

Abstract
Nearly three-quarters of a century ago, Curt Richter removed the adrenal glands from rats and noted that the animal's vitality was dependent on its increased consumption of sodium chloride. In doing so, Richter revealed an innate behavioral mechanism that serves to maintain the hydromineral balance of an animal faced with sodium deficit. This experiment and others like it, led to the development of a field of research devoted to the investigation of salt appetite. The following is a discussion of how Richter's initial observations gave birth to an evolving field that incorporates multiple approaches to examine the drive to consume sodium.
Keywords: Salt appetite, Angiotensin, Aldosterone, Corticosterone, Richter
The Discovery of Salt Appetite
At the time that Richter began his career at Johns Hopkins University much of the research examining ingestive behavior focused on the responses of individual organs or closely coordinated systems. Richter's pioneering work initiated a fundamental paradigm shift that moved the focus to adaptations of the organism as a whole. Specifically, he was interested in changes in behavior that compensated for perturbations in the internal environment. His approach toward investigation of such behaviors was as thorough as it was fundamental. Richter manipulated diet, interfered with the nervous system, and removed or replaced glandular secretions in attempt to understand the nutritional, neural, and endocrine signals that contribute to behavior.
Richter created disturbances in the nutritional or mineral content of the internal environment with dietary deficiency. Subsequently, “self selection” experiments were performed where choices of different minerals and nutrients were presented for consumption. More often than not, animals would consume minerals and nutrients in amounts that would alleviate their experimentally-induced deficiencies. One of the most striking behaviors that Richter observed was that of rats voluntarily ingesting salt when faced with sodium deficit. Sodium chloride was removed from the rats' food and they were given access to water and 3 % NaCl solution. The intake of the NaCl solution was approximately 1% of the total diet, which remarkably, is the amount of salt that was calculated as necessary for healthy development of the rat (Richter, 1956). These results suggested that the rats deliberately drank NaCl to effectively compensate for the loss of salt in their food.
Experiments like these led to other studies that examined the behavioral responses to sodium deficiency when physiological mechanisms aimed at sodium conservation were surgically eliminated. This approach is well demonstrated in Richter's 1936 publication Increased Salt Appetite in Adrenalectomized Rats. While it was previously known that adrenalectomy produced sodium insufficiency (Rubin & Krick, 1933), Richter found that immediately after adrenalectomy the consumption of NaCl solution was augmented while water intake was reduced. The increased consumption of NaCl was robust and occurred the first time the rats were given access to the solutions, suggesting that the behavior was innate because there was little time to learn of the post-ingestive consequences of the enhanced intake. Importantly, adrenalectomized rats did not increase intake of chloride supplied as magnesium chloride or aluminum chloride, but did readily ingested different sodium compounds indicating that the appetite was specific for sodium (Richter, 1956). As a result of having access to saline, the survival rate of the adrenalectomized rats was greatly increased, and except for the propensity to consume large volumes of salt solutions there were no indications of deficiency and the animals gained weight normally. However, when the salt solutions were removed, almost ninety percent of the animals died within five days, thereby providing further evidence that an increased appetite for salt was keeping these animals alive. In his discussion section Richter wrote, “The fact that the salt appetite of adrenalectomized rats has such a close relationship to the salt deficiency indicates that appetite may be used as a measure of the deficiencies produced by endocrine disturbances, or by pathological changes in other parts of the body… Thus, it may be that even in man a closer study of the patients may throw more light on the actual needs and deficiencies present in such conditions as pregnancy or in acute or chronic disturbances of the endocrine system.”
This discussion foreshadowed a subsequent case study by Wilkins and Richter (1940) that described a 3 ½-year old boy admitted to the hospital with symptoms of early-onset puberty. Tragically, seven days after admittance the child died and it was only after his death that it was determined that he was unable to retain sodium due to adrenal pathology. Interviews with the child's parents revealed that at an early age the boy had an insatiable appetite for salt, which likely kept him alive until the relatively low sodium diet at the hospital resulted in his death.
While Richter's initial studies demonstrated that sodium deficiency was accompanied by an increased salt appetite that promotes vitality, the signals relating to the arousal of the appetite were poorly understood. Richter found that grafting adrenal tissue into the eye of adrenalectomized rats decreased sodium intake to that of intact rats within 10 days, suggesting that the increased intake is due to the loss of some adrenal secretion (Richter 1956). Consequently, it was hypothesized that salt appetite was dependent on the presence of some salt retaining hormone, possibly aldosterone, which might be produced in the adrenal glomerulosa. Richter treated adrenalectomized rats with the aldosterone precursor, deoxycorticosterone (DOC), and NaCl intake returned to normal, presumably, because excessive amounts of sodium were no longer lost in urine (Richter & Eckert, 1938). However, subsequent studies revealed that treating intact rats with DOC elicited a salt appetite similar to that of adrenalectomized rats, which at the time seemed paradoxical (Rice & Richter, 1943). Adrenalectomized rats lacking endogenous mineralocorticoids had a robust salt appetite that could be attenuated by DOC, but administration of this agonist in normal rats increased NaCl intake with increasing doses. Subsequent research would attempt to resolve this conundrum by investigating the mechanisms underlying DOC-induced NaCl intake and evaluating other factors contributing to salt appetite.
Although removal of the adrenals produced a sodium intake that could be studied in the laboratory, Richter understood that under normal circumstances behavior and the corresponding physiology worked in concert to maintain hydromineral balance. His conceptualization of this interaction is shown in Figure 1. In his previous work Richter found that animals tended to over consume salt when faced with sodium deficit. Therefore, Richter's model of hydromineral balance starts with dietary salt deficiency that is over-corrected for by salt appetite. Consequently, physiological mechanisms compensate for this over-consumption and after a few cycles through this circuit blood salt settles at the appropriate level. Note how decreased blood salt is detected by centers in the hypothalamus, which stimulate adrenal secretions that conserve sodium at the level of the kidney and elicit a salt appetite that is dependent on altered gustatory processing. The increased blood salt resulting from ingestion feeds back to the hypothalamus to inhibit adrenal secretions while also returning gustatory processing to normal, which inhibits further intake. Throughout the course of our discussion we will refer back to Figure 1 to demonstrate how subsequent research confirmed or modified Richter's model.
Figure 1.
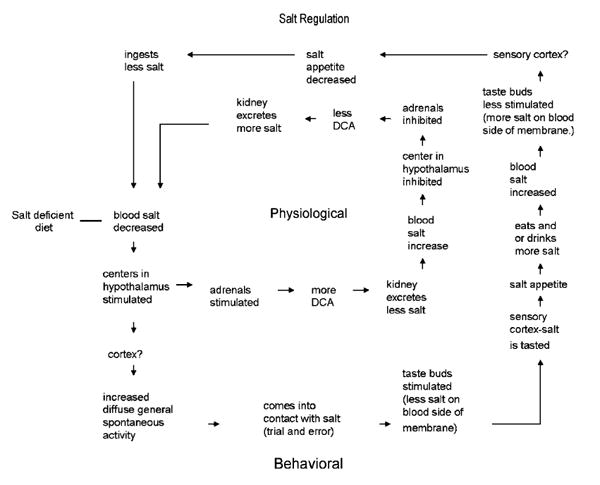
Diagram showing various possible steps in the operation of physiological and behavioral feed-back circuits for the regulation of an animal on a salt deficient diet. Recreated from Richter, L'instinct dans le comportement des animaux et de l'homme (1953).
Arousal of Salt Appetite: Contributions of Aldosterone
Several decades later, researchers revisited Richter's model of adrenalectomy to elucidate the role of adrenal steroids in the arousal of salt appetite. In this regard, Wolf (1965) expanded on Richter's initial observations by examining the NaCl intake of intact and adrenalectomized rats after administration of various doses of DOC. As expected, intact rats increased NaCl intake with elevating doses of DOC. In contrast, the dose-response curve of the adrenalectomized rats resembled an inverted U-shape, with DOC diminishing intakes at low doses, but increasing ingestion at higher doses. Interestingly, pre-treating adrenalectomized rats with corticosterone, an endogenous product of the adrenal cortex, decreased the doses of DOC required to produce NaCl intake. In addition, Wolf and others found that aldosterone, an endogenous adrenal steroid, specifically augmented the intake of sodium salts (Weisinger & Woods, 1971, Wolf & Handal, 1966). Taken together, these studies suggested that endogenous adrenal steroids contributed to the arousal of salt appetite; however, the mechanism underlying the natrorexigenic effects of these hormones remained unknown.
Early investigation of adrenal steroid regulation of salt appetite focused on the central actions of corticosterone and aldosterone because certain brain lesions inhibited the salt intake that follows DOC administration (Nitabach et al., 1989; Wolf, 1964). Brain regions that readily took up these hormones were identified with the use of receptor autoradiography. Specifically, corticosterone and aldosterone were found to compete for binding sites in the hippocampus, septum and amygdala (McEwen, 1986; Birmingham et al.,1979; Gerlach & McEwen, 1972). However, the relative contribution of adrenal steroids to salt appetite in specific brain areas was uncertain until specific antagonists for glucocorticoid (GR) and mineralcorticoid (MR) receptors were developed. In this regard, McEwen et al (1986) administered RU 28318, a MR-specific antagonist, to adrenalectomized rats and observed that the same dose of RU 28318 that inhibited aldosterone binding in the hippocampus, septum and amygdala also blunted the hormone's effects on salt intake. These results provided indirect evidence that aldosterone regulation of salt appetite involves, in part, the actions of aldosterone in the brain, but advances in the field of molecular genetics would be required before a specific mechanism and site of action could be determined.
Aldosterone was believed to activate central pathways mediating salt appetite via two mechanisms (Fluharty & Sakai, 1995). Aldosterone, like other steroid hormones, binds to cytosolic receptors that act as transcription factors, which initiate changes in gene expression. This mode of action takes hours to days and was thought to be a factor in the long-term regulation of sodium intake. In addition, aldosterone can also induce rapid changes in NaCl consumption through actions at the cell surface. Specifically, when converted into its tetrahydro metabolites, aldosterone interacts with ligand-gated ion channels to influence neuronal firing. This non-genomic mechanism was thought to contribute to the rapid ingestion of sodium that accompanies DOC administration. Thus, it was reasoned that aldosterone regulates short and long-term consumption of sodium through genomic as well as non-genomic actions within the brain.
Sakai and his colleagues (2000) provided evidence supporting this hypothesis by using antisense oligodeoxynulceotides (ASDNs), single-stranded DNA molecules that inhibit the expression of targeted genes, to evaluate the role of MR in DOC-induced salt intake. Antisense oligodeoxynulceotides directed against the GR or MR were injected into the amygdala, thereby inhibiting the expression of these receptors locally within this brain region. Subsequently, DOC was administered and salt intake was measured. Administration of ASDNs directed against MR significantly reduced DOC-induced sodium intake, where as ASDNs directed against the GR had no effect. Interestingly, the same study reported that salt intake could be elicited 15 min after direct injection of tetrahydro-aldosterone into the amygadala, and this effect was not blocked by administration of MR antagonists that inhibit the genomic actions of this receptor. Collectively, these results suggested that salt appetite could be aroused through both genomic and non-genomic effects of mineralocorticoids. However, the fact that adrenalectomized rats lacking endogenous adrenal steroids demonstrated a robust salt appetite made it unlikely that aldosterone was the only signal capable of eliciting sodium intake.
Arousal of Salt Appetite: Contributions of Angiotensin II
Angiotensin II is the effector peptide of the renin-angiotensin-system (RAS), an endocrine system that is activated in response to decreased blood pressure or volume. Prior to being implicated in salt appetite, it was known that angiotensin II infused intravenously or injected into certain brain regions increased water intake and blood pressure. Although increased water intake is a compensatory response to hypovolemia, consuming only water will not alleviate deficits in vascular volume because water is stored mostly within intracellular compartments. A more advantageous response would be to ingest NaCl and water to produce a mixture near the concentration of isotonic saline, which would more effectively replace the volume lost from extracellular fluid compartments. In this regard, it was known that angiotensin II contributed to the elicitation of salt appetite indirectly by stimulating the release of aldosterone (Davis & Speilman, 1974) and some studies suggested that angiotensin did not play a significant role in salt appetite (Stricker, 1971). Thus, there was little evidence for direct involvement of angiotensin II as a stimulator of sodium intake.
Buggy and Fisher (1974) subsequently redefined the role that angiotensin II played in the maintenance of body fluid homeostasis, finding that intracranial administration of angiotensin II increased water and sodium intake. Importantly, the elevated consumption of NaCl was specific to angiotensin II because administration of carbachol, another centrally acting dipsogen, elicited only water intake. Subsequent studies further implicated the peptide in the arousal of salt appetite and provided potential sites of actions for this effect. For example, salt appetite resulting from the natriuretic-diuretic furosemide was attenuated by pharmacological reduction of circulating angiotensin II and was restored by intravenous administration of this peptide (Weisinger et al., 1997, 1987).
Circulating angiotensin II acts centrally by binding to angiotensin type 1 (AT1) receptors in circumventricular organs, specialized brain regions that lack a normal blood brain barrier (McKinley et al., 2003). In particular, the subfornical organ (SFO) and the organum vasculosum of the lamina terminalis (OVLT) are circumventricular organs that express AT1 receptors and initiate central responses to hypotension and hypovolemia. Accordingly, investigators considered the SFO and OVLT potential sites of action for angiotensin II arousal of salt appetite. In this regard, ablation of the SFO decreased sodium intake elicited by administration of the furosemide (Thunhorst et al., 1999; Weisinger et al., 1990). Similarly, lesion of the OVLT significantly attenuated NaCl intake induced by sodium depletion (Chiaraviglio & Perez, 1984), and site-specific injection of angiotensin II into the OVLT stimulated NaCl consumption (Fitts & Masson, 1990).
Although these studies suggested that angiotensin II acted centrally to elicit salt appetite, the biological relevance of this phenomenon was called into question because supra-physiological doses of angiotensin II were required to produce this behavior. Large doses of angiotensin II administered centrally or intravenously elevate blood pressure and can cause natriuresis (Fluharty & Manaker, 1983; Findlay & Epstein, 1980). This complicated interpretation of studies examining angiotensin II-induced salt appetite because it was possible that the NaCl intake caused by angiotensin II treatment was the consequence of renal sodium excretion. However, the recognition that circulating levels of angiotensin II and aldosterone increased under normal conditions of sodium depletion (Stricker et al., 1979) led to a new understanding of hormonal regulation of salt appetite.
Arousal of Salt Appetite: The Synergy Hypothesis
Sodium depletion stimulates renin release from the kidney, leading to increased circulating levels of angiotensin II and aldosterone (Davis & Speilman, 1974). Because sodium depletion leads to the concurrent elevations in both aldosterone and angiotensin II, it was hypothesized that these two hormones worked in concert to arouse sodium appetite (Epstein, 1982) and Fluharty and Epstein (1983) found that angiotensin II and mineralcorticoids acted synergistically to elicit salt appetite. Specifically, simultaneous administration of angiotensin II and DOC produced a salt intake that was greater than the sum of the amounts consumed when the same doses of angiotensin II or DOC were separately administered. Furthermore, low doses of angiotensin II and DOC that were previously ineffective at evoking salt appetite when given independently became potent at inducing sodium intake when given at the same time. This synergistically-enhanced salt intake was not accompanied by pressure-induced natriuresis, indicating that renal sodium excretion did not contribute to the augmented ingestion of NaCl.
Ensuing research provided further support for the “synergy hypothesis”. For example, central administration of RU-28318 reduced, but did not eliminate, the salt appetite elicited by sodium depletion; however, when the formation of endogenous angiotensin II was inhibited by captopril administration and RU-28318 was given centrally, depletion-induced sodium intake was completely abolished (Sakai et al., 1986). Pharmacological inhibition of angiotensin II formation and mineralcorticoid receptors did not affect urinary sodium excretion or food intake, indicating that effects were not due to altered renal sodium handling or general inhibition of ingestive behaviors (Sakai et al., 1986). Thus, it was concluded that angiotensin II and mineralcorticoids worked synergistically to arouse salt appetite.
Central Interactions of Aldosterone and Angiotensin II
Because blocking the effects of angiotensin II and aldosterone in the brain, but not in the periphery, eliminated salt appetite of furosemide treated rats (Sakai et al., 1990; Sakai et al., 1986), researchers began examining central interactions between these two hormones. Chronic administration of DOC increased angiotensin II-receptor binding sites in tissue homogenates including the hypothalamus, thalamus and septum (Wilson et al., 1986). Additionally, drinking stimulated by central injection of angiotensin II was increased after DOC administration, demonstrating the behavioral consequences of mineralcorticoid-mediated increases in angiotensin II binding sites (Wilson et al., 1986). Subsequent studies identified increased angiotensin II receptor binding in specific brain regions following DOC administration (De Nicola et al., 1993; Gutkind et al., 1988) as well as within cultured neurons (Sumners & Fregly, 1989). Whether the up-regulation of angiotensin II receptors was due to the actions of mineralcorticoids, glucocorticoids, or a combination of both remained to be determined.
Deoxycorticosterone is a precursor for both aldosterone and corticosterone, and consequently, administration of DOC leads to elevations in both of these adrenal steroids. Because previous studies examined the affects of DOC on angiotensin II receptor binding, the function of specific adrenal steroids in the regulation of angiotensin II receptors was uncertain. However, a role for glucocorticoids was proposed because DOC increased angiotensin II-receptor binding to a greater degree than did aldosterone (Wilson et al., 1986) and because pretreatment with dexamethasone, a glucocorticoid agonist, potentiated drinking stimulated by angiotensin II (Ganesan & Sumners, 1989). In addition, intravenous infusion of dexamethasone, corticosterone, or the glucocorticoid receptor agonist RU28362, increased aldosterone-induced sodium consumption, further suggesting that glucocorticoids contribute to the arousal of salt appetite (Ma et al., 1993).
The strongest evidence for glucocorticoid regulation of salt appetite came from studies examining NaCl intake and angiotensin II-receptor binding after administration of DOC and dexamethasone (Shelat et al., 1999b). Specifically, rats treated with DOC and dexamethasone consumed more NaCl than when given DOC alone. Furthermore, co-administration of DOC and dexamethasone increased angiotensin II receptor binding in the paraventricular nucleus, subfornical organ and area postrema, but administration of only DOC had no such effect (Shelat et al., 1999a). Accordingly, new models of hormonal regulation of salt appetite incorporate the contributions of mineralcorticoids as well as glucocorticoids.
Other Contributors : Baroreceptors and Osmoreceptors
Baroreceptors are mechanoreceptors located in blood vessels near the heart that provide the brain with information pertaining to blood pressure and volume by detecting stretch on vascular walls. As blood volume increases the vessels are stretched and the firing rate of the receptors increases. Conversely, as blood volume decreases the vessels contract and the receptors decrease firing. It was reasoned that baroreceptors were involved in the regulation of salt appetite because sodium insufficiency is often accompanied by changes in blood volume.
Evidence supporting baroreceptor mediation of salt appetite comes from studies that surgically manipulate baroreceptors and subsequently examine sodium consumption. For example, Toth et al (1987) implanted small balloons into the superior vena caval-right atrial junction of rats. After the rats recovered from the surgery the balloons were inflated, stretching the baroreceptors and signaling volume expansion to the brain. During this time NaCl intake elicited either by peritoneal dialysis or administration of DOC was greatly attenuated and intakes returned to normal when the balloons were deflated. Similarly, when baroreceptor input was eliminated by sinoaortic denervation, NaCl intake elicited by furosemide was significantly reduced (Thunhorst et al., 1994). Collectively, these studies suggest that baroreceptors provide the brain with neural signals pertaining to blood volume that are critical for the normal expression of salt appetite.
While cardiac baroreceptors are thought to regulate salt appetite by detecting changes in blood volume, cerebral osmoreceptors were believed to influence sodium consumption by monitoring the [Na+] of cerebrospinal fluid (CSF). Support for osmoreceptor regulation of salt appetite comes from studies that manipulated the sodium concentration of CSF and subsequently examined sodium consumption. Intraventricular injection of CSF [Na+, 500 mM] rapidly decreased deprivation-induced sodium intake in sheep (Weisinger et al., 1979). However, when osmolality of the CSF was increased by intraventricular injection of mannitol, which decreased [Na+], sodium intake was augmented (Weisinger et al., 1979). These results suggest that the brain contains sodium-sensitive neurons or osmoreceptors that monitor the [Na+] of CSF and regulate the ingestion of sodium accordingly.
Sodium Deficit and Gustatory Processing
Taste is the sensory modality most important for the detection of sodium. Richter's previous studies suggested that adrenalectomy and the resulting sodium insufficiency created a clear preference for NaCl solutions that were previously indistinguishable from water. It is perhaps not surprising that Richter hypothesized that sodium depletion led to changes in the oral cavity that contributed to the increased salt intake. Accordingly, he designed experiments to investigate whether sodium deficiency enhanced the detection threshold for NaCl solutions.
These experiments are described in Richter's manuscript Salt Taste Thresholds of Normal and Adrenalectomized Rats (1939). Richter used a two-bottle preference test to determine the concentration at which intact or adrenalectomized rats prefer a NaCl solution over water. Intact rats preferred the NaCl solution when the concentration was about 0.055%; however, immediately after adrenalectomy rats drank more of the NaCl solution when it was at a concentration of 0.0037% or nearly fifteen fold less concentrated than before. These results demonstrate that sodium insufficiency decreases the threshold at which rats prefer NaCl solutions over water, possibly by increasing the sensitivity for the taste of sodium. Richter suggested that the enhanced preference for the NaCl was likely innate because the rats revealed this preference despite having no knowledge of the beneficial consequences of the intake. In his discussion section he postulated, “The results indicate that adrenalectomized rats ingest more salt, not because they learn that salt relieves their deficiency discomforts, but because of chemical changes in the taste mechanisms in the oral cavity, giving rise to an enhanced salt discrimination.”
Subsequent research would provide further support for Richter's interpretation. Examination of the ingestion of saline solutions at various concentrations revealed that intact rats have a preference curve that resembles an inverted-U shape with maximum consumption occurring near the concentration of isotonic saline (Richter, 1956). However, sodium deficiency arising from adrenalectomy or dietary deprivation dramatically changes this preference curve by elevating intake of NaCl at low and high concentrations (Curtis et al., 2001; Nachman, 1962; Epstein & Stellar, 1955). One study using short-term intake tests demonstrated that sodium deficient rats develop a preference for sodium within 15 sec after being given a choice between sodium and non-sodium solutions (Nachman, 1962). Short-term intake is thought to be taste-mediated because there is little time for the animal to learn of the consequences of the ingestion. It has been hypothesized that the depletion-induced preference for sodium is the result of deficiency rendering the taste of sodium more palatable. In this regard, sodium depletion altered orofacial patterns of rats in response to intra-oral infusions of high concentrations of NaCl. Specifically, the high concentrations of NaCl that elicited aversive orofacial behaviors in normal rats, elicit ingestive behaviors when rats are salt deficient (Berridge et al., 1984). Thus, behavioral studies suggest that a sodium deficit enhances the gustatory processing of sodium, which in turn, facilitates the likelihood that sodium will be detected and ingested.
Peripheral Gustatory Processing
Although there is strong behavioral evidence that sodium depletion alters the preference for sodium salts, whether this effect results from changes in the afferent gustatory signals or from the central processing of these signals remained to be determined. Gustatory information reaches the brain via three cranial nerves: vagus (X), glossopharyngeal (IX), and facial (VII). In rats transection of all three of these cranial nerves disrupts salt ingestion (Richter, 1956, 1942-43), indicating that intact gustatory afferents are critical for sodium consumption. In particular, the chorda tympani, a branch of the facial nerve, is the gustatory afferent most heavily implicated in the detection of sodium taste (Frank et al., 1983; Pfaffmann et al., 1979). However, contrary to their behavioral correlates, electrophysiological studies suggested that sodium deficiency did not affect the threshold for chorda tympani responses to NaCl (Nachman & Pfaffmann, 1963; Pfaffman & Bare, 1950). Consequently, researchers believed that the preference for sodium that accompanies depletion was due to altered central processing of gustatory signals rather than to changes in the afferent signals themselves.
Subsequent work by Contreras (1977) shifted the focus back to peripheral gustatory signals. Contreras recorded from single fibers and discovered that dietary sodium deprivation blunted chorda tympani responses to 0.1 M NaCl and that responses to other taste stimuli were not affected. Subsequent studies found that single fiber and whole nerve responses to NaCl were decreased in sodium deprived or adrenalectomized rats (Contreras & Frank, 1979; Kosten & Contreras, 1985). Interestingly, the largest decrease in gustatory responsiveness occurred at suprathreshold concentrations of NaCl (Kosten & Contreras, 1985), which may explain the tendency for sodium-deficient rats to readily consume concentrated NaCl solutions that were previously rejected. These studies validated Richter's hypothesis that sodium deprivation produces “changes in the taste mechanisms in the oral cavity”, but contradicted his claim that sodium deficiency would make the taste responses to NaCl more sensitive. In fact, it is estimated that sodium deficient rats would have to be stimulated with ten-fold higher concentrations of NaCl to achieve chorda tympani responses similar to those of control rats, indicating that sodium deficit renders the taste receptors less sensitive to sodium (Contreras & Frank, 1979).
While electrophysiological studies clearly demonstrate that sodium deficiency alters the responsiveness of peripheral and central gustatory neurons, the underlying “chemical changes in the taste mechanism” remain to be elucidated. In rodents, treating the anterior tongue with amiloride, an epithelial Na+ channel blocker, disrupts performance on sodium discrimination tasks (Spector et al., 1996) and inhibits chorda tympani responses to NaCl (Heck et al., 1984). Consequently, it is believed that sodium taste is transduced via the influx of Na+ through epithelial sodium channels (ENaC) in the anterior tongue. Although it is possible that sodium deficiency alters gustatory neural responses to NaCl by decreasing the expression of ENaC through a genomic mechanism, this does not appear to be the case because studies using immunohistochemistry demonstrate that dietary sodium restriction does not effect ENaC expression in the anterior tongue (Stewart et al., 1995).
Alternatively, the activity of the ENaC may be regulated by endocrine signals associated with sodium deficit and an obvious candidate is aldosterone. Aldosterone elevates amiloride suppression of chorda tympani responses to NaCl (Herness, 1992) and intensifies apical immunoreactivity for subunits of the ENaC located in the taste cells on the anterior tongue (Lin et al., 1999). These studies suggest that aldosterone amplifies gustatory responses to NaCl by increasing the activity of the ENaC (Herness, 1992). However, it is unlikely that aldosterone contributes to the blunted chorda tympani responses that accompany sodium insufficiency because such a response is present in adrenalectomized rats that lack aldosterone (Kosten & Contreras, 1985). Consequently, other endocrine signals involved in the regulation of hydromineral balance are being considered as potential modulators of chorda tympani responses (Lundy, 1998).
Central Gustatory Processing
Chorda tympani fibers relay taste-information to the brain via synapses in the rostral portion of the nucleus of the solitary tract (NTS; Hamilton & Norgren, 1984). Both dietary sodium deprivation (Jacobs et al., 1988; Nakamura & Norgren, 1995) and furosemide (Tamura & Norgren, 1997) influence NTS responses to NaCl administered on the tongue. Such responses are indicative of altered sensory coding of salt taste and this information is conveyed to the second central gustatory relay center, the pontine parabrachial nucleus (PBN). Bilateral lesion of the PBN eliminates salt appetite stimulated by a variety of methods (Hill & Almli 1983, Flynn et al. 1991, Spector et al. 1993, Scalera et al. 1995). However, it is the connections distal to the PBN that are believed to be critical for the expression of salt appetite because decerebrate rats, which have an intact PBN but severed connections to the forebrain, do not express a salt appetite (Grill et al. 1986).
Deficit-induced changes in gustatory processing of salt broaden the range of acceptable concentrations and increase the likelihood that sodium will be detected and ingested. However, such a change can not fully explain the increased ingestion that follows sodium deficiency. Sodium deficiency is a condition that rapidly changes an animal's behavior with the detection and consumption of sodium becoming the animal's primary focus. This type of behavior can be categorized as a reward-seeking behavior and is similar to that observed during reproduction, drug addiction, and feeding. Consequently, it has been hypothesized that salt taste information is relayed to brain circuits mediating motivated behaviors, which in turn, drive sodium ingestion by providing positive feedback.
The PBN has direct projections to the amygdala, bed nucleus of the stria terminalis, and the lateral hypothalamus, all of which are reciprocally connected to each other and to other nuclei which collectively comprise a forebrain reward circuit (MacDonald 1991, Swanson 2003, Morgane et al. 2005). The ability of the PBN to influence areas such as the pre-frontal cortex, ventral tegmantal area and nucleus accumbens allow signals from the brainstem to be integrated with reward-relevant information.
Researchers have investigated the effects of sodium deficit on the morphology of the nucleus accumbens, an integrative center within the forebrain reward circuit. Specifically, it was found that furosemide administration increased dendritic branching and spines of neurons within the shell of nucleus accumbens (Roitman et al., 2002). Such changes in morphology are similar to those that occur in the nucleus accumbens after repeated use of amphetamines (Robinson and Kolb 1997). Interestingly, repeated use of amphetamines increases or sensitizes locomotor responses (Robinson and Kolb 1997), similar to the way that prior treatments with furosemide increases or sensitizes the salt intake that follows sodium depletion (Sakai et al 1987, 1989). The similar morphological and behavioral changes caused by furosemide and amphetamines led to subsequent studies examining cross-sensitization. Prior episodes of furosemide administration sensitized the locomotor responses to amphetamines, while prior experience with amphetamines sensitized salt intake after sodium depletion (Clark and Bernstein 2004). These effects appear to be dependent on dopamine binding to D2 receptors because administration of the D2 receptor antagonist, raclopride, blocked the cross-sensitization between amphetamines and salt appetite (Clark and Bernstein 2006).
The dynamics of dopamine transmission in nucleus accumbens has been studied in a variety of motivated behaviors and salt appetite is no exception. Dopamine transporter activity is decreased in the nucleus accumbens of furosemide treated rats resulting in elevated dopamine concentrations within this nucleus (Roitman et al 1999). Furosemide treatment elevates plasma aldosterone and in vitro administration of aldosterone decreased dopamine transporter activity in nucleus accumbens (Roitman et al 1999). These studies suggest that the secretion of aldosterone that follows sodium deficit inhibits dopamine transporter activity in the nucleus accumbens, thereby elevating extracellular dopamine concentration in this nucleus.
The binding of dopamine at both the D2 and D3R receptors have been reported to be necessary for the positive feedback associated with the ingestion of palatable solutions (Davis et al 2006, Roitman 2001). Therefore, it is likely that furosemide-induced secretion of aldosterone facilitates activation of D2/D3 receptors in nucleus accumbens, thereby causing further ingestion of sodium. However, pharmacological blockade of D2 receptors did not affect furosemide-induced salt intake (Roitman et al 1997, Lucas et al 2003). Rather, the effects of D2 receptor antagonism were only present when post-ingestional signals were eliminated with the sham drinking preparation. Specifically, pretreatment with raclopride blunted furosemide-induced sham drinking of NaCl (Roitman et al 1997). Consequently, it was concluded that the dopamineric system contributes to, but cannot account for, salt appetite produced by sodium deficit.
An alternative perspective is that sodium depletion modulates dopamine tone in nucleus accumbens by influencing activity of other neurotransittmers in the forebrain reward circuit. In this regard, furosemide-induced salt intake increases mRNA expression of the endogenous opioid, enkephalin, in the shell of nucleus accumbens (Lucas et al 2003). The same study showed that administration of s-opioid antagonist, naltrindole, suppressed furosemide-induced salt intake, strongly suggesting that endogenous opioids mediate salt appetite (Lucas et al 2003). However, the mechanism by which sodium deficit produces changes in opioid activity and whether such changes affect dopamine transmission is unknown.
The emergence of salt appetite is dependent on the relay of salt taste information from the PBN to a forebrain reward circuit that is heavily implicated in mediating reward and motivation. Sodium depletion and the resulting ingestion produces morphological and neurochemical changes in this circuit that are similar that those that occur after use of drugs of abuse. While sodium depletion alters dopamine and opioid neurochemistry within the nucleus accumbens, it remains unclear how this occurs as well as how such changes contribute to the stimulation, maintenance, or satiation of salt appetite.
Satiation and Inhibition of Salt Appetite
Richter was primarily concerned with the neural and endocrine signals that stimulate and maintain salt appetite and regarded the satiation or inhibition of the intake, simply, as a reduction in stimulatory drive (see Figure 1). However, plasma sodium, aldosterone and angiotensin II are not returned to basal levels at the time that rats cease depletion-induced intake of NaCl solutions (Sakai unpublished observations) and studies examining satiation of salt appetite suggest that taste and post-ingestional factors contribute to the cessation of this behavior. For example, early studies of satiation signals demonstrated that direct delivery of NaCl into the stomach did not significantly suppress the NaCl intake of sodium deficient rats (DiCara & Wilson, 1974; Nachman & Valentino, 1966). These results were interpreted to mean that gustatory cues arising from the natural act of ingestion are critical for reducing the drive to consume sodium. In contrast, Wolf and colleagues (1984) proposed that post-ingestional signals contributed to the satiation of salt appetite because when gustatory signals were bypassed by gavaging NaCl into the stomach, deprivation-induced sodium intake was attenuated. However, the onset of gavage-induced satiation was much slower than that observed during the natural consumption of NaCl, which incorporates gustatory signals, indicating that gustatory and post-ingestional factors likely contribute to the satiation of salt appetite (Wolf et al., 1984).
With regard to post-ingestional factors, it was believed that neural signals originating from the liver provided the brain with information pertaining to the ingestion of sodium because electrophysiological studies demonstrated that changes in osmotic pressure modulated the firing pattern of the hepatic branch of the vagus nerve (Adachi et al., 1976) and that ablation of this nerve reduced 24-h sodium intakes when compared to that of sham-operated controls (Contreras & Kosten, 1981). Subsequent studies by Tordoff et al (1986 and 1987) found that NaCl intake elicited by dietary sodium deprivation or furosemide was decreased by infusion of NaCl into the hepatic-portal vein. Tordoff's studies suggested that the hepatic vagus nerve carried afferent signals pertaining to the sodium content of the hepatic portal vein and that such signals contributed to the satiation of salt appetite. However, research examining salt appetite after section of the hepatic vagus cast uncertainty on this interpretation. Specifically, Frankmann and Smith (1993) found that the hepatic branch of the vagus nerve was not necessary for the satiation of sodium appetite because surgical removal of this nerve did not affect NaCl intake elicited by sodium depletion. Consequently, the neural signals related to the satiation of salt appetite remain unclear.
Similar to studies investigating endocrine signals that stimulate salt appetite, research evaluating endocrine-mediated inhibition of salt intake examined the hormonal milieu at the time of sodium depletion to identify potential signals for inhibition. Stricker and Verbalis (1987) demonstrated that plasma levels of oxytocin were low after treatments that elicited salt appetite and that salt appetite was suppressed when pituitary secretion of oxytocin was stimulated. Consequently, it was hypothesized that pituitary secretion of oxytocin influenced salt appetite, but when this hypothesis was tested by systemic administration of oxytocin or oxytocin antagonist, neither affected sodium consumption (Stricker et al., 1987). Therefore, this hypothesis was revised to focus on the effects of centrally released oxytocin on salt appetite.
Oxytocinergic neurons are located within the supraoptic and parventricular nuclei of the hypothalamus and it was hypothesized that sodium appetite is suppressed when these neurons are stimulated to secrete oxytocin. In fact, when endogenous oxytocin secretion was stimulated by systemic naloxone, sodium appetite was eliminated (Blackburn et al., 1992b). Similarly, central administration of oxytocin abolished the NaCl intake that accompanies hypovolemia-induced by polyethylene glycol (Blackburn et al., 1992b). These findings provided evidence for oxytocin regulation of salt appetite, but whether such a role for oxytocin fit within the framework of stimulatory signals for sodium consumption remained to be determined. Accordingly, experiments were designed to examine interactions between oxytocin and angiotensin II.
Previous studies had shown that central administration of angiotensin II elicited a robust water intake that was followed by a modest salt appetite (Bryant et al., 1980; Buggy & Fisher 1974). Central administration of angiotensin II had also been shown to cause activation of oxytocinergic neurons in the hypothalamus (Ferguson & Kastings, 1988). Accordingly, Blackburn and colleagues (1992a) hypothesized that central administration of angiotensin II elicited both excitatory and inhibitory signals for salt appetite, resulting in a relatively small sodium intake. If this hypothesis were correct, then inhibition of central oxytocin pathways within the hypothalamus would remove the inhibitory signal and potentiate the stimulatory effects of angiotensin II on salt appetite. Consistent with this, central antagonism of oxytocin receptors greatly increased the NaCl intake that follows intracerebroventricular injection of angiotensin II (Blackburn et al., 1992a). Importantly, angiotensin II-elicited water intake was unaffected by oxytocin antagonism and oxytocin given alone did not elicit NaCl intake (Blackburn et al., 1992a). While these results suggest that removal of oxytocin-mediated inhibition permits the emergence of salt appetite, studies from other laboratories have been unable to replicate these results. Fitts and colleagues (2003) found that pretreatment with an oxytocin antagonist did not increase NaCl intake after central injection of angiotensin II. Moreover, they also found that an oxytocin antagonist, given by itself, had diposogenic and natrorexigenic effects (Fitts et al., 2003). These results are contrary to those of Blackburn et al (1992a) and differences in rat strains and drug doses may account for these discrepancies. However, what is certain is that further investigation is required to adequately evaluate the role of oxytocin in angiotensin II-induced salt appetite.
Sex Differences
Although much of the research examining salt appetite has been done in male animals, there is evidence that salt intake is a sexually dimorphic behavior. Several studies have found that need-free consumption of 3% NaCl is significantly larger in females than in males (Chow et al., 1992; Sakai et al., 1989; Wolf, 1982; Krecek et al., 1972). This augmented intake maybe the result of sex differences in gustatory processing and studies using short-term intake tests suggest that this is the case. Specifically, Curtis and colleagues (2004) measured lick rates during 10-sec access to varying concentrations of NaCl solutions and observed that females licked at a higher rate to concentrated NaCl solution than did males. This suggests that female rats are less sensitive to concentrated NaCl solutions and may explain why females ingest more NaCl in the need-free condition.
In contrast to the studies examining need-free NaCl intake, research investigating sex differences in salt appetite stimulated by deprivation, depletion or pharmacological manipulations have produced conflicting results. Female rats ingested more salt than male rats during 2-h intake tests when salt appetite was stimulated by sodium deprivation/depletion (Chow et al., 1992). However, when intake tests were extended to 7-h, male rats ingested more NaCl than their female counterparts (Scheidler et al., 1994). Although these studies differ in regard to the direction of their effects, they indicate that male and female rats respond differently to sodium insufficiency.
In either case, sexual dimorphism of salt intake appears to be the result of organizational effects of sex steroids. Male rats castrated at postnatal Day 1 drank similar amounts of 3% NaCl in adulthood as did intact females (Chow et al., 1992), and pre-pubescent male and female rats had comparable salt appetites after eight days of sodium deprivation (Scheidler et al., 1994). Female rats castrated on postnatal Day 1 and subsequently given testosterone drank as much 3% NaCl on a need-free basis as male rats (Chow et al., 1992). Interestingly, salt intake was not suppressed in females castrated on postnatal Day 1 and treated with the non-aromatizable androgen, dihydrotestosterone, suggesting that sexual dimorphism of salt appetite is due to the conversion of testosterone to estrogen within the brain (Chow et al., 1992). Thus, the sexual dimorphism of salt intake is likely a secondary sexual characteristic produced by the actions of sex steroids within the brain.
Summary and Interpretation
Richter discovered salt appetite, an innate behavioral mechanism that restores hydromineral balance when faced with sodium deficit. This discovery led him to investigate the endocrine signals mediating this behavior and the changes in gustatory processing that accompany sodium depletion.
In regards to endocrine signals, Richter found that DOC administration inhibited the salt appetite that follows adrenalectomy, but also stimulated salt appetite in intact rats. These results presented Richter with a conundrum, adrenalectomized rats lacking endogenous aldosterone had a robust salt appetite that could be attenuated by DOC, but administration of DOC in normal rats increased NaCl intake with increasing doses. Although Richter considered renin as a potential contributor to the emergence of salt appetite (Richter 1956), the conundrum was not resolved until studies demonstrated that angiotensin II and aldosterone worked synergistically to elicit salt appetite. Adrenalectomy eliminated the salt retaining hormone, aldosterone, but the resulting sodium insufficiency stimulated salt appetite by causing the synthesis of angiotensin II. Administering DOC to adrenalectomized rats allowed the retention of sodium, which inhibited angiotensin II synthesis and decreased salt intake. However, administration of DOC in the intact rat stimulated salt intake because aldosterone, by itself, arouses salt appetite through genomic and non-genomic actions in the brain. In regards to gustatory processing, Richter found that sodium deficit broadened the range of concentrations at which rats preferred NaCl to water. He concluded that sodium deficiency produced changes in the oral cavity that make taste responses to sodium more sensitive. He turned out to be half right. Sodium depletion does change the peripheral coding of salt taste at the level of the oral cavity, but does not make taste responses to sodium more sensitive. In fact, sodium deficiency renders the taste receptors less sensitive to sodium. Thus, subsequent research extended Richter's initial discernment of the endocrine signals mediating salt appetite and the gustatory changes that occur as a consequence of sodium insufficiency.
Richter understood that salt appetite is a complex behavior that is ultimately dependent on the central integration of peripheral signals aimed at maintaining hydromineral balance. Because the central pathways controlling salt appetite were unknown at the time, Richter speculated that, “There can be little doubt that they will shortly be localized by one or more of the many workers now interested in this aspect of the problem (Richter 1956).” He was right, ensuing researchers would use techniques ranging from behavioral to molecular to define the central pathways mediating the drive to consume sodium. Richter's initial model proposed that sodium depletion led to stimulation in centers in the hypothalamus (Figure 1), consistent with early studies showing that lesion of the dorsolateral hypothalamus blunted salt appetite elicited by DOC or acute hyponatremia. Subsequent studies determined that corticosterone and aldosterone competed for binding sites in the hippocampus, septum and amygdala, while circulating angiotensin II was shown to act at AT1 receptors in circumventricular organs to elicit salt appetite. Brain regions influenced by aldosterone, corticosterone, and angiotensin II are thought to regulate the arousal of salt appetite. However, once sodium is detected, hindbrain nuclei responsible for gustatory processing relay salt taste information to forebrain nuclei implicated in motivated behaviors, which in turn, provide positive feedback that drives the ingestion. In this regard, sodium insufficiency alters dendritic morphology and increased enkephalin mRNA expression in the nucleus accumbens, an integrative center within the forebrain reward circuit. Enkephalin, an endogenous opioid, is believed to mediate the positive feedback that stimulates deficit-induced salt intake because administration of the opioid antagonist, naltrindole, inhibited salt intake elicited by furosemide.
While Richter and others considered the neural and endocrine signals that stimulate salt intake when faced with deficit, few have pursued mechanisms mediating the satiation or inhibition of salt appetite. The scant research investigating neural or hormonal signals regulating the satiation of salt appetite have produced mixed results. It was believed that neural signals originating from the liver provided the brain with information pertaining to the ingestion of sodium because electrophysiological studies demonstrated that changes in osmotic pressure modulated the firing pattern of the hepatic branch of the vagus nerve and NaCl intake elicited by sodium deprivation/depletion was decreased by infusion of NaCl into the hepatic-portal vein. However, section of the hepatic vagus did not affect NaCl intake elicited by sodium depletion, casting uncertainty on the potential of this satiation mechanism. In regards to endocrine signals, stimulation of endogenous oxytocin secretion abolished the NaCl intake that accompanies hypovolemia-induced by polyethylene glycol. However, other laboratories have been unable to clearly define a role for oxytocin in the inhibition of salt appetite, once again casting uncertainty on the potential of this satiation mechanism.
Finally, much of the research examining salt appetite has been done in male animals, but there is evidence that salt intake is a sexually dimorphic behavior. However, like studies examining satiation, research investigating sex differences in salt appetite have produced conflicting results. Deficit-induced NaCl intakes of female rats was greater than male rats during 2-h intake tests, but when intake tests were extended to 7-h, male rats ingested more NaCl than their female counterparts. These studies indicate that male and female rats respond differently to sodium insufficiency and sexual dimorphism of salt intake is likely a secondary sexual characteristic produced by the actions of sex steroids within the brain.
Proposed Model and Remaining Questions
The field of salt appetite has evolved considerably since the time that Richter examined the salt intake of adrenalectomized rats. While several of the unknowns generated by Richter's initial studies have been addressed, there are still remaining questions for future researchers to answer. The model of salt appetite that we propose presents our current understanding of the neural and endocrine mechanisms underlying this behavior and speculates about remaining unknowns (Figure 2).
Figure 2.
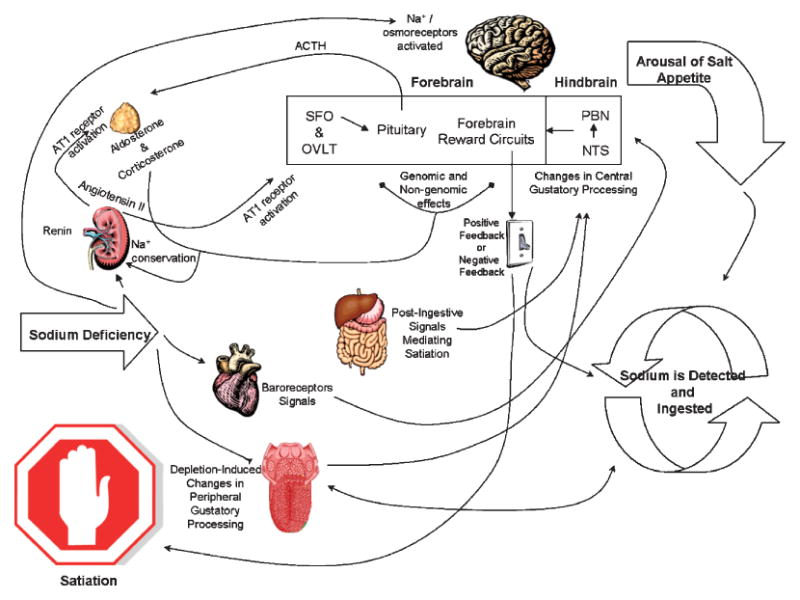
Proposed Model of Salt Appetite. Sodium deficit induces the release of renin from the kidney leading to the biosynthesis of angiotensin II. Circulating angiotensin II binds to AT1 receptors on the adrenal to cause the secretion of aldosterone, which acts at the kidney to promote sodium conservation. Angiotensin II also binds to AT1 receptors in forebrain circumventricular organs to cause the release of ACTH from the pituitary, which in turn, increases aldosterone and corticosterone secretion from the adrenal. Aldosterone, corticosterone, and angiotensin II work synergistically in the forebrain to cause the arousal of salt appetite, which is contributed to by activation of baroreceptors and central Na+ / osmoreceptors. The arousal of salt appetite leads to the initial detection and ingestion of sodium, which is facilitated by deficit-induced changes in peripheral gustatory processing. Salt taste information is relayed to the NTS in the hindbrain, where gustatory processing has also changed as a function of deficit. The next relay of salt taste occurs in the PBN, which has projections to forebrain reward circuits. Activity in the reward circuits is modulated by aldosterone and corticosterone and provides positive feedback, which in turn, elicits further ingestion of sodium. Consumption of sodium continues until postingestional signals converge in the NTS/PBN to alter activity in the forebrain reward circuits leading to negative feedback, and consequently, satiation.
Sodium insufficiency is signaled to the brain by stimulation of central Na+/osmoreceptors, cardiac baroreceptors, increased circulating angiotensin II, aldosterone, and corticosterone. The actions of angiotensin II are potentiated by aldosterone and corticosterone, which increase angiotensin II binding to AT1 receptors through genomic actions in hypothalamic nuclei. In addition, aldosterone and corticosterone influence extra-hypothalamic nuclei via genomic and non-genomic mechanisms that arouse salt appetite and prime the forebrain reward circuit to provide positive feedback once sodium is detected. Current research has only scratched the surface as to how these steroid hormones influence the activity of extra-hypothalamic brain nuclei to 1) cause the arousal of salt appetite and 2) prime brain circuits mediating reward to provide the positive feedback that drives the intake once sodium is detected.
The detection of sodium is eased by changes in peripheral gustatory processing that broaden the range of acceptable sodium concentrations. Despite numerous studies demonstrating that sodium insufficiency decreases chorda tympani responsiveness to NaCl applied to the tongue, the molecular mechanism underlying this change has remained elusive. Sodium taste is transduced via the influx of Na+ through epithelial ENaC in the anterior tongue and it is possible that hormones regulating hydromineral balance render these channels less sensitive to sodium. While a role for adrenal steroids is unlikely, angiotensin II and adrenocorticotropin hormone (ACTH) are intriguing potential moderators of chorda tympani responsiveness. ENaC activity is modulated by G-protein linked second messenger systems in the non-taste epithelium (Benos et al 1996, Ismailov et al 1995) and angiotensin II and ACTH can influence the activity of these second messenger pathways (Edwards et al 1993, Underwood et al 1989). This, in conjunction with that fact that plasma levels of angiotensin II and ACTH are elevated in sodium depleted rats make it possible that these hormones may also regulate ENaC activity on the tongue.
The salt intake that follows sodium insufficiency is a very interesting phenomenon from the psychological perspective. When faced with sodium deficit NaCl solutions that were previously aversive become rewarding and are readily ingested. However, when the deficit is alleviated the intake quickly ceases and once again the NaCl becomes aversive. This switch from rewarding to aversive and the consequent satiation of the intake is likely dependent on a mechanism that detects the restoration of sodium balance and conveys this information to the central pathways mediating salt appetite. Although the site of detection of the restoration of sodium balance is currently unknown, studies manipulating post-ingestional signals and gustatory afferents provide insight. Sham drinking preparations eliminate post-ingestional signals distal to the stomach and sodium depleted rats greatly over consume NaCl in this condition (Roitman et al 1997, Frankmann et al 1996, Tordoff et al 1987). Moreover, section of the chorda tympani nerve, the gustatory afferent most heavily implicated in the detection of sodium taste, does not significantly attenuate furosemide-elicited NaCl intake (Frankmann et al 1996). Taken together, these studies suggest that post-ingestional signals originating beyond the stomach are critical for the satiation of sodium appetite; however, further research is required to determine the exact location and underlying mechanism of such signals. Once sodium balance has been returned, neural activity in the brain must change to initiate the cessation of salt intake. We propose that post-ingestional signals converge in the NTS/PBN to alter the gustatory processing of salt taste and projections from the PBN apprise the forebrain reward circuit that sodium balance has been restored. Consequently, activity in this circuit is altered to switch the positive feedback that stimulates the ingestion to negative feedback that inhibits it. Once again, future research is required to determine the validity of this satiation mechanism.
Richter provided researchers with a testable of model of salt appetite that has subsequently been confirmed and modified. Yet after decades of investigation there are still questions remaining about how sodium appetite is aroused, maintained, and satiated. The model that we propose speculates on these unknowns and hopefully inspires subsequent research to determine its inaccuracies.
Acknowledgments
We would like to thank Dr. Gerard P. Smith for his editorial advice. We also thank Drs. Jon F. Davis and Stephen C. Woods for their thoughtful interactions that influenced the writing of the manuscript. Support: DK 48061 and DK 66596.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Adachi A, Niijima A, Jacobs HL. An hepatic osmoreceptor mechanism in the rat: Electrophysiological and behavioral studies. American Journal of Physiology. 1976;231(4):1043–1049. doi: 10.1152/ajplegacy.1976.231.4.1043. [DOI] [PubMed] [Google Scholar]
- Benos DJ, Awayda MS, Berdiev BK, Bradford AL, Fuller CM, Senyk O, Ismailov II. Diversity and regulation of amiloride-sensitive Na+ channels. Kidney International. 1996;49(6):1632–7. doi: 10.1038/ki.1996.237. Review. [DOI] [PubMed] [Google Scholar]
- Berridge KC, Flynn FW, Schulkin J, Grill HJ. Sodium depletion enhances salt palatability in rats. Behavioral Neuroscience. 1984;98(4):652–660. doi: 10.1037//0735-7044.98.4.652. [DOI] [PubMed] [Google Scholar]
- Birmingham MK, Stumpf WE, Sar M. Nuclear localization of aldosterone in rat brain cells assessed by autoradiography. Experientia. 1979;35(9):1240–1241. doi: 10.1007/BF01963313. [DOI] [PubMed] [Google Scholar]
- Blackburn RE, Hoffman GE, Stricker EM, Verbalis JG. Central oxytocin inhibition of angiotensin-induced salt appetite in rats. American Journal of Physiology. 1992a;263(6 pt 2):R1347–1353. doi: 10.1152/ajpregu.1992.263.6.R1347. [DOI] [PubMed] [Google Scholar]
- Blackburn RE, Stricker EM, Verbalis JG. Central oxytocin mediates inhibition of sodium appetite by naloxone in hypovolemic rats. Neuroendocrinology. 1992b;56(2):255–263. doi: 10.1159/000126236. [DOI] [PubMed] [Google Scholar]
- Bryant RW, Epstein AN, Fitzsimons JT, Fluharty SJ. Arousal of a specific and persistent sodium appetite in the rat with continuous intracerebroventricular infusion of angiotensin ii. Journal of Physiology. 1980;301:365–382. doi: 10.1113/jphysiol.1980.sp013211. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Buggy J, Fisher AE. Evidence for a dual central role for angiotensin in water and sodium intake. Nature. 1974;250(5469):733–735. doi: 10.1038/250733a0. [DOI] [PubMed] [Google Scholar]
- Chiaraviglio E, Perez Guaita MF. Anterior third ventricle (a3v) lesions and homeostasis regulation. Journal of Physiology (Paris) 1984;79(6):446–452. [PubMed] [Google Scholar]
- Chow SY, Sakai RR, Witcher JA, Adler NT, Epstein AN. Sex and sodium intake in the rat. Behavioral Neuroscience. 1992;106(1):172–180. doi: 10.1037//0735-7044.106.1.172. [DOI] [PubMed] [Google Scholar]
- Clark JJ, Bernstein IL. Reciprocal cross-sensitization between amphetamine and salt appetite. Pharmacology Biochemistry and Behavior. 2004;78(4):691–8. doi: 10.1016/j.pbb.2004.05.002. [DOI] [PubMed] [Google Scholar]
- Clark JJ, Bernstein IL. A role for D2 but not D1 dopamine receptors in the cross-sensitization between amphetamine and salt appetite. Pharmacology Biochemistry and Behavior. 2006;83(2):277–84. doi: 10.1016/j.pbb.2006.02.008. [DOI] [PubMed] [Google Scholar]
- Contreras RJ. Changes in gustatory nerve discharges with sodium deficiency: A single unit analysis. Brain Research. 1977;121(2):373–378. doi: 10.1016/0006-8993(77)90162-7. [DOI] [PubMed] [Google Scholar]
- Contreras RJ, Frank M. Sodium deprivation alters neural responses to gustatory stimuli. Journal of General Physiology. 1979;73(5):569–594. doi: 10.1085/jgp.73.5.569. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Contreras RJ, Kosten T. Changes in salt intake after abdominal vagotomy: Evidence for hepatic sodium receptors. Physiology and Behavior. 1981;26(4):575–582. doi: 10.1016/0031-9384(81)90127-x. [DOI] [PubMed] [Google Scholar]
- Curtis KS, Johnson AL, Therrien KL, Contreras RJ. Sex differences in behavioral taste responses to and ingestion of sucrose and nacl solutions by rats. Physiology and Behavior. 2004;80(5):657–664. doi: 10.1016/j.physbeh.2003.11.007. [DOI] [PubMed] [Google Scholar]
- Curtis KS, Krause EG, Contreras RJ. Altered nacl taste responses precede increased nacl ingestion during na(+) deprivation. Physiology and Behavior. 2001;72(5):743–749. doi: 10.1016/s0031-9384(01)00422-x. [DOI] [PubMed] [Google Scholar]
- Davis JF, McQuade JA, Drazen DL, Woods SC, Seeley RJ, Benoit SC. Role for dopamine-3 receptor in the hyperphagia of an unanticipated high-fat meal in rats. Pharmacology Biochemistry and Behavior. 2006;85(1):190–7. doi: 10.1016/j.pbb.2006.07.035. [DOI] [PubMed] [Google Scholar]
- Davis JO, Speilman WS. The renin-angiotensin system in the control of aldosterone secretion in the rat. Acta physiológica latino americana. 1974;24(5):399–404. [PubMed] [Google Scholar]
- De Nicola AF, Seltzer A, Tsutsumi K, Saavedra JM. Effects of deoxycorticosterone acetate (doca) and aldosterone on sar1-angiotensin ii binding and angiotensin-converting enzyme binding sites in brain. Cellular Molecular Neurobiology. 1993;13(5):529–539. doi: 10.1007/BF00711461. [DOI] [PubMed] [Google Scholar]
- Dicara LV, Wilson LM. Role of gustation in sodium appetite. Physiological Psychology. 1974;2:43–44. [Google Scholar]
- Edwards RM, Stack EJ. Angiotensin II inhibits glomerular adenylate cyclase via the angiotensin II receptor subtype 1 (AT1) The Journal of Pharmacology and Experimental Therapeutics. 1993;266(2):506–10. [PubMed] [Google Scholar]
- Epstein A. Mineralocorticoids and cerebral angiotensin may act together to produce sodium appetite. Peptides. 1982;3(3):493–494. doi: 10.1016/0196-9781(82)90113-9. [DOI] [PubMed] [Google Scholar]
- Epstein AN, Stellar E. The control of salt preference in the adrenalectomized rat. The Journal of Comparative and Physiological Psychology. 1955;48(3):167–172. doi: 10.1037/h0045626. [DOI] [PubMed] [Google Scholar]
- Ferguson AV, Kastings NW. Angiotensin acts at the subfornical organ to increase plasma oxytocin concentrations in the rat. Regulatory Peptides. 1988;23(3):343–352. doi: 10.1016/0167-0115(88)90235-2. [DOI] [PubMed] [Google Scholar]
- Findlay AL, Epstein AN. Increased sodium intake is somehow induced in rats by intravenous angiotnesin ii. Hormones and Behavior. 1980;14(1):86–92. doi: 10.1016/0018-506x(80)90018-5. [DOI] [PubMed] [Google Scholar]
- Fitts DA, Masson DB. Preoptic angiotensin and salt appetite. Behavioral Neuroscience. 1990;104(4):643–650. doi: 10.1037//0735-7044.104.4.643. [DOI] [PubMed] [Google Scholar]
- Fitts DA, Thornton SN, Ruhf AA, Zierath DK, Johnson AK, Thunhorst RL. Effects of central oxytocin receptor blockade on water and saline intake, mean arterial pressure, and c-fos expression in rats. American Journal of Physiology Regulatory Integrative Comparative Physiology. 2003;285(6):R1331–1339. doi: 10.1152/ajpregu.00254.2003. [DOI] [PubMed] [Google Scholar]
- Fluharty SJ, Epstein AN. Sodium appetite elicited by intracerebroventricular infusion of angiotensin ii in the rat: II. Synergistic interaction with systemic mineralocorticoids. Behavioral Neuroscience. 1983;97(5):746–758. doi: 10.1037//0735-7044.97.5.746. [DOI] [PubMed] [Google Scholar]
- Fluharty SJ, Manaker S. Sodium appetite elicited by intracerebroventricular infusion of angiotensin ii in the rat: I. Relation to urinary sodium excretion. Behavioral Neuroscience. 1983;97(5):738–745. doi: 10.1037//0735-7044.97.5.738. [DOI] [PubMed] [Google Scholar]
- Fluharty SJ, Sakai RR. Behavioral and cellular analysis of adrenal steroid and angiotensin interactions mediating salt appetite. In: Fluharty SJ, Morrison AR, Sprague J, Stellar E, editors. Progressions in Psychobiology, Physiology and Psychology. Vol. 16. San Diego: Academic Press; 1995. pp. 177–212. [Google Scholar]
- Flynn FH, Grill H, Schulkin J, Norgren R. Central gustatory lesions II. Effects on salt appetite, taste aversion learning, and regulatory challenges. Behavioral Neuroscience. 1991;97:746–758. doi: 10.1037//0735-7044.105.6.944. [DOI] [PubMed] [Google Scholar]
- Frank ME, Contreras RJ, Hettinger TP. Nerve fibers sensitive to ionic taste stimuli in chorda tympani of the rat. Journal of Neurophysiology. 1983;50(4):941–960. doi: 10.1152/jn.1983.50.4.941. [DOI] [PubMed] [Google Scholar]
- Frankmann SP, Smith GP. Hepatic vagotomy does not disrupt the normal satiation of nacl appetite. Physiology and Behavior. 1993;53(2):337–341. doi: 10.1016/0031-9384(93)90214-z. [DOI] [PubMed] [Google Scholar]
- Frankmann SP, Sollars SI, Bernstein IL. Sodium appetite in the sham-drinking rat after chorda tympani nerve transection. American Journal of Physiology. 1996;271(2 Pt 2):R339–45. doi: 10.1152/ajpregu.1996.271.2.R339. [DOI] [PubMed] [Google Scholar]
- Ganesan R, Sumners C. Glucocorticoids potentiate the dipsogenic action of angiotensin II. Brain Research. 1989;499(1):121–130. doi: 10.1016/0006-8993(89)91141-4. [DOI] [PubMed] [Google Scholar]
- Gerlach J, McEwen BS. Rat brain binds adrenal steroid hormone: Radioautography of hippocampus with corticosterone. Science. 1972;175(26):1133–1136. doi: 10.1126/science.175.4026.1133. [DOI] [PubMed] [Google Scholar]
- Grill HJ, Schulkin J, Flynn FW. Sodium homeostasis in chronic decerebrate rats. Behavioral Neuroscience. 1986;112:160–171. doi: 10.1037//0735-7044.100.4.536. [DOI] [PubMed] [Google Scholar]
- Gutkind JS, Kurihara M, Saavedra JM. Increased angiotensin II receptors in brain nuclei of doca-salt hypertensive rats. American Journal of Physiology. 1988;255(3 pt 2):H646–650. doi: 10.1152/ajpheart.1988.255.3.H646. [DOI] [PubMed] [Google Scholar]
- Hamilton RB, Norgren R. Central projections of gustatory nerves in the rat. Journal of Comparative Neurology. 1984;222(4):560–577. doi: 10.1002/cne.902220408. [DOI] [PubMed] [Google Scholar]
- Heck GL, Mierson S, DeSimone JA. Salt taste transduction occurs through an amiloride-sensitive sodium transport pathway. Science. 1984;223(4634):403–405. doi: 10.1126/science.6691151. [DOI] [PubMed] [Google Scholar]
- Herness MS. Aldosterone increases the amiloride-sensitivity of the rat gustatory neural response to nacl. Comparative Biochemistry Physiology Comparative Physiology. 1992;103(2):269–273. doi: 10.1016/0300-9629(92)90578-e. [DOI] [PubMed] [Google Scholar]
- Hill D, Almli CR. Parabrachial nuclei damage in infant rats produces residual deficits in gustatory preferences/aversions and sodium appetite. Developmental Psychobiology. 1983;16:519–533. doi: 10.1002/dev.420160608. [DOI] [PubMed] [Google Scholar]
- Ismailov II, Berdiev BK, Benos DJ. Regulation by Na+ and Ca2+ of renal epithelial Na+ channels reconstituted into planar lipid bilayers. Journal of General Physiology. 1995;106(3):445–66. doi: 10.1085/jgp.106.3.445. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jacobs KM, Mark GP, Scott TR. Taste responses in the nucleus tractus solitarius of sodium-deprived rats. Journal of Physiology. 1988;406:393–410. doi: 10.1113/jphysiol.1988.sp017387. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kosten T, Contreras RJ. Adrenalectomy reduces peripheral neural responses to gustatory stimuli in the rat. Behavioral Neuroscience. 1985;99(4):734–741. doi: 10.1037//0735-7044.99.4.734. [DOI] [PubMed] [Google Scholar]
- Krecek J, Novakova V, Stibral K. Sex differences in the taste preference for a salt solution in the rat. Physiology and Behavior. 1972;8(2):183–188. doi: 10.1016/0031-9384(72)90358-7. [DOI] [PubMed] [Google Scholar]
- Lin W, Finger T, Rossier BC, Kinnamon SC. Epithelial na+ channel subunits in rat taste cells: Localization and regulation by aldosterone. Journal of Comparative Neurology. 1999;405(3):406–420. doi: 10.1002/(sici)1096-9861(19990315)405:3<406::aid-cne10>3.0.co;2-f. [DOI] [PubMed] [Google Scholar]
- Lucas LR, Grillo CA, McEwen BS. Involvement of mesolimbic structures in short-term sodium depletion: In situ hybridization and ligand-binding analyses. Neuroendocrinology. 2003;77(6):406–415. doi: 10.1159/000071312. [DOI] [PubMed] [Google Scholar]
- Lundy RF., Jr Potential mechanisms for functional changes in taste receptor cells following sodium deficiency in mammals. Neuroscience and Biobehavioral Reviews. 1998;23(1):103–109. doi: 10.1016/s0149-7634(98)00037-2. [DOI] [PubMed] [Google Scholar]
- Ma LY, McEwen BS, Sakai RR, Schulkin J. Glucocorticoids facilitate mineralocorticoid-induced sodium intake in the rat. Hormones and Behavior. 1993;27(2):240–250. doi: 10.1006/hbeh.1993.1018. [DOI] [PubMed] [Google Scholar]
- MacDonald AJ. Topographical organization of amydaloid projections to the caudatoputamen, nucleus accumbens, and related striatal-like areas of the rat brain. Neuroscience. 1991;44:15–33. doi: 10.1016/0306-4522(91)90248-m. [DOI] [PubMed] [Google Scholar]
- McEwen BS, Lambdin LT, Rainbow TC, De Nicola AF. Aldosterone effects on salt appetite in adrenalectomized rats. Neuroendocrinology. 1986;43(1):38–43. doi: 10.1159/000124506. [DOI] [PubMed] [Google Scholar]
- McKinley MJ, Davern P, Giles ME, Penschow J, Sunn N, Uschakov A, Oldfield BJ. The sensory circumventricular organs of the mammalian brain. Advances in Anatomy, Embryology and Cell Biology. 2003;172:1–122. doi: 10.1007/978-3-642-55532-9. [DOI] [PubMed] [Google Scholar]
- Morgane PJ, Galler JR, Mokler DJ. A review of systems and networks of the limbic forebrain/limbic midbrain. Progressions in Neurobiology. 2005;75:143–69. doi: 10.1016/j.pneurobio.2005.01.001. [DOI] [PubMed] [Google Scholar]
- Nachman M. Taste preferences for sodium salts by adrenalectomized rats. Journal of Comparative and Physiological Psychology. 1962;55:1124–1129. doi: 10.1037/h0041348. [DOI] [PubMed] [Google Scholar]
- Nachman M, Valentino DA. Roles of taste and postingestional factors in the satiation of sodium appetite in rats. Journal of Comparative and Physiological Psychology. 1966;62(2):280–283. doi: 10.1037/h0023667. [DOI] [PubMed] [Google Scholar]
- Nachman M, Pfaffmann C. Gustatory nerve discharge in normal and sodium-deficient rats. Journal of Comparative and Physiological Psychology. 1963;56:1007–1011. doi: 10.1037/h0044428. [DOI] [PubMed] [Google Scholar]
- Nakamura K, Norgren R. Sodium-deficient diet reduces gustatory activity in the nucleus of the solitary tract of behaving rats. American Journal of Physiology. 1995;269(3 Pt 2):R647–661. doi: 10.1152/ajpregu.1995.269.3.R647. [DOI] [PubMed] [Google Scholar]
- Nitabach MN, Schulkin J, Epstein AN. The medial amygdala is part of a mineralocorticoid-sensitive circuit controlling NaCl intake in the rat. Behavioral Brain Research. 1989;35(2):127–134. doi: 10.1016/s0166-4328(89)80113-5. [DOI] [PubMed] [Google Scholar]
- Pfaffmann C, Frank M, Norgren R. Neural mechanisms and behavioral aspects of taste. Annual Review of Psychology. 1979;30:283–325. doi: 10.1146/annurev.ps.30.020179.001435. [DOI] [PubMed] [Google Scholar]
- Pfaffmann C, Bare JK. Gustatory nerve discharges in normal and adrenalectomized rats. Journal of Comparative and Physiological Psychology. 1950;43(4):320–324. doi: 10.1037/h0059248. [DOI] [PubMed] [Google Scholar]
- Rice KK, Richter CP. Increased sodium chloride and water intake of normal rats treated with desoxycorticosterone acetate. Endocrinology. 1943;33:106–115. [Google Scholar]
- Richter CP. Increased salt appetite in adrenalectomized rats. American Journal of Physiology. 1936;115:155–161. [Google Scholar]
- Richter CP. Salt taste thresholds of normal and adrenalectomized rats. Endocrinology. 1939;24:367–371. [Google Scholar]
- Richter CP. Total self regulatory functions in animals and human beings. Harvey Lecture Series. 19423;38:63–103. [Google Scholar]
- Richter CP. L'instinct dans le comportement des animaux et de l'homme. Paris, France: 1956. Salt appetite of mammals: Its dependence on instinct and metabolism; pp. 577–632. [Google Scholar]
- Richter CP, Eckert JF. Mineral metabolism of adrenalectomized rats studied by the appetite method. Endocrinology. 1938;22:214–224. [Google Scholar]
- Robinson TE, Kolb B. Persistent structural modifications in nucleus accumbens and prefrontal cortex neurons produced by previous experience with amphetamine. Journal of Neuroscience. 1997;17(21):8491–7. doi: 10.1523/JNEUROSCI.17-21-08491.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roitman MF, Na E, Anderson G, Jones TA, Bernstein IL. Induction of a salt appetite alters dendritic morphology in nucleus accumbens and sensitizes rats to amphetamine. Journal of Neuroscience. 2002;22(11):RC225. doi: 10.1523/JNEUROSCI.22-11-j0001.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roitman MF, Patterson TA, Sakai RR, Bernstein IL, Figlewicz DP. Sodium depletion and aldosterone decrease dopamine transporter activity in nucleus accumbens but not striatum. American Journal of Physiology. 1999;276(5 Pt 2):R1339–45. doi: 10.1152/ajpregu.1999.276.5.R1339. [DOI] [PubMed] [Google Scholar]
- Roitman MF, Schafe GE, Thiele TE, Bernstein IL. Dopamine and sodium appetite: antagonists suppress sham drinking of NaCl solutions in the rat. Behavioral Neuroscience. 1997;111(3):606–11. doi: 10.1037//0735-7044.111.3.606. [DOI] [PubMed] [Google Scholar]
- Roitman MF, van Dijk G, Thiele TE, Bernstein IL. Dopamine mediation of the feeding response to violations of spatial and temporal expectancies. Behavioral Brain Research. 2001;122(2):193–9. doi: 10.1016/s0166-4328(01)00189-9. [DOI] [PubMed] [Google Scholar]
- Rubin MI, Krick ET. Proceedings of the Society for Experimental Biology and Medicine. Vol. 31. 1933. p. 228. [Google Scholar]
- Sakai RR, Fine W, Frankmann S, Epstein A. Salt appetite is enhanced by one prior episode of sodium depletion in the rat. Behavioral Neuroscience. 1987;101:724–731. doi: 10.1037//0735-7044.101.5.724. [DOI] [PubMed] [Google Scholar]
- Sakai RR, Fluharty SJ, Fine WB, Epstein AN. Prior episodes of sodium depletion increase the need-free sodium intake of the rat. Behavioral Neuroscience. 1989;103(1):186–192. doi: 10.1037//0735-7044.103.1.186. [DOI] [PubMed] [Google Scholar]
- Sakai RR, McEwen BS, Fluharty SJ, Ma LY. The amygdala: Site of genomic and nongenomic arousal of aldosterone-induced sodium intake. Kidney International. 2000;57(4):1337–1345. doi: 10.1046/j.1523-1755.2000.00972.x. [DOI] [PubMed] [Google Scholar]
- Sakai RR, Nicolaidis S, Epstein AN. Salt appetite is suppressed by interference with angiotensin ii and aldosterone. American Journal of Physiology. 1986;251(4 Pt 2):R762–768. doi: 10.1152/ajpregu.1986.251.4.R762. [DOI] [PubMed] [Google Scholar]
- Scalera G, Spector AC, Norgren R. Excitotoxic lesions of the parabrachial nuclei prevent conditioned taste aversions and sodium appetite in rats. Behavioral Neuroscience. 1995;109:997–1008. [PubMed] [Google Scholar]
- Scheidler MG, Verbalis JG, Stricker EM. Inhibitory effects of estrogen on stimulated salt appetite in rats. Behavioral Neuroscience. 1994;108(1):141–150. [PubMed] [Google Scholar]
- Shelat SG, Flanagan-Cato LM, Fluharty SJ. Glucocorticoid and mineralocorticoid regulation of angiotensin ii type 1 receptor binding and inositol triphosphate formation in wb cells. Journal of Endocrinology. 1999a;162(3):381–391. doi: 10.1677/joe.0.1620381. [DOI] [PubMed] [Google Scholar]
- Shelat SG, King JL, Flanagan-Cato LM, Fluharty SJ. Mineralocorticoids and glucocorticoids cooperatively increase salt intake and angiotensin ii receptor binding in rat brain. Neuroendocrinology. 1999b;69(5):339–351. doi: 10.1159/000054436. [DOI] [PubMed] [Google Scholar]
- Spector AC, Grill H, Norgren R. Concentration-dependent licking of sucrose and sodium chloride in rats with parabrachial gustatory lesions. Physiology and Behavior. 1993;53:277–283. doi: 10.1016/0031-9384(93)90205-t. [DOI] [PubMed] [Google Scholar]
- Spector AC, Guagliardo NA, St John SJ. Amiloride disrupts nacl versus kcl discrimination performance: Implications for salt taste coding in rats. Journal of Neuroscience. 1996;16(24):8115–8122. doi: 10.1523/JNEUROSCI.16-24-08115.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stewart RE, Lasiter P, Benos DJ, Hill DL. Immunohistochemical correlates of peripheral gustatory sensitivity to sodium and amiloride. Acta Anatomica (Basel) 1995;153(4):310–319. doi: 10.1159/000147740. [DOI] [PubMed] [Google Scholar]
- Stricker EM. Effects of hypovolemia and-or caval ligation on water and nacl solution drinking by rats. Physiology and Behavior. 1971;6(4):299–305. doi: 10.1016/0031-9384(71)90159-4. [DOI] [PubMed] [Google Scholar]
- Stricker EM, Hosutt JA, Verbalis JG. Neurohypophyseal secretion in hypovolemic rats: Inverse relation to sodium appetite. American Journal Physiology. 1987;252(5 Pt 2):R889–896. doi: 10.1152/ajpregu.1987.252.5.R889. [DOI] [PubMed] [Google Scholar]
- Stricker EM, Vagnucci AH, McDonald RH, Jr, Leenen FH. Renin and aldosterone secretions during hypovolemia in rats: Relation to nacl intake. American Journal of Physiology. 1979;237(1):R45–51. doi: 10.1152/ajpregu.1979.237.1.R45. [DOI] [PubMed] [Google Scholar]
- Stricker EM, Verbalis JG. Central inhibitory control of sodium appetite in rats: Correlation with pituitary oxytocin secretion. Behavioral Neuroscience. 1987;101(4):560–567. doi: 10.1037//0735-7044.101.4.560. [DOI] [PubMed] [Google Scholar]
- Sumners C, Fregly MJ. Modulation of angiotensin ii binding sites in neuronal cultures by mineralocorticoids. American Journal of Physiology. 1989;256(1 Pt 1):C121–129. doi: 10.1152/ajpcell.1989.256.1.C121. [DOI] [PubMed] [Google Scholar]
- Swanson LW. The amygdala and its place in the cerebral hemisphere. Annals of the New York Academy of Sciences. 2003;985:174–184. doi: 10.1111/j.1749-6632.2003.tb07081.x. [DOI] [PubMed] [Google Scholar]
- Tamura R, Norgren R. Repeated sodium depletion affects gustatory neural responses in the nucleus of the solitary tract of rats. American Journal of Physiology. 1997;273(4 Pt 2):R1381–1391. doi: 10.1152/ajpregu.1997.273.4.R1381. [DOI] [PubMed] [Google Scholar]
- Thunhorst RL, Beltz TG, Johnson AK. Effects of subfornical organ lesions on acutely induced thirst and salt appetite. American Journal Physiology. 1999;277(1 Pt 2):R56–65. doi: 10.1152/ajpregu.1999.277.1.r56. [DOI] [PubMed] [Google Scholar]
- Thunhorst RL, Lewis SJ, Johnson AK. Effects of sinoaortic baroreceptor denervation on depletion-induced salt appetite. American Journal of Physiology. 1994;267(4 Pt 2):R1043–1049. doi: 10.1152/ajpregu.1994.267.4.R1043. [DOI] [PubMed] [Google Scholar]
- Tordoff MG, Schulkin J, Friedman MI. Hepatic contribution to satiation of salt appetite in rats. American Journal of Physiology. 1986;251(6 Pt 2):R1095–1102. doi: 10.1152/ajpregu.1986.251.6.R1095. [DOI] [PubMed] [Google Scholar]
- Tordoff MG, Schulkin J, Friedman MI. Further evidence for hepatic control of salt intake in rats. American Journal of Physiology. 1987;253(3 Pt 2):R444–449. doi: 10.1152/ajpregu.1987.253.3.R444. [DOI] [PubMed] [Google Scholar]
- Toth E, Stelfox J, Kaufman S. Cardiac control of salt appetite. American Journal of Physiology. 1987;252(5 Pt 2):R925–929. doi: 10.1152/ajpregu.1987.252.5.R925. [DOI] [PubMed] [Google Scholar]
- Underwood RH, Menachery AI, Williams GH. Effect of sodium intake on phosphoinositides and inositol trisphosphate response to angiotensin II, K+ and ACTH in rat glomerulosa cells. The Journal of Endocrinology. 1989;122(1):371–7. doi: 10.1677/joe.0.1220371. [DOI] [PubMed] [Google Scholar]
- Weisinger RS, Blair-West JR, Burns P, Denton DA, Tarjan E. Role of brain angiotensin in thirst and sodium appetite of rats. Peptides. 1997;18(7):977–984. doi: 10.1016/s0196-9781(97)00077-6. [DOI] [PubMed] [Google Scholar]
- Weisinger RS, Considine P, Denton DA, McKinley MJ. Rapid effect of change in cerebrospinal fluid sodium concentration on salt appetite. Nature. 1979;280(5722):490–491. doi: 10.1038/280490a0. [DOI] [PubMed] [Google Scholar]
- Weisinger RS, Denton DA, Di Nicolantonio R, Hards DK, McKinley MJ, Oldfield B, Osborne PG. Subfornical organ lesion decreases sodium appetite in the sodium-depleted rat. Brain Research. 1990;526(1):23–30. doi: 10.1016/0006-8993(90)90245-7. [DOI] [PubMed] [Google Scholar]
- Weisinger RS, Denton DA, Di Nicolantonio R, McKinley MJ, Muller AF, Tarjan E. Role of angiotensin in sodium appetite of sodium-deplete sheep. American Journal of Physiology. 1987;253(3 Pt 2):R482–488. doi: 10.1152/ajpregu.1987.253.3.R482. [DOI] [PubMed] [Google Scholar]
- Weisinger RS, Woods SC. Aldosterone-elicited sodium appetite. Endocrinology. 1971;89(2):538–544. doi: 10.1210/endo-89-2-538. [DOI] [PubMed] [Google Scholar]
- Wilkins L, Richter CP. A great craving for salt by a child with corticoadrenal insufficiency. JAMA : the journal of the American Medical Association. 1940;114:866–868. [Google Scholar]
- Wilson KM, Hathaway S, Fregly MJ. Mineralocorticoids modulate central angiotensin ii receptors in rats. Brain Research. 1986;382(1):87–96. doi: 10.1016/0006-8993(86)90114-9. [DOI] [PubMed] [Google Scholar]
- Wolf G. Effect of dorsolateral hypothalamic lesions on sodium appetite elicited by deoxycorticosterone and by acute hyponatremia. Journal of Comparative and Physiological Psychology. 1964;58(3):396–402. doi: 10.1037/h0048232. [DOI] [PubMed] [Google Scholar]
- Wolf G. Effect of deoxycorticosterone on sodium appetite of intact and adrenalectomized rats. American Journal of Physiology. 1965;208:1281–1285. doi: 10.1152/ajplegacy.1965.208.6.1281. [DOI] [PubMed] [Google Scholar]
- Wolf G. Refined salt appetite methodology for rats demonstrated by assessing sex differences. Journal of Comparative Physiology and Psychology. 1982;96(6):1016–1021. [PubMed] [Google Scholar]
- Wolf G, Handal PJ. Aldosterone-induced sodium appetite: Dose response and specificity. Endocrinology. 1966;78:1120–1124. doi: 10.1210/endo-78-6-1120. [DOI] [PubMed] [Google Scholar]
- Wolf G, Schulkin J, Simson PE. Multiple factors in the satiation of salt appetite. Behavioral Neuroscience. 1984;98(4):661–673. doi: 10.1037//0735-7044.98.4.661. [DOI] [PubMed] [Google Scholar]