-catalyzed microreactors using capillary electrophoresis. Electrophoresis 1998; 19: 420-. [DOI] [PubMed]
- 23.Van Dyck S, Vissers S, Van Schepdael A, Hoogmartens J. Kinetic study of angiotensin converting enzyme activity by capillary electrophoresis after in-line reaction at the capillary inlet. J Chromatogr A 2003; 986: 303-1. [DOI] [PubMed]
- 24.Van Dyck S, Novakova S, Van Schepdael A, Hoogmartens J. Inhibition study of angiotensin converting enzyme by capillary electrophoresis after enzymatic reaction at capillary inlet. J Chromatogr, A 2003; 1013: 149-6. [DOI] [PubMed]
- 25.Kegel B, Braun N, Heine P et al. An ecto-ATPase and an ecto-ATP diphosphohydrolase are expressed in rat brain. Neuropharmacology 1997; 36: 1189-00. [DOI] [PubMed]
- 26.Zimmermann H, Pelletier J, Sevigny J, Braun N. Cloning and characterization of the ecto-nucleotidase NTPDase3 from rat brain: Predicted secondary structure and relation to other members of the E-NTPDase family and actin. Purinergic Signalling 2005; in press. [DOI] [PMC free article] [PubMed]
- 27.Lanzetta PA, Alvarez LJ, Reinach PS, Candia OA. An improved assay for nanomole amounts of inorganic phosphate. Anal Biochem 1979; 100: 95-. [DOI] [PubMed]
- 28.Spector T. Refinement of the coomassie blue method of protein quantitation. A simple and linear spectrophotometric assay for less than or equal to 0.5 to 50 microgram of protein. Anal Biochem 1978; 86: 142-. [DOI] [PubMed]
- 29.Chen BC, Lee CM, Lin WW. Inhibition of ecto-ATPase by PPADS, suramin and reactive blue in endothelial cells, C6 glioma cells and RAW 264.7 macrophages. Br J Pharmacol 1996; 119: 1628-4. [DOI] [PMC free article] [PubMed]
- 30.Bigonnesse F, Levesque SA, Kukulski F et al. Cloning and characterization of mouse nucleoside triphosphate diphosphohydrolase-8. Biochemistry 2004; 43: 5511-. [DOI] [PubMed]
- 31.Zimmermann H. Ectonucleotidases: Some recent developments and a note on nomenclature. Drug Dev Res 2001b; 52: 44–56. [DOI]
- 32.Kukulski F, Lévesque SA, Lavoie ÉG et al. Comparative hydrolysis of P2 receptor agonists by NTPDases 1, 2, 3 and 8. Purinergic Signalling 2005; in press. [DOI] [PMC free article] [PubMed]
- 33.Heine P, Braun N, Heilbronn A, Zimmermann H. Functional characterization of rat ecto-ATPase and ecto-ATP diphosphohydrolase after heterologous expression in CHO cells. Eur J Biochem 1999; 262: 102-. [DOI] [PubMed]
- 34.Hoffmann C, Heine P, Pradel G et al. Inhibition of ecto-apyrase and ecto-ATPase by pyridoxal phosphate-related compounds. Drug Dev Res 2000; 51: 153-. [DOI] [PMC free article] [PubMed]
- 35.Heine P, Braun N, Sevigny J et al. The C-terminal cysteine-rich region dictates specific catalytic properties in chimeras of the ectonucleotidases NTPDase1 and NTPDase2. Eur J Biochem 2001; 268: 364-3. [DOI] [PubMed]
- 36.Mihaylova-Todorova ST, Todorov LD, Westfall DP. Enzyme kinetics and pharmacological characterization of nucleotidases released from the guinea pig isolated vas deferens during nerve stimulation: Evidence for a soluble ecto-nucleoside triphosphate diphosphohydrolase-like ATPase and a soluble ecto-5-nucleotidase-like AMPase. J Pharmacol Exp Ther 2002; 302: 992–1001. [DOI] [PubMed]
- 37.Knowles AF, Nagy AK. Inhibition of an ecto-ATP-diphosphohydrolase by azide. Eur J Biochem 1999; 262: 349-7. [DOI] [PubMed]
- 38.Knowles AF, Chiang WC. Enzymatic and transcriptional regulation of human ecto-ATPase/E-NTPDase 2. Arch Biochem Biophys 2003; 418: 217-7. [DOI] [PubMed]
- 39.Mateo J, Harden TK, Boyer JL. Functional expression of a cDNA encoding a human ecto-ATPase. Br J Pharmacol 1999; 128: 396–402. [DOI] [PMC free article] [PubMed]
- 40.Smith TM, Kirley TL. Site-directed mutagenesis of a human brain ecto-apyrase: Evidence that the E-type ATPases are related to the actin/heat shock 70/sugar kinase superfamily. Biochemistry 1999; 38: 321-. [DOI] [PubMed]
- 41.Lavoie EG, Kukulski F, Levesque SA et al. Cloning and characterization of mouse nucleoside triphosphate diphosphohydrolase-3. Biochem Pharmacol 2004; 67: 1917-6. [DOI] [PubMed]
- 42.Wang TF, Ou Y, Guidotti G. The transmembrane domains of ectoapyrase (CD39) affect its enzymatic activity and quaternary structure. J Biol Chem 1998; 273: 24814-1. [DOI] [PubMed]
- 43.Hourani SM, Chown JA. The effects of some possible inhibitors of ectonucleotidases on the breakdown and pharmacological effects of ATP in the guinea-pig urinary bladder. Gen Pharmacol 1989; 20: 413-. [DOI] [PubMed]
- 44.Beukers MW, Kerkhof CJ, van Rhee MA et al. Suramin analogs, divalent cations and ATP gamma S as inhibitors of ecto-ATPase. Naunyn–Schmiedeberg's Arch Pharmacol 1995; 351: 523-. [DOI] [PubMed]
- 45.Meghji P, Burnstock G. Inhibition of extracellular ATP degradation in endothelial cells. Life Sci 1995; 57: 763-1. [DOI] [PubMed]
- 46.Bultmann R, Wittenburg H, Pause B et al. P2-purinoceptor antagonists: III. Blockade of P2-purinoceptor subtypes and ecto-nucleotidases by compounds related to suramin. Naunyn–Schmiedeberg's Arch Pharmacol 1996; 354: 498–504. [DOI] [PubMed]
- 47.Bonan CD, Roesler R, Quevedo J et al. Effects of suramin on hippocampal apyrase activity and inhibitory avoidance learning of rats. Pharmacol Biochem Behav 1999; 63: 153-. [DOI] [PubMed]
- 48.Dowd FJ, Li LS, Zeng W. Inhibition of rat parotid ecto-ATPase activity. Arch Oral Biol 1999; 44: 1055-2. [DOI] [PubMed]
- 49.Yegutkin GG, Burnstock G. Inhibitory effects of some purinergic agents on ecto-ATPase activity and pattern of stepwise ATP hydrolysis in rat liver plasma membranes. Biochim Biophys Acta 2000; 1466: 234-4. [DOI] [PubMed]
- 50.Tuluc F, Bultmann R, Glanzel M et al. P2-receptor antagonists: IV. Blockade of P2-receptor subtypes and ecto-nucleotidases by compounds related to reactive blue 2. Naunyn–Schmiedeberg's Arch Pharmacol 1998; 357: 111-0. [DOI] [PubMed]
- 51.Drakulich DA, Spellmon C, Hexum TD. Effect of the ecto-ATPase inhibitor, ARL 67156, on the bovine chromaffin cell response to ATP. Eur J Pharmacol 2004; 485: 137-0. [DOI] [PubMed]
- 52.Kukulski F, Komoszynski M. Purification and characterization of NTPDase1 (ecto-apyrase) and NTPDase2 (ecto-ATPase) from porcine brain cortex synaptosomes. Eur J Biochem 2003; 270: 3447-4. [DOI] [PubMed]