

Abstract
Enteropathogenic Escherichia coli (EPEC) triggers a large release of adenosine triphosphate (ATP) from host intestinal cells and the extracellular ATP is broken down to adenosine diphosphate (ADP), AMP, and adenosine. Adenosine is a potent secretagogue in the small and large intestine. We suspected that ecto-5′-nucleotidase (CD73, an intestinal enzyme) was a critical enzyme involved in the conversion of AMP to adenosine and in the pathogenesis of EPEC diarrhea. We developed a nonradioactive method for measuring ecto-5′-nucleotidase in cultured T84 cell monolayers based on the detection of phosphate release from 5′-AMP. EPEC infection triggered a release of ecto-5′-nucleotidase from the cell surface into the supernatant medium. EPEC-induced 5′-nucleotidase release was not correlated with host cell death but instead with activation of phosphatidylinositol-specific phospholipase C (PI-PLC). Ecto-5′-nucleotidase was susceptible to inhibition by zinc acetate and by α,β-methylene-adenosine diphosphate (α,β-methylene-ADP). In the Ussing chamber, these inhibitors could reverse the chloride secretory responses triggered by 5′-AMP. In addition, α,β-methylene-ADP and zinc blocked the ability of 5′-AMP to stimulate EPEC growth under nutrient-limited conditions in vitro. Ecto-5′-nucleotidase appears to be the major enzyme responsible for generation of adenosine from adenine nucleotides in the T84 cell line, and inhibitors of ecto-5′-nucleotidase, such as α,β-methylene-ADP and zinc, might be useful for treatment of the watery diarrhea produced by EPEC infection.
Keywords: Adenosine, Diarrheal disease, Extracellular nucleotides, Zinc
Introduction
Ecto-5′-nucleotidase (CD73, EC 3.1.3.5) is a key extracellular enzyme which catalyzes the hydrolysis of extracellular nucleoside monophosphates to their corresponding nucleosides, with 5′-AMP being the preferred substrate. Ecto-5′-nucleotidase is highly expressed in endothelium, liver, and intestinal mucosa and at varying levels in other tissues including lymphocytes, kidney, and certain cancers. Ecto-5′-nucleotidase is tethered to the extracellular surface of mammalian cells by a glycosylphosphatidylinositol (GPI) lipid anchor. Most of the research on this enzyme has been in the cardiovascular system because of its role in the production of adenosine, which protects cells against ischemia by multiple mechanisms including vasodilation [1, 2].
Enteropathogenic Escherichia coli (EPEC) is a common cause of watery diarrhea in children in developing countries. While some aspects of EPEC infection, such as adherence, are well studied, the way that EPEC triggers watery diarrhea has been obscure, since EPEC produces no toxins. We recently proposed a theory that release of adenine nucleotides from host intestinal cells, followed by breakdown to adenosine, could trigger watery diarrhea by activation of adenosine receptors on intestinal cells [3].
In the gastrointestinal tract, ecto-5′-nucleotidase was found to be a key enzyme necessary for enterocyte responses to 5′-AMP, the neutrophil-derived secretagogue [4–6]. At that time the significance of ecto-5′-nucleotidase was felt to be in the setting of invasive, inflammatory pathogens because polymorphonuclear neutrophils (PMNs) release 5′-AMP during the process of chemotaxis into areas of infection or inflammation. Since that time, however, we have discovered that a noninvasive pathogen, EPEC, triggers a large release of ATP directly from host intestinal cells without any requirement for PMNs to be present [3]. Other diarrheal pathogens not considered classically invasive, such as Aeromonas hydrophila, also cause ATP release from enterocytes [7]. Extracellular ATP is broken down to ADP, AMP, and then adenosine, where it triggers a vigorous fluid secretory response in intestinal epithelium. We suspected that ecto-5′-nucleotidase also played a role in the diarrheal fluid secretion induced by EPEC and initiated this study to determine if it is the case, and, if so, whether inhibitors of ecto-5′-nucleotidase could block fluid secretion triggered by adenine nucleotides and EPEC infection. We used the T84 colon carcinoma cell line as a model because we have used these cells to study EPEC infection, they express ecto-5′-nucleotidase, and they can be studied in the Ussing chamber to measure chloride secretion. In order to facilitate our study we also developed a nonradioactive, non-high-pressure liquid chromatography (non-HPLC) method for measuring ecto-5′-nucleotidase in living T84 cell monolayers.
Materials and methods
Bacterial strains used E. coli strains used included laboratory E. coli strain HB101 (O: rough), commensal strain HS (O9: H4), and classic human EPEC strains E2348/69 (serotype O127: H6), B171-8 (O111: NM), and JCP88 (O119: B14) as described in several publications [8–12]. EPEC mutants included JPN15, an E2348 derivative which has lost the EPEC adherence factor (EAF) plasmid [13], UMD874, the espF mutant derived from E2348, which is deficient in host cell killing [3, 14], and SE1010, with a mutation in sepZ (also called espZ), which is defective in type III secretion [15]. Bacteria were added to yield a multiplicity of infection (MOI) of 100:1.
Materials The following reagents were obtained from Sigma-Aldrich Chemicals: α,β-methylene-ADP, adenosine, adenosine 5′-monophosphate (AMP), tetramisole (also called levamisole), polymyxin B, neomycin, purified phosphatidylinositol-specific phospholipase C (PI-PLC, from Bacillus cereus), and zinc acetate. BIOMOL (Plymouth Meeting, PA, USA) was the source of the BIOMOL GREEN reagent used in the phosphate release assay for nucleotidase activity and of U73122, a PI-PLC inhibitor. U73122 is 1-(6-[17 beta-3-methoxyestra-1,3,5- (10) triene-17-yl] amino/hexyl) 1H-pyrroledione. A cell permeant PI-PLC activator, 3M3-FBS, was from the Calbiochem Division of EMD Biosciences (La Jolla, CA, USA). 3M3-FBS is 2,4,6-trimethyl-N-(m-3-trifluoromethylphenyl)benzenesulfonamide. Phosphate-free DMEM medium was purchased from MP Biomedicals (formerly ICN Biomedicals, Aurora, OH, USA). UNIFILTER plates were from Whatman (Clifton, NJ, USA).
Bacterial culture E. coli strains were grown overnight in Luria-Bertani (LB) broth at 37°C with 300 rpm shaking, then subcultured for 2 h in serum-free DMEM/F12 medium supplemented with 18 mM NaHCO3, 25 mM hydroxyethylpiperazine ethanesulfonic acid (HEPES) buffer, pH 7.4, and 1% D-mannose as previously described [3]. For experiments with bacteria in minimal medium, bacteria were subcultured at a dilution of 1:2,000 into minimal medium (M9 salts plus casamino acids supplemented with 2 mM glucose). For convenience we used M9-CA liquid broth packets (E. coli Fast Media, MBI-Fermentas, Hanover, MD, USA) and added 2 mM glucose before use.
Cell culture T84 colon cancer cells were grown in DMEM/F12 medium supplemented with 7.5% fetal bovine serum (Gibco/Invitrogen, Grand Island, NY, USA), 18 mM NaHCO3, 20 μg/ml vancomycin, and 15 μg/ml gentamicin as previously described [16]. Ussing chamber studies of secretion were performed on T84 cell monolayers grown in Snapwell inserts (Corning Costar, Corning, NY, USA). The Snapwell inserts, which had a 0.4 μm pore size, were coated with 32 μg collagen per well by applying 0.16 ml of 0.2 mg/ml type III collagen (Sigma; dissolved in warm 0.2 M acetic acid) to the Snapwell and allowing it to dry in the tissue culture hood under UV light. T84 cells were seeded onto the Snapwell inserts at ~1.2 × 106 cells per well and allowed to grow to confluency for 7–9 days. At this time the monolayers had transepithelial electrical resistances (TER) of 400–1,000 Ω · cm2.
Assay for ecto-5′-nucleotidase by phosphate release An assay for ecto-5′-nucleotidase activity in living cells was developed based on the ability to detect inorganic phosphate (Pi) released from 5′-AMP. This method has been used to detect activity of protein phosphatases such as PTEN [17] and lipid phosphatases [18] and is based on sensitive detection of low levels of Pi using the BIOMOL GREEN reagent, an enhanced and stabilized formulation of malachite green. To carry out the assay a phosphate-free buffer was used consisting of (in mM): NaCl, 154; KCl, 2: MgCl2, 4; NaHCO3, 18: HEPES, pH 7.4, 25; and glucose, 10. This buffer is referred to as nucleotidase buffer. To measure ecto-5′-nucleotidase activity, the cell monolayer was rinsed once with sterile normal saline, then the medium was replaced with warm nucleotidase buffer. For cells in a 48-well plate, 0.25 ml of nucleotidase buffer were added per well, and the cells were allowed to rewarm to 37°C in the CO2 incubator. During pipetting the multiwell plate was kept warm using a metal heating block set at 37°, and a stopwatch was used to time the AMP addition and to terminate the assay. The procedure used for measuring monolayer activity was slightly different from that used to measure 5′-nucleotidase activity released into the supernatant, as described below.
Cell-bound or monolayer activity To measure cell-bound ecto-5′-nucleotidase activity in cell monolayers, 5′-AMP was added to yield a final concentration of 0.2 mM to quadruplicate wells. Two other wells were left without addition of AMP (the “no AMP blank”). After a 10-min incubation at 37° an aliquot (usually 50 μl) was removed and quickly transferred to a well of a 96-well plate to terminate the reaction.
E. coli-induced release of nucleotidase activity into the supernatant medium For nucleotidase release experiments, the cell monolayer was changed to warm, phosphate-free DMEM, then infected with an E. coli strain for 35 min to allow adherence, then the medium was changed to nucleotidase buffer and the infection was allowed to continue for 2 or 3 h. Note that in this procedure any nucleotidase activity that is released in the first 35 min is discarded and not detected by our method. However, this two-stage procedure with the medium change was necessary because EPEC bacteria did not adhere normally if they first encountered the host cell in nucleotidase buffer. After a period of infection, supernatant medium was collected with a multichannel pipettor and transferred to the wells of a Whatman UNIFILTER plate (a 96-well with 0.45-μm membrane for sterile filtration). Sterile filtrates were prepared by centrifugation with collection of the filtered medium into another 96-well plate placed beneath the UNIFILTER as previously described [19]. Once again, experimental conditions were usually done in groups of six, with two wells not receiving any AMP (no AMP blanks) and four wells receiving 0.2 mM AMP. Again, the usual assay condition was 10 min at 37° before the reaction was stopped by addition of 10 μl of 1 M HCl (“stop solution”).
BIOMOL GREEN detection of phosphate released from AMP Stopped samples in a 96-well plate were brought to 100 or 110 μl volume with water if necessary, then treated with 100 μl of BIOMOL GREEN reagent. A standard curve of inorganic phosphate was prepared and run with every experiment; standards and unknown samples were incubated at room temperature for 20 min to allow a green color to develop, then the 96-well plate was read on a multiwell plate spectrophotometer at 620 nm. Unknown values were calculated from the standard curve using a hyperbolic curve fit using GraphPad Prism software, version 4.0. Results of monolayer activity were expressed as nmol of Pi produced/min per 106 cells. For experiments showing nucleotidase release, the results were often expressed as nmol Pi released/ min per well since the assay was done on a cell-free filtrate and because we often noted some detachment of cells during the longer incubations of 2–3 h needed to observe release.Although we believed we were developing a new method for assay of ecto-5′-nucleotidase by phosphate release, during the course of this work another group reported using a virtually identical method, also based on detection of phosphate released from 5′-AMP [1].
Detection of CD73 by Western immunoblot To prove that the released 5′-nucleotidase activity we measured was of host cell rather than bacterial origin, we performed immunoblots on the supernatants of infected T84 cells with antibodies against CD73. Initial attempts at immunoblotting using a commercially available monoclonal anti-CD73 antibody (Abnova Corp., Taipei, Taiwan) were unsuccessful. Dr. Linda F. Thompson, Oklahoma Medical Research Foundation, kindly sent us mouse monoclonal antibodies against human CD73 which had been generated by Dr. Wolf Gutensohn several years earlier. Of these, the two antibodies that gave the best results were designated CD73.4 and CD73.6 by Dr. Gutensohn; both were of isotype IgG2b and were used at a concentration of 1 μg/ml. After washings, the secondary antibody was goat anti-mouse IgG2b conjugated to peroxidase at a dilution of 1:3,000 (Roche Molecular Biochemicals, Indianapolis, IN, USA). Blots were developed by chemiluminescence as previously described [20].
Ussing chamber studies A Snapwell insert containing a monolayer of T84 cells was placed in the plexiglass “slider” and inserted into the Ussing chamber (Physiologic Instruments, San Diego, CA, USA) at 37°C and continuously short-circuited by a four electrode, automatic voltage-clamp apparatus which measured short-circuit current (Isc) and transepithelial resistance (TER); chamber fluid resistance was automatically subtracted. Transepithelial resistance was determined by passing 10-s 10-mV current pulses through the tissues. Short-circuit current was measured by passing sufficient current through the tissues via Ag/AgCl electrodes to reduce the spontaneous transepithelial potential to zero. The composition of the tissue bathing solution was (in mM): 140 Na+, 124 Cl−, 21 HCO3-, 5.4 K+, 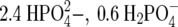 , 1.2 Mg2+, 1.2 Ca2+, and 10 glucose. Raw short-circuit current (Isc) values were converted to μA per cm2 by dividing by the area of the Snapwell monolayer (1.13 cm2). Other details of the Ussing chamber methods were exactly as described [16].
, 1.2 Mg2+, 1.2 Ca2+, and 10 glucose. Raw short-circuit current (Isc) values were converted to μA per cm2 by dividing by the area of the Snapwell monolayer (1.13 cm2). Other details of the Ussing chamber methods were exactly as described [16].
Protein assay Protein assay was by the Coomassie blue dye binding assay method of Bradford, using a Bio-Rad kit [21].
Expression of ecto-5′-nucleotidase RNA by reverse transcription and real-time polymerase chain reaction (PCR) T84 cells grown in 24-well plates were infected with EPEC for 35 min, then the medium was changed to remove unbound bacteria. Three hours after the medium change, ciprofloxacin was added to 25 μg/ml to kill EPEC and the incubation was continued 1 more hour. Old medium was removed, and the cell monolayer was lysed in extraction buffer with 10% β-mercaptoethanol (RNeasy Kits, Qiagen, Valencia, CA, USA). RNA was subjected to reverse transcription using Invitrogen Superscript III reverse transcriptase; 5 μl of purified RNA was used per 50 μl reaction, and gene-specific primers at 0.2 μM were used. Reverse transcription reaction was at 55° for 1 h. Copy DNA from reverse transcription was diluted 100-fold, then analyzed by quantitative real-time PCR using the same oligonucleotide primers. For ecto-5′-nucleotidase the primers used were 5′-TTC CAC CCT GAA GAA GGC CTT TGA-3′ (forward) and 5′-ATA ACT GGG CAC TCG ACA CTT GGT-3′ (reverse). As a normalizing gene we used glyceraldehyde phosphate dehydrogenase (GAPDH) as described by Khan et al. [22] except that we redesigned longer primers which were 5′-TCG ACA GTC AGC CGC ATC TTC TTT-3′ and 5′-ACC AAA TCC GTT GAC TCC GACC CTT-3′. PCR was carried out using a MyiQ Single-Color qRT-PCR machine from Bio-Rad (Hercules, CA, USA) using SYBR Green as the dye to monitor the amplification. Relative expression was calculated by the ΔΔCt (“Livak”) method as described [23], where Ct is the number of cycles to threshold. SYBR Green PCR reagents were from Bio-Rad and to reduce the cost, the PCR reaction volume was reduced to 25 μl. PCR was performed using a two-step protocol with an annealing temperature of 58.7° and denaturation at 95° for 30 s each (i.e, no extension step) for 35 cycles. Thermal melt curve analysis was performed at the end of the PCR amplification and showed a single sharp peak for the genes analyzed.
Data analysis and presentation All error bars shown in graphs and error values reported in the text are standard deviations. Significance was tested by one-way analysis of variance (ANOVA) with the Tukey-Kramer post-test for multiple comparisons, using InStat software for the Macintosh from GraphPad software (San Diego, CA, USA). Graphs were prepared using Prism 4.0 software, also from GraphPad. Asterisks shown on graphs indicate a p value of < 0.05.
Results
Ecto-5′-nucleotidase activity was readily measurable in T84 cell monolayers using the phosphate release assay method developed in this study. Figure 1 shows the characteristics of the ectoenzyme in this system. Figure 1a shows a curve of activity vs substrate concentration, demonstrating that the enzyme shows Michaelis-Menten kinetics. In four experiments similar to that shown in Fig. 1a, the KM was 229 ± 30 μM and the Vmax was 2.7 ± 1.8 nmol/min per 106 cells (mean ± SD of 4 experiments). While the KM was very consistent over time and from experiment to experiment, the Vmax was more variable as we compared experiments separated by months or years. We noted that highly T84 passaged cells (passage number of 70 or greater) showed lower levels of ecto-5′-nucleotidase activity than less passaged cells from the same source (experiments not shown). The KM we observed in intact T84 cells was higher than that reported for the purified enzyme derived from a colon carcinoma cell line, BCS-TC2 cells [24]. Figure 1b shows the raw data of Pi generated over time in the presence and absence of added AMP substrate and demonstrates that the assay was linear for 10 min; therefore, 10 min was chosen as the standard assay duration.
Fig. 1.
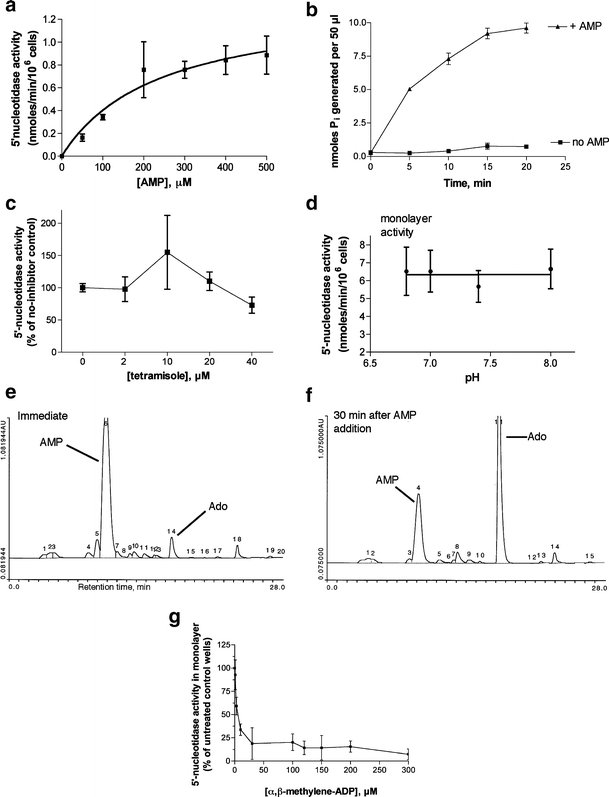
Characterization of ecto-5′-nucleotidase activity in T84 cell monolayers. 5′-Nucleotidase activity was measured by the release of inorganic phosphate (Pi) from AMP in phosphate-free buffer using a colorimetric method as described in “Materials and methods.” For a, c, and d, the spectrophotometric reading in the absence of AMP (“no AMP blank”) was subtracted from the total reading. In b the “no AMP blank” was not subtracted and is shown in b as labeled. a Michaelis-Menten curve of enzyme velocity vs concentration of AMP substrate. b Time course of Pi release vs time. c Lack of inhibition by tetramisole, an inhibitor of alkaline phosphatase. d pH dependence of 5′-nucleotidase activity. e, f HPLC traces showing conversion of AMP (e) to adenosine (Ado, f) in T84 cell monolayers. g Sensitivity of the AMP-hydrolyzing activity to the inhibitor α,β-methylene-ADP, confirming it as ecto-5′-nucleotidase
Alkaline phosphatase is another abundant intestinal ectoenzyme which can hydrolyze 5′-AMP [25]. Alkaline phosphatase can be distinguished from 5′-nucleotidase by its alkaline pH optimum and sensitivity to inhibition by tetramisole. Figure 1c shows that the ectonucleotidase we measured on T84 cells was not inhibited by tetramisole (also known as levamisole). Furthermore, its activity did not increase with increasing pH (Fig. 1d) providing evidence that the activity being measured was not alkaline phosphatase. In addition, we used HPLC to confirm that 5′-AMP was being converted to adenosine in the supernatant medium of T84 cells (Fig. 1e and f). Morover, the 5′-nucleotidase activity we measured by phosphate release was sensitive to inhibition by α,β-methylene-ADP, a known inhibitor of ecto-5′-nucleotidase (Fig. 1g and see also Fig. 4). Lastly, after we initiated this study, other researchers also reported using phosphate release from 5′-AMP as their assay for ecto-5′-nucleotidase [1]. We have found that the increased throughput of the phosphate release assay increases its usefulness compared to the much slower HPLC methods for ecto-5′-nucleotidase.
Fig. 4.
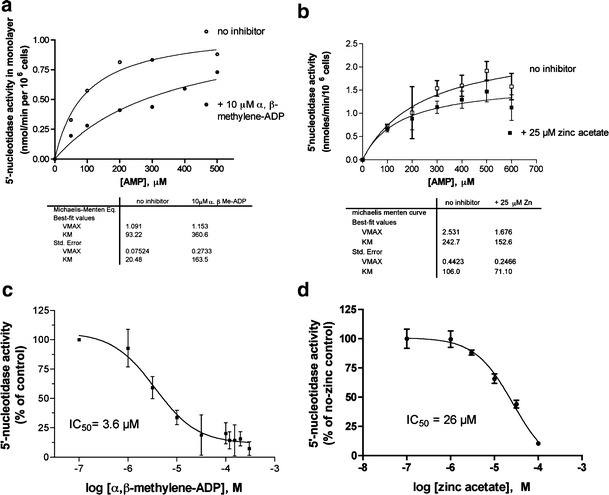
Effect of the 5′-nucleotidase inhibitors on enzyme activity in T84 cell monolayers. a, b Michaelis-Menten curves of enzyme activity vs substrate concentration in the absence (open symbols) and presence (closed symbols) of the inhibitor shown. Enzyme parameters were calculated by nonlinear curve fitting using GraphPad Prism and the fitted parameters of Vmax and KM are shown below the graphs. c, d Dose-response curves for inhibition of ecto-5′-nucleotidase activity by α,β-methylene-ADP (c) and zinc acetate (d) using a 0.2 mM concentration of AMP substrate. In all parts of Fig. 4 the inhibitor was added before the AMP, and in c and d the curves were fitted to a sigmoidal inhibitory dose-response curve and the inhibitory concentration 50% (IC50) value derived from the curve fit is shown
Figure 2 shows the effect of EPEC infection on ecto-5′-nucleotidase activity released into the supernatant medium during an experimental EPEC infection of cultured T84 cells. Wild-type EPEC strains E2348/69 and JCP88 triggered a release of 5′-nucleotidase into the supernatant medium while nonpathogenic E. coli strains HB101 and HS did not. The supernatant medium was subjected to sterile filtration prior to assay to remove bacterial cells as well as any detached host cells, so that the activity reflects a soluble and not a cell-bound form. In experiments where we determined both the supernatant release and the monolayer activity in the same experiment, supernatant release by wild-type EPEC was 30–40% of monolayer activity 3 h after the medium change (Fig. 2a and data not shown). To determine if the 5′-nucleotidase could possibly be derived from the E. coli bacteria rather than from the host we also assayed sterile filtrates of E. coli for 5′-nucleotidase activity and it was virtually absent from the culture filtrates of all E. coli strains tested, including EPEC (Fig. 2b, light gray bars). Even after treatment with a concentration of polymyxin B sufficient to cause > 99% bacterial cell lysis the amount of bacterial nucleotidase-like activity was far less than the amounts we observed in the supernatant in Fig. 2a. In addition, our protocol included a medium change step in order to remove unbound bacteria. Therefore we concluded that the 5′-nucleotidase activity being released in Fig. 2a was of host cell origin.
Fig. 2.
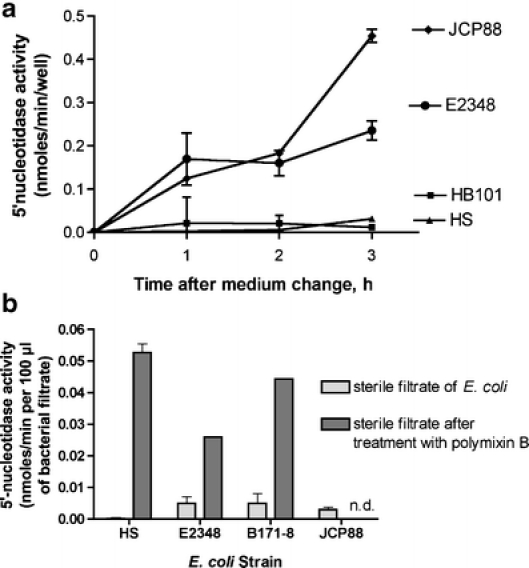
Effect of E. coli infection on 5′-nucleotidase activity in the supernatant medium. a Nonpathogenic E. coli strains (HB101 and HS) or EPEC strains E2348/69 and JCP88 were subcultured in DMEM medium for 2 h, then used to infect T84 cells at a multiplicity of infection of 100:1 in phosphate-free DMEM. After 35 min to allow adherence, the medium was changed to nucleotidase buffer and aliquots were collected at various times after the medium change, filtered through a 0.45-μm filter to remove bacterial cells, then assayed for 5′-nucleotidase activity. Since the activity was measured in the cell-free sterile filtrates the activity is expressed as nmol/min per well. bE. coli suspensions were subjected to sterile filtration without treatment (light gray bars) or following treatment with 50 μg/ml polymyxin B, a lytic antibiotic (dark gray bars). Then the nucleotidase activity of a 100-μl aliquot was measured. The 100 μl volume was chosen because this was the volume of inoculum needed to achieve an MOI of 100:1 for the slowest growing strain (E2348/69) in a typical 48-well plate of T84 cells
To determine if this hypothesis was correct and to try to determine the mechanism of EPEC-induced 5′-nucleotidase release, we compared the 5′-nucleotidase release triggered by wild-type EPEC strain E2348/69 with the plasmid-cured derivative, JPN15, and its espF mutant, UMD874 (Fig. 3a). The espF mutant is defective in host cell killing and in inducing damage to monolayer integrity [14, 26]. Interestingly, the espF mutant was not attenuated in its ability to trigger 5′-nucleotidase release, but instead it consistently outperformed the wild-type strain in this regard. In six experiments 5′-nucleotidase release induced by the espF mutant exceeded that of the wild-type E2348/69 by an average of 2.3 ± 0.14 fold (p = 0.03 by paired t-test), suggesting that host cell death or damage is not the mechanism of EPEC-induced nucleotidase release.
Fig. 3.
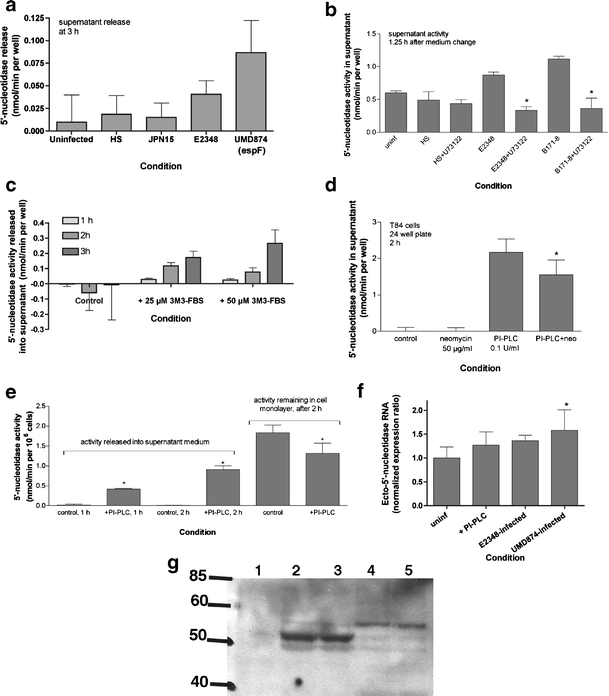
a Effect of EPEC mutants and PI-PLC inhibitors and activators on induced 5′-nucleotidase release into supernatant. Release of 5′-nucleotidase activity into supernatant medium was measured as described in “Materials and methods” and in the legend to Fig. 2. Once again, activity in the cell-free sterile filtrates is expressed as nmol/min per well. However, for purposes of comparison 48-well plates contain ~0.25 × 106 T84 cells per well at confluency. b U73122, an inhibitor of PI-PLC, was used at a concentration of 2 μm and was re-added after the medium change; *significantly decreased compared to the EPEC strain alone, p < 0.05. cm-3M3-FBS, a cell-permeant sulfonamide activator of PI-PLC, was added at the concentrations and for the times indicated. d Inhibitory effect of neomycin on PI-PLC-induced 5′-nucleotidase release; this experiment was performed on cells grown in a 24-well plate, with ~0.8 × 106 cells per well; *significantly decreased compared to PI-PLC alone. e PI-PLC was again added to a final concentration of 0.1 U/ml for the times shown; *significantly different from the corresponding control. This figure is a composite of experiments that were separated in time and with cells of different passage number; therefore the absolute amount of activity varies among the figure parts. f Effect of EPEC infection on expression of RNA encoding ecto-5′-nucleotidase in T84 cells, by reverse transcription and real-time PCR. T84 cells were infected for 35 min, then the supernatant medium was changed to remove unattached bacteria, then the T84 cell monolayer harvested for RNA extraction 4 h after the medium change. Reverse transcription and PCR conditions were as described in “Materials and methods.” Expression of ecto-5′-nucleotidase was normalized to that of glyceraldehyde 3-phosphate dehydrogenase (GAPDH). f Normalized expression of a single representative experiment (mean ± SD of 6 PCR wells). g Immunoblot analysis of the proteins released into the supernatant after 2 h of infection with EPEC or an EPEC mutant (SE1010, sepZ/espZ), using monoclonal antibody against CD73. Lane 1, supernatant from uninfected, control T84 cells. Lanes 2 and 3, supernatant from cells infected with wild-type E2348/69. Lanes 4 and 5, supernatants from cells infected with the sepZ mutant. GPI-linked CD73 has an apparent molecular size of 72 kDa (not seen in this blot) and the GPI-cleaved soluble portion runs at 51 kDa (heavy band in lanes 2 and 3)
Ecto-5′-nucleotidase is tethered to the extracellular surface of the cell by a glycosylphosphatidylinositol (GPI) lipid anchor [24, 27]. GPI lipid anchors are cleaved by phosphatidylinositol-specific phospholipases C (PI-PLC), and EPEC has been shown to activate at least one type of PI-PLC in the host cell, PI-PLC-γ [28]. Therefore we considered whether PI-PLC activation by EPEC could be a nonlethal mechanism by which EPEC triggered 5′-nucleotidase release from host intestinal cells.
Figure 3b shows an experiment in which we tested whether a PI-PLC inhibitor could block EPEC-induced nucleotidase release. Indeed, 2 μM U73122 did reverse the nucleotidase release triggered by wild-type EPEC strains. Conversely, a newly described cell-permeable activator of PI-PLC, called m-3M3-FBS [29], triggered nucleotidase release from uninfected cells in a time- and concentration-dependent manner (Fig. 3c). Experiments to test if m-3M3-FBS would enhance EPEC-induced release were inconsistent, with some experiments showing a mild enhancement or additive effect and others showing no enhancement or mild antagonism (results not shown).
In addition we also tested whether purified PI-PLC added to the apical surface of the T84 cell monolayer induced 5′-nucleotidase release and it did (Fig. 3d and e). Figure 3d also shows that 50 μg/ml neomycin could partially block 5′-nucleotidase release by purified PI-PLC. In addition to its antimicrobial effects, neomycin is an inhibitor of several types of PLC enzymes including PI-PLC. Figure 3e shows that after a 2-h treatment with PI-PLC not only was 5′-nucleotidase activity increased in the supernatant, but that monolayer activity was depleted as well (right portion of Fig. 3e). In experiments similar to Fig. 3e done with EPEC infection, we could not detect any decrease in monolayer activity 2–3 h after infection, despite the liberation of 5′-nucleotidase activity into the supernatant. We considered the possibility that increased host cell synthesis of ecto-5′-nucleotidase might be occurring in response to the cleavage of the enzyme from the cell surface. We analyzed the abundance of RNA encoding ecto-5′-nucleotidase by reverse transcription and real-time quantitative PCR in response to EPEC infection. Figure 3f and Table 1 show that infection with wild-type EPEC and the EPEC espF mutant (UMD874) increased the abundance of ecto-5′-nucleotidase RNA compared to uninfected control cells. The increase in expression of ecto-5′-nucleotidase RNA by wild-type EPEC was 33% above control. While this is not a large increase, it does explain the maintenance of normal levels of ecto-5′-nucleotidase on the surface of EPEC-infected cells, because under the most optimum conditions the release of enzyme activity into the supernatant is about 30–40% of monolayer activity (e.g., Fig. 3e).
Table 1.
Effect of EPEC infection on expression of RNA encoding ecto-5′-nucleotidase in T84 cells, by reverse transcription and real-time PCR
| Condition | Normalized expression ratioa |
|---|---|
| Uninfected control | 1.0, by definition |
| +1 U/ml PI-PLC | 1.23 ± 0.26 |
| HS-infectedb | 0.66 ± 0.22 |
| E2348/69-infectedb | 1.33 ± 0.21c |
| UMD874 (espF)-infectedb | 1.40 ± 0.19c |
aCells were infected for 35 min, then the supernatant medium was changed to remove unattached bacteria, then the T84 cell monolayer harvested for RNA extraction 4 h after the medium change. Expression of ecto-5′-nucleotidase was normalized to that of glyceraldehyde 3-phosphate dehydrogenase (GAPDH). Data shown are the means ± SD of 5 similar experiments
bBacteria were added with the intent of achieving a multiplicity of infection (MOI) of 100:1; actual MOIs in the 5 experiments ranged from 71 to 144
cSignificantly increased compared to uninfected control by paired t-test (p = 0.04)
The 5′-nucleotidase released into the supernatant appeared to be of host cell origin, because it could be triggered by chemical stimuli in the absence of EPEC infection (Fig. 3c–e). The host origin was also proven using a mouse monoclonal antibody against human CD73. Wild-type EPEC infection of T84 cells triggered the release into the supernatant of a 51 kDa protein recognized by a mAb against CD73 (Fig. 3g, lanes 2 and 3). In contrast, the EPEC sepZ/espZ mutant, which is defective in type III secretion, triggered the appearance of a fainter band which was of ~55 kDa in size. The origin of this faint band is unknown, but it is possible it is generated by proteolysis rather than PI-PLC activation. For example, E2348/69 expresses EspC, an autotransporter protease whose export is independent of type III secretion.
Figure 4 shows that 5′-nucleotidase activity in living T84 cells was susceptible to the same inhibitors as the purified enzyme studied by others. Figure 4a shows that α,β-methylene-ADP inhibited ecto-5′-nucleotidase activity and that its inhibition has the characteristics of a competitive inhibitor. In the presence of α,β-methylene-ADP the enzyme shows an apparent increase in KM but no increase in the Vmax, and the inhibition can be overcome at high concentrations of AMP substrate. In contrast in Fig. 4b one observes that zinc acetate behaves as a noncompetitive inhibitor of the enzyme, with a decrease in the Vmax but no increase in the KM (in fact there is a small, paradoxical decrease in KM in the presence of zinc). Figures 4c and d show the inhibitory dose-response curves at a single 0.2 mM concentration of AMP substrate. For both inhibitors, when concentration is expressed on a logarithmic scale, the curves are well fit by a sigmoidal, single-site model, allowing determination of the inhibitory concentration 50% (IC50) as shown in Figs. 4c and d. In addition to α,β-methylene-ADP and zinc acetate, ecto-5′-nucleotidase activity was also inhibited by 3–10 mM sodium fluoride as reported for the purified enzyme (data not shown). 5′-Nucleotidase activity released into the supernatant was also sensitive to inhibition by α,β-methylene-ADP and zinc acetate with similar potencies (data not shown).
In Fig. 4 the inhibitor was always added to the cell monolayer prior to the AMP substrate. Using the Ussing chamber apparatus to measure chloride secretion, we realized we could monitor the effects of inhibition of 5′-nucleotidase activity dynamically and assess the ability of inhibitors to reverse a secretory response already underway. In preliminary experiments we found that to reverse an ongoing secretory stimulus we had to add inhibitors at concentrations considerably above their IC50 values. The requirement for addition of inhibitors well above the IC50 is due to the fact that adenosine is a very potent secretagogue in intestinal tissues and a very high degree of inhibition of ecto-5′-nucleotidase is needed to reduce the production of adenosine below the secretory threshold.
Figure 5a shows that secretion triggered by 10 μM AMP is reversed by theophylline, an adenosine receptor antagonist. Figure 5a emphasizes the fact that 5′-AMP has no secretory ability unless it is converted into adenosine. Adenosine then acts via apical adenosine A2b receptors to trigger chloride secretion toward the apical side of the monolayer. Figure 5b shows that chloride secretion triggered by 1 μM AMP is partially antagonized by 2 mM zinc acetate even when the inhibitor is added after secretion has started. Addition of 1 μM adenosine restored the short-circuit current (Isc) to its previous trend, showing that zinc does not block adenosine receptors or “poison” the secretory machinery. Under the same experimental conditions, 200 μM α,β-methylene-ADP produced a much larger degree of reversal of Isc than did zinc (Fig. 5c). As with zinc, α,β-methylene-ADP did not prevent a subsequent, normal Isc response to addition of adenosine (Fig. 5c, right-hand portion), showing that it is acting as a 5′-nucleotidase inhibitor and not an adenosine antagonist. The efficacy of inhibition by α,β-methylene-ADP in this paradigm, however, was strongly dependent on the concentration of AMP used to trigger secretion. At higher concentrations of AMP (e.g., 20 μM), the degree of inhibition by α,β-methylene-ADP was markedly less (Fig. 5d) than when 1 μM AMP was used as the stimulus. In contrast, zinc acetate produced the same, modest inhibition of Isc regardless of the concentration of AMP used. Figure 5e summarizes the effectiveness of the two inhibitors, zinc and α,β-methylene-ADP, when tested against various different concentrations of AMP as the initial stimulus. Figure 5e shows that the effectiveness of α,β-methylene-ADP declines significantly with increasing concentrations of AMP used, whereas zinc’s inhibitory effects are fairly constant over a range of AMP concentrations. The results of Fig. 5 (b–e) vividly illustrate, at the level of of an electrophysiologic response, the difference between a competitive and a noncompetitive mode of action of an enzyme inhibitor (compare with Fig. 4a and b).
Fig. 5.
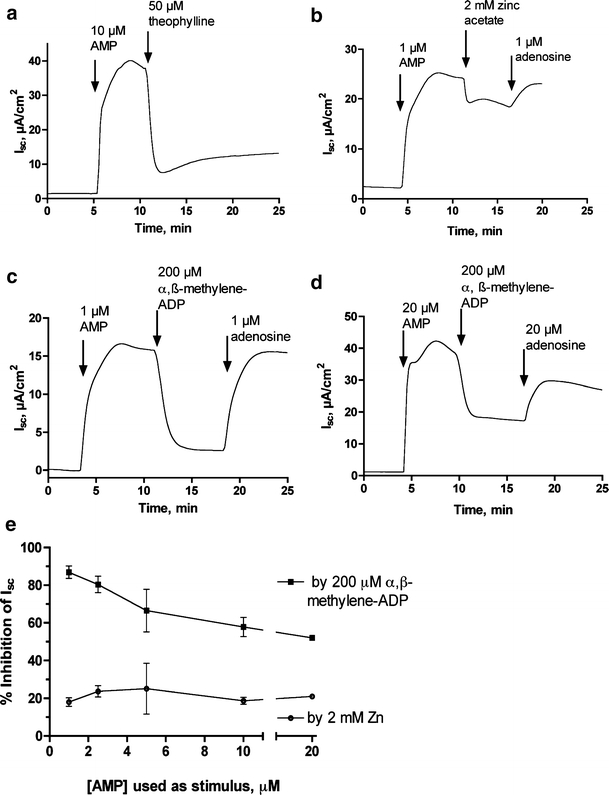
Effect of 5′-nucleotidase inhibitors on the chloride secretory response to AMP in T84 cells in the Ussing chamber. Short-circuit current (Isc) was measured in T84 cells grown in Snapwell inserts as described in “Materials and methods”; in this cell line and electrode configuration, a positive Isc (upward deflection on the graphs) represents chloride secretion in the basolateral to apical direction. All of the additions of agonists and inhibitors in these parts of the figure was on the apical (mucosal) side of the monolayers only. a Reversal of AMP-induced secretory response by theophylline, an adenosine receptor antagonist. b Partial inhibition of AMP-induced secretion by 2 mM zinc acetate, with restoration of the tracing to its previous trend by adenosine, demonstrating that adenosine receptors are not blocked by zinc. c Substantial reversal of Isc by α,β-methylene-ADP when a low concentration of AMP (1 μM) is used as the stimulus. Note that adenosine fully restores secretion to its previous level. d Lesser degree of reversal of Isc by α,β-methylene-ADP when a higher concentration of AMP (20 μM) is used as stimulus. e Summary of many experiments in which fixed concentrations of zinc and α,β-methylene-ADP were tested for inhibitory efficacy against varying concentrations of AMP as test stimulus. In Fig. 5e the slope of the α,β-methylene-ADP curve was −1.73 ± 0.5 by linear regression (95% confidence limits of the slope value −3.27 to −0.2, significantly different from zero, p < 0.04). In contrast the slope of the zinc curve was −0.04 ± 0.23 and did not differ significantly from zero
Since the discovery of EPEC-induced ATP release we have hypothesized that EPEC bacteria may gain a nutritional advantage by this inflicting kind of damage on the host cell [3]. The lumen of the gastrointestinal tract is a purine-limited environment, as demonstrated by in vivo gene expression screens such as in vivo expression technology (IVET) [30] and signature-tagged mutagenesis (STM) [31]. Nucleotides and nucleosides enhance growth of EPEC strains in vitro, and these growth-promoting effects are most obvious when adenosine is added to minimal medium (Fig. 6). Compared to minimal medium, addition of 30 μM adenosine markedly accelerated growth of EPEC strains such as E2348/69 (Fig. 6). Addition of 5′-AMP also increased EPEC growth, although not as much as adenosine. Alpha-β-methylene-ADP alone has no effect, stimulatory or inhibitory, on EPEC growth, but it did completely reverse the growth-promoting effects of 5′-AMP (Fig. 6a and b, right-hand bars). Alpha-β-methylene-ADP did not block the enhanced growth seen with adenosine (not shown). Similarly, 100 μM zinc acetate did not affect growth in minimal medium (Fig. 6c, left-hand bars) or the enhanced growth seen with adenosine (Fig. 6c, two middle bars), but zinc did abolish the enhancement of growth by AMP. E. coli 5′-nucleotidase is susceptible to inhibition by α,β-methylene-ADP and zinc; in fact, the crystal structure of the E. coli enzyme was solved with α,β-methylene-ADP bound in the catalytic pocket [32]. Bacterial utilization of extracellular nucleotides is usually by dephosphorylation followed by uptake of the nucleoside (in this case adenosine) and inorganic phosphate [33]. Although we began studying α,β-methylene-ADP and zinc as inhibitors of the host cell enzyme, CD73, a side effect (or side benefit) of these compounds may be inhibition of bacterial 5′-nucleotidase activity as well, an effect that is able to block the nutritional benefits that E. coli bacteria derive from extracellular AMP (Fig. 6).
Fig. 6.
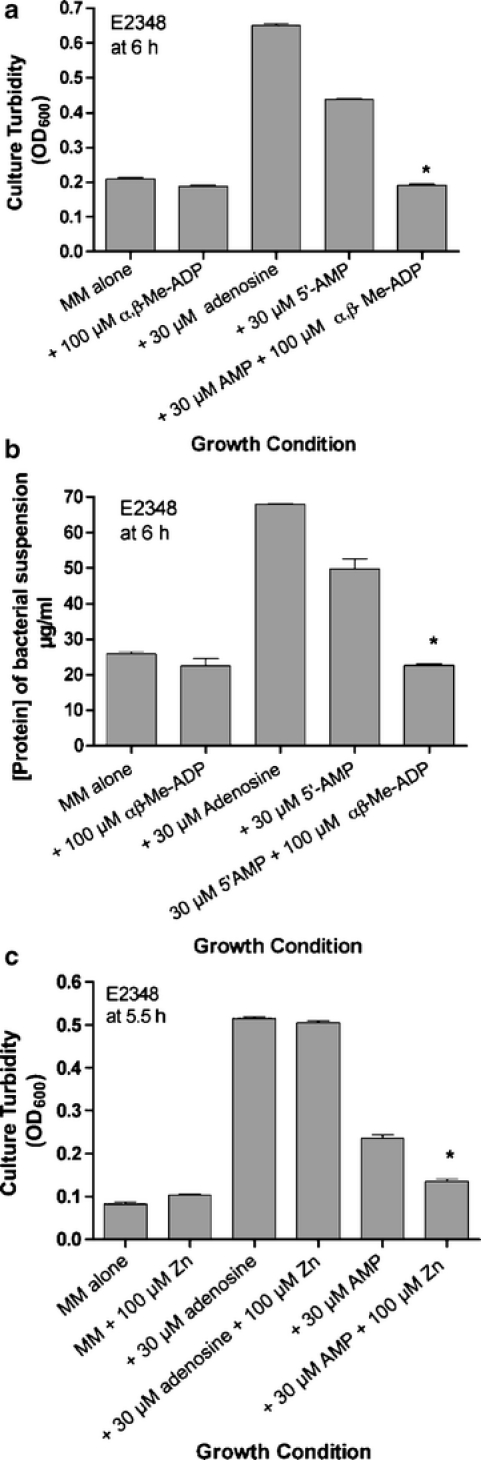
Effect of 5′-nucleotidase inhibitors on growth of EPEC strain E2348/69 in minimal medium supplemented with 30 μM adenosine or AMP. Minimal medium here refers to M9 salts, 2% casamino acids, and 2 mM glucose. Growth was at 37°C with 300 rpm shaking after a 1: 2,000 dilution from an overnight culture in LB medium. a, c Culture turbidity by spectrophotometric absorbance at 600 nm. b Bacterial protein content. a, b Growth-enhancing effects of AMP are blocked by α,β-methylene-ADP. c Growth-enhancing effects of AMP are blocked by zinc acetate; *significantly decreased compared to AMP alone (p < 0.05)
Discussion
We became interested in ecto-5′-nucleotidase (CD73) because of its likely role in the generation of adenosine from adenine nucleotides released during EPEC infection. This host enzyme has not received a great deal of scrutiny in the context of microbial pathogenesis, and when it was studied its role was felt to be in the setting of a polymorphonuclear neutrophil (PMN) response, since activated PMNs release 5′-AMP during migration into an infected area [4, 34]. EPEC behaves clinically as a noninvasive pathogen and does not trigger an influx of fecal leukocytes, but nevertheless triggers ATP release from the host. Therefore our results suggest that ecto-5′-nucleotidase plays a role in the response to a broader range of pathogens, including noninvasive enteric pathogens, than was previously thought.
One important finding of our study is that EPEC infection triggers a release of ecto-5′-nucleotidase from a cell-bound to a soluble, free form (Figs. 2 and 3). The significance of this finding is not clear at present but there are several possibilities to be considered. First, studies by other investigators have shown that cleavage of ecto-5′-nucleotidase from its GPI lipid anchor increases its activity significantly via an increase in the Vmax [27]. This could result in an increase in the activity of 5′-nucleotidase present in an EPEC-infected portion of the intestine, which could be an advantage to EPEC (see Fig. 6 and additional discussion below). The observed increase in transcription of ecto-5′-nucleotidase in response to EPEC infection (Fig. 3f and Table 1), if accompanied by a similar increase in protein synthesis and surface expression of the enzyme, would replace ecto-5′-nucleotidase lost due to cleavage of the lipid anchor and also contribute to an increase in total activity. Third, release of nucleotidase from its lipid anchor would change its distribution from a mucosal location to a soluble form in the intestinal lumen, where it could possibly be more accessible to substrate. This could be important in the human or animal EPEC infection because expression of ecto-5′-nucleotidase is highest in the intestinal crypts [35], whereas EPEC adheres to villus tips as well as crypts. Last, after many days of infection the mucosa could be stripped of ecto-5′-nucleotidase activity, leading to malabsorption of nucleotides in the diet. Indeed, malabsorption of electrolytes has been demonstrated after 7 days of infection in a rabbit model of EPEC infection [36]. In addition to ecto-5′-nucleotidase, several other important digestive enzymes have GPI lipid anchors, including alkaline phosphatase and aminopeptidase N (CD13) and these also could be liberated by EPEC-induced activation of PI-PLC. Another GPI-linked protein of interest in the intestinal tract is decay-accelerating factor (CD55), the cellular receptor for adhesion by diffuse adherent E. coli [37, 38] and which is also involved in internalization of Salmonella into vacuoles.
Several virulent pathogens have been found to secrete their own microbial ATPases or nucleotidases into the extracellular medium, including Entamoeba histolytica [39], Trichinella spiralis [40], and Vibrio cholerae [41]. Researchers have speculated that secreted microbial nucleotidases may serve in protection against killing by host immune cells, acquisition of nutrients from the host, or dampening of host immune responses. EPEC does not appear to secrete nucleotidase activity under any conditions we have encountered, but may achieve the same goal by triggering the release of 5′-nucleotidase from the host cell.
Ecto-5′-nucleotidase acts on nucleoside monophosphates, with 5′-AMP as the preferred substrate, and is inactive toward ATP. Therefore, the generation of AMP from ATP requires another enzyme or enzymes. CD39, or ectonucleoside triphosphate diphosphohydrolase, is the major enzyme responsible for this conversion in the cardiovascular system and probably in the gastrointestinal tract as well [42, 43]. CD39 is also deserving of further study in the context of microbial pathogenesis.
One surprising finding of our study was that the espF EPEC mutant was not attenuated but in fact better than the wild-type EPEC in release of ecto-5′-nucleotidase (Fig. 3a). Although this mutant is defective in host cell killing, its phenotype has been instructive in many ways. For example, the ability of the espF mutant to trigger a robust ATP release led to us the discovery of a new pathway for ATP release from the host cell; this “second pathway” is not dependent on cell death but is dependent on the cystic fibrosis transmembrane regulator (CFTR) for ATP efflux [19]. The competence of the espF mutant in release of ATP and of ecto-5′-nucleotidase from the host cell may help explain why this mutant is only modestly attenuated compared to wild-type in some animal models of EPEC infection, such as Citrobacter rodentium in mice [44]. On the other hand, the sepZ/espZ mutant is attenuated in its ability to trigger ecto-5′-nucleotidase release (Fig. 3f), showing that type III secretion is necessary for PI-PLC activation and release of the enzyme from its lipid anchor.
The role of ecto-5′-nucleotidase in triggering EPEC-induced fluid secretion is highlighted in the Ussing chamber studies shown in Fig. 5. Even after AMP has induced a chloride secretory response, addition of the nucleotidase inhibitor α,β-methylene-ADP is able to reverse the short-circuit current in a manner similar to that of an adenosine receptor antagonist, theophylline (compare Fig. 5a and c). Tracings such as those in Fig. 5 seem to indicate that to sustain a secretory response there must be on ongoing production of adenosine from AMP. In other words, there seems to be a “sink” for depletion of adenosine from the extracellular medium, either by cellular reuptake or by adenosine deaminase. The rapid turnover of adenosine implied in these experiments suggested that inhibitors of ecto-5′-nucleotidase might be able to block EPEC-induced fluid secretion in animal models of infection.
In discussions of zinc on ecto-5′-nucleotidase it is worthwhile to note that zinc supplements have been shown to reduce the duration and severity of watery diarrhea in children in developing countries around the world [45–48]. In most of these trials the microbial etiology of the diarrhea was not carefully investigated, so it is unknown if zinc is particularly efficacious in diarrhea due to certain pathogens, such as EPEC, or if it beneficial “across the board” for multiple agents. Likewise, the mechanism by which zinc exerts its beneficial effects has not been carefully studied in the field trials. In earlier reports it was assumed that zinc was acting by correcting a zinc deficiency, but this assumption has been called into question by the finding that zinc supplements have a beneficial effect on diarrhea even in children with normal serum zinc levels at the outset [49]. Kelleher et al. showed that zinc provided some added benefit beyond a probiotic (Lactobacillus rhamnosus) in rhesus monkeys infected with the human EPEC strain E2348/69; the monkeys studied were not zinc deficient [50]. Inhibition of ecto-5′-nucleotidase or K+ channels [51] by zinc could slow diarrhea but this should be considered a pharmacological effect of the mineral rather than repletion of a deficiency. Inhibition of 5′-nucleotidase also blocks the growth-enhancing effects of AMP on EPEC bacteria (Fig. 6); since this is a direct effect on bacterial cells this action of zinc would not depend on the nutritional status of the host. The effects of zinc on EPEC infection and on diarrheal diseases in non-zinc-deficient children deserve further study.
Acknowledgements
We thankfully acknowledge the support of Research for Health in Erie County (for beginning stages of this work) and of NIAID, National Institutes of Health, via grants R21 AI 66055 and RO1 AI 50652 (to J.K.C.), which allowed completion of this project. We thank Dr. Michael E. Duffey, Dept. of Physiology and Biophysics, Univ. at Buffalo, for teaching us the Ussing chamber methods used in this study. We thank Drs. Linda F. Thompson and Wolf Gutensohn for providing the valuable monoclonal Ab used for immunodetection of CD73.
References
- 1.Koszalka P, Ozuyaman B, Huo Y, Zernecke A, Flogel U, Braun N, Buchheiser A et al (2004) Targeted disruption of cd73/ecto-5′-nucleotidase alters thromboregulation and augments vascular inflammatory response. Circ Res 95(8):814–821 [DOI] [PubMed]
- 2.Olsson RA (2004) Cardiovascular ecto-5′-nucleotidase: an end to 40 years in the wilderness? Circ Res 95(8):752–753 [DOI] [PubMed]
- 3.Crane J, Olson R, Jones H, Duffey M (2002) Release of ATP during host cell killing by enteropathogenic E. coli and its role as a secretory mediator. Am J Physiol Gastrointest Liver Physiol 282:G74–G86 [DOI] [PubMed]
- 4.Nash S, Parkos C, Nusrat A, Delp C, Madara J (1991) In vitro model of intestinal crypt abscess: a novel neutrophil-derived secretagogue activity. J Clin Invest 87:1474–1477 [DOI] [PMC free article] [PubMed]
- 5.Nusrat A, Parkos CA, Liang TW, Carnes DK, Madara JL (1997) Neutrophil migration across model intestinal epithelia: monolayer disruption and subsequent events in epithelial repair. Gastroenterology 113(5):1489–1500 [DOI] [PubMed]
- 6.Strohmeier GR, Lencer WI, Patapoff TW, Thompson LF, Carlson SL, Moe SJ, Carnes DK, Mrsny RJ, Madara JL (1997) Surface expression, polarization, and functional significance of CD73 in human intestinal epithelia. J Clin Invest 99(11):2588–2601 [DOI] [PMC free article] [PubMed]
- 7.Fujii Y, Nomuar T, Yokoyama R, Shinoda S, Okamoto K (2003) Studies on the mechanism of action of the aerolysin-like hemolysin of Aeromonas sobria in stimulating T84 cells to produce cyclic AMP. Infect Immun 71:1557–1560 [DOI] [PMC free article] [PubMed]
- 8.Levine M, Nalin D, Hornick R, Bergquist E, Waterman D, Young C, Sotman S, Rowe B (1978) Escherichia coli strains that cause diarrhoea but do not produce heat-labile or heat-stable enterotoxins and are non-invasive. Lancet 1:1119–1122 [DOI] [PubMed]
- 9.Bieber D, Ramer S, Wu C-Y, Murray W, Tobe T, Fernandez R, Schoolnik G (1998) Type IV pili, transient bacterial aggregates, and virulence of enteropathogenic Escherichia coli. Science 280:2114–2118 [DOI] [PubMed]
- 10.Elliott SJ, Wainwright LA, McDaniel TK, Jarvis KG, Deng Y-K, Lai L-C, McNamara BP, Donnenberg MS, Kaper JB (1998) The complete sequence of the locus for enterocyte effacement (LEE) from enteropathogenic E. coli E2348/69. Mol Microbiol 28:1–4 [DOI] [PubMed]
- 11.Rothbaum R, McAdams A, Giannella R, Partin J (1982) A clinicopathologic study of enterocyte-adherent Escherichia coli: a cause of protracted diarrhea in infants. Gastroenterology 83:441–454 [PubMed]
- 12.Rothbaum R, Partin J, Saalfield K, McAdams A (1983) An ultrastructural study of enteropathogenic Escherichia coli infection in human infants. Ultrastruct Pathol 4:291–304 [DOI] [PubMed]
- 13.Levine M, Nataro J, Karch H, Baldini M, Kaper J, Black R, Clements M, O’Brien A (1985) The diarrheal response of humans to some classic serotypes of enteropathogenic Escherichia coli is dependent on a plasmid encoding an enteroadhesiveness factor. J Infect Dis 152:550–559 [DOI] [PubMed]
- 14.Crane J, McNamara B, Donnenberg M (2001) Role of EspF in host cell death due to enteropathogenic Escherichia coli. Cell Microbiol 3:197–211 [DOI] [PubMed]
- 15.Devinney R, Nisan I, Ruschkowski S, Rosenshine I, Finlay BB (2001) Tir tyrosine phosphorylation and pedestal formation are delayed in enteropathogenic Escherichia coli sepZ:TnphoA mutant 30-5-1(3). Infect Immun 69(1):559–563 [DOI] [PMC free article] [PubMed]
- 16.Crane J, Choudhari S, Naeher T, Duffey M (2006) Mutual enhancement of virulence by enterotoxigenic and enteropathogenic Escherichia coli. Infect Immun 74:1505–1515 [DOI] [PMC free article] [PubMed]
- 17.Xu J, Yeon JE, Chang H, Tison G, Chen GJ, Wands J, de la Monte S (2003) Ethanol impairs insulin-stimulated neuronal survival in the developing brain: role of PTEN phosphatase. J Biol Chem 278(29):26929–26937 [DOI] [PubMed]
- 18.Oganesian A, Poot M, Daum G, Coats SA, Wright MB, Seifert RA, Bowen-Pope DF (2003) Protein tyrosine phosphatase RQ is a phosphatidylinositol phosphatase that can regulate cell survival and proliferation. Proc Natl Acad Sci U S A 100(13):7563–7568 [DOI] [PMC free article] [PubMed]
- 19.Crane JK, Naeher TM, Choudhari SS, Giroux EM (2005) Two pathways for ATP release from host cells in enteropathogenic Escherichia coli infection. Am J Physiol Gasterointest Liver Physiol 289:G407–G417 [DOI] [PubMed]
- 20.Crane J, Vezina C (2005) Externalization of host cell protein kinase C during enteropathogenic Escherichia coli infection. Cell Death Differ 12:115–127 [DOI] [PubMed]
- 21.Bradford M (1976) A rapid and sensitive method for the quantitation of microgram quantities of protein by dye binding. Anal Biochem 72:248–254 [DOI] [PubMed]
- 22.Khan M, Caixia M, Knodler L, Valdez Y, Rosenberger C, Deng W, Finlay B, Vallance B (2006) Toll-like receptor 4 contributes to colitis development but not to host defense during Citrobacter rodentium infection in mice. Infect Immun 74:2522–2536 [DOI] [PMC free article] [PubMed]
- 23.Anonymous (2005) Real-time PCR Applications Guide. Bio-Rad Laboratories, Hercules, CA
- 24.Navarro J, Olmo N, Turnay J, Lopez-Conejo M, Lizarbe M (1998) Ecto-5′-nucleotidase from a human colon adenocarcinoma cell line. Correlation between enzyme activity and levels in intact cells. Mol Cell Biochem 187:121–131 [DOI] [PubMed]
- 25.Ohkubo S, Kimuar J, Matsuoka I (2000) Ecto-alkaline phosphatase in NG108-15 cells: a key enzyme mediating P1 antagonist-sensitive ATP response. Br J Pharmacol 131:1667–1672 [DOI] [PMC free article] [PubMed]
- 26.McNamara B, Koutsouris A, O’Connell C, Nougayrede J-P, Donnenberg M, Hecht G (2001) Translocated EspF protein of enteropathogenic Escherichia coli (EPEC) disrupts host intestinal barrier function. J Clin Invest 107:621–629 [DOI] [PMC free article] [PubMed]
- 27.Lehto MT, Sharom FJ (1998) Release of the glycosylphosphatidylinositol-anchored enzyme ecto-5′-nucleotidase by phospholipase C: catalytic activation and modulation by the lipid bilayer. Biochem J 332(Pt 1):101–109 [DOI] [PMC free article] [PubMed]
- 28.Kenny B, Finlay BB (1997) Intimin-dependent binding of enteropathogenic Escherichia coli to host cells triggers novel signaling events, including tyrosine phosphorylation of phospholipase C-gamma1. Infect Immun 65:2528–2536 [DOI] [PMC free article] [PubMed]
- 29.Bae Y-S, Lee T, Park J, Hur J, Kim Y, Heo K, Kwak J-Y, Suh P-G, Ryu S (2003) Identification of a compound that directly stimulates phospholipase C activity. Mol Pharmacol 63:1043–1050 [DOI] [PubMed]
- 30.Heithoff D, Sinsheimer R, Low D, Maham M (2000) In vivo gene expression and the adaptive response: from pathogenesis to vaccines and antimicrobials. Philos Trans R Soc Lond B Biol Sci 355:633–642 [DOI] [PMC free article] [PubMed]
- 31.Chiang S, Mekalanos J (1998) Use of signature-tagged mutagenesis to identify Vibrio cholerae genes critical for colonization. Mol Microbiol 27:797–805 [DOI] [PubMed]
- 32.Knöfel T, Sträter N (2001) Mechanism of hydrolysis of phosphate esters by the dimetal center of 5′-nucleotidase based on crystal structures. J Mol Biol 309:239–254 [DOI] [PubMed]
- 33.Zalkin H, Nygaard P (1996) Biosynthesis of purine nucleotides. In: Neidhardt F (ed) Escherichia coli and Salmonella, 2nd edn. ASM Press, Washington, D.C., pp 575–576
- 34.McCormick BA, Parkos CA, Colgan SP, Carnes DK, Madara JL (1998) Apical secretion of a pathogen-elicited epithelial chemoattractant activity in response to surface colonization of intestinal epithelia by Salmonella typhimurium. J Immunol 160(1):455–466 [PubMed]
- 35.Colgan S, Eltzschig H, Eckle T, Thompson L (2006) Physiological roles for ecto-5′-nucleotidase (CD73). Purinergic Signalling 2:351–360 [DOI] [PMC free article] [PubMed]
- 36.Tai Y-H, Gage T, McQueen C, Formal S, Boedeker E (1989) Electrolyte transport in rabbit cecum. I. Effect of RDEC-1 infection. Am J Physiol 256:G721–G726 [DOI] [PubMed]
- 37.Peiffer I, Servin AL, Bernet-Camard MF (1998) Piracy of decay-accelerating factor (CD55) signal transduction by the diffusely adhering strain Escherichia coli C1845 promotes cytoskeletal F-actin rearrangements in cultured human intestinal INT407 cells. Infect Immun 66(9):4036–4042 [DOI] [PMC free article] [PubMed]
- 38.Bernet-Camard MF, Coconnier MH, Hudault S, Servin AL (1996) Pathogenicity of the diffusely adhering strain Escherichia coli C1845: F1845 adhesin-decay accelerating factor interaction, brush border microvillus injury, and actin disassembly in cultured human intestinal epithelial cells. Infect Immun 64(6):1918–1928 [DOI] [PMC free article] [PubMed]
- 39.Barros FS, De Menezes LF, Pinheiro AA, Silva EF, Lopes AH, De Souza W, Meyer-Fernandes JR (2000) Ectonucleotide diphosphohydrolase activities in Entamoeba histolytica. Arch Biochem Biophys 375(2):304–314 [DOI] [PubMed]
- 40.Gounaris K (2002) Nucleotidase cascades are catalyzed by secreted products of the parasitic nematode Trichinella spiralis. Infect Immun 70:4917–4924 [DOI] [PMC free article] [PubMed]
- 41.Puni V, Zaborina O, Dhiman N, Falzari K, Bagdasarian M, Chakrabarty A (2000) Phagocytic cell killing by secreted cytotoxic factors of Vibrio cholerae. Infect Immun 68:4930–4937 [DOI] [PMC free article] [PubMed]
- 42.Guckelberger O, Sun XF, Sevigny J, Imai M, Kaczmarek E, Enjyoji K, Kruskal JB, Robson SC (2004) Beneficial effects of CD39/ecto-nucleoside triphosphate diphosphohydrolase-1 in murine intestinal ischemia-reperfusion injury. Thromb Haemost 91(3):576–586 [DOI] [PubMed]
- 43.Synnestvedt K, Furuta GT, Comerford KM, Louis N, Karhausen J, Eltzschig HK, Hansen KR, Thompson LF, Colgan SP (2002) Ecto-5′-nucleotidase (CD73) regulation by hypoxia-inducible factor-1 mediates permeability changes in intestinal epithelia. J Clin Invest 110(7):993–1002 [DOI] [PMC free article] [PubMed]
- 44.Deng W, Puente J, Gruenheid S, Li Y, Vallance B, Vasquez A, Barba J et al (2004) Dissecting virulence: systematic and functional analyses of a pathogenicity island. Proc Natl Acad Sci U S A 101:3597–3602 [DOI] [PMC free article] [PubMed]
- 45.Darmon N, Briend A, Desjeux JF (1997) Zinc in the treatment of diarrhea. J Pediatr Gastroenterol Nutr 25(3):363–365 [DOI] [PubMed]
- 46.Dutta P, Mitra U, Datta A, Niyogi SK, Dutta S, Manna B, Basak M, Mahapatra TS, Bhattacharya SK (2000) Impact of zinc supplementation in malnourished children with acute watery diarrhoea. J Trop Pediatr 46(5):259–263 [DOI] [PubMed]
- 47.Gupta DN, Mondal SK, Ghosh S, Rajendran K, Sur D, Manna B (2003) Impact of zinc supplementation on diarrhoeal morbidity in rural children of West Bengal, India. Acta Paediatr 92(5):531–536 [DOI] [PubMed]
- 48.Penny ME, Peerson JM, Marin RM, Duran A, Lanata CF, Lonnerdal B, Black RE, Brown KH (1999) Randomized, community-based trial of the effect of zinc supplementation, with and without other micronutrients, on the duration of persistent childhood diarrhea in Lima, Peru. J Pediatr 135(2 Pt 1):208–217 [DOI] [PubMed]
- 49.Sazawal S, Black R, Bhan M, Bhandari N, Sinha A, Jalla S (1995) Zinc supplementation in young children with acute diarrhea in India. N Engl J Med 333:839–844 [DOI] [PubMed]
- 50.Kelleher S, Casa I, Carbajal N, Lonnerdal B (2002) Supplementation of infant formula with the probiotic Lactobacillus reuteri and zinc: impact on enteric infection and nutrition in infant rhesus monkeys. J Pediatr Gastroenterol Nutr 35:162–168 [DOI] [PubMed]
- 51.Hoque K, Rajendran V, Binder H (2005) Zinc inhibits cAMP-stimulated Cl secretion via basolateral K-channel blockade in rat ileum. Am J Physiol Gastrointest Liver Physiol 288:G956–G963 [DOI] [PubMed]