

1. Introduction
Androgen receptor (AR) is a member of the steroid and nuclear receptor superfamily,1 which is composed of over 100 members and continues to grow. Among this large family of proteins, only five vertebrate steroid receptors—estrogen, progesterone, androgen, glucocorticoid, and mineralocorticoid receptors—are known. Two subtypes of estrogen receptor have been identified, estrogen receptor α and estrogen receptor β.2 Like other steroid receptors, AR is a soluble protein that functions as an intracellular transcriptional factor. AR function is regulated by the binding of androgens, which initiates sequential conformational changes of the receptor that affect receptor–protein interactions and receptor–DNA interactions.
AR-regulated gene expression is responsible for male sexual differentiation and male pubertal changes. AR ligands are widely used in a variety of clinical applications (i.e., agonists are employed for hypogonadism, while antagonists are used for prostate cancer therapy). Fang et al.3 recently summarized a large number of chemicals that bind to the AR. The current review focuses on well-characterized AR ligands that bind to the AR with high affinity and integrates discussion regarding the biology, metabolism, and structure-activity relationships for therapeutic and emerging classes of AR ligands. The known AR ligands can be classified as steroidal or nonsteroidal based on their structure or as agonist or antagonist based on their ability to activate or inhibit transcription of AR target genes. Synthetic AR ligands were first developed by modifying the steroidal structure of endogenous androgens. The structure–activity relationship of these steroidal AR ligands is well documented4-6 and will only be briefly summarized in this review. However, low oral bio-availability, poor pharmacokinetic properties, and side effects have limited the use of many steroidal AR ligands. Until recently, it was considered impossible to separate the androgenic and anabolic effects of AR ligands due to their reliance on a single AR. However, newly discovered nonsteroidal AR ligands may provide a new strategy to achieve tissue selectivity, as is possible with estrogen receptor ligands. Novel nonsteroidal pharmacophores are summarized in this review with discussion of the emerging structure–activity relationships and examples of their tissue selectivity included.7
1.1. Physiologic Roles and Clinical Application of Androgens
AR is mainly expressed in androgen target tissues, such as the prostate, skeletal muscle, liver, and central nervous system (CNS), with the highest expression level observed in the prostate, adrenal gland, and epididymis as determined by real-time polymerase chain reaction (PCR).8 AR can be activated by the binding of endogenous androgens, including testosterone and 5α-dihydrotestosterone (5α-DHT). Physiologically, functional AR is responsible for male sexual differentiation in utero and for male pubertal changes. In adult males, androgen is mainly responsible for maintaining libido, spermato-genesis, muscle mass and strength, bone mineral density, and erythropoisis.7,9 The actions of androgen in the reproductive tissues, including prostate, seminal vesicle, testis, and accessory structures, are known as the androgenic effects, while the nitrogen-retaining effects of androgen in muscle and bone are known as the anabolic effects.
Numerous and varied site mutations in AR have been identified (The Androgen Receptor Gene Mutations Database World Wide Web Server, http://www.androgendb.mcgill.ca/). The majority of these mutations are associated with diseases, like Androgen Insensitivity Syndrome and prostate cancer. The androgen withdrawal syndrome observed in prostate cancer therapy also appeared to be related to certain AR mutations, such as T877A and W741C mutations, which convert some AR antagonists into agonists (see more discussion in section 3.1.1). Besides the site mutations documented, AR gene polymorphism has also been identified, particularly, the poly-Q (CAG)n at exon I. The polymorphic (CAG)10–35 triplet repeat sequence, starting from codon 58, codes for polyglutamine. The length of the repeat is inversely correlated with the transactivation activity of AR. The correlation between the length of the CAG repeat and disease stage was recently reviewed by Oettel.10
Classically, testosterone is used to treat male hypogonadism, Klinefelter's syndrome, anemia secondary to chronic renal failure, aplastic anemia, protein wasting diseases associated with cancer, burns, traumas,acquiredimmunodeficiencysyndrome(AIDS), etc., short stature, breast cancer (as an anti-estrogen), and hereditary angioedema.7 Recently, hormone replacement therapy in aging males has also been proposed to improve body composition, bone and cartilage metabolism, and certain domains of brain function and even decrease cardiovascular risk.10
1.2. Gene and Protein Structure and Function
1.2.1. Androgen Receptor Gene and Protein Structure
In 1981, Migeon et al.11 first localized the AR gene to the human X chromosome. In 1998, Lubahn et al.12 cloned human AR genomic DNA from a human X chromosome library using a consensus nucleotide sequence from the DNA-binding domain of the nuclear receptor family. In the same year, several groups, including Chang et al.,13 Lubahn et al.,14 and Trapman et al.,15 cloned human AR cDNAs. To date, only one AR gene has been identified in humans.
The AR gene is more than 90 kb long and codes for a protein of 919 amino acids that has three major functional domains, as illustrated in Figure 1. The N-terminal domain (NTD), which serves a modulatory function, is encoded by exon 1 (1586 bp). The DNA-binding domain (DBD) is encoded by exons 2 and 3 (152 and 117 bp, respectively).16 The ligand-binding domain (LBD) is encoded by five exons, which vary from 131 to 288 bp in size. There is also a small hinge region between the DNA-binding domain and ligand-binding domain. Two transactivation functions have been identified. The N-terminal activation function 1 (AF1) is constitutively active in truncated receptor that does not contain the ligand-binding domain and is not conserved in sequence compared to other steroid receptors (Figure 2), whereas the C-terminal activation function 2 (AF2) functions in a ligand-dependent manner and is relatively more conserved in sequence as compared to other steroid hormone receptors, particularly with regard to the charge-clamp residues.17 A nuclear localization signal (NLS) spans the region between the DNA-binding domain and the hinge region.
Figure 1.

Structural organization of the AR gene and protein.
Figure 2.

Amino acid sequence identity among members of the steroid receptor family (adapted from ref 19). Sequence alignment was performed using William Pearson's LALIGN program.20
The human AR amino acid sequence is very similar to the rat AR amino acid sequence with identical sequences in the DNA- and ligand-binding domains and an overall sequence identity of 85% 14 (Figure 2). All steroid receptors share a similar organization with an individual N-terminal domain, conserved DNA-binding domain, and C-terminal ligand-binding domain. The N-terminal domain of different steroid receptors shows the least conservation of sequence (less than 25% identity), while the central DNA-binding domain is well conserved for all steroid receptors (59–82%), reflecting the common need to bind to DNA, while the variation is responsible for the selection of different target sequences. The ligand-binding domain of different steroid receptors shows sequence identity ranging from 22% to 55%, reflecting receptor specificity for individual hormones. Among all the steroid receptors, the human AR ligand-binding domain shares more sequence identity with the human progesterone receptor, glucocorticoid receptor, and mineralocorticoid receptor ligand-binding domains (all around 50%)18 with an overall sequence homology of 88% to the progesterone receptor ligand-binding domain when conservative mutations are included. Even though the ligand-binding domains of steroid receptors share relatively low sequence identity, they all assume a similar three-dimensional structure with certain highly conserved structural features, including a “charge clamp” and helical features (see subsequent sections for more detailed discussion). These similarities in conformation provide the structural basis for the cross reactivity that is commonly observed with synthetic steroids.
1.2.2. Androgen Receptor Protein Conformation and Function
Similar to the other steroid receptors, unbound AR is mainly located in the cytoplasm and associated with a complex of heat shock proteins (HSPs) through interactions with the ligand-binding domain.21 Upon agonist binding,1 AR goes through a series of conformational changes: the heat shock proteins dissociate from AR, and the transformed AR undergoes dimerization, phosphorylation, and translocation to the nucleus, which is mediated by the nuclear localization signal. Translocated receptor then binds to the androgen response element (ARE), which is characterized by the six-nucleotide half-site consensus sequence 5′-TGTTCT-3′ spaced by three random nucleotides and is located in the promoter or enhancer region of AR gene targets. Recruitment of other transcription co-regulators (including co-activators and co-repressors)22 and transcriptional machinery23 further ensures the transactivation of AR-regulated gene expression. All of these complicated processes are initiated by the ligand-induced conformational changes in the ligand-binding domain.
Currently, ligand-binding domain and DNA-binding domain crystal structures of many nuclear receptors are solved, but no crystal structure of a full-length receptor is available yet. The first crystal structure of the AR ligand-binding domain was solved by Matias et al. in 2000.24 The AR ligand-binding domain shares similar three-dimensional structure with other agonist-bound steroid receptors (e.g., estrogen receptor) (Figures 3 and 4A). The protein contains 11 α-helices (H) and two short β-turns, which are arranged in three layers to form an antiparallel “α-helical sandwich”. Different from other steroid receptors, H2 is not present in the AR ligand-binding domain. However, it is important to note that the same numbering of the other helices was retained for easy comparison. Helices H1 and H3 form one face of the ligand-binding domain, while helices H4 and H5, the first β-turn, and helices H8 and H9 form the central layer of the structure, and helices H6, H7, H10, and H11 constitute the second face. H5, the N-terminal region of H3 and the C-terminal region of H10 and H11 form the main part of the hydrophobic ligand-binding pocket (LBP). Upon agonist binding, H12 is repositioned and serves as the “lid” of the ligand-binding pocket to stabilize the ligand, and the very end of the C-terminal region of the ligand-binding domain forms the second β-turn (next to H8 and H10), which works as a “lock” to further stabilize the “lid” (H12) conformation. The agonist-induced conformational change in the ligand-binding domain allows the formation of a functional activation function 2 (AF2) region on the surface of ligand-binding domain (Figure 3, highlighted in green), which is crucial for both the amino/carboxyl-terminal (N/C) interaction of AR and co-regulator recruitment during transcriptional activation (Figure 5A,B). Unlike other steroid receptors, agonist-bound AR prefers N-terminal and C-terminal interaction,25 which could further stabilize the agonist-bound ligand-binding domain.
Figure 3.
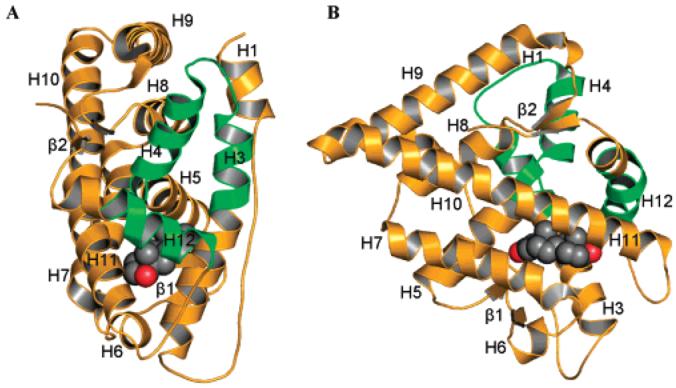
Crystal structures of wild-type AR ligand-binding domain bound with DHT (1I37.pdb): (A) front view; (B) ligand view. Space filled atoms are (black) carbon and (red) oxygen. The activation function 2 region (helices 3, 4, and 12) is highlighted in green.
Figure 4.
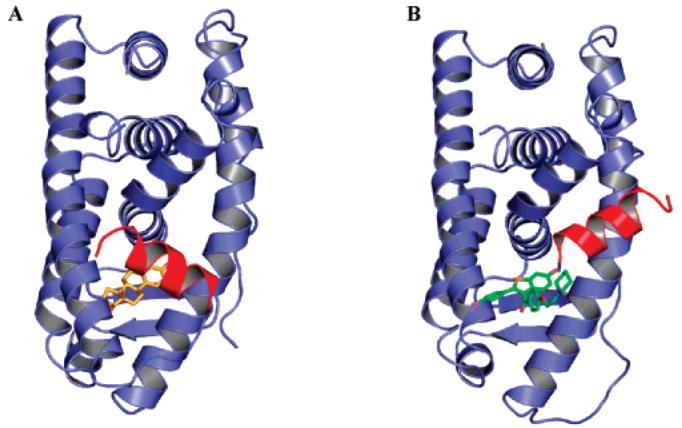
AF2 antagonist model, as illustrated by crystal structures of wild-type estrogen receptor α ligand-binding domain bound with (A) estradiol (1ERE.pdb) and (B) raloxifene (1ERR.pdb). Estradiol is shown in yellow; raloxifene is shown in green. Helix 12 is highlighted in red. Helix 12 folds over the activation function 2 (AF2) region when antagonist (raloxifene) binds to the estrogen receptor α ligand-binding domain.
Figure 5.
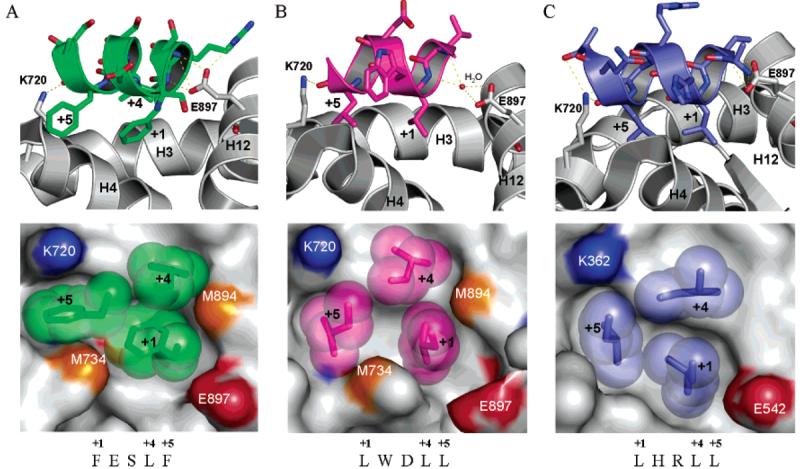
Interactions of FxxLF or LxxLL motifs with the AR or estrogen receptor ligand-binding domain: upper panels, hydrogen-bonding interactions (shown as yellow dotted line) between peptide and activation function 2 region residues; lower panels, surface view of the activation function 2 interface, in which side chains of the hydrophobic residues of FxxLF and LxxLL motifs are shown as spheres; (A) FxxLF bound to AR activation function 2 interface (1T7R.pdb); (B) LxxLL bound to AR activation function 2 interface (1T7F.pdb); (C) LxxLL bound to estrogen receptor α activation function 2 interface (1GWQ.pdb).
The activation function 2 region is a surface hydrophobic groove (Figure 5A,B) formed by the C-terminal region of H3, loop 3-4, H4, and H12, a region that covers the highly conserved nuclear receptor ligand-binding domain signature motif 26 and the activation function 2 core, which is similar to the structure that is also observed in agonist-bound estrogen receptor 27 (Figure 5C). A functional activation function 2 region is believed to be crucial for co-activator recruitment, because the so-called nuclear receptor box “LxxLL” motif 28 from the nuclear-receptor-interacting domain (NID) of co-activators specifically binds to this surface.29 On the other hand, similar motifs from the AR N-terminal domain, 23FxxLF27 and 433WxxLF437 (Figure 1), can also interact with the activation function 2 region.25 Therefore, both interactions could compete for the activation function 2 region upon agonist binding. The detailed topology change induced by different ligands within this region might provide the structural basis for recognition of specific binding motifs.30,31
The steroidal androgen-bound AR ligand-binding domain was cocrystallized with short peptides that contain the LxxLL motif or the FxxLF motif (Figure 4A,B). Hydrophobic interactions between the leucine residues in the LxxLL motif or phenylalanine residues in the FxxLF motif and the hydrophobic groove hold the peptide in place, while the hydrogen bonds between peptide backbone atoms and two well conserved residues, a lysine (K720) at the C-terminus of H3 and a glutamate (E897) in H12, form a charge clamp (Figure 4A,B) to further stabilize the interaction.30,32 Very similar interactions were also observed in the LxxLL motif bound estrogen receptor α ligand-binding domain activation function 2 region (Figure 5C).33
Despite the overall similarity in peptide binding modes, DHT-bound AR ligand-binding domain prefers the binding of the FxxLF motif to that of the LxxLL motif, suggesting that N/C interaction is preferred over co-activator recruitment in DHT-bound AR. In contrast, agonist-bound estrogen receptor α ligand-binding domain prefers the binding of the LxxLL motif to that of the FxxLF motif.32 As shown in Figure 5, DHT-bound AR formed a deeper hydrophobic groove in activation function 2 region (region located between the charge clamp residues, as illustrated in Figure 5A,B lower panels), which could accommodate the bulky side chain of phenylalanine residues. However, the hydrophobic groove in agonist-bound estrogen receptor α ligand-binding domain is more shallow and accommodates the side chain of leucine residues better. On the other hand, when the LxxLL motif binds to the AR ligand-binding domain (Figure 5B),30 the peptide backbone only forms a hydrogen bond with K720; a shift in peptide position prevents the direct hydrogen bonding with E897, which could explain the relatively lower affinity of AR for this LxxLL motif.
The distinct preferences of AR for N/C interaction could become targets for new drug discovery.34 Studies35,36 have shown that ligand binding induced AR N/C interaction correlates with its ability to activate transcription, where disruption of the N/C interaction might become an effective strategy to develop antagonists. In comparison, although estrogen receptor α does not share a similar N/C interaction as AR, estrogen receptor α antagonists have been developed to disrupt co-activator recruitment by the activation function 2 (AF2) region by blocking LxxLL motif binding. The “AF2 antagonist model” is shown in Figure 4. When agonist (estradiol) binds estrogen receptor α (Figure 4A), H12 (shown in red) adopts a conformation that helps form a functional activation function 2. However, when antagonist (tamoxifene) binds estrogen receptor α (Figure 4B), H12 is displaced from agonist conformation and folds over the AF2 region. The 540LLEML544 motif in H12 binds to the AF2 region in a similar way as the LxxLL motif and blocks the binding of co-activators.
In AR, the 895MAEII899 motif in H12 may also mimic the LxxLL motif of co-activators. In fact, one train of thought is that the AF2-antagonist model might apply to AR as well, and H12 is repositioned to bind to the activation function 2 (AF2) region upon antagonist binding with the MAEII motif blocking the interaction with other binding motifs.29 However, this hypothesis has not been proved by crystallography studies. Besides the AF2 antagonist model, other mechanisms have been proposed to explain AR antagonist activity. Although the agonist-induced conformational changes in the ligand-binding domain result in the dissociation of the chaperone protein complex from AR,37 the dissociation does not seem to happen upon antagonist binding in AR,38 which might also account for the antagonist activity by simply blocking access to the activation function 2 region. On the other hand, some evidence39 suggested that AR antagonist bicalutamide could stimulate AR nuclear translocation and specific DNA binding, but it could not mediate co-activator recruitment, instead, bicalutamide binding mediated the recruitment of co-repressor, NCoR.40 Also, similar ligand-specific (agonist vs antagonist) recruitment of coregulators has been characterized with selective estrogen receptor modulators, which appears to be related to the tissue selectivity of selective estrogen receptor modulators.41-43 Therefore, differential recruitment of co-regulators is also considered as a possible mechanism of action for AR antagonist. Despite all the theories proposed above, the precise mechanism of action for AR antagonists remains unclear.
Ligand-induced AR conformational changes provide the structural basis for the recruitment of cofactor proteins and transcriptional machinery, which is also required for the assembly of AR-mediated transcription complexes.23 Shang et al.23 showed that the formation of an activation complex involves AR, co-activators, and RNA polymerase II recruitment to both the enhancer and promoter regions of the prostate-specific antigen (PSA) gene, whereas the formation of a repression complex involves factors bound only at the promoter but not the enhancer. Since the formation of a functional activation function 2 region provides a structural basis for ligand-induced protein-protein interaction, ligand-specific recruitment of co-regulators might be crucial for the agonist or antagonist activity of AR ligands.40 On the other hand, as has been demonstrated with bicalutamide, possible ligand-specific interactions could be directly regulated by the surface topology of the activation function 2 region as shown by Sathya et al.31 Due to the subtle change in surface topology, formation of a functional activation function 2 does not guarantee effective N/C interaction. In other words, lack of N/C interaction does not correlate with the loss of activation function 2 functionality.
DNA binding is also required for AR-regulated gene expression, which is known as the classic genomic function of AR and has been characterized by DNA microarray studies.44-46 The androgen response element half-site sequence can be arranged as either inverted repeats or direct repeats,47,48 and AR recognizes and binds to the ARE site through two zinc fingers located in the DNA-binding domain. Like other steroid receptors, ligand-bound AR forms homodimers and appears to form “head-to-head’ dimers 49 even when it is bound to the direct repeats of androgen response element. Selective recognition of specific androgen response element sequences could be regulated by ligand binding50 or the presence of other transcriptional factors, which bind to their own DNA binding sites as well (combinatorial regulation), or both.51
1.2.3. Nongenomic Pathway
Besides the genomic pathway, the nongenomic pathway of AR has also been reported in oocytes,52 skeletal muscle cells,53 osteoblasts,54,55 and prostate cancer cells.56,57 As compared to the genomic pathway, the nongenomic actions of steroid receptors are characterized by the rapidity of action, which varies from seconds to an hour or so, and interaction with plasma membrane-associated signaling pathways.58 Nevertheless, the structural basis for nongenomic action is direct interactions between AR and cytosolic proteins from different signaling pathways,59 which could be closely related to the ligand-induced conformational change of the ligand-binding domain or, indirectly, the N-terminal domain. However, the detailed structural basis for these interactions is unclear. Functionally, the nongenomic action of androgen involves either rapid activation of kinase-signaling cascades or modulation of intracellular calcium levels, which could be related to stimulation of gap junction communication, neuronal plasticity, and aortic relaxation.60 Separation of the genomic and nongenomic functions of steroid receptors using specific ligands was also proposed as a new strategy to achieve tissue selectivity.58,61 However, structural features that are essential for achieving the separation have not been determined.
1.3. Androgen Biochemistry, Endogenous Agonists
1.3.1. Testosterone Synthesis
Endogenous AR ligands include testosterone and its active metabolite, 5α-DHT. Testosterone is primarily synthesized from cholesterol (Figure 6) in Leydig cells in the testes. It is also synthesized in adrenal cortex, liver, and ovary in women. The rate-limiting step in testosterone synthesis, cholesterol side chain cleavage by P450scc, is regulated by luteinizing hormone (LH) from the pituitary, which is controlled by gonadotropin releasing hormone (GnRH) from the hypothalamus and the feedback regulation of testosterone at both the pituitary and hypothalamus levels. Although dehydroepiandrosterone (DHEA) also has weak agonist activity, test-osterone and 5α-DHT are the major endogenous androgens. Besides AR, testosterone also cross-reacts with other steroid receptors at low affinity, such as progesterone receptor and estrogen receptor. In comparison, 5α-DHT binds more specifically to AR.
Figure 6.
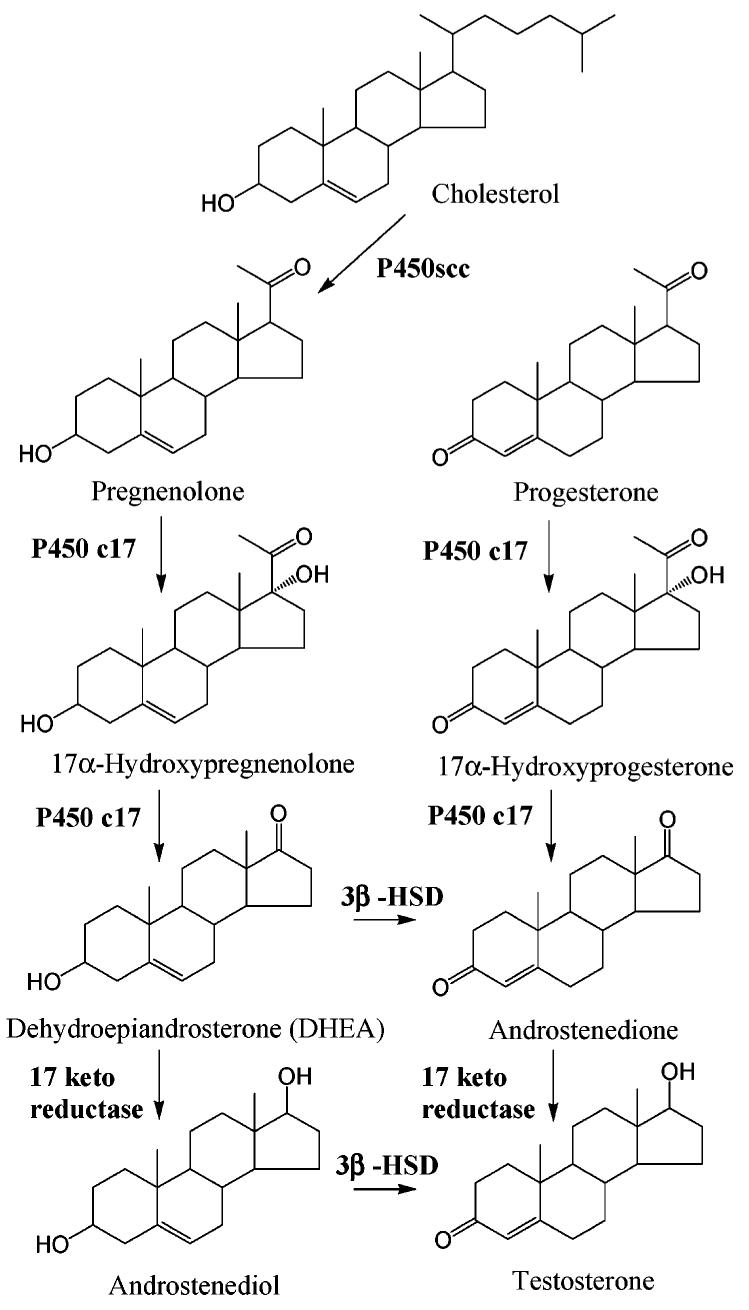
Testosterone synthesis (adapted from ref 62). Abbreviations are as follows: P450scc, cholesterol side-chain cleavage enzyme; HSD, hydroxy steroid dehydrogenase.
Healthy adult men typically produce approximately 3–10 mg of testosterone per day with circulating levels ranging from 300 to 700 ng/dL in eugonadal men. Due to the pulsatile release of gonadotropin releasing hormone, endogenous testosterone secretion is pulsatile and diurnal, the highest concentration occurring at about 8:00 a.m. and the lowest at about 8:00 p.m. Average serum concentrations and diurnal variation in testosterone diminish as men age.63
Testosterone is highly bound to plasma proteins. About 40% is sequestered with high affinity to sex hormone-binding globulin (SHBG), while almost 60% is bound with low affinity to albumin, leaving only about 2% as free, unbound hormone. 5α-DHT has even greater binding affinity to sex hormone-binding globulin than does testosterone, although 5α-DHT is only about 5% as abundant in the blood as testosterone and is largely derived from peripheral metabolism of testosterone. The unbound testosterone concentration determines its metabolic clearance rate; therefore the amount of sex hormone-binding globulin in plasma affects the half-life of circulating testosterone.
Endogenous testosterone levels decline in aging males, while circulating levels of sex hormone-binding globulin increase, which further decreases free testosterone levels.64 Despite the decrease in free testosterone concentrations, the incidence of prostate cancer and benign prostate hyperplasia (BPH) increases with age, which could be related to the fact that testosterone is almost completely converted to 5α-DHT in the prostate by 5α-reductase. Although there is no direct evidence to suggest that testosterone causes the disease, early-stage prostate cancer is clearly dependent on androgen. Evidence is also accumulating to suggest that residual or adrenal androgens and the AR play a role in “hormone-refractory” prostate cancer.65
1.3.2. Testosterone Metabolism
Testosterone can be metabolized in either its target tissues or the liver 4,66,67 (Figure 7). In androgen target tissues, testosterone can be converted to physiologically active metabolites. In the prostate gland, skin, and liver,68 testosterone is reduced to 5α-DHT by 5α-reductase (type 1 or type 2)69 in the presence of NADPH. 5α-DHT is the most potent endogenous androgen. On the other hand, a small amount of testosterone (0.2%) can also be converted to estradiol by aromatase through the cleavage of the C19 methyl group and aromatization of ring A, which mainly occurs in adipose tissue. This process also occurs in the ovaries of women. In men, approximately 80% of the circulating estrogen arises from aromatization of testosterone in the adipose tissue9 with the other 20% secreted by the Leydig cells in the testes.70
Figure 7.

Testosterone metabolism (adapted from ref 72). Abbreviationsare as follows: G, glucuronide; HSD, hydroxy steroid dehydrogenase; UGT, UDP-glucuronosyltransferase.
Both 5α-reduction and aromatization are irreversible processes. Besides these pathways, testosterone can also be further inactivated in the liver through reduction and oxidation, followed by glucuronidation and renal excretion. It can be metabolized to androstenedione through oxidation of the 17β-OH group and androstanedione with 5α-reduction of ring A. Androstanedione can be further converted to androsterone after 3-keto group reduction. Alternatively, androstenedione can also be converted to etiocholanolone through 5β- and 3-keto reduction. Similarly, 5α-DHT can be converted to androstanedione, androsterone, and androstanediol.71
After the administration of radiolabeled testosterone, about 90% of the radioactivity is found in the urine and 6% is recovered in the feces through enterohepatic circulation.7 Major urinary metabolites include androsterone and etiocholanolone. Both are inactive metabolites and are excreted mainly as glucuronide conjugates or to a lesser extent as sulfate conjugates.4 Most of the other metabolites mentioned above undergo extensive glucuronidation of the 3α-or 17β-OH groups as well, either in the target tissues or the liver,72 and are further excreted in the urine. Therefore, following oral administration, the plasma testosterone half-life is less than 30 min due to the extensive metabolism. Approximately 90% of an oral dose of testosterone is metabolized before it reaches the systemic circulation. To improve the bioavailability, most of the testosterone preparations are delivered through transdermal patch or intramuscular injections. Alkylation or esterification at the 17 position was widely used in structural modification to markedly slow the hepatic metabolism and increase the oral bioavailability or duration of action of testosterone.
1.3.3. Testosterone Tissue Disposition and Function
Animal studies73 showed that, after intravenous administration, radiolabeled androgens demonstrate higher tissue uptake in androgen target tissues, like the prostate where AR is highly expressed. The tissue uptake efficiency and selectivity of different ligands seemed to be related to their binding affinity to AR and their resistance to metabolism.
As mentioned above, there are three modes of action of testosterone. It may directly act through AR in target tissues where 5α-reductase is not expressed, be converted to 5α-DHT (5–10%) by 5α-reductase before binding to AR, or be aromatized to estrogen (0.2%) and act through the estrogen receptor.7 The formation of 5α-DHT is a natural way for the “DHT-dependent” tissues, such as prostate and seminal vesicle, to amplify the androgenic activity of testosterone. 5α-DHT is a more potent AR ligand than testosterone. It binds to AR with higher affinity (Table 1) and has 2–10-fold higher potency than testosterone in androgen-responsive tissues.10 On the other hand, estrogen plays a major role in regulating metabolic process,74,75 mood and cognition,76 cardiovascular disease,77,78 sexual function including libido,79 and bone turnover in men.80,81 Besides these active metabolites, testosterone is the major androgen that acts in the “DHT-independent” tissues, such as skeletal muscle, where 5α-reductase is not expressed or is expressed at a very low level.68 It directly regulates skeletal muscle growth, bone formation, fat distribution, and sexual function. Free testosterone is considered the most “biologically active” form, so the circulating level of sex hormone-binding globulin also affects the biologic effects of testosterone.
Table 1.
Steroidal AR Agonists


In addition, although bone tissue used to be considered as a DHT-independent tissue, it was recently observed that 5α-reductase was expressed in human osteoblast-like cells,82 suggesting that bone tissue could be a DHT-dependent tissue as well, which needs to be further confirmed by clinical evidence.
2. Steroidal Androgen Receptor Ligands and Related Clinical Applications
Generally, novel AR ligands are first identified by in vitro receptor binding assay, using either rat prostate cytosolic AR, recombinant AR protein, or cells that express AR, as summarized by Fang et al.3 Although the binding affinity of the ligands were determined in different research groups using slightly different methods (detailed binding assay methods can be retrieved from corresponding references), the AR binding affinity of all ligands discussed in this review will be presented as the relative binding affinity (RBA) compared to the synthetic ligand R1881 (Tables 1–8), which has a Kd value of 0.53 nM as determined by Kelce et al.83 and Waller et al.84 using rat prostate cytosolic AR. The agonist or antagonist activity of the ligand is often examined in vitro using cotransfection assays in which the recombinant AR and a hormone-dependent reporter gene are transiently expressed in a receptor-negative cell line. Sometimes, the antagonist activity of the ligand is also tested as its ability to suppress androgen-dependent prostate cancer cell growth. Further in vivo testing of the androgenic and anabolic activities or antagonist activity is mainly done by measuring the prostate (measure of androgenic activity) and levator ani muscle (measure of anabolic activity) sizes in castrated rat models after treatment85 with the compound alone or in combination with testosterone preparations. The antiandrogen activity can also be tested in the androgen-sensitive prostate cancer xenograft model, including LNCaP xenografts in immunodeficient mice or the Dunning rat model.
Table 8.
Selective Androgen Receptor Modulators (SARMs)



2.1. Steroidal Androgens
2.1.1. Testosterone Esters
Testosterone esters (Figure 8) are often used for clinical management of disease and are usually administered parenterally for prolonged activity. The modification enhances the lipid solubility of the steroid and permits the formation of a local depot after intramuscular injection. However, since the esters are eventually hydrolyzed, testosterone is the actual active species in vivo.
Figure 8.
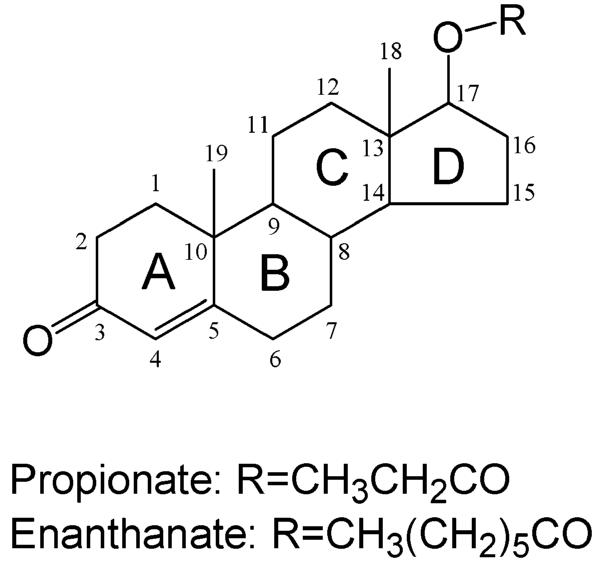
Testosterone esters.
2.1.2. 17α-Alkylated Androgens
Some of the earlier studies (1950s) with androgens focused on structural modification of the naturally occurring hormones, which maintained the steroidal structure. Mainly, the 17α-position substitution was modified to block the metabolism of the 17β-hydroxyl group, which greatly improved the oral bioavailability of these compounds, such as 17α-methyltestosterone (Table 1). 17α-Methyltestosterone has similar binding affinity to AR and similar androgenic and anabolic activities as testosterone. The addition of the 17α-alkyl group slowed metabolism at the 17-position, improved the oral bioavailability,87 and prolonged the in vivo half-life (2–3 h)88 of 17α-methyltestosterone. However, increasing the length of the alkyl side chain resulted in decreased activity. As revealed by recent AR crystal structures, the longer side chain appears to interfere with hydrogen bonding of the 17β-OH group to the receptor24 (see discussion in section 4.1.1).
Further structural modification of 17α-methyltestosterone led to more potent and orally active steroids, like oxymesterone, methandrostenolone, and fluoxymesterone. Fluoxymesterone, particularly, has a prolonged half-life of 9 h; however, it causes sodium and water retention that could lead to edema, which may be caused by its cross-reactivity with glucocorticoid receptor.89
Although these synthetic androgens show improved pharmacokinetic profiles while maintaining the androgenic and anabolic activities of testosterone, long term use of these steroids is associated with severe hepatotoxicity,90,91 which has greatly limited the clinical applications of these agents. On the other hand, due to the lack of tissue selectivity, the intrinsic androgenic activity of the steroidal androgens can cause undesirable side effects such as the virilizing and masculinizing actions in women and children, suppression of gonadotropin secretion, and salt and water retention resulting in edema.
2.1.3. Anabolic Steroids
Another class of synthetic androgens was developed as anabolic steroids as a result of efforts to minimize the undesirable androgenic side effects of testosterone and its esters. These agents appear to demonstrate stronger anabolic than androgenic activities. Although complete separation of the androgenic and anabolic activities has not been achieved with steroidal ligands, a large number of anabolic steroids are available on the market, including oxandrolone, oxymetholone, stanozolol, and nandrolone (Table 2).
Table 2.
Anabolic Steroids



For 19-norandrogens, removal of the 19-methyl group seemed to result in the reduction of the androgenic activity but retention of the anabolic activity, compared to testosterone propionate (TP). Since 19-nortestosterone showed some separation of anabolic and androgenic activities, related analogues were also developed: nandrolone, the 17α-ethyl analogue norethandrolone, and ethylestrenol.
Similar to testosterone, nandrolone is usually prepared and administered as an ester. Phenpropionate or decanoate esters of nandrolone are administered intramuscularly. Slow hydrolysis of the ester in vivo releases free nandrolone over a prolonged period of time. Nandrolone decanoate is the long acting ester used for the treatment of anemia associated with renal insufficiency. Nandrolone phenpropionate has a longer duration of action than the decanoate and is used to treat metastatic breast cancer. Although nandrolone is used as an anabolic agent, the androgenic and progestational side effects of nandrolone are still observed.4
Even with the presence of the 19-methyl group, replacement of one of the methylene groups with an oxygen atom also resulted in the favorable separation of the androgenic and anabolic activity. The most successful example of the series is oxandrolone, a 2-oxasteroid analogue of 17α-methyltestosterone that contains a lactone in the A ring. As an orally active anabolic steroid, oxandrolone has a prolonged terminal half-life of 9 h in humans.94 It demonstrates strong anabolic activity but slight androgenic activity and is clinically used to treat diseases related to muscle wasting.95 Another heterocyclic compound of the class is the pyrazole derivative, stanozolol. Similar to oxandrolone, 17α-alkylation reduces the hepatic metabolism of this agent and makes it orally available, while the pyrazole ring in the structure seems to enhance the anabolic activity of the ligand.
Another widely used anabolic steroid is oxymetholone, which is primarily used to stimulate production of erythropoietin in the treatment of anemas resulting from bone marrow failure as recently reviewed by Pavlatos et al.96 Very similar to oxandrolone, oxymetholone is also orally available and has a prolonged elimination half-life of 8 hours.97
As oral anabolic steroids, oxandrolone, stanozolol, and oxymetholone show lesser androgenic activity. However, the shared 17α-methyl group of these agents still causes potential hepatotoxicity, which is similar to that observed with 17α-alkylated steroids.
2.1.4. Structure–Activity Relationship
For steroidal androgens, the structure–activity relationships were first developed based on the results of extensive structural modifications. 3-Keto or 3α-OH groups appear to enhance the androgenic activity of steroidal AR ligands, while the 17β-hydroxyl group is essential for the ligand–receptor interaction. Removal of the 19-methyl group seems to be favorable for the separation of androgenic and anabolic activity. 17α-Alkylation significantly slows the hepatic metabolism of the 17β-OH group, increases the oral bioavailability, and prolongs the elimination half-life of these ligands. However, 17α-alkylated androgens are more likely to cause hepatotoxicity, the most serious side effect of the synthetic steroids. Similar to endogenous androgens, reduction of the 3-keto and 4,5 double bond are common routes of metabolism. Both metabolites and parent drug are eliminated either directly or as glucuronidate or sulfate conjugate.
Complete separation of androgenic and anabolic activity has not been accomplished with synthetic steroids. The androgenic activities of the synthetic steroids often cause undesirable side effects during therapy. On the other hand, due to the structural similarity in the steroid skeleton, steroidal AR ligands also tend to cross-react with other steroid receptors, which will cause adverse effects as well.
2.2. Steroidal Antiandrogens
Antiandrogens, by definition, antagonize the actions of testosterone or 5α-DHT by competing for AR binding sites. Such compounds have therapeutic potential in the treatment of prostate cancer, BPH, acne, virilization in women, and male contraception.
A few steroidal ligands have been used as antiandrogens, including cyproterone acetate, oxendolone, and spironolactone (Table 3). Only oxendolone was originally developed as an antiandrogen. After intramuscular administration, it has a prolonged half-life of 5–6 days, although it has considerably lower binding affinity to AR than cyproterone acetate.
Table 3.
Steroidal Antiandrogen

Cyproterone acetate and spironolactone, however, were originally developed for other therapeutic purposes. When they are used for their originally intended uses, the antiandrogen activity manifests as undesired side effects. Cyproterone acetate is a progestin that suppresses gonadotropin release.98 It is a weak antiandrogen even though it binds to AR with relatively high affinity compared to other antiandrogens.99 When used clinically as an antiandrogen, it is moderately effective in reducing hirsutism alone or in combination with an oral contraceptive.100 However, it is not approved for use in the United States. Spironolactone is a mineralocorticoid receptor (MR) antagonist that cross-reacts with AR with a binding affinity much lower than that of 5α-DHT.101 As a weak antiandrogen, it is approved by the US Food and Drug Administration (FDA) to treat hirsutism in women,102 although it may cause irregular menses. As an aldosterone inhibitor, spironolactone is used for the treatment of fluid retention and hypertension in men, but gynecomastia is a common side effect,103 which also limited its application as antiandrogen therapy in men.
3. Nonsteroidal Androgen Receptor Ligands
As discussed above, the clinical application of steroidal AR ligands has been limited by poor oral bioavailability, potential hepatotoxicity, lack of tissue selectivity, and occasionally, cross-reaction with other steroid receptors. Also, structural modification of the steroidal ligands is somewhat limited by the steroid skeleton. Therefore, nonsteroidal ligands were proposed to achieve high AR specificity, improve oral bioavailability, and allow more flexible structural modifications if necessary. Unlike steroidal ligands, nonsteroidal antiandrogen was first developed.
3.1. Nonsteroidal Antiandrogens
3.1.1. Toluidide Derivatives
Substituted toluidides were first developed as nonsteroidal antiandrogens, such as bicalutamide, flutamide, and nilutamide (Table 4). Unlike the steroidal antiandrogens, these toluidides are considered pure antiandrogens since they possess little if any intrinsic androgenic activity when bound to wild-type AR nor cross-react with any of the other steroid receptors. As such, the nonsteroidal antiandrogens are mainly used to treat androgen-sensitive prostate cancer or hyperplasia (BPH). Besides their pure antagonist activity, these ligands are orally available with in vivo half-lives ranging from 8 h to 6 days in humans.
Table 4.
Nonsteroidal Antiandrogens–Toluidides
| Compounds | Compound Structure | RBA (%) |
Ref |
|---|---|---|---|
| R-Bicalutamide | 0.4 | 86 | |
| Flutamide | 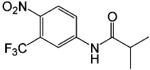 |
0.01 | 86 |
| Hydroxyflutamide | 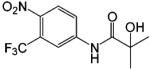 |
0.1 | 86 |
| Nilutamide | 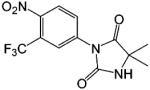 |
0.08 | 104 |
| 3-trifluoromethyl-4-nitroaniline | 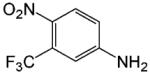 |
a |
Does not bind AR.
After oral administration, flutamide is completely absorbed from the gastrointestinal tract and undergoes extensive first pass metabolism to its major metabolite 2-hydroxyflutamide and hydrolysis product 3-trifluoromethyl-4-nitroaniline. 2-Hydroxyflutamide is a more powerful antiandrogen in vivo, with higher binding affinity for the AR than flutamide.104 In humans, hydroxyflutamide has an elimination half-life of about 8 hours.105 Hydrolysis of the amide bond represents the major metabolic pathway for this active metabolite.106 To achieve complete AR blockage in therapy, flutamide is generally used at doses of 750 mg/day. Extensive hepatic metabolism of the drug generates a large number of hydrolysis product, 3-trifluoromethyl-4-nitroaniline, which might be related to the hepatotoxicity of flutamide.107 Therefore, the clinical application of flutamide has been limited by its hepatotoxicity after long-term administration.
As a hydantoin analogue of flutamide, nilutamide is also eliminated exclusively by metabolism,108 mainly reduction of the aromatic nitro group. Although the hydrolysis of one of the carbonyl functions of the imidazolinedione was also identified, it is less susceptible to hepatic metabolism than the amide bond in hydroxyflutamide, which results in a much longer half-life of 2 days in humans. Even so, the nitro anion free radical formed during nitro reduction might still be associated with hepatotoxicity109,110 in humans, especially when the relatively high dosage (150–300 mg/day) employed for androgen blockage is used.
Bicalutamide has replaced flutamide and nilutamide as the antiandrogen of choice for prostate cancer treatment since it has less hepatotoxicity and longer half-life (6 days in humans),111 which allows once a day administration at a relatively lower dosage (50 mg/day). As a structural analogue, bicalutamide shares the amide bond structure with flutamide. However, the amide bond hydrolysis was observed in rat but not in humans,112,113 which could explain the prolonged half-life of bicalutamide in humans. Bicalutamide is mainly metabolized by hydroxylation and glucuronidation. Also, the replacement of the nitro group with a cyano group avoids the nitro reduction observed in nilutamide and hydroxyflutamide. With the presence of the chiral carbon in the structure, bicalutamide is administered as racemate. However, the in vivo antiandrogenic activity of bicalutamide arises almost entirely from its R-isomer, which has approximately 30-fold greater binding affinity and is cleared at a rate 1/100th of the S-isomer. Although bicalutamide appeared to be peripherally selective in rats114 with less antiandrogen activity in the pituitary, which could be related to its low tissue distribution in central nervous system, similar tissue selectivity was not observed in humans.
The greatly improved specificity and favorable pharmacokinetic profile of nonsteroidal antiandrogens, as compared to steroidal antiandrogens, affords much more efficient androgen blockage for prostate cancer treatment. At therapeutic doses, due to the competitive blockage of AR in both prostate and pituitary, these drugs often trigger significant increases in luteinizing hormone release, which further stimulates higher serum testosterone concentrations. Therefore, they are used primarily in combination with a gonadotropin releasing hormone analogue, which shuts down the testicular but not adrenal testosterone production. The treatments are similarly effective as surgical castration.115 However, this so-called “chemical castration” also abolishes libido and the anabolic activity of androgens in the muscle and bone, causing undesirable side effects.
3.1.2. Antiandrogen Withdrawal Syndrome
Antiandrogens are particularly useful for the treatment of prostate cancer during its early stages. However, prostate cancer often advances to a “hormone-refractory” state in which the disease progresses in the presence of continued androgen ablation or antiandrogen therapy, suggesting the development of androgen-independent prostate cancer cells or the ability of adrenal androgens to support tumor growth. Instances of antiandrogen withdrawal syndrome have also been reported after prolonged treatment with antiandrogens.116 Antiandrogen withdrawal syndrome is commonly observed clinically and is defined in terms of the tumor regression or symptomatic relief observed upon cessation of antiandrogen therapy. AR mutations that result in receptor promiscuity and the ability of these antiandrogens to exhibit agonist activity might at least partially account for this phenomenon. For example, hydroxyflutamide and bicalutamide act as AR agonists in T877A and W741L/W741C AR mutants,117,118 respectively. Therefore, more research efforts have been devoted to the development of new generation of “pure antiandrogens” that would work in both wild-type and mutant AR.
3.1.3. Hydantoin Derivatives
A variety of investigational antiandrogens are in development. These compounds have not yet been evaluated clinically but demonstrate potent antiandrogenic activity in in vitro and preclinical models. Structural modification of nilutamide led to the development of a series of bicyclic-1H-isoindole-1,3-(2H)-dione analogues that act as AR antagonists (Table 5).119-122 Lead compounds BMS-1122 and BMS-15121 bind to the wild-type AR with high affinity. The 4-nitro-naphthyl structure is also favorable for high-affinity binding of the ligand, as shown in compounds BMS-337143119 and BMS-434681.120,122 Similar to hydroxyflutamide, compounds BMS-15, BMS-337143, and BMS-434681 behaved as antagonists in wild-type AR but acted as agonists in mutant AR (T877A).122 However, BMS-1 and BMS-501949122 (structure not listed) maintained antagonist activity even in the mutant AR. Furthermore, BMS-501949 also demonstrated strong antiandrogenic activity in androgen-dependent prostate cancer xenograft models and high specificity for AR compared to other members of the steroid receptor family.
Table 5.
Nonsteroidal Antiandrogens—Hydantoin Analogs
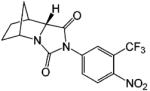
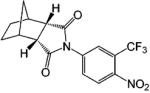
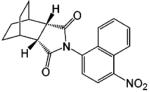
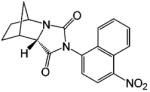


X-ray crystallography and molecular modeling studies122 suggested that the hydantoin ring nitrogen of BMS-1 and BMS-434681 serves as an H bond acceptor for N705, which improves the binding affinity of the ligand. On the other hand, increasing the size of the 2.2.2 ring system (BMS-337143 vs BMS-434681) seemed to introduce more steric interactions and help maintain the antagonist activity in mutant AR at the cost of reducing binding affinity to wild-type AR.
Another strategy to help maintain the antiandrogen activity in the mutant AR combined the structural features of both bicalutamide and BMS-434681, which led to the discovery of BMS-5.120 Although the original design intended to displace H12, a similar mechanism as proposed for bicalutamide based on a docking model using 5α-DHT bound wild-type AR ligand-binding domain crystal structure, recently solved bicalutamide bound mutant AR ligand-binding domain (W741L) crystal structure (see discussion in 4.1) suggests that this may not be the case.
In addition to their use for prostate disease, antiandrogens are also effective for the treatment of alopecia,124 hirsutism,100,125 and acne.126 Other hydantoin derivatives, like RU58841 (Table 5), were developed in Europe for topical treatment of acne and alopecia.123,127 For example, RU58841 has a short half-life of less than 1 h. Thus, topical application not only avoids the extensive hepatic metabolism (N-dealkylation) but also provides for effective regional treatment without systemic antiandrogen activity due to the formation of active metabolite.128
3.1.4. Phthalimide Derivatives
Another class of investigational nonsteroidal AR ligands is the phthalimide derivatives (Table 6). Lead compound N-[(3,5-dimethyl-4-isoxazolyl)methyl] phthalimide (DIMP) showed similar antiandrogenic activities in the castrated rat model after oral and subcutaneous administration at 4 mg/day with 2 μg/day of TP.129,130 Also, DIMP is free of any androgenic, estrogenic, antigonadotropic, and antiestrogenic activities.
Table 6.
Nonsteroidal Antiandrogens—Phthalimide Derivatives
| Compounds | Compound Structure | RBA (%) |
Ref |
|---|---|---|---|
| DIMP | 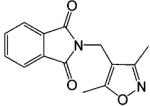 |
0.1 | 104 |
| S-FPTN |  |
a | 131 |
| R-FPTH |  |
a | 131 |
| Thalidomide |  |
a | 131 |
| ISOP-4 |  |
0.6 | 131 |
| ISOB |  |
0.5 | 131 |
Not available.
Many years after the discovery of DIMP, Hashimoto132 further developed the phthalimide analogues utilizing the structural similarities between DIMP and thalidomide, a hypnotic/sedative drug that has been used to treat prostate cancer due to its regulatory effects on cytokine production (see review by Hashimoto132,133). Representative ligands include S-FPTN, R-FPTH, ISOP-4, and ISOB. S-FPTN and R-FPTH showed potent antiandrogenic activity in suppressing the growth of an androgen-dependent cell line,133 while ISOP-4 and ISOB showed high binding affinity to AR.131 The in vivo activities of these ligands have not been reported. It is unclear whether these ligands possess any intrinsic androgenic activity.
3.1.5. Quinolinone Derivatives
As mentioned earlier, complete blockage of the AR at both the prostate and pituitary results in elevated plasma testosterone levels, which may cause breast tenderness and gynecomastia due to increased conversion of testosterone to estrogen. Therefore, selective AR antagonists (i.e., compounds that act as antagonists in the prostate but not in the pituitary) were proposed as a better antiandrogens to treat prostate cancer. A series of quinolone derivatives (Table 7) with a linear tricyclic pharmacophore, 2(1H)-piperidino[3,2-g]quinolinone, bind to AR in the nanomolar range and work as AR antagonists. In intact male rats, lead compound LG120907 showed antagonist activity in the prostate and seminal vesicle without raising the plasma levels of luteinizing hormone and testosterone.134 However, the tissue selectivity observed has not yet been demonstrated in humans. Further, it is important to note that, although it may be possible to selectively avoid feedback regulation, the anabolic effects of androgens in the muscle and bone will still likely be abolished.
Table 7.
Nonsteroidal Ligand—Quinolinone Derivatives

3.2. Nonsteroidal Androgens
Several pharmacophores possessing high binding affinity to AR were identified during the development of nonsteroidal antiandrogens. Further structural modifications of these pharmacophores led to the discovery of several classes of nonsteroidal androgen receptor agonists, including the quinolones, tetrahydroquinolone, hydantoin, and bicalutamide derivatives.
3.2.1. Quinolone Derivatives
The quinolone derivative (Table 7) LG121071 was identified as an orally active AR full agonist135 with binding affinity in the nanomolar range (Ki = 17 nM). LG121071 successfully suppressed LH release in castrated male rats, suggesting that it works as an agonist in the pituitary. However, its in vivo androgenic and anabolic activities were not discussed in published data. Computer modeling136 suggested that the A-ring keto group and C-ring ethyl group in LG121071 mimic the A-ring keto group and the 17β-OH group in testosterone, respectively, which could explain the relatively high binding affinity of LG121071. Comparing the structure of LG120907 (antagonist) and LG121071 (agonist), it is clear that C-ring substituents play a important role in determining the agonist or antagonist activity in this class of molecules.137
3.2.2. Selective Androgen Receptor Modulators
In the past several years, the successful development and marketing of selective estrogen receptor modulators (SERMs) has raised the possibility of developing selective ligands for other members of the nuclear receptor superfamily. The concept of selective androgen receptor modulators (SARMs)138,139 also emerged: a compound that is an antagonist or weak agonist in the prostate but agonist in the pituitary and muscle and orally available with low hepatotoxicity. For an ideal selective androgen receptor modulator, the antagonist or weak agonist activity in the prostate will reduce concern for the potential to stimulate nascent or undetected prostate cancer; while the strong agonist activity in the muscle and bone can be used to treat muscle-wasting conditions, hypogonadism, or age-related frailty.
Structural modifications of bicalutamide led to the discovery of the first generation of selective androgen receptor modulators (Table 8). Lead compounds S1 and S4 not only bind AR with high affinity (low nanomolar range) but also demonstrate tissue selectivity in animal models.140,141 In castrated rats, both ligands prevented castration caused tissue weight loss and behaved as partial agonists in the prostate (ED50 = 2 (mg/kg)/day) but full agonists in the levator ani muscle (ED50 = 0.6 (mg/kg)/day). On the other hand, prolonged treatment (8 weeks) with S4 also restored tissue weight loss three months after castration. At the dose rate of 3 (mg/kg)/day, S4 only partially restored the prostate weight to less than 20% of intact level, but fully restored the levator ani muscle weight to control level. Besides these androgen-dependent tissues, S4 (3 (mg/kg)/day) also demonstrated strong anabolic effects in restoring skeletal muscle (i.e., soleus muscle) strength, total body bone mineral density, and lean mass; and agonist activity in the pituitary by suppressing luteinizing hormone and follicle stimulating hormone (FSH) release.142 Furthermore, it also prevented orchidectomy induced bone loss in a female rat model for osteoporosis.143
In intact male rats,141 S1 and S4 worked as antagonists in the prostate without abolishing the anabolic effects of androgens in the levator ani muscle or increasing gonadotropin release and plasma testosterone concentrations, suggesting that selective androgen receptor modulators with low intrinsic activity in the prostate might serve as an alternative therapy for benign prostate hyperplasia (BPH) or even prostate cancer. Furthermore, suppressive effects of this class of selective androgen receptor modulators as demonstrated in both intact141 and castrated rats144 suggest that such compounds might be used for male contraception.
More importantly, both ligands are orally available145,146 with in vivo half-lives of about 4 h in rats. Metabolism studies have shown that, similar to hydroxyflutamide and nilutamide, amide bond hydrolysis and nitro reduction are the major metabolic pathway observed in vivo.147 These derivatives are eliminated exclusively through hepatic metabolism. This is the first class of nonsteroidal AR ligands that demonstrated the desired tissue selectivity of selective androgen receptor modulators.
Both the ether linkage and B-ring para-position substitution are critical for the agonist activity of the bicalutamide derivatives.150 Based on available crystal structures, compounds with the ether linkage might adopt a more compact conformation than bicalutamide due to the establishment of an intramolecular H bond (see discussion later), allowing the B-ring to avoid steric conflict with the side chain of W741 in the wild-type AR (as is observed with bicalutamide) and potentially explaining the agonist activity observed in compounds incorporating ether or thioether linkages.
Another important class of selective androgen receptor modulators that have shown tissue selectivity is the hydantoin derivatives (Table 8).148 Lead compound BMS-564929 binds AR with high affinity (RBA 3%) and high specificity. Comparing the structural features of BMS-1 (an antagonist, Table 5) and BMS-564929 (an agonist, Table 8), it is clear that the presence of the 2,2,2 ring system provides the steric hindrance necessary for antagonist activity. Likewise, crystallography and molecular modeling studies suggest that the five-membered ring in BMS-564929 creates the optimal geometry for the hydroxyl group to H bond with T877, which could contribute to the high binding affinity and agonist activity of the ligand. In castrated rats, BMS-564929 also demonstrated tissue selectivity, with ED50 = 141 (μg/kg)/ day in the prostate, ED50) 0.9 (μg/kg)/day in the levator ani muscle, and ID50 = 8 (μg/kg)/day in suppressing LH release. The compound is orally available in humans, with an in vivo half-life of 8–14 h. Compared to the bicalutamide derivatives, the prolonged in vivo half-life of these ligands could explain the lower dose needed to achieve its pharmacological activities in animal models, since the in vivo activities seem to be more related to the tissue exposure of the ligands when they share similar binding affinity and intrinsic activity. However, the potent suppression of luteinizing hormone observed with these compounds may have implications for their use in androgen replacement therapy. Studies regarding the effects of BMS-564929 on bone or other androgenic and anabolic tissues have not been reported to date.
In comparison, Hanada et al.149,151 developed a series of tetrahydroquinolin (THQ) derivatives as tissue selective AR agonists for bone. However, these compounds also showed strong agonist activity in the prostate and levator ani muscle with little tissue selectivity between androgenic and anabolic tissues. Although these compounds showed relatively high binding affinity to AR (Table 8), significant in vivo pharmacological activity was only observed at higher subcutaneous doses, about 30 (mg/kg)/day, which could be related to the pharmacokinetic profiles of these ligands. It is unclear whether these ligands are orally bioavailable.
In summary, for all three classes of compounds described above, slight structural modifications change the ligand from AR antagonist to agonist. Despite the detailed structure–activity relationships that have been elucidated for these compounds, it remains unclear what kind of ligand–receptor interaction determines the agonist or antagonist activity of the ligand, largely due to the limited knowledge about the receptor conformation upon antagonist binding. As a whole, favorable H bonding between ligand and the T877 side chain, structural features that mimic the 3-keto group of testosterone, and hydrophobic interactions seem to be critical for the ligand to bind with high affinity to the AR.
4. Structure and Function
4.1. Crystallography and Molecular Modeling
By far, crystallography remains the best way to illustrate the molecular binding mechanism of different pharmacophores. Molecular modeling becomes very useful to guide further structural modifications once knowledge of the crystal structure is available. However, predicting or modeling the binding mechanism across different structural (e.g., steroidal versus nonsteroidal ligands) or functional classes (e.g., antagonists verus agonists) is problematic.
4.1.1. Steroidal Agonist Bound Androgen Receptor Ligand-Binding Domain
The crystal structure of both wild-type and mutant AR ligand-binding domain bound with a steroidal ligand, 5α-DHT, was solved.30,152 The binding mechanism of testosterone was also proposed on the basis of molecular modeling using the 5α-DHT bound ligand-binding domain structure.18 Hydrophobic interactions play an essential role in AR ligand binding, because the steroid skeleton interacts with the ligand-binding pocket largely through hydrophobic interactions. In addition to hydrophobic interactions, hydrogen bonding in some regions of the ligand-binding pocket (Figure 9A,C) also plays a critical role in steroidal ligand binding. Mainly, the 3-keto group on ring A forms H bonds with the side chains of residues Q711 and R752 directly or indirectly (through a H2O molecule), ring C is in close contact with the main chain of L704 and side chain of N705, and the 17β-OH group H bonds with the side chains of N705 and T877. The interactions between the ligand and residues N705 (N-terminal region of H3) and T877 (C-terminal region of H11) appear to be crucial to the conformational changes that pull the N-terminal region of H3 and C-terminal region of H11 toward the ligand-binding pocket, which serves as part of the mechanism to close the hydrophobic ligand-binding pocket upon ligand binding.29
Figure 9.
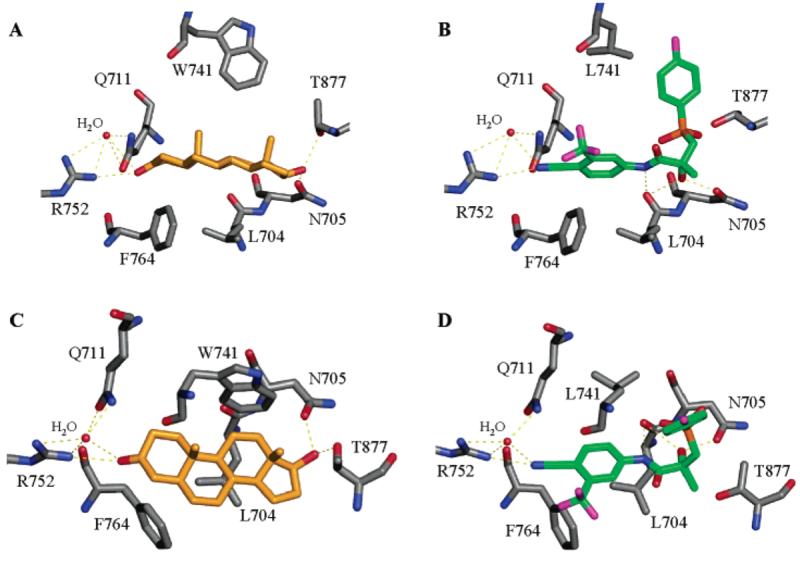
Steroidal (DHT) and nonsteroidal (R-bicalutamide) ligand interactions with AR ligand-binding domain binding pocket. R-Bicalutamide (green) W741L complex (1Z95.pdb) and DHT (gold) WT complex (1T7R.pdb) are shown as side views (A,B) and top views (C,D) of the steroidal plane. H bonds are labeled as yellow dotted lines.
In the mutant form of the AR ligand-binding domain (T877A), the mutation introduces more space around the D-ring of 5α-DHT to accommodate a larger substituent at the 17-position, which allows ligands with greater steric bulk at this position, like progesterone and cortisol,153 to bind AR as well. Also, absence of the threonine hydroxyl group (as observed in the T877A mutant) abolishes the hydrogen bond between the 17α-OH group of steroids and the receptor, significantly reducing the binding affinity of 5α-DHT to the mutant AR. Another steroidal androgen, R1881 (methyltrienolone), which binds to AR with even higher affinity than 5α-DHT, binds to the AR with a very similar mechanism as 5α-DHT.24
As discussed in section 2.1, most of the synthetic steroids, whether agonists or antagonists, incorporate an A-ring 3-keto group, which forms H bonds with Q711 and R752 and maintains the high binding affinity of the ligand. On the other hand, modification of the 17β-OH group of ring D interferes with the H bonding to N705 and T877, which not only decreases the binding affinity of the ligand but may also be related to the antagonist activity of the ligand, considering the close proximity of these residues to H12 and the activation function 2 region. However, it is still not clear how these ligand–receptor interactions would affect receptor conformation or surface topology.
4.1.2. Bicalutamide Bound Androgen Receptor Ligand-Binding Domain (W741L)
Although several structural classes of nonsteroidal compounds demonstrate high AR binding affinity 154 (nanomolar range), the binding mechanism for these compounds was unknown until just recently.122,148 The first nonsteroidal ligand bound AR ligand-binding domain crystal structure, bicalutamide bound ligand-binding domain mutant (W741L), was solved recently in our laboratory.155 Surprisingly, the molecular conformation of bicalutamide is completely different from those proposed previously based on molecular modeling using homology models.18,120 The ligand adopts a greatly bent conformation (Figure 9B). Although the majority of the bicalutamide molecule (A-ring and the amide bond) overlaps the steroidal plane (Figure 9C,D), the B-ring of the molecule folds away from the plane, pointing to the top of the ligand-binding pocket, which forms a unique structural feature of this class of ligands. The A-ring cyano group forms H bonds with Q711 and R752, similar to the 3-keto group in 5α-DHT. The chiral hydroxyl group forms H bonds with L704 and N705, mimicking the 17β-OH group in 5α-DHT. These H bonding interactions are believed to be critical for high binding affinity of the ligand.
However, different from 5α-DHT, there was no H bonding between bicalutamide and the T877 side chain, and the B-ring folded into the position of the W741 side chain (not present in this mutant) and made direct contact with H12. The absence of a favorable H bond with T877 might partially explain the relatively lower binding affinity of bicalutamide to wild-type AR, since the interaction between the DHT 17β-OH group and the T877 side chain is believed to be crucial for the bending of the C-terminus of H11 (Figure 3A and 9C). Lack of this interaction could result in a less folded overall conformation of the ligand-binding domain that might be related to the less efficient dissociation of the HSP90 complex upon antagonist (i.e., bicalutamide) binding in wild-type receptor.38 On the other hand, the absence of the W741 side chain in the W741L AR mutant exposed more space to accommodate the B-ring of bicalutamide, which favors the formation of a functional activation function 2 region and explains the agonist activity of bicalutamide in this mutant. However, assuming similar bicalutamide geometry in the wild-type AR, the B-ring of bicalutamide would displace the W741 side chain from the ligand-binding pocket (Figure 9), sterically disrupting the formation of activation function 2 region and providing a plausible mechanism for the antagonist activity of bicalutamide.
Furthermore, intramolecular hydrogen bonding in bicalutamide was also observed between the amide bond hydrogen, the chiral hydroxyl group, and the sulfonyl linkage oxygen, likely helping to achieve the ligand geometry shown in Figure 9. Due to the similarities in structure, the bicalutamide derivatives, as listed in Table 8, might adopt similar ligand geometry. Considering the flexibility of the ether linkage in these ligands, the molecule might adopt an even more bent conformation as compared to bicalutamide, which could allow the accommodation of the B-ring without displacing the bulky side chain of W741.
4.1.3. Hydantoin Derivative Bound Androgen Receptor Ligand-Binding Domain (T877A)
The hydantoin derivatives share similar A-ring structure as bicalutamide. As such, the cyano or nitro group is thought to interact with Q711 and R752 122 in a similar way as bicalutamide, although the pdb file is not available in the public database. The benzene ring or the naphthyl group, together with the hydantoin ring, overlap the steroid plane as well. On the other side of the ligand, the hydantoin ring nitrogen forms a H bond with N705. As a result, ligands without the nitrogen atom, like BMS-15 and BMS-337143 (Table 5), showed relatively lower binding affinity compared to analogues (BMS-1 and BMS-434681) with the hydantoin ring nitrogen. The 2,2,2 or 2,2,1 ring system in the antagonist structures (Table 5) was accommodated in the T877A mutant, suggesting that it might form steric interactions with the T877 side chain in wild-type receptor, which could explain the antagonist activity of these ligands. In comparison, ligands lacking the steric interaction (BMS-564929) would likely be better accommodated in the ligand-binding pocket,148 allowing either the hydantoin nitrogen or the hydroxyl group to form a favorable interaction with T877 and contribute to the agonist activity of BMS-564929.
As shown in the crystal structures described above, most of the nonsteroidal ligands, regardless of whether they act as agonist or antagonist, possess structural features that appear to be important to achieve high binding affinity, including structural features that mimic the steroid plane and maintain the hydrophobic interactions similar to those observed with steroidal ligands and structural features that mimic the 3-keto or 17β-OH groups and help form H bonds with Q711 and R752. The antagonist activity of nonsteroidal ligands seems to be related to other structural features extruding beyond the steroidal plane and disrupting H bonding with T877, such as those observed in bicalutamide and hydantoin antagonists, which are better accommodated in a mutant AR that allows more space in critical regions of the ligand-binding pocket. More importantly, only agonist bound AR ligand-binding domain was crystallized, since the antagonist bound ligand-binding domain was also solved with mutant AR in which they behaved as “agonist”. To date, no AR ligand-binding domain crystal structure in “antagonist” form has been solved; the exact molecular mechanism underlying antagonism of AR ligands remains unclear.
4.2. Mechanisms of Tissue Selectivity
As discussed in the Introduction, for certain clinical applications, it is desirable to separate the androgenic and anabolic effects of AR ligands; selective androgen receptor modulators have been proposed to achieve this goal. So far, several possible mechanisms 41-43 have been introduced to explain the tissue selectivity of nuclear receptor modulators, which are mainly based on the knowledge learned from selective estrogen receptor modulators. First of all, ligand binding induces specific conformational changes in the ligand-binding domain, which could further modulate the surface topology of the protein and subsequent protein–protein interactions between the receptor and other cellular proteins, either cytosolic proteins involved in different signaling pathways (nongenomic pathway) or coregulators involved in transcriptional activation (genomic pathway). Furthermore, ligand-specific receptor conformation and protein–protein interactions could also result in ligand-specific gene regulation, due to potential changes in recognition of the androgen response element or interactions with coregulators, other transcription factors, or both.
Ligand-induced protein–protein interaction is known to contribute to N/C interaction 36 or co-activator recruitment with the AR.156 Both interactions seem to be mediated by the interaction between the activation function 2 region and the FxxLF or LxxLL binding motifs.157 The hydrophobic groove present in the activation function 2 region of AR ligand-binding domain appears to be more favorable for the phenylalanine side chain binding, which suggests that the N/C interaction is preferred over co-activator recruitment upon agonist binding. In comparison, antagonist-binding does not initiate N/C interaction in AR,31 and it recruits corepressor,40 NCoR, instead of co-activators, demonstrating dramatic differences in receptor conformation. On the other hand, although nonsteroidal selective androgen receptor modulator bound AR ligand-binding domain conformation has not been well characterized, Sathya et al.31 have investigated some proposed steroidal selective androgen receptor modulators in vitro. It is obvious that synthetic AR agonists can induce an “activating” conformational change in AR without facilitating N/C interactions of the receptor, which suggests that ligand-specific conformational change is achievable with synthetic ligands and provides the structural basis for possible differences in protein–protein interactions.
Beyond the ligand–receptor interactions, the tissue selectivity of selective androgen receptor modulators could also be related to the tissue distribution of the ligand, potential interactions with 5α-reductase or aromatase, or tissue-specific expression of coregulators.22 However, total body autoradiography studies with bicalutamide and hydantoin derivatives148 showed that preferential accumulation of these drugs in anabolic tissues does not occur. As discussed earlier, the action of testosterone in androgenic tissues is amplified through its conversion to 5α-DHT by 5α-reductase. Studies141,148 have shown that nonsteroidal selective androgen receptor modulators are not substrates for 5α-reductase and cannot be converted to 5α-reduced metabolites, which indicates that the action of these ligands in androgenic tissues is not amplified in a similar way as testosterone. Since selective androgen receptor modulators are not as potent as 5α-DHT in the androgenic tissues, they are more likely to behave as partial agonist. However, in the anabolic tissues wherein androgen action is DHT-independent, selective androgen receptor modulators perfectly mimic the direct action of testosterone and work as full agonists. Similarly, in the presence of 5α-reductase inhibitors, testosterone also demonstrated tissue selectivity in intact rats.158 Therefore, the tissue selectivity of selective androgen receptor modulators might be simply related to the tissue-specific expression of 5α-reductase or coregulatory proteins.
5. Future Perspectives
Although the agonism and antagonism of estrogen receptor ligands are well established27 and seem to apply to some of the other nuclear receptors as well,41,159 it is unclear whether the “AF2 antagonist model” (H12 displacement) also applies to AR antagonists, since little is known about the conformation of antagonist bound AR. Nevertheless, even if the molecular mechanism underlying antagonism of AR ligands is different from that of the estrogen receptor, both appear to be related to the disruption of the formation of a functional activation function (AF2) region. Several new approaches targeting the activation function 2 region have been proposed34 to block the agonist activity of AR for the treatment of prostate cancer, including small peptides that contain the LxxLL binding motif160 or small ligands that can introduce conformational changes to interrupt activation function 2 function.31 Therefore, the ligand-binding domain activation function 2 region has become a popular drug target site.
As opposed to pure agonists and antagonists, it seems probable that many drug discovery and development efforts for the AR will also be devoted to selective androgen receptor modulators. Due to their flexibility in structural modification, selective androgen receptor modulators represent an interesting new class of AR ligands that could potentially regulate multiple aspects of AR activation: ligand-induced receptor conformation changes, protein–protein interactions, and protein–DNA interactions. “Selective ARE (androgen response element)”50 was proposed a few years ago as a mechanism for ligand-specific regulation of AR function. Selective androgen receptor modulators with various intrinsic activities could provide a promising opportunity to better understand the role of receptor–protein or receptor–DNA interaction or both in the tissue selectivity of AR ligands.
More and more evidence has emerged to suggest that nongenomic pathways also play an important role in AR function. Tissue selectivity may also be achieved by developing ligands that specifically activate the nongenomic pathway, as demonstrated for the estrogen receptor ligand, estren.54,61 The separation of the genomic and nongenomic pathways is likely dependent on ligand-specific regulation of receptor–protein interaction. However, better understanding of the molecular pharmacology and structural basis of the process will be necessary to identify potential targeting sites.
In summary, the development of AR ligands will continue, particularly focusing on the search for ligands that are AR specific, metabolically stable, safe, and tissue selective and discriminate between genomic and nongenomic pathways. A better understanding of the protein chemistry of the AR will help design ligands that could specifically target important processes during AR activation. This new generation of AR ligands appears poised to provide new drugs for the treatment of many diseases.
Biography

Wenqing Gao (right) received her Ph.D. from The Ohio State University in 2004. Her dissertation research focused on the pharmacology of selective androgen receptor modulators. She is currently working with Dr. James T. Dalton as a postdoctoral fellow. Her research interests include structural biology and molecular pharmacology of selective nuclear receptor modulators.
Casey E. Bohl (left) received a B.S. in Biochemistry at the Ohio State University in 2001. He completed a Ph.D. in 2005 on structural interactions of nonsteroidal ligands with the androgen receptor. His current research interests include crystallography of receptor-drug complexes and receptor-based drug design.
James T. Dalton (middle), Ph.D., is Professor of Pharmaceutics in the College of Pharmacy at The Ohio State University. He is currently on leave of absence from the university and serving as Vice-President of Preclinical Research and Development at GTx, Inc., Memphis, TN (www.gtxinc.com), a men's health biotech company leading clinical development of nonsteroidal selective androgen receptor modulators. Dr. Dalton is a co-inventor on over 50 US and international patents and patent applications on SARMs. His research interests include the molecular, preclinical, and clinical pharmacology of novel drugs with an emphasis on selective nuclear receptor modulators and anticancer agents. E-mail: jdalton@gtxinc.com or dalton.1@osu.edu. Office telephone: 901-523-9700, ext 110.
6. References
- 1.Freedman LP. Molecular biology of steroid and nuclear hormone receptors. Birkhauser; Boston, MA: 1998. [Google Scholar]
- 2.Mosselman S, Polman J, Dijkema R. FEBS Lett. 1996;392:49. doi: 10.1016/0014-5793(96)00782-x. [DOI] [PubMed] [Google Scholar]
- 3.Fang H, Tong W, Branham WS, Moland CL, Dial SL, Hong H, Xie Q, Perkins R, Owens W, Sheehan DM. Chem. Res. Toxicol. 2003;16:1338. doi: 10.1021/tx030011g. [DOI] [PubMed] [Google Scholar]
- 4.Burger A, Abraham DJ. Burger's medicinal chemistry and drug discovery. 6th ed. Wiley; Hoboken, NJ: 2003. [Google Scholar]
- 5.Foye WO, Williams DA, Lemke TL. Foye's principles of medicinal chemistry. 5th ed. Lippincott Williams & Wilkins; Philadelphia, PA: 2002. [Google Scholar]
- 6.Singh SM, Gauthier S, Labrie F. Curr. Med. Chem. 2000;7:211. doi: 10.2174/0929867003375371. [DOI] [PubMed] [Google Scholar]
- 7.Goodman LS, Hardman JG, Limbird LE, Gilman AG. Goodman & Gilman's the pharmacological basis of therapeutics. 10th ed. McGraw-Hill Medical Pub. Division; New York: 2001. [Google Scholar]
- 8.Keller ET, Ershler WB, Chang C. Front. Biosci. 1996;1:d59. doi: 10.2741/a116. [DOI] [PubMed] [Google Scholar]
- 9.Johansen KL. Semin. Dial. 2004;17:202. doi: 10.1111/j.0894-0959.2004.17307.x. [DOI] [PubMed] [Google Scholar]
- 10.Oettel M. Aging Male. 2003;6:230. doi: 10.1080/13685530312331309772. [DOI] [PubMed] [Google Scholar]
- 11.Migeon BR, Brown TR, Axelman J, Migeon CJ. Proc. Natl. Acad. Sci. U.S.A. 1981;78:6339. doi: 10.1073/pnas.78.10.6339. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Lubahn DB, Joseph DR, Sullivan PM, Willard HF, French FS, Wilson EM. Science. 1988;240:327. doi: 10.1126/science.3353727. [DOI] [PubMed] [Google Scholar]
- 13.Chang CS, Kokontis J, Liao ST. Science. 1988;240:324. doi: 10.1126/science.3353726. [DOI] [PubMed] [Google Scholar]
- 14.Lubahn DB, Joseph DR, Sar M, Tan J, Higgs HN, Larson RE, French FS, Wilson EM. Mol. Endocrinol. 1988;2:1265. doi: 10.1210/mend-2-12-1265. [DOI] [PubMed] [Google Scholar]
- 15.Trapman J, Klaassen P, Kuiper GG, van der Korput JA, Faber PW, van Rooij HC, Geurts van Kessel A, Voorhorst MM, Mulder E, Brinkmann AO. Biochem. Biophys. Res. Commun. 1988;153:241. doi: 10.1016/s0006-291x(88)81214-2. [DOI] [PubMed] [Google Scholar]
- 16.McEwan IJ. Endocr. Relat. Cancer. 2004;11:281. doi: 10.1677/erc.0.0110281. [DOI] [PubMed] [Google Scholar]
- 17.Chawnshang C. Androgens and androgen receptor: mechanisms, functions, and clinical applications. Kluwer Academic Publishers; Boston, MA: 2002. [Google Scholar]
- 18.Marhefka CA, Moore BM, 2nd, Bishop TC, Kirkovsky L, Mukherjee A, Dalton JT, Miller DD. J. Med. Chem. 2001;44:1729. doi: 10.1021/jm0005353. [DOI] [PubMed] [Google Scholar]
- 19.Zhou ZX, Wong CI, Sar M, Wilson EM. Recent Prog. Horm. Res. 1994;49:249. doi: 10.1016/b978-0-12-571149-4.50017-9. [DOI] [PubMed] [Google Scholar]
- 20.Myers EW, Miller W. Bull. Math. Biol. 1989;51:5. doi: 10.1007/BF02458834. [DOI] [PubMed] [Google Scholar]
- 21.Pratt WB, Toft DO. Endocr. Rev. 1997;18:306. doi: 10.1210/edrv.18.3.0303. [DOI] [PubMed] [Google Scholar]
- 22.Heinlein CA, Chang C. Endocr. Rev. 2002;23:175. doi: 10.1210/edrv.23.2.0460. [DOI] [PubMed] [Google Scholar]
- 23.Shang Y, Myers M, Brown M. Mol. Cell. 2002;9:601. doi: 10.1016/s1097-2765(02)00471-9. [DOI] [PubMed] [Google Scholar]
- 24.Matias PM, Donner P, Coelho R, Thomaz M, Peixoto C, Macedo S, Otto N, Joschko S, Scholz P, Wegg A, Basler S, Schafer M, Egner U, Carrondo MA. J. Biol. Chem. 2000;275:26164. doi: 10.1074/jbc.M004571200. [DOI] [PubMed] [Google Scholar]
- 25.He B, Minges JT, Lee LW, Wilson EM. J. Biol. Chem. 2002;277:10226. doi: 10.1074/jbc.M111975200. [DOI] [PubMed] [Google Scholar]
- 26.Wurtz JM, Bourguet W, Renaud JP, Vivat V, Chambon P, Moras D, Gronemeyer H. Nat. Struct. Biol. 1996;3:87. doi: 10.1038/nsb0196-87. [DOI] [PubMed] [Google Scholar]
- 27.Brzozowski AM, Pike AC, Dauter Z, Hubbard RE, Bonn T, Engstrom O, Ohman L, Greene GL, Gustafsson JA, Carlquist M. Nature. 1997;389:753. doi: 10.1038/39645. [DOI] [PubMed] [Google Scholar]
- 28.Heery DM, Kalkhoven E, Hoare S, Parker MG. Nature. 1997;387:733. doi: 10.1038/42750. [DOI] [PubMed] [Google Scholar]
- 29.Bourguet W, Germain P, Gronemeyer H. Trends Pharmacol. Sci. 2000;21:381. doi: 10.1016/s0165-6147(00)01548-0. [DOI] [PubMed] [Google Scholar]
- 30.Hur E, Pfaff SJ, Payne ES, Gron H, Buehrer BM, Fletterick RJ. PLoS Biol. 2004;2:E274. doi: 10.1371/journal.pbio.0020274. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Sathya G, Chang CY, Kazmin D, Cook CE, McDonnell DP. Cancer Res. 2003;63:8029. [PubMed] [Google Scholar]
- 32.He B, Gampe RT, Jr., Kole AJ, Hnat AT, Stanley TB, An G, Stewart EL, Kalman RI, Minges JT, Wilson EM. Mol. Cell. 2004;16:425. doi: 10.1016/j.molcel.2004.09.036. [DOI] [PubMed] [Google Scholar]
- 33.Warnmark A, Treuter E, Gustafsson JA, Hubbard RE, Brzozowski AM, Pike AC. J. Biol. Chem. 2002;277:21862. doi: 10.1074/jbc.M200764200. [DOI] [PubMed] [Google Scholar]
- 34.Chang CY, McDonnell DP. Trends Pharmacol. Sci. 2005;26:225. doi: 10.1016/j.tips.2005.03.002. [DOI] [PubMed] [Google Scholar]
- 35.Langley E, Kemppainen JA, Wilson EM. J. Biol. Chem. 1998;273:92. doi: 10.1074/jbc.273.1.92. [DOI] [PubMed] [Google Scholar]
- 36.Kemppainen JA, Langley E, Wong CI, Bobseine K, Kelce WR, Wilson EM. Mol. Endocrinol. 1999;13:440. doi: 10.1210/mend.13.3.0255. [DOI] [PubMed] [Google Scholar]
- 37.Veldscholte J, Berrevoets CA, Zegers ND, van der Kwast TH, Grootegoed JA, Mulder E. Biochemistry. 1992;31:7422. doi: 10.1021/bi00147a029. [DOI] [PubMed] [Google Scholar]
- 38.Kuil CW, Berrevoets CA, Mulder E. J. Biol. Chem. 1995;270:27569. doi: 10.1074/jbc.270.46.27569. [DOI] [PubMed] [Google Scholar]
- 39.Masiello D, Cheng S, Bubley GJ, Lu ML, Balk SP. J. Biol. Chem. 2002;277:26321. doi: 10.1074/jbc.M203310200. [DOI] [PubMed] [Google Scholar]
- 40.Hodgson MC, Astapova I, Cheng S, Lee LJ, Verhoeven MC, Choi E, Balk SP, Hollenberg AN. J. Biol. Chem. 2005;280:6511. doi: 10.1074/jbc.M408972200. [DOI] [PubMed] [Google Scholar]
- 41.Smith CL, O'Malley BW. Endocr. Rev. 2004;25:45. doi: 10.1210/er.2003-0023. [DOI] [PubMed] [Google Scholar]
- 42.Gronemeyer H, Gustafsson JA, Laudet V. Nat. Rev. Drug Discov. 2004;3:950. doi: 10.1038/nrd1551. [DOI] [PubMed] [Google Scholar]
- 43.Katzenellenbogen BS, Katzenellenbogen JA. Science. 2002;295:2380. doi: 10.1126/science.1070442. [DOI] [PubMed] [Google Scholar]
- 44.Nantermet PV, Xu J, Yu Y, Hodor P, Holder D, Adamski S, Gentile MA, Kimmel DB, Harada S, Gerhold D, Freedman LP, Ray WJ. J. Biol. Chem. 2004;279:1310. doi: 10.1074/jbc.M310206200. [DOI] [PubMed] [Google Scholar]
- 45.Nelson PS, Clegg N, Arnold H, Ferguson C, Bonham M, White J, Hood L, Lin B. Proc. Natl. Acad. Sci. U.S.A. 2002;99:11890. doi: 10.1073/pnas.182376299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.DePrimo SE, Diehn M, Nelson JB, Reiter RE, Matese J, Fero M, Tibshirani R, Brown PO, Brooks JD. Genome Biol. 2002;3 doi: 10.1186/gb-2002-3-7-research0032. RESEARCH0032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Claessens F, Verrijdt G, Schoenmakers E, Haelens A, Peeters B, Verhoeven G, Rombauts W. J. Steroid Biochem. Mol. Biol. 2001;76:23. doi: 10.1016/s0960-0760(00)00154-0. [DOI] [PubMed] [Google Scholar]
- 48.Haelens A, Verrijdt G, Callewaert L, Christiaens V, Schauwaers K, Peeters B, Rombauts W, Claessens F. Biochem. J. 2003;369:141. doi: 10.1042/BJ20020912. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Shaffer PL, Jivan A, Dollins DE, Claessens F, Gewirth DT. Proc. Natl. Acad. Sci. U.S.A. 2004;101:4758. doi: 10.1073/pnas.0401123101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Hsiao PW, Thin TH, Lin DL, Chang C. Mol. Cell. Biochem. 2000;206:169. doi: 10.1023/a:1007024726889. [DOI] [PubMed] [Google Scholar]
- 51.Remenyi A, Scholer HR, Wilmanns M. Nat. Struct. Mol. Biol. 2004;11:812. doi: 10.1038/nsmb820. [DOI] [PubMed] [Google Scholar]
- 52.Lutz LB, Jamnongjit M, Yang WH, Jahani D, Gill A, Hammes SR. Mol. Endocrinol. 2003;17:1106. doi: 10.1210/me.2003-0032. [DOI] [PubMed] [Google Scholar]
- 53.Estrada M, Espinosa A, Muller M, Jaimovich E. Endocrinology. 2003;144:3586. doi: 10.1210/en.2002-0164. [DOI] [PubMed] [Google Scholar]
- 54.Kousteni S, Bellido T, Plotkin LI, O'Brien CA, Bodenner DL, Han L, Han K, DiGregorio GB, Katzenellenbogen JA, Katzenellenbogen BS, Roberson PK, Weinstein RS, Jilka RL, Manolagas SC. Cell. 2001;104:719. [PubMed] [Google Scholar]
- 55.Zagar Y, Chaumaz G, Lieberherr M. J. Biol. Chem. 2004;279:2403. doi: 10.1074/jbc.M309132200. [DOI] [PubMed] [Google Scholar]
- 56.Kampa M, Papakonstanti EA, Hatzoglou A, Stathopoulos EN, Stournaras C, Castanas E. FASEB J. 2002;16:1429. doi: 10.1096/fj.02-0131fje. [DOI] [PubMed] [Google Scholar]
- 57.Unni E, Sun S, Nan B, McPhaul MJ, Cheskis B, Mancini MA, Marcelli M. Cancer Res. 2004;64:7156. doi: 10.1158/0008-5472.CAN-04-1121. [DOI] [PubMed] [Google Scholar]
- 58.Norman AW, Mizwicki MT, Norman DP. Nat. Rev. Drug Discov. 2004;3:27. doi: 10.1038/nrd1283. [DOI] [PubMed] [Google Scholar]
- 59.Migliaccio A, Castoria G, Di Domenico M, de Falco A, Bilancio A, Lombardi M, Barone MV, Ametrano D, Zannini MS, Abbondanza C, Auricchio F. EMBO J. 2000;19:5406. doi: 10.1093/emboj/19.20.5406. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Simoncini T, Genazzani AR. Eur. J. Endocrinol. 2003;148:281. doi: 10.1530/eje.0.1480281. [DOI] [PubMed] [Google Scholar]
- 61.Kousteni S, Chen JR, Bellido T, Han L, Ali AA, O'Brien CA, Plotkin L, Fu Q, Mancino AT, Wen Y, Vertino AM, Powers CC, Stewart SA, Ebert R, Parfitt AM, Weinstein RS, Jilka RL, Manolagas SC. Science. 2002;298:843. doi: 10.1126/science.1074935. [DOI] [PubMed] [Google Scholar]
- 62.Miller WL. Endocr. Rev. 1988;9:295. doi: 10.1210/edrv-9-3-295. [DOI] [PubMed] [Google Scholar]
- 63.Bremner WJ, Vitiello MV, Prinz PN. J. Clin. Endocrinol. Metab. 1983;56:1278. doi: 10.1210/jcem-56-6-1278. [DOI] [PubMed] [Google Scholar]
- 64.Schulman C, Lunenfeld B. World J. Urol. 2002;20:4. doi: 10.1007/s00345-002-0258-3. [DOI] [PubMed] [Google Scholar]
- 65.Hakimi JM, Rondinelli RH, Schoenberg MP, Barrack ER. World J. Urol. 1996;14:329. doi: 10.1007/BF00184606. [DOI] [PubMed] [Google Scholar]
- 66.Dorfman RI, Ungar F. Metabolism of steroid hormones. 2nd ed. Academic Press; New York: 1965. [Google Scholar]
- 67.Verderame M. CRC handbook of hormones, vitamins, and radiopaques. CRC Press; Boca Raton, FL: 1986. [Google Scholar]
- 68.Thigpen AE, Silver RI, Guileyardo JM, Casey ML, McConnell JD, Russell DW. J. Clin. Invest. 1993;92:903. doi: 10.1172/JCI116665. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Russell DW, Berman DM, Bryant JT, Cala KM, Davis DL, Landrum CP, Prihoda JS, Silver RI, Thigpen AE, Wigley WC. Recent Prog. Horm. Res. 1994;49:275. doi: 10.1016/b978-0-12-571149-4.50018-0. [DOI] [PubMed] [Google Scholar]
- 70.Vermeulen A, Kaufman JM, Goemaere S, van Pottelberg I. Aging Male. 2002;5:98. [PubMed] [Google Scholar]
- 71.Fotherby K, James F, Atkinson L, Dumasia M. J. Endocrinol. 1972;52:v. [PubMed] [Google Scholar]
- 72.Belanger A, Pelletier G, Labrie F, Barbier O, Chouinard S. Trends Endocrinol. Metab. 2003;14:473. doi: 10.1016/j.tem.2003.10.005. [DOI] [PubMed] [Google Scholar]
- 73.Carlson KE, Katzenellenbogen JA. J. Steroid Biochem. 1990;36:549. doi: 10.1016/0022-4731(90)90172-o. [DOI] [PubMed] [Google Scholar]
- 74.Oettel M. Aging Male. 2002;5:248. [PubMed] [Google Scholar]
- 75.de Ronde W, Pols HA, van Leeuwen JP, de Jong FH. Clin. Endocrinol. (Oxford) 2003;58:529. doi: 10.1046/j.1365-2265.2003.01669.x. [DOI] [PubMed] [Google Scholar]
- 76.Barrett-Connor E, Goodman-Gruen D, Patay B. J. Clin. Endocrinol. Metab. 1999;84:3681. doi: 10.1210/jcem.84.10.6086. [DOI] [PubMed] [Google Scholar]
- 77.Van Pottelbergh I, Braeckman L, De Bacquer D, De Backer G, Kaufman JM. Atherosclerosis. 2003;166:95. doi: 10.1016/s0021-9150(02)00308-8. [DOI] [PubMed] [Google Scholar]
- 78.Mukherjee TK, Dinh H, Chaudhuri G, Nathan L. Proc. Natl. Acad. Sci. U.S.A. 2002;99:4055. doi: 10.1073/pnas.052703199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Toda K, Okada T, Takeda K, Akira S, Saibara T, Shiraishi M, Onishi S, Shizuta Y. J. Endocrinol. 2001;168:455. doi: 10.1677/joe.0.1680455. [DOI] [PubMed] [Google Scholar]
- 80.Falahati-Nini A, Riggs BL, Atkinson EJ, O'Fallon WM, Eastell R, Khosla S. J. Clin. Invest. 2000;106:1553. doi: 10.1172/JCI10942. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Orwoll ES. Osteoporosis Int. 2003;14:93. doi: 10.1007/s00198-002-1332-9. [DOI] [PubMed] [Google Scholar]
- 82.Issa S, Schnabel D, Feix M, Wolf L, Schaefer HE, Russell DW, Schweikert HU. J. Clin. Endocrinol. Metab. 2002;87:5401. doi: 10.1210/jc.2001-011902. [DOI] [PubMed] [Google Scholar]
- 83.Kelce WR, Monosson E, Gamcsik MP, Laws SC, Gray LE., Jr. Toxicol. Appl. Pharmacol. 1994;126:276. doi: 10.1006/taap.1994.1117. [DOI] [PubMed] [Google Scholar]
- 84.Waller CL, Juma BW, Gray LE, Jr., Kelce WR. Toxicol. Appl. Pharmacol. 1996;137:219. doi: 10.1006/taap.1996.0075. [DOI] [PubMed] [Google Scholar]
- 85.Hershberger LG, Shipley EG, Meyer RK. Proc. Soc. Exp. Biol. Med. 1953;83:175. doi: 10.3181/00379727-83-20301. [DOI] [PubMed] [Google Scholar]
- 86.Christiansen RG, Bell MR, D'Ambra TE, Mallamo JP, Herrmann JL, Ackerman JH, Opalka CJ, Kullnig RK, Winneker RC, Snyder BW, Batzold FH, Schane HP. J. Med. Chem. 1990;33:2094. doi: 10.1021/jm00170a008. [DOI] [PubMed] [Google Scholar]
- 87.Shinohara Y, Baba S. Steroids. 1990;55:170. doi: 10.1016/0039-128x(90)90106-l. [DOI] [PubMed] [Google Scholar]
- 88.Quincey RV, Gray CH. J. Endocrinol. 1967;37:37. doi: 10.1677/joe.0.0370037. [DOI] [PubMed] [Google Scholar]
- 89.Mayer M, Rosen F. Am J. Physiol. 1975;229:1381. doi: 10.1152/ajplegacy.1975.229.5.1381. [DOI] [PubMed] [Google Scholar]
- 90.Cabasso A. Med. Sci. Sports Exercise. 1994;26:2. [PubMed] [Google Scholar]
- 91.Petera V, Bobek K, Lahn V. Clin. Chim. Acta. 1962;7:604. doi: 10.1016/0009-8981(62)90138-9. [DOI] [PubMed] [Google Scholar]
- 92.Litwack G. Biochemical actions of hormones. Academic Press; New York: 1970. [Google Scholar]
- 93.Cunningham GR, Tindall DJ, Means AR. Steroids. 1979;33:261. doi: 10.1016/0039-128x(79)90003-5. [DOI] [PubMed] [Google Scholar]
- 94.Karim A, Ranney RE, Zagarella J, Maibach HI. Clin. Pharmacol. Ther. 1973;14:862. doi: 10.1002/cpt1973145862. [DOI] [PubMed] [Google Scholar]
- 95.Langer CJ, Hoffman JP, Ottery FD. Nutrition. 2001;17:S1. doi: 10.1016/s0899-9007(01)80001-0. [DOI] [PubMed] [Google Scholar]
- 96.Pavlatos AM, Fultz O, Monberg MJ, Vootkur A. Pharmacol. Clin. Ther. 2001;23:789. doi: 10.1016/s0149-2918(01)80070-9. [DOI] [PubMed] [Google Scholar]
- 97.Cardoso CR, Marques MA, Caminha RC, Maioli MC, Aquino Neto FRJ. Chromatogr., B: Anal. Technol. Biomed. Life Sci. 2002;775:1. doi: 10.1016/s1570-0232(02)00243-x. [DOI] [PubMed] [Google Scholar]
- 98.Neri RO. Adv. Sex Horm. Res. 1976;2:233. [PubMed] [Google Scholar]
- 99.Fang S, Liao S. Mol. Pharmacol. 1969;5:428. [PubMed] [Google Scholar]
- 100.Venturoli S, Marescalchi O, Colombo FM, Macrelli S, Ravaioli B, Bagnoli A, Paradisi R, Flamigni C. J. Clin. Endocrinol. Metab. 1999;84:1304. doi: 10.1210/jcem.84.4.5591. [DOI] [PubMed] [Google Scholar]
- 101.Rifka SM, Pita JC, Vigersky RA, Wilson YA, Loriaux DL. J. Clin. Endocrinol. Metab. 1978;46:338. doi: 10.1210/jcem-46-2-338. [DOI] [PubMed] [Google Scholar]
- 102.Cumming DC, Yang JC, Rebar RW, Yen SS. J. Am. Med. Assoc. 1982;247:1295. [PubMed] [Google Scholar]
- 103.Caminos-Torres R, Ma L, Snyder PJ. J. Clin. Endocrinol. Metab. 1977;45:255. doi: 10.1210/jcem-45-2-255. [DOI] [PubMed] [Google Scholar]
- 104.Wakeling AE, Furr BJ, Glen AT, Hughes LR. J. Steroid Biochem. 1981;15:355. doi: 10.1016/0022-4731(81)90297-1. [DOI] [PubMed] [Google Scholar]
- 105.Schulz M, Schmoldt A, Donn F, Becker H. Eur. J. Clin. Pharmacol. 1988;34:633. doi: 10.1007/BF00615229. [DOI] [PubMed] [Google Scholar]
- 106.Katchen B, Buxbaum S. J. Clin. Endocrinol. Metab. 1975;41:373. doi: 10.1210/jcem-41-2-373. [DOI] [PubMed] [Google Scholar]
- 107.Fau D, Eugene D, Berson A, Letteron P, Fromenty B, Fisch C, Pessayre D. J. Pharmacol. Exp. Ther. 1994;269:954. [PubMed] [Google Scholar]
- 108.Creaven PJ, Pendyala L, Tremblay D. Urology. 1991;37:13. doi: 10.1016/0090-4295(91)80096-p. [DOI] [PubMed] [Google Scholar]
- 109.Berson A, Wolf C, Berger V, Fau D, Chachaty C, Fromenty B, Pessayre D. J. Pharmacol. Exp. Ther. 1991;257:714. [PubMed] [Google Scholar]
- 110.Fau D, Berson A, Eugene D, Fromenty B, Fisch C, Pessayre D. J. Pharmacol. Exp. Ther. 1992;263:69. [PubMed] [Google Scholar]
- 111.Cockshott ID. Clin. Pharmacokinet. 2004;43:855. doi: 10.2165/00003088-200443130-00003. [DOI] [PubMed] [Google Scholar]
- 112.Boyle GW, McKillop D, Phillips PJ, Harding JR, Pickford R, McCormick AD. Xenobiotica. 1993;23:781. doi: 10.3109/00498259309166784. [DOI] [PubMed] [Google Scholar]
- 113.McKillop D, Boyle GW, Cockshott ID, Jones DC, Phillips PJ, Yates RA. Xenobiotica. 1993;23:1241. doi: 10.3109/00498259309059435. [DOI] [PubMed] [Google Scholar]
- 114.Furr BJ, Tucker H. Urology. 1996;47:13. doi: 10.1016/s0090-4295(96)80003-3. [DOI] [PubMed] [Google Scholar]
- 115.Schellhammer P, Sharifi R, Block N, Soloway M, Venner P, Patterson AL, Sarosdy M, Vogelzang N, Jones J, Kolvenbag G. Urology. 1995;45:745. doi: 10.1016/s0090-4295(99)80077-6. [DOI] [PubMed] [Google Scholar]
- 116.Taplin ME, Rajeshkumar B, Halabi S, Werner CP, Woda BA, Picus J, Stadler W, Hayes DF, Kantoff PW, Vogelzang NJ, Small EJ. J. Clin. Oncol. 2003;21:2673. doi: 10.1200/JCO.2003.11.102. [DOI] [PubMed] [Google Scholar]
- 117.Suzuki H, Akakura K, Komiya A, Aida S, Akimoto S, Shimazaki J. Prostate. 1996;29:153. doi: 10.1002/1097-0045(199609)29:3<153::aid-pros2990290303>3.0.co;2-5. [DOI] [PubMed] [Google Scholar]
- 118.Hara T, Miyazaki J, Araki H, Yamaoka M, Kanzaki N, Kusaka M, Miyamoto M. Cancer Res. 2003;63:149. [PubMed] [Google Scholar]
- 119.Salvati ME, Balog A, Shan W, Wei DD, Pickering D, Attar RM, Geng J, Rizzo CA, Gottardis MM, Weinmann R, Krystek SR, Sack J, An Y, Kish K. Bioorg. Med. Chem. Lett. 2005;15:271. doi: 10.1016/j.bmcl.2004.10.085. [DOI] [PubMed] [Google Scholar]
- 120.Balog A, Salvati ME, Shan W, Mathur A, Leith LW, Wei DD, Attar RM, Geng J, Rizzo CA, Wang C, Krystek SR, Tokarski JS, Hunt JT, Gottardis M, Weinmann R. Bioorg. Med. Chem. Lett. 2004;14:6107. doi: 10.1016/j.bmcl.2004.09.049. [DOI] [PubMed] [Google Scholar]
- 121.Salvati ME, Balog A, Wei DD, Pickering D, Attar RM, Geng J, Rizzo CA, Hunt JT, Gottardis MM, Weinmann R, Martinez R. Bioorg. Med. Chem. Lett. 2005;15:389. doi: 10.1016/j.bmcl.2004.10.051. [DOI] [PubMed] [Google Scholar]
- 122.Salvati ME, Attar RM, Krystek SR, Lee F, Hunt JT, Gottardis M, Jure-Kunkeld M, Sack J, Giese S, Martinez R, Mitt T, Pickering D, Shan W, Wei DD, Zhu H, Cullinan C, Geng J, Rizzo CA, Wang C, Mathur A, Leith LW, Kish K, Vite G, Dell J, Spires T, An Y, Weinmann R. Am. Assoc. Cancer Res. 2004 Abstract No. 1948. [Google Scholar]
- 123.Van Dort ME, Jung YW. Bioorg. Med. Chem. Lett. 2001;11:1045. doi: 10.1016/s0960-894x(01)00146-9. [DOI] [PubMed] [Google Scholar]
- 124.Diamanti-Kandarakis E. Curr. Pharm. Des. 1999;5:707. [PubMed] [Google Scholar]
- 125.Muderris, Bayram F, Ozcelik B, Guven M. Gynecol. Endocrinol. 2002;16:63. [PubMed] [Google Scholar]
- 126.Carmina E, Lobo RA. Clin. Endocrinol. (Oxford) 2002;57:231. doi: 10.1046/j.1365-2265.2002.01594.x. [DOI] [PubMed] [Google Scholar]
- 127.Battmann T, Bonfils A, Branche C, Humbert J, Goubet F, Teutsch G, Philibert D. J. Steroid Biochem. Mol. Biol. 1994;48:55. doi: 10.1016/0960-0760(94)90250-x. [DOI] [PubMed] [Google Scholar]
- 128.Cousty-Berlin D, Bergaud B, Bruyant MC, Battmann T, Branche C, Philibert D. J. Steroid Biochem. Mol. Biol. 1994;51:47. doi: 10.1016/0960-0760(94)90114-7. [DOI] [PubMed] [Google Scholar]
- 129.Scott JW, Boris A. J. Med. Chem. 1973;16:512. doi: 10.1021/jm00263a023. [DOI] [PubMed] [Google Scholar]
- 130.Boris A, Scott JW, DeMartino L, Cox DC. Acta Endocrinol. (Copenhagen) 1973;72:604. doi: 10.1530/acta.0.0720604. [DOI] [PubMed] [Google Scholar]
- 131.Ishioka T, Kubo A, Koiso Y, Nagasawa K, Itai A, Hashimoto Y. Bioorg. Med. Chem. 2002;10:1555. doi: 10.1016/s0968-0896(01)00421-7. [DOI] [PubMed] [Google Scholar]
- 132.Hashimoto Y. Bioorg. Med. Chem. 2002;10:461. doi: 10.1016/s0968-0896(01)00308-x. [DOI] [PubMed] [Google Scholar]
- 133.Hashimoto Y. Cancer Chemother. Pharmacol. 2003;52(Suppl 1):S16. doi: 10.1007/s00280-003-0590-3. [DOI] [PubMed] [Google Scholar]
- 134.Hamann LG, Higuchi RI, Zhi L, Edwards JP, Wang XN, Marschke KB, Kong JW, Farmer LJ, Jones TK. J. Med. Chem. 1998;41:623. doi: 10.1021/jm970699s. [DOI] [PubMed] [Google Scholar]
- 135.Hamann LG, Mani NS, Davis RL, Wang XN, Marschke KB, Jones TK. J. Med. Chem. 1999;42:210. doi: 10.1021/jm9806648. [DOI] [PubMed] [Google Scholar]
- 136.Bohl CE, Chang C, Mohler ML, Chen J, Miller DD, Swaan PW, Dalton JT. J. Med. Chem. 2004;47:3765. doi: 10.1021/JM0499007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 137.Zhi L, Tegley CM, Marschke KB, Jones TK. Bioorg. Med. Chem. Lett. 1999;9:1009. doi: 10.1016/s0960-894x(99)00119-5. [DOI] [PubMed] [Google Scholar]
- 138.Zhi L, Martinborough E. Annu. Rep. Med. Chem. 2001;36:169. [Google Scholar]
- 139.Negro-Vilar A. J. Clin. Endocrinol. Metab. 1999;84:3459. doi: 10.1210/jcem.84.10.6122. [DOI] [PubMed] [Google Scholar]
- 140.Yin D, Gao W, Kearbey JD, Xu H, Chung K, He Y, Marhefka CA, Veverka KA, Miller DD, Dalton JT. J. Pharmacol. Exp. Ther. 2003;304:1334. doi: 10.1124/jpet.102.040840. [DOI] [PubMed] [Google Scholar]
- 141.Gao W, Kearbey JD, Nair VA, Chung K, Parlow AF, Miller DD, Dalton JT. Endocrinology. 2004;145:5420. doi: 10.1210/en.2004-0627. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 142.Gao W, Reiser PJ, Coss CC, Phelps MA, Kearbey JD, Miller DD, Dalton JT. Endocrinology. 2005 August 11; doi: 10.1210/en.2005-0572. doi: 10.1210/en.2005-0572. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 143.Kearbey JD, Gao W, Miller DD, Dalton JT. AAPS J. 2003;5(4) (special issues): AAPS Pharm. Sci. Abstract R6167. [Google Scholar]
- 144.Chen J, Hwang DJ, Bohl CE, Miller DD, Dalton JT. J. Pharmacol. Exp. Ther. 2004 doi: 10.1124/jpet.104.075424. [DOI] [PubMed] [Google Scholar]
- 145.Kearbey JD, Wu D, Gao W, Miller DD, Dalton JT. Xenobiotica. 2004;34:273. doi: 10.1080/0049825041008962. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 146.Wu D, Gao W, Kearbey JD, Chung K, Miller DD, Dalton JT. AAPS J. 2003;5(4) (special issues): AAPS Pharm. Sci. Abstract W5267. [Google Scholar]
- 147.Gao W, Wu Z, Chung K, Miller DD, Dalton JT. AAPS J. 2003;5(4) (special issues): AAPS Pharm. Sci. Abstract T3337. [Google Scholar]
- 148.Hamann LG. Natl. Meet. Am. Chem. Soc., Med. Chem. Div. 2004;227:MEDI-175. [Google Scholar]
- 149.Miyakawa M, Oguro N, Hanada K, Furuya K, Yamamoto N. World Patent WO 2004013104 2004
- 150.Yin D, He Y, Perera MA, Hong SS, Marhefka C, Stourman N, Kirkovsky L, Miller DD, Dalton JT. Mol. Pharmacol. 2003;63:211. doi: 10.1124/mol.63.1.211. [DOI] [PubMed] [Google Scholar]
- 151.Hanada K, Furuya K, Yamamoto N, Nejishima H, Ichikawa K, Nakamura T, Miyakawa M, Amano S, Sumita Y, Oguro N. Biol. Pharm. Bull. 2003;26:1563. doi: 10.1248/bpb.26.1563. [DOI] [PubMed] [Google Scholar]
- 152.Sack JS, Kish KF, Wang C, Attar RM, Kiefer SE, An Y, Wu GY, Scheffler JE, Salvati ME, Krystek SR, Jr., Weinmann R, Einspahr HM. Proc. Natl. Acad. Sci. U.S.A. 2001;98:4904. doi: 10.1073/pnas.081565498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 153.Matias PM, Carrondo MA, Coelho R, Thomaz M, Zhao XY, Wegg A, Crusius K, Egner U, Donner P. J. Med. Chem. 2002;45:1439. doi: 10.1021/jm011072j. [DOI] [PubMed] [Google Scholar]
- 154.Allan GF, Sui Z. NURSA e-J. 2003;1 No. 3.09172003.1. [Google Scholar]
- 155.Bohl CE, Gao W, Miller DD, Bell CE, Dalton JT. Proc. Natl. Acad. Sci. U.S.A. 2005;102:6201. doi: 10.1073/pnas.0500381102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 156.Masiello D, Chen SY, Xu Y, Verhoeven MC, Choi E, Hollenberg AN, Balk SP. Mol. Endocrinol. 2004;18:2388. doi: 10.1210/me.2003-0436. [DOI] [PubMed] [Google Scholar]
- 157.Song LN, Herrell R, Byers S, Shah S, Wilson EM, Gelmann EP. Mol. Cell. Biol. 2003;23:1674. doi: 10.1128/MCB.23.5.1674-1687.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 158.Wright AS, Douglas RC, Thomas LN, Lazier CB, Rittmaster RS. Endocrinology. 1999;140:4509. doi: 10.1210/endo.140.10.7039. [DOI] [PubMed] [Google Scholar]
- 159.Kauppi B, Jakob C, Farnegardh M, Yang J, Ahola H, Alarcon M, Calles K, Engstrom O, Harlan J, Muchmore S, Ramqvist AK, Thorell S, Ohman L, Greer J, Gustafsson JA, Carlstedt-Duke J, Carlquist M. J. Biol. Chem. 2003;278:22748. doi: 10.1074/jbc.M212711200. [DOI] [PubMed] [Google Scholar]
- 160.Chang CY, McDonnell DP. Mol. Endocrinol. 2002;16:647. doi: 10.1210/mend.16.4.0818. [DOI] [PubMed] [Google Scholar]