

Abstract
Downstream A3 receptor signalling plays an important role in the regulation of cell death and proliferation. Therefore, it is important to determine the molecular pathways involved through A3 receptor stimulation. The phosphatidylinositide-3-OH kinase (PI3K)/Akt and the Raf/mitogen-activated protein kinase (MAPK/ERK) kinase (MEK)/mitogen-activated protein kinase (MAPK) pathways have central roles in the regulation of cell survival and proliferation. The crosstalk between these two pathways has also been investigated. The focus of this review centres on downstream mediators of A3 adenosine receptor signalling.
Key words: adenosine, A3 receptors, cell death, cell proliferation, cell signalling
Overview of the A3 receptor signalling
A3 adenosine receptors couple, via G proteins, to classical second-messenger pathways such as inhibition of adenylyl cyclase [1], stimulation of phospholipase C (PLC) [2] and calcium mobilization [3]. The activation of these signals controls important biological events, such as cell survival [4]. In particular, the A3 receptor-mediated induction of apoptosis or protection from cell death has been demonstrated in various experimental models and systems [4]. Certainly, A3 receptors have an important role in the control of neuronal cell death [5, 6]. Neuroprotective activity has been demonstrated following chronic administration of the A3 receptor agonist N6-(3-iodobenzyl)adenosine-5′-N-methyluronamide, (IB-MECA) to gerbils with cerebral ischaemia, thus supporting speculation that treatment with adenosine A3 receptor agonists may decrease the infarct size following focal brain ischaemia [7]. A3 receptors have also been involved in cardioprotection during prolonged stimulated ischaemia by rescuing injured myocytes [8]. Activation of A3 adenosine receptor protects against doxorubicin-induced cardiotoxicity and can attenuate myocyte injury during hypoxia [9–12]. Furthermore, A3 adenosine receptor activation triggers phosphorylation of protein kinase B and protects rat basophilic leukemia 2H3 mast cells from apoptosis [13]. In human melanoma cells, A3 adenosine receptor triggers a prosurvival signal, and the blockade of A3 receptors by selective antagonists induces deleterious consequences [14].
In contrast, high concentrations of the agonists for A3 receptor can induce apoptosis. Different studies have suggested the involvement of the A3 receptor in adenosine-induced apoptosis using N6(3-iodobenzyl)2-chloroadenosine-5′N-methyluronamide (Cl-IB-MECA) in rat astrocytes [15] and human astrocytoma ADF cells [16], human peripheral blood mononuclear cells, both myeloid and lymphoid cells (U937, HL60) [17, 18] and cardiac myocytes [19, 20]. However, further investigations on CHO cells transfected with the human recombinant A3 adenosine receptors (CHO-hA3) and on mast and lymphoma cell lines [13, 21–23] revealed no involvement of A3 receptors on induction of cell death, suggesting that the A3 receptor signalling is highly dependent on the cell-specific or tissuespecific context.
The molecular pathways sustaining the action of A3 receptor on cell survival have been defined. The present review will focus on the Akt-mediated signalling and the mitogen-activated protein (MAP) kinase cascades as principal pathways regulated by the A3 receptor.
The A3 receptor and the Ras-Raf-MEK-ERK signal transduction cascade
The A3 receptor signalling cascade has been demonstrated in different cellular models. Firstly, it has been suggested that A3 receptor modulates mitogenesis and the activation of ERK1/2 in human foetal astrocytes [24]. Physiological concentrations of adenosine (10–100 nM) have been demonstrated to cause an increase in phosphorylation of ERK1/2 after 5 min in CHO cells transfected with any one of the four adenosine receptors [25]. Levels of adenosine reached during ischaemia (3 µM) induce a more pronounced, but still transient, activation of ERK1/2. Thus, all the human adenosine receptors transfected into CHO cells are able to activate ERK1/2 at physiologically relevant concentrations of the endogenous agonist. Both 5′-N(ethyl)carboxamidoadenosine (NECA) and the endogenous agonist (adenosine) lead to a time- and dose-dependent increase in ERK1/2 phosphorylation, at concentrations as low as 10 nM to 30 nM [25]. Moreover, these studies show that A3 receptor signalling in CHO-hA3 cells leads to stimulation of ERK1/2 activity. In particular, A3 receptor signalling to ERK1/2 depends on βγ release from PTX-sensitive G proteins, PI3K, Ras and MEK [26]. Functional A3 receptors activating ERK1/2 have also been described in microglia cells [27]. In this cell model, by selectively stimulating the A3 receptor in both primary mouse microglia cells and in the N13 microglia cell line with the agonist Cl-IB-MECA, it has been found to be a biphasic, partly Gi protein-dependent influence on the phosphorylation of the ERK1/2.
However, different results have been obtained on this issue. In the human melanoma A375 cell line it has been demonstrated that Cl-IB-MECAwas unable to activate ERK phosphorylation, while the A3 antagonists are able to improve the activity of MEKs [14]. Similar results were obtained in murine melanoma cells [28]. Furthermore, recently, we have found that A3 adenosine receptor stimulation inhibits cell proliferation by impairment of ERK kinase activation [29]. We studied the changes in ERK1/2 phosphorylation after treatment of melanoma cells with Cl-IB-MECA 10 µM in a time-course experiment. Interestingly, prolonged inhibition of ERK1/2 phosphorylation was induced after the treatment of melanoma cells with Cl-IB-MECA, and this effect was sustained for 2 h after Cl-IBMECA treatment. The kinetics of ERK1/2 phosphorylation reduction was very rapid: Cl-IB-MECA appeared to impair ERK1/2 phosphorylation after 5 min. Furthermore, we investigated the dose-dependence of Cl-IB-MECA to inhibit ERK1/2 phosphorylation. We found that the inhibition of ERK1/2 phosphorylation was produced by Cl-IB-MECA with an EC50 of 4.3 ± 0.5 µM and 2.2 ± 0.3 µM for ERK-2 and ERK-1, respectively. The effect of Cl-IB-MECA was mediated by A3 receptor because, while the A1, A2A and A2B receptor antagonists were not able to prevent ERK1/2 inhibition induced by Cl-IB-MECA, the selective A3 receptor antagonist MRE 3008F20 (0.1 µM) abrogated the Cl-IB-MECA-induced inhibition of ERK1/2 activation. Furthermore, we found that the inhibition of A3 receptor expression is sufficient to block Cl-IB-MECAinduced inhibition of ERK1/2 phosphorylation levels. These results emphasize the role of A3 receptors as inhibitors of ERK activation and clearly show the connection between A3 receptor stimulation and MEK/MAPK signalling in melanoma cells.
To explain why different results have been obtained on this issue it could be suggested that this mechanism may be peculiar for melanoma cells, having a misregulation of proliferative pathways, as A3 receptors increased ERK1/2 phosphorylation in CHO-hA3 cells in a dose-dependent manner [26] and induced a biphasic effect on the phosphorylation levels of ERK1/2 on microglia cells [27]. Further studies in other different cell systems will enhance our understanding of the role of A3 receptors in the modulation of mitogenic signalling.
Furthermore, it has been shown that MAPK activation is involved in A3 receptor regulation, both contributing to direct phosphorylation and controlling G protein-coupled receptor (GPCR) kinase protein membrane translocation, which are involved in receptor phosphorylation. Thus, an active MAPK pathway appears to be essential for A3 receptor phosphorylation, desensitization and internalization [30].
The A3 receptor and the Akt signal transduction cascade
Another important pathway that is triggered by adenosine via A3 receptor is the PI3K/Akt pathway [4]. One of the crucial downstream targets of PI3K is the serine/threonine kinase Akt. Active Akt causes a variety of biological effects, including suppression of apoptosis by phosphorylation and inactivation of several targets along pro-apoptotic pathways [31]. In particular, activated Akt is able to phosphorylate a variety of downstream substrates, e.g., the pro-apoptotic molecule Bad, caspase-9, the forkhead family transcription factors, I-K (a kinase that regulates the NF-kB transcription factor) and Raf.
Recent studies have demonstrated that protein kinase A (PKA) and PKB/Akt phosphorylate and inactivate glycogen synthase kinase 3β (GSK-3β), a serine/threonine kinase acting as a key element in the Wnt signalling pathway. In its active form, GSK-3β suppresses mammalian cell proliferation [28]. Only one study has indicated that the A3 agonist IB-MECA is able to decrease the levels of PKA, a downstream effector of cAMP, and of the phosphorylated form of PKB/Akt in melanoma cells. This implies the deregulation of the Wnt signalling pathway, generally active during embryogenesis and tumorigenesis to increase cell cycle progression and cell proliferation [28]. In contrast, several studies have indicated the ability of A3 receptor to activate Akt [4]. There is evidence that A3 adenosine receptor activation triggers phosphorylation of PKB/Akt, protecting rat basophilic leukemia 2H3 mast cells from apoptosis by a pathway involving the βγ subunits of Gi and PI3K-β. This process is blocked by pertussis toxin and wortmannin [13]. More recently, it has been demonstrated that A3 receptors trigger increases in Akt phosphorylation in rat cardiomyocytes via a Gi/Go-protein and tyrosine kinase-dependent pathway [32]. An elegant study has recently documented a role of adenosine A3 receptor in cell survival signalling in resveratrol preconditioning of the heart. This study provides evidence that resveratrol preconditions the heart through the activation of adenosine A1 and A3 receptors, transmitting a survival signal through the PI3-kinase-Akt-Bcl2 signalling pathway [33]. Further studies indicate that A3 receptor activation can decrease IL-12 production, which is also mediated by interfering with PKB/Akt [34, 35]. Using human melanoma A375 cells we showed that A3 adenosine receptor stimulation results in PI3K-dependent phosphorylation of Akt. We found that serum-deprived A375 melanoma cells had no basal Akt phosphorylation, whereas the A3 receptor agonist Cl-IB-MECA treatment resulted in the phosphorylation of Akt at the Ser 573 phosphorylation site. In particular, we have demonstrated that the antiproliferative effect of Cl-IB-MECA is mediated by a PLC-PI3K-Akt signalling pathway [29].
Crosstalk between MAPK and PI3K/Akt signalling pathways, and modulation by the A3 receptor
Crosstalk between the PI3K and the Raf/MEK/ERK pathways has been reported on multiple levels, with some research stating that PI3K activity is essential for induction of Raf/MEK/ERK activity [31, 36]. Additional studies suggest that the PI3K pathway enhances and/or synergizes with Raf/MEK/ERK signalling to provide a more robust signal through the lower components of the MAPK cascade (i.e., ERK). However, there is conflicting evidence that states that Akt is able to phosphorylate Raf, thereby efficiently abrogating Raf activity on downstream substrates [37–41].
Considering the pleiotropic substrates and the ubiquitous expression of ERK, cell specific regulation must occur to ensure conduction of the appropriate signal.
Recently, we investigated the role of the adenosinergic system in the regulation of ERK activation in melanoma cells. We focused our attention on melanoma cells because the importance has been demonstrated of small autacoids, primarily thought to be neurotransmitters, in mediating a variety of biological activities in the skin [14, 42–45]. In particular, several studies have indicated that adenosine, via stimulation of its receptors, is involved in cell proliferation and cell death. In agreement with these studies, we also found that A3 receptor inhibits human melanoma A375 cell line proliferation [42].
The activities of Akt or MAPK, or both, are elevated in many cancer cells and are known to play critically important roles in cellular proliferation [31, 36, 46]. On the basis of this consideration, we tested the hypothesis that A3 receptor stimulation regulates cell growth signalling via the ERK1/2 and/or the Akt pathway in melanoma cells.
We found that Akt phosphorylation mediated by Cl-IB-MECA induced Raf phosphorylation at an inhibitory phosphorylation site on Ser 259 of Raf. As a consequence, Cl-IB-MECA inactivated Raf in a dose- and time-dependent manner. Interestingly, Cl-IB-MECA stimulation resulted in a time- and dose-dependent reduction in ERK1/2 phosphorylation.
We examined whether any crosstalk existed between ERK1/2 and Akt pathways in A375 melanoma cells. The classical MAPK cascade leads from the Ras kinases to the MAPKK MEK1/2. There is evidence that Akt is able to phosphorylate Raf, thereby efficiently abrogating Raf activity on downstream substrates [39, 41, 47]. We studied the effects of A3 receptor stimulation on the proliferation of melanoma cells in the presence of specific inhibitors of the PI3K and Akt signal transduction pathways. We could effectively block the Cl-IB-MECA-induced reduction of ERK1/2 phosphorylation with an inhibitor of PI3K. Indeed, application of Cl-IB-MECA in combination with PI3K inhibition resulted in a clear increase of ERK1/2 phosphorylation when compared with P-ERK1/2 in the presence of Cl-IB-MECA alone. These data suggest that the Ras-Raf-MEK-ERK pathway is normally activated by A3 receptor stimulation, as is the PI3K-Akt route. It is clear that these apparently separate routes should actually interact.
In order to investigate the functionality of A3 receptors expressed in melanoma cells we used the selective adenosine analogue Cl-IB-MECA. It is not clear whether the growth inhibitory action of micromolar concentrations of the A3 receptor agonist Cl-IB-MECA is due to its role as an extracellular ligand for cell surface receptors or whether it acts intracellularly as a second messenger. In addition, this agonist may, in high concentrations, activate A1 receptors, which, however, when compared with A3 receptors, are expressed at low level in A375 cells (BMAX = 23 ± 7 fmol mg−1 of protein and BMAX = 291 ± 50 fmol mg−1 of protein for A1 and A3, respectively) [48]. Thus, the effects we report here on melanoma cell proliferation and on ERK1/2 phosphorylation induced by Cl-IB-MECA using high (10−6 M) concentrations of Cl-IB-MECA are almost certainly due to A3 receptor stimulation. In particular, the effects of Cl-IB-MECA on cell proliferation and on ERK1/2 phosphorylation are not mediated by A1, A2A or A2B receptors. In support of this conclusion, DPCPX, SCH 58261 and MRE 2029F20, adenosine receptor antagonists highly selective for A1, A2A and A2B receptors [49, 50], respectively, did not block the inhibitory effect of A3 receptor stimulation on cell proliferation and on P-ERK1/2 modulation. On the contrary, the effects on cell proliferation and on P-ERK1/2 modulation were inhibited by the A3 receptor antagonist, MRE 3008F20. Furthermore, the Cl-IB-MECA-induced effects on cell proliferation and ERK1/2 phosphorylation in human A375 melanoma cells were abolished in cells in which A3 receptor protein was knocked down by si-RNAA3 treatment, when compared with wild-type cells. These findings, together with the specificity of the agonist used, makes us confident that the effects are due to the A3 receptor subtype.
A3 receptor stimulation inhibits the proliferation of melanoma cells partly by a PLC-sensitive mechanism. Pretreatment of cells with a PLC-γ inhibitor strongly abrogated the Cl-IB-MECA effect on cell proliferation and on ERK1/2 phosphorylation, suggesting a critical role for PLC-γ in A3 receptor signalling. Furthermore, pretreatment of A375 cells with a PI3K inhibitor and an Akt inhibitor impaired Cl-IB-MECA-induced inhibition of cell proliferation and the effects of A3 receptor stimulation on Raf, MEK1/2 and ERK1/2 phosphorylation. These data suggest that the A3 adenosine receptor signals through a pathway that includes PI3K-Akt. In contrast, Ras was not activated. These results confirm that, in A375 cells, A3 receptors decrease MEK1/2-ERK1/2 phosphorylation and cell proliferation via the inhibition of Raf by a PI3K-Akt pathway, without affecting Ras.
Our results indicate that Cl-IB-MECA acts extracellularly as a first messenger for cell surface receptors. In A375 cells the inhibition of PLC-PI3K-Akt signalling is able to block the effect of A3 receptor stimulation on cell proliferation, suggesting that, in the melanoma cell system, an inhibitory connection between PLC-PI3K-Akt and ERK1/2 is present (Figure 1).
Figure 1.
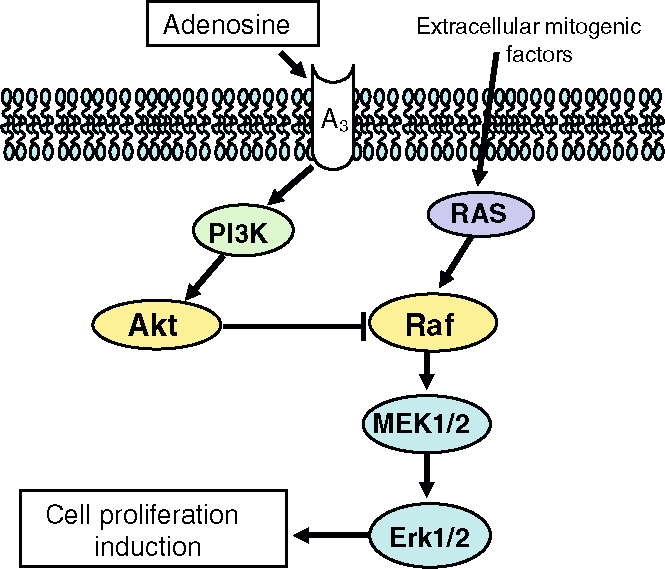
A3 adenosine receptor stimulates PI3K-dependent phosphorylation of Akt, leading to the reduction of basal levels of ERK1/2 phosphorylation that, in turn, inhibits cell proliferation
Finally, we have described the molecular mechanism sustained by Cl-IB-MECA interfering with cell proliferation. Cl-IB-MECA, via A3 adenosine receptor binding, activates PLC-PI3K-Akt signalling that, in turn, reduces P-ERK1/2 levels necessary for cell proliferation. As a consequence, cells accumulate in G0/G1 cell cycle phases, and low level of DNA incorporation is observed.
Further definition of the pathways leading to ERK1/2 inactivation, and translation into an in vivo model, are required to clarify whether adenosine signalling in vivo has characteristics similar to those observed in this in vitro model. In this scenario the regulation of ERK1/2 by A3 receptor and PI3K would represent an important aspect of adenosine signalling.
Although it is possible that the signalling pathways may be different in other cellular backgrounds, it seems likely that the biological events regulated by adenosine A3 receptors under physiological and pathophysiological conditions may depend not only on changes in cAMP and Ca2+ but also on mitogenic signalling via ERK1/2 and Akt.
Conclusions
Work by several groups, including ours, has shown a modulation of the Akt/Ras/Raf/MEK/ERK signalling pathway by adenosine. We propose that this modulation is essential to determine the final physiological response of A3 adenosine receptor activation.
Of great interest are the different effects of adenosine on the Akt/Ras/Raf/MEK/ERK signalling pathway modulation in different cells.
Interestingly, adenosine may be, on the one hand, preventing the proliferative activation of ERK1/2 by blocking Raf through Akt activation and, on the other hand, stimulating PI3K activation in order to induce cell survival.
We believe that the opposite effects of adenosine are due to the presence of more than one receptor subtype in the same cellular system, each mediating seemingly opposite actions, e.g., having conflicting actions on cell proliferation and cell death. As a consequence, the net effect may not be defined. It is clear that adenosine concentration may determine the pattern of differential activation of co-expressed receptor subtypes. One adenosine receptor may also be coupled to more than one G protein. Several studies are helping to show if such coupling is physiologically important [51].
Further work to determine the various physiological responses mediated by A3 receptor stimulation through the regulation of the Akt/Ras/Raf/MEK/ERK pathway will help us to understand the role played by adenosine in cell proliferation and cell death.
Abbreviations
- CHO-hA3
CHO cells transfected with the human recombinant A3
- Cl-IB-MECA
N >>6(3-iodobenzyl)2-chloroadenosine-5′-N-methyluronamide
- DPCPX
1,3-dipropyl-8-cyclopentylxanthine
- ERK
extracellular signal-regulated kinase
- GSK-3β
glycogen synthase kinase
- IB-MECA
N >6-(3-iodobenzyl)adenosine-5′-N-methyluronamide
- MAPK
mitogen-activated protein kinase
- MEK
mitogen-activated protein kinase kinase
- MRE 3008F20
5N-(4-methxyphenyl-carbomoyl)amino-8-propyl-2-(2-furyl)-pyrazolo-[4,3e]1,2,3-triazolo[1,5c] pyrimidine
- MRE 2029F20
[N-benzo[1,3]dioxol-5-yl-2-[5-(2,6-dioxo-1,3-dipropyl-2,3,6,7-tetrahydro-1H-purin-8-yl)-1-methyl-1H-pyrazol-3-yloxy]-acetamide]
- NECA
5′-N(ethyl)carboxamidoadenosine
- pAkt
phosphorylated Akt
- PI3K
phosphatidylinositol 3-kinase
- PLC
phospholipase C
- PKA
protein kinase A
- SCH 58261
7-(2-phenylethyl)2-(2-furyl)pyrazolo[4,3e] 1,2,3-triazolo[1,5c]pyrimidine
- siRNA
small interfering RNA
- siRNAA3
small interfering RNA that targets A3 receptor mRNA
References
- 1.Zhou QY, Li C, Olah ME, Johnson RA, Stiles GL, Civelli O (1992) Molecular cloning and characterization of an adenosine receptor: the A3 adenosine receptor. Proc Natl Acad Sci USA 89:7432′436 [DOI] [PMC free article] [PubMed]
- 2.Abbracchio MP, Brambilla R, Ceruti S, Kim HO, von Lubitz DK, Jacobson KA, Cattabeni F (1995) G protein-dependent activation of phospholipase C by adenosine A3 receptors in rat brain. Mol Pharmacol 48:1038′045 [PubMed]
- 3.Fossetta J, Jackson J, Deno G, Fan X, Du XK, Bober L, Soude-Bermejo A, de Bouteiller O, Caux C, Lunn C, Lundell D, Palmer RK (2003) Pharmacological analysis of calcium responses mediated by the human A3 adenosine receptor in monocyte-derived dendritic cells and recombinant cells. Mol Pharmacol 63:342′50 [DOI] [PubMed]
- 4.Merighi S, Mirandola P, Varani K, Gessi S, Leung E, Baraldi PG, Tabrizi MA, Borea PA (2003) A glance at adenosine receptors: novel target for antitumor therapy. Pharmacol Ther 100:31′8 [DOI] [PubMed]
- 5.Appel E, Kazimirsky G, Ashkenazi E, Kim SG, Jacobson KA, Brodie C (2001) Roles of BCL-2 and caspase 3 in the adenosine A3 receptor-induced apoptosis. J Mol Neurosci 17:285′92 [DOI] [PMC free article] [PubMed]
- 6.Abbracchio MP, Rainaldi G, Giammarioli AM, Ceruti S, Brambilla R, Cattabeni F, Barbieri D, Franceschi C, Jacobson KA, Malorni W (1997) The A3 adenosine receptor mediates cell spreading, reorganization of actin cytoskeleton, and distribution of Bcl-XL: Studies in human astroglioma cells. Biochem Biophys Res Commun 241:297′04 [DOI] [PMC free article] [PubMed]
- 7.Von Lubitz DK, Simpson KL, Lin RC (2001) Right thing at a wrong time? Adenosine A3 receptors and cerebroprotection in stroke. Ann NY Acad Sci 939:85′6 [DOI] [PubMed]
- 8.Liu GS, Richards SC, Olsson RA, Mullane K, Walsh RS, Downey JM (1994) Evidence that the adenosine A3 receptor may mediate the protection afforded by preconditioning in the isolated rabbit heart. Cardiovasc Res 28:1057′061 [DOI] [PubMed]
- 9.Shneyvays V, Mamedova L, Zinman T, Jacobson K, Shainberg A (2001) Activation of A3 adenosine receptor protects against doxorubicin-induced cardiotoxicity. J Mol Cell Cardiol 33:1249′261 [DOI] [PMC free article] [PubMed]
- 10.Shneyvays V, Mamedova LK, Korkus A, Shainberg A (2002) Cardiomyocyte resistance to doxorubicin mediated by A3 adenosine receptor. J Mol Cell Cardiol 34:493′07 [DOI] [PubMed]
- 11.Safran N, Shneyvays V, Balas N, Jacobson KA, Nawrath H, Shainberg A (2001) Cardioprotective effects of adenosine A1 and A3 receptor activation during hypoxia in isolated rat cardiac myocytes. Mol Cell Biochem 217:143′52 [DOI] [PMC free article] [PubMed]
- 12.Tracey WR, Magee W, Masamune H, Oleynek JJ, Hill RJ (1998) Selective activation of adenosine A3 receptors with N6-(3-chlorobenzyl)-5-N-methylcarboxamidoadenosine (Cl-IB-MECA) provides cardioprotection via KATP channel activation. Cardiovasc Res 40:138′45 [DOI] [PubMed]
- 13.Gao Z, Li BS, Day YJ, Linden J (2001) A3 adenosine receptor activation triggers phosphorylation of protein kinase B and protects rat basophilic leukemia 2H3 mast cells from apoptosis. Mol Pharmacol 59:76′2 [DOI] [PubMed]
- 14.Merighi S, Mirandola P, Milani D, Varani K, Gessi S, Klotz KN, Leung E, Baraldi PG, Borea PA (2002) Adenosine receptors as mediators of both cell proliferation and cell death of cultured human melanoma cells. J Invest Dermatol 119:923′33 [DOI] [PubMed]
- 15.Abbracchio MP, Ceruti S, Brambilla R, Barbieri D, Cimurri A, Franceschi C, Giammarioli AM, Jacobson KA, Cattabeni F, Malorni W (1998) Adenosine A3 receptors and viability of astrocytes. Drug Dev Res 45:379′86 [DOI] [PMC free article] [PubMed]
- 16.Barbieri D, Abbracchio MP, Salvioli S, Monti D, Cossarizza A, Ceruti S, Brambilla R, Cattabeni F, Jacobson KA, Franceschi C (1998) Apoptosis by 2-chloro-2-deoxy-adenosine and 2-chloro-adenosine in human peripheral blood mononuclear cells. Neurochem Int 32:493′04 [DOI] [PMC free article] [PubMed]
- 17.Kohno Y, Sei Y, Koshiba M, Kim HO, Jacobson KA (1996) Induction of apoptosis in HL-60 human promyelocytic leukemia cells by adenosine A3 receptor agonists. Biochem Biophys Res Commun 219:904′10 [DOI] [PMC free article] [PubMed]
- 18.Yao Y, Sei Y, Abbracchio MP, Jiang JL, Kim YC, Jacobson KA (1997) Adenosine A3 receptor agonists protect HL-60 and U-937 cells from apoptosis induced by A3 antagonists. Biochem Biophys Res Commun 232:317′22 [DOI] [PMC free article] [PubMed]
- 19.Shneyvays V, Nawrath H, Jacobson KA, Shainberg A (1998) Induction of apoptosis in cardiac myocytes by an A3 adenosine receptor agonist. Exp Cell Res 243:383′97 [DOI] [PubMed]
- 20.Shneyvays V, Jacobson KA, Li AH, Nawrath H, Zinman T, Isaac A, Shainberg A (2000) Induction of apoptosis in rat cardiocytes by A3 adenosine receptor activation and its suppression by isoproterenol. Exp Cell Res 257:111′26 [DOI] [PMC free article] [PubMed]
- 21.Brambilla R, Cattabeni F, Ceruti S, Barbieri D, Franceschi C, Kim YC, Jacobson KA, Klotz KN, Lohse MJ, Abbracchio MP (2000) Activation of the A3 adenosine receptor affects cell cycle progression and cell growth. Naunyn Schmiedebergs Arch Pharmacol 361:225′34 [DOI] [PMC free article] [PubMed]
- 22.Fishman P, Bar-Yehuda S, Ohana G, Pathak S, Wasserman L, Barer F, Multani AS (2000) Adenosine acts as an inhibitor of lymphoma cell growth: a major role for the A3 adenosine receptor. Eur J Cancer 36:1452′458 [DOI] [PubMed]
- 23.Kim SG, Ravi G, Hoffmann C, Jung YJ, Kim M, Chen A, Jacobson KA (2002) p53-Independent induction of Fas and apoptosis in leukemic cells by an adenosine derivative, Cl-IB-MECA. Biochem Pharmacol 63:871′80 [DOI] [PMC free article] [PubMed]
- 24.Neary JT, McCarthy M, Kang Y, Zuniga S (1998) Mitogenic signaling from P1 and P2 purinergic receptors to mitogen-activated protein kinase in human fetal astrocyte cultures. Neurosci Lett 242:159′62 [DOI] [PubMed]
- 25.Schulte G, Fredholm BB (2000) Human adenosine A1, A2A, A2B, and A3 receptors expressed in Chinese hamster ovary cells all mediate the phosphorylation of extracellular-regulated kinase 1/2. Mol Pharmacol 58:477′82 [PubMed]
- 26.Schulte G, Fredholm BB (2002) Signaling pathway from the human adenosine A3 receptor expressed in Chinese hamster ovary cells to the extracellular signal-regulated kinase 1/2. Mol Pharmacol 62:1137′146 [DOI] [PubMed]
- 27.Hammarberg C, Schulte G, Fredholm BB (2003) Evidence for functional adenosine A3 receptors in microglia cells. J Neurochem 86:1051′054 [DOI] [PubMed]
- 28.Fishman P, Madi L, Bar-Yehuda S, Barer F, Del Valle L, Khalili K (2002) Evidence for involvement of Wnt signaling pathway in IB-MECA mediated suppression of melanoma cells. Oncogene 21:4060′064 [DOI] [PubMed]
- 29.Merighi S, Benini A, Mirandola P, Gessi S, Varani K, Leung E, Maclennan S, Borea PA (2005) A3 adenosine receptor activation inhibits cell proliferation via phosphatidylinositol 3-kinase (PI3K)/AKT-dependent inhibition of the extracellular signal-regulated kinase (ERK)1/2 phosphorylation in A375 human melanoma cells. J Biol Chem 280:19516′9526 [DOI] [PubMed]
- 30.Trincavelli ML, Tuscano D, Marroni M, Klotz KN, Lucacchini A, Martini C (2002) Involvement of mitogen protein kinase cascade in agonist-mediated human A3 adenosine receptor regulation. Biochim Biophys Acta 1591:55′2 [DOI] [PubMed]
- 31.Vivanco I, Sawyers CL (2002) The phosphatidylinositol 3-Kinase AKT pathway in human cancer. Nat Rev Cancer 2:489′01 [DOI] [PubMed]
- 32.Germack R, Griffin M, Dickenson JM (2004) Activation of protein kinase B by adenosine A1 and A3 receptors in newborn rat cardiomyocytes. J Mol Cell Cardiol 37:989′99 [DOI] [PubMed]
- 33.Das S, Cordis GA, Maulik N, Das DK (2005) Pharmacological preconditioning with resveratrol: role of CREB-dependent Bcl-2 signaling via adenosine A3 receptor activation. Am J Physiol 288:H328–H335 [DOI] [PubMed]
- 34.Hasko G, Nemeth ZH, Vizi ES, Salzman AL, Szabo C (1998) An agonist of adenosine A3 receptors decreases interleukin-12 and interferon-gamma production and prevents lethality in endotoxemic mice. Eur J Pharmacol 358:261′68 [DOI] [PubMed]
- 35.la Sala A, Gadina M, Kelsall BL (2005) G(i)-protein-dependent inhibition of IL-12 production is mediated by activation of the phosphatidylinositol 3-kinase-protein 3 kinase B/Akt pathway and JNK. J Immunol 175:2994′999 [DOI] [PubMed]
- 36.Sebolt-Leopold JS, Herrera R (2004) Targeting the mitogen-activated protein kinase cascade to treat cancer. Nat Rev Cancer 4:937′47 [DOI] [PubMed]
- 37.Rommel C, Clarke BA, Zimmermann S, Nunez L, Rossman R, Reid K, Moelling K, Yancopoulos GD, Glass DJ (1999) Differentiation stage-specific inhibition of the Raf-MEK-ERK pathway by Akt. Science 286:1738′741 [DOI] [PubMed]
- 38.Guan KL, Figueroa C, Brtva TR, Zhu T, Taylor J, Barber TD, Vojtek AB (2000) Negative regulation of the serine/threonine kinase B-Raf by Akt. J Biol Chem 275:27354′7359 [DOI] [PubMed]
- 39.Reusch HP, Zimmermann S, Schaefer M, Paul M, Moelling K (2001) Regulation of Raf by Akt controls growth and differentiation in vascular smooth muscle cells. J Biol Chem 276:33630′3637 [DOI] [PubMed]
- 40.Moelling K, Schad K, Bosse M, Zimmermann S, Schweneker M (2002) Regulation of Raf-Akt cross-talk. J Biol Chem 277:31099′1106 [DOI] [PubMed]
- 41.Zimmermann S, Moelling K (1999) Phosphorylation and regulation of Raf by Akt (protein kinase B). Science 286:1741′744 [DOI] [PubMed]
- 42.Merighi S, Mirandola P, Varani K, Gessi S, Capitani S, Leung E, Baraldi PG, Tabrizi MA, Borea PA (2003) Pyrazolotriazolopyrimidine derivatives sensitize melanoma cells to the chemotherapeutic drugs: taxol and vindesine. Biochem Pharmacol 66:739′48 [DOI] [PubMed]
- 43.Haberberger RV, Pfeil U, Lips KS, Kummer W (2002) Expression of the high-affinity choline transporter, CHT1, in the neuronal and non-neuronal cholinergic system of human and rat skin. J Invest Dermatol 119:943′48 [DOI] [PubMed]
- 44.Slominski A, Wortsman J, Kohn L, Ain KB, Venkataraman GM, Pisarchik A, Chung JH, Giuliani C, Thornton M, Slugocki G, Tobin DJ (2002) Expression of hypothalamic-pituitary-thyroid axis related genes in the human skin. J Invest Dermatol 119:1449′455 [DOI] [PMC free article] [PubMed]
- 45.Slominski A, Pisarchik A, Semak I, Sweatman T, Szczesniewski A, Wortsman J (2002) Serotoninergic system in hamster skin. J Invest Dermatol 119:934′42 [DOI] [PubMed]
- 46.Lee JT Jr, McCubrey JA (2002) The Raf/MEK/ERK signal transduction cascade as a target for chemotherapeutic intervention in leukemia. Leukemia 16:486′07 [DOI] [PubMed]
- 47.Guan KL, Figueroa C, Brtva TR, Zhu T, Taylor J, Barber TD, Vojtek AB (2000) Negative regulation of the serine/threonine kinase B-Raf by Akt. J Biol Chem 275:27354′7359 [DOI] [PubMed]
- 48.Merighi S, Varani K, Gessi S, Cattabriga E, Iannotta V, Ulouglu C, Leung E, Borea PA (2001) Pharmacological and biochemical characterization of adenosine receptors in the human malignant melanoma A375 cell line. Br J Pharmacol 134:1215′226 [DOI] [PMC free article] [PubMed]
- 49.Varani K, Merighi S, Gessi S, Klotz KN, Leung E, Baraldi PG, Cacciari B, Romagnoli R, Spalluto G, Borea PA (2000) [3H]MRE 3008F20: a novel antagonist radioligand for the pharmacological and biochemical characterization of human A3 adenosine receptors. Mol Pharmacol 57:968′75 [PubMed]
- 50.Baraldi PG, Tabrizi MA, Preti D, Bovero A, Romagnoli R, Fruttarolo F, Zaid NA, Moorman AR, Varani K, Gessi S, Merighi S, Borea PA (2004) Design, synthesis, and biological evaluation of new 8-heterocyclic xanthine derivatives as highly potent and selective human A2B adenosine receptor antagonists. J Med Chem 47:1434′447 [DOI] [PubMed]
- 51.Klinger M, Freissmuth M, Nanoff C (2002) Adenosine receptors: G protein-mediated signalling and the role of accessory proteins. Cell Signal 14:99′08 [DOI] [PubMed]