

Abstract
Heme oxygenase-1 (HO-1) catalyzes the oxidation of heme to biologically active products: carbon monoxide (CO), biliverdin, and ferrous iron. It participates in maintaining cellular homeostasis and plays an important protective role in the tissues by reducing oxidative injury, attenuating the inflammatory response, inhibiting cell apoptosis, and regulating cell proliferation. HO-1 is also an important proangiogenic mediator. Most studies have focused on the role of HO-1 in cardiovascular diseases, in which its significant, beneficial activity is well recognized. A growing body of evidence indicates, however, that HO-1 activation may play a role in carcinogenesis and can potently influence the growth and metastasis of tumors. HO-1 is very often upregulated in tumor tissues, and its expression is further increased in response to therapies. Although the exact effect can be tissue specific, HO-1 can be regarded as an enzyme facilitating tumor progression. Accordingly, inhibition of HO-1 can be suggested as a potential therapeutic approach sensitizing tumors to radiation, chemotherapy, or photodynamic therapy.
HEME OXYGENASE-1 (HO-1)
Heme oxygenase (HO) is a microsomal enzyme catalyzing the first, rate-limiting step in degradation of heme and playing an important role in recycling of iron (103). It cleaves the α-meso carbon bridge of heme, yielding equimolar quantities of carbon monoxide (CO) and iron ions Fe2+ and biliverdin (156). CO is then exhaled from the organisms through the lung (50). Free iron induces the expression of the iron-sequestering ferritin and activates Fe-ATPase, an iron transporter, which decrease intracellular Fe2+ content. Finally, biliverdin is converted by biliverdin reductase to bilirubin (144), which can be oxidized by cytochrome P450 enzymes, such as Cyp1A1, Cyp2B1, or Cyp2a5, or glucoronidated by UDP-glucuronyl-transferase and subsequently eliminated as bilirubin glucoronides by the biliary-fecal pathway (25).
The enzymatic activity of HO results in decreased oxidative stress, attenuated inflammatory response, and a lower rate of apoptosis (Fig. 1). This is due to removal of heme, a potent prooxidant and proinflammatory agent, but also because of generation of biologically active products. Among them, CO is an important cellular messenger, with the signaling function resembling that of nitric oxide (NO). Like NO, CO induces soluble guanylyl cyclase (sGC) and thereby inhibits platelet aggregation, decreases leukocyte adhesion, and reduces endothelial cell apoptosis. In addition, it exerts antiinflammatory effects by inhibition of tumor necrosis factor (TNF), interleukin-1β (IL-1β), and macrophage inflammatory protein-1β (MIP-1β), or by upregulation of interleukin-10 (IL-10) (123). Ferrous iron, the second product of heme decomposition, can be potentially toxic, giving rise to hydroxyl free radicals. Simultaneous upregulation of ferritin and cytosolic iron efflux, however, protects cells from oxidative stress. Both biliverdin and bilirubin, which have been long regarded as toxic end products of heme metabolism, are inhibitors of the complement cascade and potent antioxidants, capable of reducing the inflammatory response and attenuating oxidative injury by scavenging peroxyl radicals and decreasing peroxidation of membrane lipids and proteins (144).
FIG. 1.

Schematic demonstration of HO-1 pathway and biologic activities of HO-1 products.
Three distinct mammalian HO isoforms (HO-1, HO-2, and HO-3) have been identified, which are the products of different genes (102). HO-1, the inducible 32-kDa isoform, is highly expressed in the liver and spleen, but can be also detected in many other tissues. HO-2 is a constitutively expressed 36-kDa protein, present in high levels in the brain, testes, or endothelial cells. HO-3 was postulated as a 33-kDa protein expressed in different organs, very similar to HO-2, but with much lower catalytic activity (109). However, HO-3 has been found only in rats, and its expression was demonstrated only at mRNA level; thus, it is supposed to be a pseudogene derived from HO-2 transcripts (53).
Unlike the constitutively expressed HO-2, HO-1 can be strongly induced in response to cellular stress and diverse oxidative stimuli, including its substrate heme (3), heat shock (151), UV irradiation, reactive oxygen species (ROS) (81), nitric oxide (NO) (38), inflammatory cytokines (157), prostaglandins (68), ethanol (174), and heavy metals (3). It can also be induced by hypoxia, but this effect appears to be tissue and species dependent (113, 141). Only in some unique types of cells does HO-1 seem to be constitutively expressed. Among them are renal inner medullary cells (46), Kupffer cells in the liver (8), Purkinje cells in the cerebellum (117), and CD4+/CD25+ regulatory T cells (124).
EXPRESSION OF HO-1 IN TUMORS
Interestingly, expression of HO-1 is usually increased in tumors, compared with surrounding healthy tissues (Table 1). This was shown in lymphosarcoma (136), adenocarcinoma (47), hepatoma (28), glioblastoma (26), melanoma (158), prostate cancers (101), Kaposi sarcoma (108), squamous carcinoma (162), pancreatic cancer (9), and in brain tumors (52). Moreover, the primary chronic myeloid leukemia cells express HO-1 in a constitutive manner, and BCR/ABL fusion protein was found to upregulate HO-1 production (107). HO-1 is also induced by viral G protein-coupled receptor (vGPCR), an oncogene encoded by HHV-8, a Kaposi sarcoma-associated herpes virus (105). Of importance, expression of HO-1 in cancer cells can be further increased in response to chemotherapy, radiation (9), or photodynamic therapy (78, 120).
Table 1.
Expression of HO-1 in Tumors. ND-No Data
| Type of cancer | Species | Type of cells | HO-1 | Ref. |
|---|---|---|---|---|
| Brain tumors | Human | ND | ↑ | 52 |
| Chronic myeloid leukemia | Human | Leukemic cells | ↑ | 107 |
| Glioblastoma | Rat | Macrophages | ↑ | 26 |
| Hepatoma | Rat | Tumor cells | ↑ | 28, 36 |
| Lymphosarcoma | Rat | Hepatocytes | ↑ | 136 |
| Melanoma | Human | Macrophages | ↑ | 158 |
| Pancreatic cancer | Human | Tumor cells, macrophages | ↑ | 9 |
| Prostate cancer | Human | Tumor cells | ↑ | 101 |
| Renal adenocarcinoma | Human | ND | ↑ | 47 |
| Squamous carcinoma | Human | Tumor cells | ⇅ | 162 |
The exact location of HO-1 is not so clear and may depend on the type of lesion. In many specimens, HO-1 was specifically expressed in macrophages, but only slightly in tumor cells, as found in human melanomas (158) or in rat and human gliomas (26). Prominent accumulation of HO-1-expressing macrophages can be characteristic for perinecrotic areas of tumors (26). In other cases, for example, in rat hepatoma, HO-1 was found only in tumor cells, not in macrophages (28). Finally, in human pancreatic cancer, cancer tissues revealed marked HO-1 immunoreactivity in tumor cells and in tumor-associated immunocytes (9).
HO-1 AND TUMOR GROWTH
HO-1 is very often upregulated in quickly proliferating cells such as epithelium within the wounded skin or psoriatic lesions (51) and cancer cells in growing tumors (101). Interestingly, HO-1 has been shown to influence the cell cycle, although its effects are cell-type specific and can be opposite in different tissues (Fig. 2). In epidermal keratinocytes (22) and in vascular endothelium (67, 105), increased expression of HO-1 stimulated proliferation, whereas inhibition of HO-1 enzymatic activity by administration of tin protoporphyrin (SnPPIX) or targeted knockdown of HO-1 gene expression by siRNA reversed this pro-proliferative effect (22, 103). In contrast, HO-1 is a negative regulator of growth in epithelium (2), astroglia (147), T lymphocytes (146), fibroblasts (126), and smooth muscle cells (29, 122). Here, the upregulation of HO-1 decreases cell proliferation, and appropriately, the antiproliferative effect is reversed by HO-1 inhibition (29).
FIG. 2. Schematic demonstration of the tissue-specific role of the HO-1 pathway in regulation of cell proliferation.
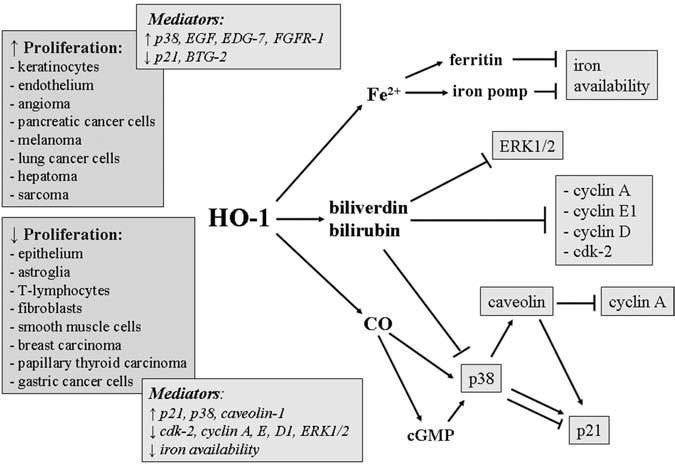
Suggested mediators are shown in the boxes.
Influence of HO-1 on proliferation can be also distinct in different types of cancer cells (Table 2). Direct pro-proliferative action has been shown in pancreatic tumor cell lines, in which downregulation of HO-1 by means of siRNA was associated with a significant inhibition of growth (9), and in murine and human melanoma cell lines, in which overexpression of HO-1 resulted in significantly augmented proliferation (167). Similarly, enhanced proliferation of the transformed endothelial cells infected with HHV-8 could be dependent on increased expression of HO-1, as suggested by abolishing effects of chromium mesoporphyrin IX (CrMPIX), an HO-1 inhibitor (108).
Table 2.
Effects of Activation of HO-1 in Tumor Cells
| Cancer | Species | Major effects of HO-1 activity | Ref. |
|---|---|---|---|
| Adenocarcinoma | Human | ↓ Oxidative stress | 47 |
| Angioma | Mouse | ↑ Tumor growth | 105 |
| Astroglia | Rat | ↓ Proliferation | 147 |
| Bladder carcinoma | Human | ↓ Apoptosis | 78 |
| Breast carcinoma | Rat, Human | ↓ Proliferation | 57 |
| Chronic myeloid leukemia | Human | ↓ Apoptosis | 107 |
| Colon carcinoma | Mouse, human | ↓ Apoptosis | 12, 120 |
| Gastric cancer cell line | Human | ↓ Apoptosis | 92 |
| Glioblastoma | Rat | ↑ Tumor vascularization | 26 |
| Hepatoma | Rat | ↑ Proliferation | 28, 59, 155 |
| ↓ apoptosis | |||
| Kaposi sarcoma | ↑ Proliferation, ↑ angiogenic potential | 105, 108 | |
| Leukemia | ↓ Apoptosis | 89 | |
| Lung cancer | Mouse | ↑ Tumor growth, ↑ angiogenic potential | 59 |
| ↓ apoptosis | |||
| Melanoma | Human | ↑ Proliferation, ↑ angiogenic potential, ↑ resistance to oxidative stress | 158, 167 |
| ↑vascularization of tumors | |||
| Melanoma | Mouse | ↑ Proliferation, ↑ angiogenic potential, ↑ resistance to oxidative stress | 167 |
| ↑ tumor vascularization, ↑ metastasis, ↓ inflammatory of infiltration of tumors | |||
| ↓ survival of tumor bearing mice | |||
| Pancreatic cancer | Human | ↑ Proliferation, ↑ tumor growth | 9, 152 |
| ↑ vascularization of tumors | |||
| Sarcoma | Mouse | ↑ Tumor growth | 28, 35, 59 |
| ↓ apoptosis | |||
| Squamous carcinoma | Human | ↑ Metastasis | 162 |
| Thyroid carcinoma | Human | ↓ Apoptosis | 18 |
Conversely, the opposite, antiproliferative results of HO-1 activity were shown for breast cancer cells. In this case, HO-1 inhibition with SnPPIX led to a small but significant increase in proliferation of rat and human cell lines, whereas induction of HO-1 with cobalt protoporphyrin IX (CoPPIX) drastically decreased cell-cycle progression. A less dramatic but still significant result was obtained with the other HO-1 inducer, heme, or on transduction of breast cancer cells with lentiviral vectors coding for HO-1 (57).
Importantly, the influence of HO-1 on the proliferation of cancer cells has been supported by results of experiments performed in animal models. Reports published hitherto confirm the permissive role of HO-1 in tumor growth. High levels of HO-1 expression resulting either from pharmacologic or genetic manipulation were associated with faster growth of tumors, as indicated by bigger volumes of nodules or by more numerous cancer cells. This has been shown for pancreatic cancer (152), angioma (105), and melanoma (167). Accordingly, inhibition of HO-1 resulted in decreased growth of tumors, as demonstrated in pancreatic cancer (9, 152), lung cancer (59), angioma (105), hepatoma, and sarcoma (28).
Mechanisms involved in modulation of cell divisions by HO-1 still require elucidation (see Fig. 2). Microarray analysis of murine melanoma transcriptome showed that although the underlying signal-transduction pathways remain unknown, the increased proliferation of HO-1 overexpressing cells can be associated with lower levels of two negative regulators of the cell cycle, p21 and B-cell translocation gene-2 (BTG2) (167). The cyclin-dependent kinase (Cdk) inhibitor p21, a major player in cell-cycle control, is often downregulated in tumors, and its binding to Cdk2 inhibits cell-cycle progression (7). Decreased p21 expression resulting from HO-1 activation was observed in endothelial cells (86) and a colon cancer cell line (12). Similarly BTG2, a tumor suppressor, is reduced in many types of cancers (150). It blocks proliferation through inhibition of retinoblastoma protein (pRb) pathway, decreased expression of cyclin D1, and by direct binding to Cdk2 (87). Importance of downregulation of p21 has been already indicated in clinical studies, in cutaneous malignant melanoma growth (70), whereas inhibition of BTG2 was postulated as an essential step in renal cancer development (150).
Additionally, overexpression of HO-1 in melanoma cells was accompanied by increased production of epidermal growth factor (EGF) and by higher levels of two mitogenic receptors: endothelial differentiation gene-7 (EDG-7), a specific receptor for lysophosphatidic acid, and FGFR1, a receptor for fibroblast growth factors (167). Earlier experiments done in ovarian cancer and melanoma cells have shown that EDG7 or FGFR1 may increase proliferation (149), but the mechanism responsible for regulation of EGF, EDG7, or FGFR1 by HO-1 are not recognized.
Among the products of HO-1 enzymatic activity, the most possible mediator of the pro-proliferative effects seems to be CO. Its significance was shown in endothelial cells (67), and the mechanism responsible for increased proliferation was the downregulation of p21 (86). Confusingly, p21 was also suggested as a protein mediating an HO-1-dependent inhibition of proliferation. In this case, increased activity of HO-1 was associated with augmented expression of p21. Such a relation was demonstrated in epithelial cells (46), vascular smooth muscle cells (29), papillary thyroid carcinoma cells (18), and the gastric cancer cell line (92). Again, the postulated effector molecule in antiproliferative action of HO-1 is CO, which stimulates the p38 mitogen-activated protein kinase (p38-MAPK) through induction of sGC and elevation of cGMP. Activation or overexpression of p38-MAPK can result in permanent cell-cycle arrest and premature cell senescence (62).
An interesting recent study suggests that the antiproliferative effect of CO can to be governed by caveolin-1 (75), the principal structural component of caveolae, potentially regulating many downstream signaling processes that originate in the plasma membrane (139). It has been shown, for example, that overexpression of caveolin-1 in mouse fibroblasts arrests these cells in the G0/G1 phase of the cell cycle through a pathway dependent on p53 and p21 (41). Interestingly, caveolin-1 negatively regulates the enzymatic activity of HO-1 in endothelium (74).
Elevated levels of caveolin-1 appear in aged animals and in fully differentiated or senescent cells (165). Conversely, its expression is downregulated in tumors or in oncogene-transformed cell lines (32). Activation of the p38-MAPK and sGC/cGMP signaling pathways by CO may lead to an enhanced expression of caveolin-1, as demonstrated in fibroblasts and smooth muscle cells. This in turn results in an increased expression of p21, downregulation of cyclin A, and consequently, in growth arrest (75). No data, however, show the effect of CO on caveolin-1 expression in cancer cells.
It can be supposed that antiproliferative activity of HO-1 can be also mediated by biliverdin/bilirubin, as shown in vascular smooth muscle cells. Bilirubin can suppress expression of cyclin A, D1, and E, as well as cdk-2, and inhibit hyperphosphorylation of the pRb (122). Additionally, bilirubin significantly inhibits phosphorylation of p38-MAPK. Despite results showing the negative effect of p38-MAPK activation in cell proliferation (62). p38-MAPK can be also an important positive mediator in cellular mitogenesis, necessary for cell-cycle progression (175). Thus, inhibition of p38-MAPK by bilirubin may block proliferation. The discrepancies in opinions on the role of p38 in proliferation are still not clarified, and the proposed role for p38 as an effector kinase in bilirubin- or CO-mediated regulation of cell cycle appears to be contradictory, even in the same cell type studied (75, 122). One can suppose that such differences in interpretation arise because the specific roles of individual isotypes of p38 MAPKs (α, β, γ, δ) remain incompletely understood (75).
Finally, bilirubin was shown to inhibit phosphorylation of extracellular signal-regulated protein kinase-1/2 (ERK1/2) and thereby decrease mitogenic response of airway smooth muscle cells (154). The mechanisms responsible for role of biliverdin/bilirubin in cancer cells are yet to be explored. It should also be kept in mind that decrease in cellular heme content and resulting iron scarcity in cells with a very high level of HO-1 expression may be also a cause of inhibition of tumor growth (40).
The well-documented, but contradictory effects of HO-1 on cell proliferation suggest that the final output does not depend on HO-1 alone, but is rather determined by the mutual balance of several players, including HO-1, biliverdin reductase (BvR), P450 cytochromes, ferritin, iron-regulated proteins (IRPs), caveolin-1, p38-MAPK isoforms, and possibly other, still not recognized mediators.
HO-1 AND CELL DIFFERENTIATION
The role of HO-1 in cancer cell differentiation is not well established. Significant changes in HO-1 expression were described in erythroleukemia, in which the onset of erythroid differentiation by treatment with dimethyl sulfoxide (DMSO) is associated with a rapid decline in mRNAs for HO-1, heat-shock protein-70 (Hsp-70), and nonspecific δ-aminolevulinate synthase (ALAS). These alterations are followed by sequential increases in mRNAs for enzymes in the heme biosynthetic pathway, such as erythroid-specific ALAS, δ-aminolevulinate dehydratase, porphobilinogen deaminase and uroporphyrinogen decarboxylase (40).
Changes in HO-1 expression were also detected in differentiating the myeloid leukemia cell line. These cells can differentiate bidirectionally (i.e., to erythrocytes after treatment with hemin) and to monocytes in response to 12-O-tetradecanoylphorbol 13-acetate (TPA). Both compounds can upregulate HO-1; however, inhibition of HO-1 activity by SnPPIX suppresses TPA-induced maturation to monocytes. Thus, HO-1 could be suggested as a directional switch and permissive enzyme in monocytic differentiation program of myeloid leukemia (79). Conversely, pharmacologic activation of HO-1 can block maturation of dendritic cells (17) and prevent differentiation of osteoclast precursors to osteoclasts (176).
Many cell-differentiation programs involve the activation of p38-MAPK, including neuronal differentiation (111), maturation of erythroid precursors (114), as well as myogenesis (24) and adipogenesis (31). Keeping in mind the regulation of p38-MAPK by HO-1-derived CO and bilirubin, one can expect that HO-1 would contribute to these processes. Such an involvement, including potential induction of differentiation of cancer cells is, however, still mostly speculative and not supported by strong experimental data.
HO-1 AND TUMOR RESISTANCE TO STRESS AND APOPTOSIS
Studies on HO-1 knockout mice strongly support the cytoprotective role of HO-1 in various cells and organs. HO-1 deficiency results in sustained oxidative stress, enhanced lipid peroxidation, accentuated oxidative damage in cardiovascular system, and progressive chronic inflammation in the kidney and liver. Cultured HO-1-/- embryonic fibroblasts are also highly sensitive to heme- or H2O2-mediated cytotoxicity (130). Accordingly, expression of HO-1 provides an effective protection for cells and tissues under stressful conditions (Fig. 3).
FIG. 3. Schematic demonstration of the role of the HO-1 pathway in regulation of cell apoptosis.
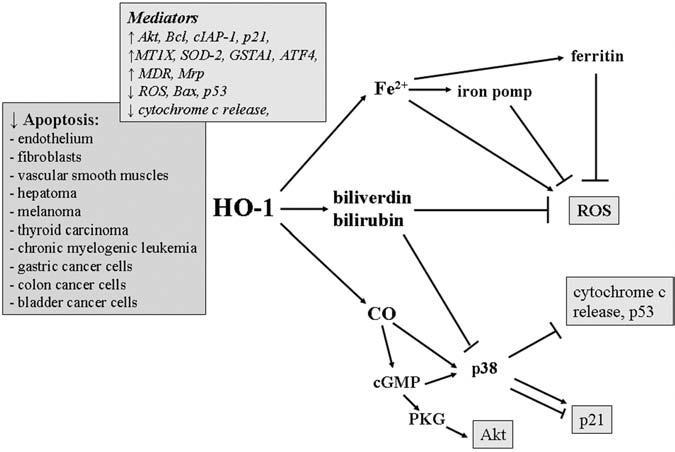
Suggested mediators are shown in the boxes.
Many studies have convincingly shown that HO-1 is a cytoprotective and antiapoptotic enzyme also in tumor cells exposed to oxidative stress, hypoxia, serum deprivation, or toxic compounds. Pharmacologic or genetic upregulation of HO-1 significantly improves survival of hepatoma (28), melanoma (167), thyroid carcinoma (18), chronic myelogenous leukemia (107), gastric cancer (92), and colon cancer cell lines (12). Decreased viability resulting from HO-1 inhibition was found in colon carcinoma (12) and in chronic myelogenous leukemia cells (107). Importantly, HO-1 inhibitors or targeted knockdown of HO-1 expression makes the cultured tumor cell lines much more sensitive to anticancer therapy, whereas HO-1 activation can efficiently protect them. Thus, reduced survival after HO-1 inhibition was demonstrated in pancreatic cells subjected to radiotherapy and chemotherapy (9), or in colon carcinoma (36) and chronic myelogenous leukemia (107) treated with chemotherapy. In some cases, the efficacy of the treatment was increased by more than 80% (36). Conversely, induction of HO-1 makes tumor cells more tolerant to the therapies, as shown in leukemia cells (89) and in bladder carcinoma cells subjected to photodynamic therapy (78), or chronic myelogenous leukemia treated with chemotherapeutic drugs (107). Interestingly, tumor cell lines known to express HO-1 constitutively, such as human lung adenocarcinoma A549 cells, are resistant to some toxic compounds [for example, to epigallocatechin 3-gallate (EGCG), the major polyphenol found in green tea, which itself upregulates HO-1 and exerts antiproliferative or proapoptotic effects in many cancer cells (80)]. Interestingly, inhibition of proliferation by EGCG is observed also in melanoma cells, in which upregulation of HO-1 is a pro-proliferative event (116). This indicates that HO-1 is relatively weak modulator of proliferation, and its effect can be outweighed by cytostatics.
The most important observations seem to be, however, those made in vivo, in tumor-bearing animals subjected to anticancer therapies. Generally they confirm conclusions from data obtained in vitro: induction of HO-1 protects tumor cells, whereas its inhibition increases the efficacy of anticancer therapies. Thus, administration of zinc protoporphyrin-IX (ZnPPIX) or ZnPPIX-polyethylene glycol (ZnPPIX-PEG) resulted in a significant increase in apoptosis of hepatoma cells in rats (155), as well as sarcoma and lung tumor cells in mice. In contrast, CoPPIX treatment increased HO-1 expression and reduced apoptosis of hepatoma (59).
Surprisingly, some clinical data suggested that expression of HO-1 in tumor tissues was associated with a higher sensitivity of patients to radiotherapy, whereas lack of HO-1 in tumors was much more common in the nonresponders. As a consequence, the authors postulated that expression of HO-1 may be an useful indicator of radiosensitivity, at least for esophageal cancer patients (173). More clinical data, including other types of tumors, are required to understand this relation and to explain the inconsistency with animal models.
The mechanism underlying the cytoprotective activity of HO-1 in tumor cells is still not clarified equivocally. One possible explanation is the increase in biliverdin/bilirubin levels, because supplementation of cultured cells with biliverdin or bilirubin can mimic the effects of HO-1 induction, as shown for hepatoma (155) and colon canceroma (12). Some other results suggest that cytoprotection of HO-1 is mediated by CO (155). Many studies have reported that exogenous administration of CO augmented the viability of different cell types, including fibroblasts, endothelial cells, and vascular smooth muscle cells (20, 126). Finally, indications were noted that the major cytoprotective compound may be ferritin, which is upregulated by many HO-1 inducers, such as hemin, and then by iron released from the HO-1-mediated degradation of heme. Thus, it has been demonstrated that sensitivity to oxidants in tumor cell lines is inversely correlated with ferritin protein levels (14). Increased expression of ferritin was suggested as a mechanism responsible for the protective effect of hemin in leukemia cells subjected to photodynamic therapy (89). Evaluations of ferritin levels in tumor tissue itself have revealed, however, a complex, perhaps disease-specific picture: in some cases, such as colon cancer and breast cancer, increase in ferritin in tumor tissue versus comparable normal tissue have been reported; in other cases, including liver cancer, a decrease in ferritin is seen (159).
Aside from its antioxidative action, HO-1 can directly affect cell viability by blocking apoptosis. This property was shown first in fibroblasts (126) and then was confirmed in various cell types (11, 145), as well as in animal models of inflammation, ischemia-reperfusion injury, hypoxia, and organ transplantation (102). Apparently, protection against apoptosis plays an important role in the cancer-supportive environment (140). Moreover, it may lead to a considerable resistance of tumors to chemotherapy. Here, the meaning of HO-1 can be significant, as its antiapoptotic efficacy has been already demonstrated both in tumor cells cultured in vitro and in solid tumors growing in animals (12, 18, 92, 107, 155).
Phosphorylation of Akt/protein kinase B has been reported in many carcinomas as a pathway involved in blocking apoptosis (49). It may contribute also to antiapoptotic effects of HO-1 in cancer cells, as shown in a colon cancer cell line, in which activation of Akt was accompanied by a higher B-cell lymphoma-2 (Bcl-2) to Bcl-2-associated X protein (Bax) ratio (12). In addition, elevated HO-1 can be related to increased cellular caspase inhibitory protein-2 (cIAP2) and decreased activity of caspase-3 in gastric cancer cells (92). Finally, induction of p21, observed in some HO-1-overexpressing cell lines, may result in improved resistance to apoptotic stimuli (31, 92). It seems, however, that no direct relation exists between resistance to apoptosis and expression of a single gene. For example, melanoma cells or colon cancer cells overexpressing HO-1 were more resistant to stressful conditions despite downregulated p21 (12). Instead, analysis of the transcriptome of melanoma suggested that the cytoprotective effect of HO-1 overexpression could be associated with upregulation of metallothionein-1X (MT1X), superoxide dismutase-2 (SOD2), and glutathione S-transferase A1 (GSTA1), which are known to enhance cancer resistance to radiotherapy, and with increased expression of activating transcription factor-4 (ATF4), which interacts with Nrf2 and increases production of enzymes of antioxidant and xenobiotic detoxification functions (167).
Antiapoptotic effects of HO-1 can be directly associated with its antioxidative potency, as ROS are the known inducers of apoptosis in many cell types (65). On the one hand, it may result from decrease in the cellular levels of iron, as shown in embryonic fibroblasts (37). On the other, the important role may be played by the antioxidant bilirubin. The latter is suggested on the basis of experiments performed in rat hepatoma, in which supplementation with bilirubin was able to reverse proapoptotic effects of ZnPPIX-induced HO-1 inhibition (155). Similarly, bilirubin and biliverdin were indicated as antiapoptotic mediators in colon cancer cells (12).
Another molecule potentially involved in the inhibition of apoptosis by HO-1 in tumors can be CO, which may act both through activation of p38-MAPK (75) and by activation of soluble guanylyl cyclase (20). Importantly, CO was showed not only to block release of the mitochondrial cytochrome c, the crucial event for induction of apoptosis, but also to inhibit expression of the proapoptotic protein p53 (36). Actually, the exposure to CO has been shown to reduce the TNF-induced apoptosis in cultured fibroblasts (126), endothelium (11), vascular smooth muscle cells (91), and hepatocytes (161). However, in colon carcinoma, gastric cancer cells, and chronic myelogenous leukemia cells, CO did not seem to play a role (12, 107).
The induction of HO-1 is not always sufficient to protect the cells. As demonstrated in breast carcinoma and B-lymphoblasts, HO-1 did not protect the cells from chemotherapy-induced apoptosis (5). Moreover, some observations showed that overexpression of HO-1 increased oxidative damage to mitochondria and resulted in augmented apoptosis in breast carcinoma and astroglia (147). Detrimental effects of HO-1 overexpression have been reported also in other cell types, such as smooth muscle cells (91). Nevertheless, the vast majority of experiments performed in different tumor cells, such as hepatoma (155), colon adenocarcinoma (12, 120, 155), thyroid carcinoma (18), gastric carcinoma (92), melanoma (167), and myelogenous leukemia cells (107), indicate that HO-1 is a potent cytoprotective and antiapoptotic enzyme, which improves survival of cancer cells subjected to different kinds of therapy.
Interestingly, some observations point to concomitant expression of HO-1 and multidrug resistance (MDR) or multidrug resistance-associated (Mrp) proteins, which might additionally decrease sensitivity of cancer cells to chemotherapy (118). However, no data exist on the regulation of MDR or Mrp by HO-1, and these genes can be induced independently, in response to the same stressors. As HO-1 is upregulated with the panel of other cytoprotective agents, it is always necessary to use proper inhibitors or activators to clarify the specific role of HO-1, and reliable conclusions cannot be drawn on the basis of analysis of HO-1 expression only.
HO-1 AND TUMOR ANGIOGENESIS
Apart from the cytoprotective activities, HO-1 has been recognized as a proangiogenic enzyme. Angiogenesis, the formation of new blood vessels from preexisting ones, plays a crucial role during growth and spreading of tumors; thus, proangiogenic action of HO-1 may further support tumor progression (20).
The role of HO-1 in angiogenesis (Fig. 4) has been convincingly demonstrated in both in vitro and in vivo experiments. HO-1-deficient endothelial cells produced less vascular endothelial growth factor (VEGF) than did the wild-type counterparts, and their response to exogenous VEGF and basic fibroblast growth factor (bFGF) was weaker (21). Accordingly, transduction of the HO-1 gene into endothelial cells increased production of VEGF and facilitated VEGF-induced activities. Thus, overexpression of HO-1 led to more-efficient cell proliferation and migration, improved formation of capillary-like tubular structures in a Matrigel matrix, and augmented outgrowth of capillaries from endothelial spheroids in a collagen gel (67). Stimulatory effects of HO-1 on the expression of VEGF, one of the most important inducers of angiogenesis, were also demonstrated in vascular smooth muscle cells (29) and keratinocytes (63). At least in microvascular endothelium and vascular smooth muscle cells, this effect appears to be mediated by CO (30, 67, 86). Importantly, the results of experiments performed in vitro have been confirmed in vivo in the rat ischemic hindlimb model, in which HO-1 gene transfer increased VEGF synthesis, facilitated angiogenesis, and improved the blood flow in the ischemic muscles (153).
FIG. 4. Schematic demonstration of the role of the HO-1 pathway in regulation of angiogenesis in tumors.

Suggested mediators are shown in the boxes.
The influence of HO-1 on angiogenesis in tumors was suggested first on the basis of observations that expression of HO-1 in macrophages infiltrating the tumors seems to correlate with increased vascular density (26, 152, 158), as shown in human gliomas (117) and human vertical growth melanomas (158). Then it was reported that HO-1 accelerates pancreatic cancer growth by promoting tumor angiogenesis in mice (152). Moreover, targeted knockdown gene expression of HO-1 by siRNA or chemical inhibition of HO-1 enzymatic activity by SnPPIX impaired vGPCR oncogene-induced survival and VEGF-A expression in the transformed endothelial cells (105). A similar decrease in VEGF synthesis after treatment with HO-1 inhibitor was found in the lung carcinoma (59).
A proangiogenic role of HO-1 (Fig. 5) was also directly evidenced in murine melanoma (167) and murine lung cancer (59). Interestingly, HO-1 overexpression in the melanoma cell line did not lead to increased production of VEGF in vitro, and VEGF did not contribute significantly to the increased angiogenic potential of media harvested from such cells. Instead, the transcriptome analysis demonstrated that overexpression of HO-1 was accompanied by upregulation of EGF, thymosin-β4 (Tβ4), hyaluronidase-1 (HYAL-1), and malignant T cell-amplified sequence-1 (MCT-1) (167).
FIG. 5. HO-1 overexpression in murine B16(F10) melanoma cells increases vascularization of tumors grown subcutaneously in mice.
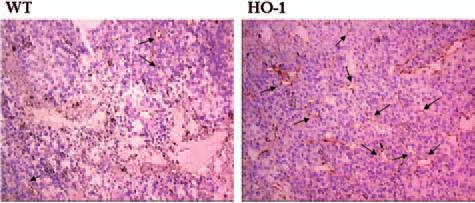
Blood vessels were stained in the frozen sections with anti-CD31 antibodies, visualized by peroxidase reaction. Tissues were counterstained with hematoxylin. Representative pictures. Endothelial cells labeled by brownish reaction product are indicated by arrows. WT, tumors formed by the wild-type cells; HO-1, tumors formed by HO-1-overexpressing cells (after 167).
The involvement of these proteins in proangiogenic activity of HO-1 has not been proven; however, their role can be hypothesized. An especially interesting candidate seems to be Tβ4, a small peptide present in the cytoplasm and nucleus of cells and circulating in the blood, known for its G-actin-sequestering capacities. Several observations suggested that Tβ4 plays a role in angiogenesis. First, Tβ4 can induce synthesis of VEGF (15) and is a potent chemotactic agent for endothelial cells (148). Second, transfection with a Tβ4-expression vector, similar to the overexpression of the HO-1 transgene, was associated with increased endothelial cell migration and stronger tube formation on Matrigel (104). Of importance, the effects of overexpression of Tβ4 in murine melanoma cells greatly resembled those of HO-1, as in both cases, augmented angiogenesis in tumors was observed (15).
Similarly, it was found that overexpression of HYAL1, an endoglycosidase, which degrades hyaluronic acid into small angiogenic fragments, resulted in a higher microvessel density in bladder cancer (94) and in prostate cancer (143), whereas MCT-1 was shown to induce angiogenesis in breast cancer (85). Finally, comparison of endothelium originating from healthy tissues and from tumors indicated that tumor-derived endothelial cells, but not normal endothelial cells respond efficiently to EGF. This suggested that gain of sensitivity to mitogenic signals of EGF by endothelium may constitute a switch that promotes tumor neovascularization (4). Thus, the observed increase in EGF production in the HO-1-overexpressing melanoma cells, together with upregulation of HYAL1 or MCT-1, can potentially contribute to the observed increase in angiogenesis. Tumors formed by such cells produced higher amounts of VEGF. Keeping in mind the lack of correlation of HO-1 and VEGF expression in melanoma cells themselves, one can suppose that an indirect influence of HO-1 present in tumor cells may exist on production of VEGF in stroma, especially in macrophages. This effect could be possibly mediated by diffused CO (167).
It seems that induction of HO-1 can be also a pathway responsible for proangiogenic effects of thymidyne phosphorylase (TP), the enzyme that is often upregulated in tumor tissues. Increased formation of capillaries in vitro as well as augmented vascularization in the tumors in vivo was observed as a result of TP expression in endothelial cells and in tumor-infiltrating macrophages, respectively (104, 158). TP directly induces HO-1, and coexpression of these two proteins was detected especially in the melanoma tumors of advanced stages. The importance of HO-1 in TP-stimulated activities was confirmed by observations that some effects of TP, such as regulation of proliferation of vascular smooth muscle cells, were blocked by inhibitors of both TP and HO-1 (82).
For many years, tumor vascularization was explained solely by the ingrowth of new vessels into the tumor tissue from preexisting ones. However, in recent years, additional mechanisms have been recognized, and at present, great interest exists in evaluating the role of endothelial progenitor cells and vasculogenesis in tumor neovascularization (48). The importance of homing and engraftment of bone marrow-derived vascular progenitors remains undefined. It seems, however, that at least in some types of tumors, vasculogenesis may be meaningful (1).
One of the crucial factors governing vasculogenesis is VEGF, regulation of which is influenced by HO-1. The second very important agent is stroma-derived growth factor-1 (SDF-1), a chemokine that plays a major role in migration, recruitment, and retention of endothelial progenitor cells to sites of ischemic injury and contributes to neovascularization. Analyses of intracranial murine gliomas evidenced the production of SDF-1 in the vasculogenic tumors. Furthermore, enforced expression of SDF-1 augmented, whereas blocking SDF-1 receptor, C-X-C motif receptor-4 (CXCR4), reduced homing and engraftment of vascular progenitors (1). These observations confirm an involvement of SDF-1 in tumor vasculogenesis.
No data are available on the contribution of HO-1 in this process. Such an influence, however, is very possible, in light of the very recent experiments demonstrating an important role of HO-1 in SDF-1 activities. Thus, nanomolar concentrations of SDF-1 potently activated HO-1 expression in human and murine aortic endothelial cells and murine EPC. Pharmacologic inhibition of HO activity or genetic ablation of HO-1 resulted in the loss of SDF-1-mediated endothelial tube formation in vitro and sprouting from aortic rings ex vivo, effects that were restored by the addition of CO, but not bilirubin. HO-1 was also required for the effects of SDF-1 on migration of mature endothelial cells and circulating endothelial precursors in vitro, as well as in vivo in Matrigel-plug, wound-healing, and retinal ischemia models of angiogenesis (27). It would be worthwhile to clarify the role of HO-1 in tumor vasculogenesis.
HO-1 AND METASTASIS
Angiogenesis is critical not only for tumor growth but also for metastasis. The invasion and metastasis of tumors is a highly complex and multistep process that requires a tumor cell to modulate its ability to adhere, degrade the surrounding extracellular matrix, migrate, proliferate at a secondary site, and stimulate angiogenesis. The progression of a tumor from benign and delimited growth to invasive and metastatic growth is the major cause of poor clinical outcome in cancer patients (131).
Because of stimulatory role of HO-1 in angiogenesis, one can expect that HO-1 can also facilitate metastasis (Fig. 6). Melanoma cells overexpressing HO-1 formed many more nodules in the lungs after the intravenous injection than did their wild-type counterparts (167). Similarly, overexpression of HO-1 in pancreatic cancer cells potentiated cancer aggressiveness and increased the colonization of the lungs, whereas inhibition of HO activity by stannous mesoporphyrin completely inhibited the occurrence of metastasis (152).
FIG. 6. HO-1 overexpression in the murine B16(F10) melanoma line increases the metastatic potential of cells.

Number of metastatic lung nodules in mice 3 weeks after intravenous inoculation with the wild-type (WT) and HO-1-overexpressing (HO-1) melanoma cells. (A) Representative photos showing the lungs fixed with Bouin solution. (B) Quantitative analysis: bars represent the mean ± SEM of 10 values. *p < 0.05 in comparison with B16-WT animals (after 167).
The mechanisms responsible for prometastatic activity of HO-1 are not recognized. It can be supposed that the same mediators that can augment angiogenesis can also improve colonization of lung and growth of metastatic nodules. Thus, Tβ4 and HYAL1, which were upregulated in HO-1-overexpressing melanoma cells, have been already shown to play a role in tumor metastasis. Expression of Tβ4 was demonstrated in metastatic oral squamous cell carcinoma lines (164), melanomas (167), fibrosarcomas (77), and in samples coming from patients with non-small cell lung cancer (64). Enforced overexpression was associated with an increase in melanoma cell migration and with stronger vascularization of solid tumors, although it had no effect on cell invasion, proliferation, or matrix metalloproteinase activities (15). In colon carcinoma cells, overexpression of Tβ4 increased the growth rate, motility, and colony formation in soft agar, all good indicators for malignant progression (166).
The second candidate, HYAL1, is the major hyaluronidase expressed and secreted by the head and neck, prostate, and bladder cancer cells (39, 97, 98). In patients with bladder cancers, increased HYAL1 level can serve as a marker of the high-grade tumors (95), which illustrates its significance. HYAL1 is also increased in metastatic breast cancer cells (100). Enzymatic activity of HYAL1 reduces contact inhibition and promotes tumor cell migration (55). It has been shown that overexpression of HYAL1 in prostate cancer cells causes an increase in lung metastasis (125). Accordingly, blocking HYAL1 expression in an invasive bladder cancer cell line decreased tumor growth, decreased microvessel density, and inhibited tumor infiltration (94). Taken together, these observations show that HYAL1 is a tumor promoter and can be hypothesized as a potential mediator of the prometastatic action of HO-1.
However, it should be kept in mind that HO-1 not always increases tumor invasiveness. It has been demonstrated that inhibition of HO-1 in colon carcinoma by treatment with zinc deuteroporphyrin 2,4-bis glycol (ZnDPBG) does not have any effect on metastasis to the lung and even increases metastasis to the liver (61). Furthermore, low-level HO-1 was associated with an increased risk of developing lymph node metastasis in oral and tongue squamous cell carcinoma. It could be associated with the lower expression of HO-1 in more undifferentiated cells (172). Thus, the influence of HO-1 on the metastatic potential of cancer cells is not univocal and may depend on the type of cancer or other, still not defined factors.
HO-1 AND INFLAMMATION
HO-1 is commonly regarded as a potent antiinflammatory enzyme (Fig. 7). Numerous reports demonstrated that activation of HO-1 diminishes inflammation and may result in immunosuppression (170). The importance of HO-1 is elegantly illustrated by studies in HO-1-/- mice, in which HO-1 deficiency results in strongly increased generation of proinflammatory cytokines, including IL-1β, interferon-γ (IFN-γ), TNF, and interleukin-6 (IL-6), and, in general, Th1-weighted shift in immune activity (69). Such animals develop progressive inflammatory disease characterized by splenomegaly, lymphadenopathy, leukocytosis, as well as hepatic and renal inflammation (130). Moreover, a constitutive expression of HO-1 observed in CD4+/CD25+ regulatory T cells indicates its crucial role in modulation of the immune response (124). Accordingly, elevated HO-1 activity may lead to clonal deletion of CD4+ T cells, resulting in a specific immunomodulation and in prolongation of transplanted organ survival (110).
FIG. 7. Schematic demonstration of the role of HO-1 pathway in the regulation of inflammatory reactions in tumors.

Suggested mediators are shown in the boxes.
Many data suggest that overexpression of HO-1 may defend tissues and organs from immune-mediated injury, either through protection against oxidative damage or via a local immunomodulatory influence on infiltrating inflammatory cells (123, 124). The exact mechanisms underlying the antiinflammatory functions of the HO-1 have not been fully elucidated. However, the signaling action of CO combined with the antioxidant properties of biliverdin/bilirubin and the sequestration of iron by ferritin could all contribute to suppression of inflammation.
CO has been described as an efficient antiinflammatory mediator in several models of inflammation and tissue injury. Thus, in lipopolysaccharide (LPS)-stimulated macrophages, it decreased significantly the generation of inducible nitric oxide synthase (iNOS)-derived NO and production of TNF or IL-6 (123, 135). These effects of CO can be caused by interfering with AP-1 activity via a c-Jun N-terminal kinase (JNK)-dependent pathway (112). Conversely, CO increases production of antiinflammatory cytokine, IL-10, in macrophages (84, 123).
Similarly, biliverdin/bilirubin production and Fe2+ chelation can diminish inflammatory response. For example, they reduce leukocyte adhesion to vascular endothelium via changes in expression of various adhesion molecules, such as E-selectin, intracellular adhesion molecule-1 (ICAM-1), and vascular cell adhesion molecule-1 (VCAM-1), the effects depending on inhibition of nuclear factor-κB (NF-κB) activation (54). The importance of biliverdin/bilirubin was also confirmed in HO-1-mediated reduction in monocyte chemotaxis in response to oxidized low-density lipoprotein (LDL) (60), whereas the role of iron was demonstrated in regulation of TNF in a monocytic cell line (142).
It is well known that inflammation can be involved in tumorigenesis; thus, the modulation of an inflammatory response by HO-1 may influence the progression of tumors. However, the precise role of inflammation in tumor development is not fully understood. On the one hand, the infiltration of leukocytes into tumor tissue can be considered an antitumor response, which results in reduction of tumor growth (88). On the other, many data suggest that the infiltrating macrophages and lymphocytes are the major sources of proinflammatory cytokines, growth factors, and angiogenic mediators. All these products can influence cell proliferation and survival, promote angiogenesis, stimulate migration of tumor cells into the stroma, and increase metastasis (23).
Tumors are infiltrated by many types of leukocytes, including T and B lymphocytes, NK cells, macrophages, dendritic cells, neutrophils, mastocytes, and eosinophils. Most of them, however, are lymphocytes, and the antitumor effects depend mainly on the lymphocyte-derived cytokines (88). Macrophages play an important role. They are able to destroy tumor cells by releasing lysosomal enzymes, TNF, or macrophage-activating factor (MAF) and function as the antigen-presenting cells, thereby activating lymphocytes (56). Simultaneously, however, macrophages can promote growth of tumors by production of angiogenic factors such as IL-1β, IL-8, bFGF, VEGF, or EGF (33), and by releasing proteolytic enzymes, which degrade extracellular matrix and stimulate tumor cell migration. Many clinical data suggest that leukocyte infiltration within the tumor is a negative prognostic factor for the patients (23). Moreover, infiltration by early-phase inflammatory cells, both macrophages and neutrophils, is associated with increased progression of many types of tumors (119). This suggests that the antiinflammatory action of HO-1 could be potentially beneficial for patients.
Unfortunately, knowledge of the role of HO-1 in a tumor-associated inflammation is limited. Results obtained from murine melanoma suggest that, at least in some models, HO-1 overexpression can inhibit inflammation, as indicated by less-pronounced leukocyte infiltration of the tumors. In melanomas growing subcutaneously, in which leukocyte infiltrates consist mostly of neutrophils, this inhibition was accompanied by better vascularization of tumors and worse survival of the tumor-bearing mice (167). Because CO, the important mediator of the antiinflammatory activity of HO-1, can diffuse through cell membranes, overexpression of HO-1 in melanoma cells can directly modulate the activity of infiltrating immunocytes through increased generation of CO. In this way, CO could reduce the release of proinflammatory cytokines. Conversely, the biliverdin/bilirubin system could protect melanoma cells from oxidative stress mediators generated by activated neutrophils.
Mice bearing melanoma cells with a high level of HO-1 had lower concentrations of TNF and higher levels of soluble TNF-R1 in the blood and tumor tissues, which could contribute to the observed reduction of the inflammatory reaction. Additionally, it has been suggested that reduction of TNF availability can promote vascularization of tumors, as the toxic effects of high doses of TNF on tumor vasculature are well documented (167). The endothelial cells of the tumor vessels seem to have a stronger expression of TNF-R1, a receptor crucial for induction of apoptosis, than do those of the healthy vessels. Therefore, tumor vessels could be more vulnerable to TNF-induced apoptosis. Injury of vessels leads to increased permeability and pronounced tissue edema (163). Actually, higher density of vessels and smaller edema were both observed in melanomas overexpressing HO-1, which could result from decreased TNF activity.
Additionally, melanoma with enforced HO-1 expression generated more Tβ4 (167), which can also decrease an inflammatory reaction (148), and produced less leukocyte-specific protein-1 (LSP-1), an intracellular molecule involved in leukocyte migration and recruitment to sites of inflammation (167). However, the mechanism(s) responsible for the antiinflammatory effects of HO-1 in melanoma have not been proven. Similarly, it is difficult to assess the real contribution of inhibition of inflammation in promoting the tumor development observed in HO-1-overexpressing melanoma, and to clarify whether these are causative or coincidental events.
HO-1 INHIBITION AS A POTENTIAL THERAPEUTIC STRATEGY
Investigations of the role of HO-1 seem to be important not only for better understanding of tumor-growth regulation but also for clinical practice. Although some reports describe a selective diminishing of HO-1 in malignant cancer cells, such as adenocarcinoma or tongue squamous carcinoma (172), the majority of analyses indicate that the expression of HO-1 is strongly upregulated in various tumors (26, 28, 47, 56, 101, 158, 162), especially in those treated with chemotherapeutics or exposed to radiation or photodynamic therapy (56, 101).
This upregulation in response to therapeutic procedures may have important consequences. For example, in the chronic myeloid leukemia-derived cell line K562, induction of HO-1 was found to counteract Gleevec-induced apoptosis (107). Similarly, increased expression of HO-1 in mice harboring adenocarcinoma and treated with photodynamic therapy resulted in much faster regrowth of tumors (120). These results support the idea that HO-1 may be a potential target in antitumor therapy. Thus, pharmacologic inhibition of HO-1 has been suggested as a new therapeutic option and potential sensitizer to chemotherapy, radiotherapy, or photodynamic therapy for chronic myeloid leukemia (107), colon carcinoma (36), adenocarcinoma (120), pancreatic cancer (9), and melanoma (167). The efficacy of such treatments has been proven in animal models. Thus, administration of ZnPPIX significantly suppressed the growth of hepatoma in rats (28), and sarcoma (35) or lung cancer in mice (59).
However, ZnPPIX must be administrated in nonphysiologic solutions. Therefore, especially interesting are the water-soluble HO-1 inhibitors, such as polyethyleneglycol (PEG)-conjugated ZnPPIX (PEG-ZnPPIX). PEG can be bound to ZnPPIX through newly introduced amino groups, in which ethylenediamine residues are added at C6 and C7 of protoporphyrin. The resulting compound becomes highly water soluble and forms multimolecular associations in aqueous media (133).
It has been demonstrated that PEG-ZnPPIX administered intravenously has a circulation time in blood that is 40 times longer than that for nonpegylated ZnPPIX. More important, it preferentially accumulates in solid-tumor tissues and produces tumor-selective suppression of HO activity. The major reason for tumor-selective targeting of PEG-ZnPPIX is attributed to the enhanced permeability and retention effect that is observed commonly in solid tumors for biocompatible macromolecular drugs (35). Similar accumulation within the tumor tissue was demonstrated for PEG-conjugated xanthine oxidase (PEG-XO), which mediates anticancer activity because of its ability to generate cytotoxic reactive oxygen species (134).
Intravenous administration of PEG-ZnPPIX induced apoptosis and suppressed the growth of sarcoma tumors in mice, without any apparent side effects (35). Similar promising results were obtained in the inhibition of growth of murine colon carcinoma (36). Therefore, possibly this type of compound could be regarded as a potential anticancer drug, sensitizing tumors to the different types of therapies.
It must be kept in mind, however, that many effects of pharmacologic HO-1 inhibitors [e.g., SnPPIX, ZnPPIX, zinc mesoporphyrin (ZnMPIX), ZnDPIX], as well as HO-1 activators (e.g., heme, CoPPIX, CoCl2), are HO-1 independent, because all these compounds display strong, unspecific functions (66, 93). For example, exposure of Hep3B cells to heme, CoCl2, and SnPPIX resulted in a potent, HO-1-independent induction of erythropoietin (99). Similar effects have been observed in endothelial cells, in which CoPPIX upregulated synthesis of proangiogenic and proinflammatory cytokine IL-8. This upregulation was not influenced by SnPPIX and therefore was possibly not associated with increased HO-1 expression (93). Finally, in THP-1 monocyte heme, ZnMPIX, ZnDPPIX, and CoPPIX, regardless of their effects on HO-1, reduced the transduction of the IFN-γ signal, whereas SnPPIX enhanced it and elevated the expression of MHC-II (169). It was shown that CoPPIX, SnPPIX, and ZnPPIX are direct inhibitors of caspase-3 and caspase-8, and thereby, they decrease the rate of apoptosis, independent of the HO-1 pathway (10).
Similarly, in vivo experiments showed that protoporphyrins significantly decreased tumor blood flow in rats, but this effect was apparently independent of HO-1, as both ZnPPIX (HO-1 inhibitor) and copper propoporphyrin-IX (CuPPIX, a compound that does not modulate HO-1 activity) were similarly efficient (72, 160). Finally, even a relatively high dose of ZnPPIX (45 μmol/kg) injected i.p. to the rats may be unable to inhibit the activity of HO-1 in tumors, although it could produce HO-1-independent effects (160).
HO-1 AND CARCINOGENESIS
HO-1 can protect tumor cells and thereby contribute to the progression of the disease. However, it acts as a cytoprotective agent also in healthy tissues exposed to harmful stimuli, including carcinogens. Therefore, the important question is whether HO-1 can be involved in protection of cells from induction of carcinogenesis or can promote this process. Unfortunately, no data directly address that subject. It can be only hypothesized that HO-1 may increase the resistance of cells to carcinogenesis, at least to that induced by compounds acting through generation of oxidative stress. Conversely, because of inhibiting apoptosis, HO-1 might facilitate cell transformation in response to mutagens. Most possibly, HO-1 can be an efficient protective enzyme against oxidative injury caused by heme. Heme is genotoxic, as shown in the human colon cell line and primary human colonocytes (44), and it acts as a carcinogen, especially to the colon (128). However, even in such experimental settings, the role of HO-1 in carcinogenesis has not been definitely proven.
Almost nothing is known, as well, about the effect of modulation of HO-1 on the expression or activity of the most important oncogenes or cancer-related genes. Only one report concerns the telomerase activity. It demonstrates that telomerase and telomerase reverse transcriptase (TERT), the major regulator of telomerase function, are not influenced by HO-1 or by tin mesoporphyrin-IX (SnMPIX) (43).
Nevertheless, many studies convincingly showed that treatment of animals with carcinogens is very often associated with induction of HO-1. For example, HO-1 was upregulated and maintained elevated expression in rats exposed to alachlor, the inducer of olfactory mucosal tumors (42). Similarly, induction of HO-1 was increased in hepatic carcinogenesis induced by dietary p-dimethylaminoazobenzene (DAB). Here, the high level of HO-1 activity was suggested to protect hepatocytes against reactive oxygen species produced from DAB (132). HO-1 expression increased during the experimental trial in morphologically normal hepatocytes in DAB-treated animals, whereas it diminished in altered hepatic foci and early preneoplastic lesions. Thus, in this experimental model, the downmodulation of HO-1 expression correlated with malignancy progression (13). Unfortunately, it is impossible to discriminate whether HO-1 downregulation is a cause or consequence of transformation.
Divergent levels of HO-1 expression have been observed in the rat strains of different sensitivities to cancerogenesis. Thus, carcinogen-resistant DRH rats show a remarkably lower incidence of liver tumors than did the carcinogen-sensitive Donryu strain when they are fed diets containing hepatocarcinogens such as DAB. In DRH rats exposed to DAB, much weaker inductions of HO-1 and hepatocyte growth factor (HGF) were observed. This was not regarded, however, as a mechanism responsible for carcinogen resistance, but rather as a marker of milder injury of tissues. Weaker induction of HO-1 would be an indicator of lower degradation of heme proteins, whereas a lower level of HGF would result from a reduced rate of cell death and less pronounced regeneration of the liver. Instead, the suggested reason underlying the different susceptibility of DRH and Donryu rats to carcinogens were the distinct levels of expression of cytochrome P-450 2E, which may contribute to the generation of reactive oxygen species, and glutathione S-transferase or γ-glutamyltranspeptidase, which can be marker enzymes of preneoplastic changes of hepatocytes (171).
Accordingly, the high levels of expression of HO-1 were detected in the Long-Evans with a cinnamon-like color (LEC) rats, a mutant strain that spontaneously develops acute hepatitis and hepatoma. Upregulation of HO-1 in the LEC rat livers was, however, not due to the actual cancer lesion but, rather, due to the surrounding uninvolved tissues, including hepatocytes. Again, it was suggested that the physiologic relevance of HO-1 induction might be an adaptive response to oxidative stress (106).
Expression of HO-1 can be increased in response not only to carcinogens, but also to some chemopreventive compounds. One of them is diallyl sulfide (DAS), which can reduce chemically induced hepatotoxicity, mutagenesis, and carcinogenesis. It has been shown that DAS can induce the expression of HO-1 because of elevated production of ROS and subsequent increase in activity of Nrf2 transcription factor, as well as ERK and p38 kinases. The increase in HO-1 produced by DAS protected the HepG2 cells against toxicity by hydrogen peroxide or arachidonic acid. Therefore, it has been proposed that induction of HO-1 may play a role also in the anticarcinogenic effects of DAS (45). Conversely, it has been implied that HO-1 expression and subsequent iron accumulation may be involved in enhancement of oxidative DNA damage in epithelium of small bile ducts induced by infection with Opisthorchis viverrini, a risk factor of cholangiocarcinoma in hamsters (129).
The role of HO-1 in carcinogenesis has not been elucidated. Data available can support either protective or detrimental effects. Experiments performed in HO-1-deficient animals or at least in animals treated with pharmacologic inhibitors are necessary to distinguish the primary effect of HO-1 activity on induction of cancer from the secondary influence of carcinogenesis on expression of HO-1.
HO-1 POLYMORPHISM AND TUMORS
Some light can be shed on the role of HO-1 in carcinogenesis by analysis of clinical data and comparison of HO-1 allele frequency distribution in the healthy people and the cancer patients. Importantly, the human HO-1 gene promoter is highly polymorphic, mainly because of variation in the number of GT repeats in a microsatellite DNA fragment ranging from 15 to 40 (76). This purine-pyrimidine-alternating sequence can negatively affect transcription, especially as it is located between the regulatory elements and TATA box of the HO-1 gene (121). Indeed, transient transfections with reporter plasmids showed that basal transcriptions from the short promoter constructs (GT, 16-20) were 2.5 times higher than those from long ones (GT, 29-38) and that only short promoters were induced in response to oxidative stress (19).
These results are meaningful because the expression of HO-1 is regulated predominantly at the transcriptional level (102). Their importance was confirmed in lymphoblastoid cell lines derived from human subjects possessing short (S/S) or long (L/L) HO-1 genotypes. After H2O2 stimulation, the S/S cells showed much higher activity of HO-1 enzyme than did their L/L counterparts. Furthermore, only S/S promoters ensured protection of lymphoblastoid cells from H2O2-induced apoptosis (58).
Most important, HO-1 promoter polymorphism is of clinical relevance. As demonstrated in many reports, the absence of the short allele significantly augments the inflammatory reaction in the vessel wall and increases the risk for restenosis in patients undergoing balloon angioplasty (34) or stenting (137). It also potentiates the oxidative stress, leading to the higher risk of coronary artery disease in diabetic or hypercholesterolemic patients (19) and correlates with increased probability of development of abdominal aortic aneurysms (138) and with decreased survival time of kidney transplants (6). Finally, the L/L genotype carriers have a higher extent of lipid peroxidation in serum, supporting the genetic linkage of HO-1 with oxidative stress (19).
Finally, recent reports indicate that lack of short allele and resulting lower activity of HO-1 can be associated with a higher incidence of cancers. One of studies was performed in the Japanese population. In this case, the proportion of class L allele frequencies was significantly higher in patients with lung adenocarcinoma than in control subjects. Furthermore, the risk of lung adenocarcinoma for L allele carriers versus non-L allele carriers was much increased in the group of male smokers. However, in the female nonsmokers, the proportion of L allele carriers did not differ between patients and control subjects (73).
A second study concerned the oral squamous cell carcinoma (OSCC) caused by betel chewing in an Asian population. Also in this case, the longer (GT)n repeat allele in the HO-1 promoter is associated with the increased risks of areca-related OSCC, whereas the shorter (GT)n repeat allele may have protective effects for OSCC (16). Additionally, an insertion/deletion polymorphism (-94ins/delATTG) in NF-κB promoter, which may drive the ins allele twofold increase in NF-κB transcription relative to the del allele, was recently found. Interestingly, subjects carried both NF-κB ins and HO-1 L alleles had significant risks for various subsets of OSCC, including those of advanced stage or with node metastasis (90). Thus, one can expect that lower resistance to oxidative stress, especially when combined with increased inflammatory reactions, results in a higher risk of cancers, especially in smokers and betel chewers.
However, the role of HO-1 promoter polymorphism may depend on the type of tumor. Differently than for OSCC or lung cancers, the calculated risk for acquiring primary malignant melanoma in L-allele carriers was twofold lower compared with those with S/S types. Additionally, the S/S genotype was significantly associated with primary tumors with deeper Breslow thickness compared with L-allele carriers. These data suggest that higher levels of HO-1 expression might facilitate the pathogenesis and growth of malignant melanoma (121), which confirms data obtained in the murine melanoma model (167).
FINAL REMARKS
Some results suggest that HO-1 can act as a protective enzyme, decreasing the risk of development of some kinds of tumors. However, the actual role of HO-1 in transformation of cells and induction of carcinogenesis has not been clarified, and the data available hitherto are not conclusive. Much more is known of the function of HO-1 in the progression of tumors (Fig. 8). Procancerogenic effects of HO-1 seem to be associated with its cytoprotective and antiapoptotic activities, which result in better tumor cell survival and higher resistance to different types of therapies. Moreover, HO-1 is a proangiogenic mediator, which augments vascularization of tumors and increases the metastatic potential of cancer cells. In general, HO-1 seems to facilitate tumor growth and metastasis, although the exact effects can depend on the type of disease.
FIG. 8.
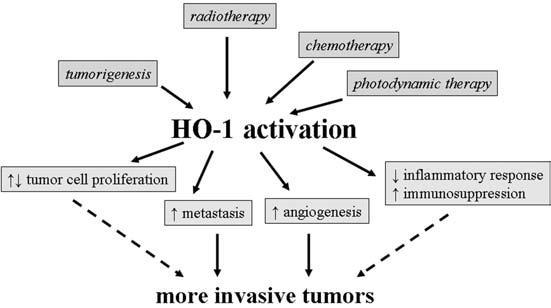
Schematic summary of possible effects of HO-1 activation on tumor growth.
Thus, we propose that HO-1 can be considered a “friend” protecting healthy tissues from induction of some types of cancers. However, if, despite this protection, the disease starts to develop, HO-1 turns into a “false friend,” as it will protect the tumor cells, improving their survival and resistance to treatments. Therefore, the inhibition of HO-1 activity can be proposed as a potential therapeutic strategy that could sensitize cancer cells to chemotherapy, radiation, or photodynamic therapy.
ACKNOWLEDGMENTS
This work was supported by grants from the Polish Ministry for Education and Science, PBZ KBN 107 P04 2004, 2 P04 016 26, 106 P05 01, and N301 080 32/3156. A.J. is the International Senior Research Fellow of The Wellcome Trust.
ABBREVIATIONS
- ALAS
aminolevulinate synthase
- AP-1
activator protein-1
- ATF4
activating transcription factor-4
- Bax
Bcl-2-associated X protein
- Bcl-2
B-cell lymphoma-2
- bFGF
basic fibroblast growth factor
- BTG2
B-cell translocation gene-2
- BvR
biliverdin reductase
- Cdk
cyclin-dependent kinase
- c-IAP-2
cellular caspase inhibitory protein-2
- CoPPIX
cobalt protoporphyrin IX
- CrMPIX
chromium mesoporphyrin IX
- CuPPIX
copper protoporphyrin-IX
- CXCR-4
C-X-C motif receptor-4
- DAB
p-dimethylaminoazobenzene
- DAS
diallyl sulfide
- DMSO
dimethyl sulfoxide
- EDG7
endothelial differentiation gene-7
- EGCG
epigallocatechin 3-gallate
- EGF
epidermal growth factor
- ERK
extracellular signal-regulated protein kinase
- FGFR-1
fibroblast growth factor receptor-1
- GSTA1
glutathione S-transferase A1
- HGF-1
hepatocuyte growth factor
- HO-1
heme oxygenase-1
- HO-2
heme oxygenase-2
- Hsp
heat-shock protein
- HYAL1
hyaluronidase-1
- ICAM-1
intracellular adhesion molecule-1
- IL-1β
interleukin-1β
- IL-6
interleukin-6
- IL-8
interleukin-8
- IL-10
interleukin-10
- IFN-γ
interferon-γ
- iNOS
inducible nitric oxide synthase
- IRP
ironregulated proteins
- JNK
c-Jun N-terminal kinase
- LDL
lowdensity lipoprotein
- LEC
Long-Evans with a cinnamon-like color
- LPS
lipopolysaccharide
- LSP-1
leukocyte-specific protein-1
- MAF
macrophage-activating factor
- MAPK
mitogenactivated protein kinase
- MCT-1
malignant T cell-amplified sequence-1
- MDR
multidrug resistance
- MHC-II
major histocompatibility complex-II
- Mrp
multidrug resistance-associated proteins
- MT1X
metallothionein-1X
- MIP-1β
macrophage inflammatory protein-1β
- NF-κB
nuclear factor κB
- Nrf-2
NF-E2-related factor-2
- OSCC
oral squamous cell carcinoma
- PEG
polyethylene glycol
- pRb
protein retinoblastoma
- ROS
reactive oxygen species
- SDF-1
stromal cell-derived growth factor-1
- sGC
soluble guanylyl cyclase
- SnMPIX
tin mesoporphyrin-IX
- SnPPIX
tin protoporphyrin-IX
- SOD-2
superoxide dismutase-2
- Tβ4
thymosin-β4
- TERT
telomerase reverse transcriptase
- TNF
tumor necrosis factor
- TNF-R1
tumor necrosis factor receptor-1
- TP
thymidine phosphorylase
- TPA
12-O-tetracanoylphorbol 13-acetate
- VCAM-1
vascular cell adhesion molecule-1
- VEGF
vascular endothelial growth factor
- ZnDPBG
zinc deuteroporphyrin 2,4-bis glycol
- ZnMPIX
zinc mesoporphyrin-IX
- ZnPPIX
zinc protoporphyrin-IX
REFERENCES
- 1.Aghi M, Cohen KS, Klein RJ, Scadden DT, Chiocca EA. Tumor stromal-derived factor-1 recruits vascular progenitors to mitotic neovasculature, where microenviroment influences their differentiated phenotypes. Cancer Res. 2006;66:9054–9064. doi: 10.1158/0008-5472.CAN-05-3759. [DOI] [PubMed] [Google Scholar]
- 2.Aizawa T, Ishizaka N, Kurokawa K, Nagai R, Nakajima H, Taguchi J, Ohno M. Different effects of angiotensin II and catecholamine on renal cell apoptosis and proliferation in rats. Kidney Int. 2001;59:645–653. doi: 10.1046/j.1523-1755.2001.059002645.x. [DOI] [PubMed] [Google Scholar]
- 3.Alam J, Shibahara S, Smith A. Transcriptional activation of the heme oxygenase gene by heme and cadmium in mouse hepatoma cells. J Biol Chem. 1989;264:6371–6375. [PubMed] [Google Scholar]
- 4.Amin DN, Hida K, Bielenberg DR, Klagsbrun M. Tumor endothelial cells express epidermal growth factor receptor (EGFR) but not ErbB3 and are responsive to EGF and to EGFR kinase inhibitors. Cancer Res. 2006;66:2173–2180. doi: 10.1158/0008-5472.CAN-05-3387. [DOI] [PubMed] [Google Scholar]
- 5.Andreadi CK, Howells LM, Atherfold PA, Manson MM. Involvement of Nrf2, p38, B-Raf, and nuclear factor-kappaB, but not phosphatidylinositol 3-kinase, in induction of hemeoxygenase-1 by dietary polyphenols. Mol Pharmacol. 2006;69:1033–1040. doi: 10.1124/mol.105.018374. [DOI] [PubMed] [Google Scholar]
- 6.Baan C, Peeters A, Lemos F, Uitterlinden A, Doxiadis I, Claas F, Ijzermans J, Roodnat J, Weimar W. Fundamental role for HO-1 in the self-protection of renal allografts. Am J Transplant. 2004;4:811–818. doi: 10.1111/j.1600-6143.2004.00420.x. [DOI] [PubMed] [Google Scholar]
- 7.Ball KL. p21: Structure and functions associated with cyclin-CDK binding. Prog Cell Cycle Res. 1997;3:125–134. doi: 10.1007/978-1-4615-5371-7_10. [DOI] [PubMed] [Google Scholar]
- 8.Bauer I, Rensing H, Florax A, Ulrich C, Pistorius G, Redl H, Bauer M. Expression pattern and regulation of heme oxygenase-1/heat shock protein 32 in human liver cells. Shock. 2003;20:116–122. doi: 10.1097/01.shk.0000075568.93053.fa. [DOI] [PubMed] [Google Scholar]
- 9.Berberat PO, Dambrauskas Z, Gulbinas A, Giese T, Giese N, Kunzli B, Autschbach F, Meuer S, Buchler MW, Friess H. Inhibition of heme oxygenase-1 increases responsiveness of pancreatic cancer cells to anticancer treatment. Clin Cancer Res. 2005;11:3790–3798. doi: 10.1158/1078-0432.CCR-04-2159. [DOI] [PubMed] [Google Scholar]
- 10.Blumenthal SB, Kiemer AK, Tiegs G, Seyfried S, Holtje M, Brandt B, Holtje HD, Zahler S, Vollmar AM. Metalloporphyrins inactivate caspase-3 and -8. FASEB J. 2005;19:1272–1279. doi: 10.1096/fj.04-3259com. [DOI] [PubMed] [Google Scholar]
- 11.Brouard S, Otterbein LE, Anrather J, Tobiasch E, Bach FH, Choi AM, Soares MP. Carbon monoxide generated by heme oxygenase 1 suppresses endothelial cell apoptosis. J Exp Med. 2000;192:1015–1026. doi: 10.1084/jem.192.7.1015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Busserolles J, Megias J, Terencio MC, Alcaraz MJ. Heme oxygenase-1 inhibits apoptosis in Caco-2 cells via activation of Akt pathway. Int J Biochem Cell Biol. 2006;38:1510–1517. doi: 10.1016/j.biocel.2006.03.013. [DOI] [PubMed] [Google Scholar]
- 13.Caballero F, Meiss R, Gimenez A, Batlle A, Vazquez E. Immunohistochemical analysis of heme oxygenase-1 in preneoplastic and neoplastic lesions during chemical hepatocarcinogenesis. Int J Exp Pathol. 2004;85:213–222. doi: 10.1111/j.0959-9673.2004.00391.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Cermak J, Balla J, Jacob HS, Balla G, Enright N, Nath K, Vercellotti GM. Tumor cell heme uptake induces ferritin synthesis resulting in altered oxidant sensitivity: possible role in chemotherapy efficacy. Cancer Res. 1993;53:5308–5313. [PubMed] [Google Scholar]
- 15.Cha HJ, Jeong MJ, Kleinman HK. Role of thymosin beta4 in tumor metastasis and angiogenesis. J Natl Cancer Inst. 2003;95:1674–1680. doi: 10.1093/jnci/djg100. [DOI] [PubMed] [Google Scholar]
- 16.Chang KW, Lee TC, Yeh WI, Chung MY, Liu CJ, Chi LY, Lin SC. Polymorphism in heme oxygenase-1 (HO-1) promoter is related to the risk of oral squamous cell carcinoma occurring on male areca chewers. Br J Cancer. 2004;91:1551–1555. doi: 10.1038/sj.bjc.6602186. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Chauveau C, Remy S, Royer PJ, Hill M, Tanguy-Royer S, Hubert FX, Tesson L, Brion R, Beriou G, Gregoire M, Josien R, Cuturi MC, Anegon I. Heme oxygenase-1 expression inhibits dendritic cell maturition and proinflammatory function but conserves IL-10 expression. Blood. 2005;106:1694–1702. doi: 10.1182/blood-2005-02-0494. [DOI] [PubMed] [Google Scholar]
- 18.Chen GG, Liu ZM, Vlantis AC, Tse GM, Leung BC, van Hasselt CA. Heme oxygenase-1 protects against apoptosis by tumor necrosis factor-alpha and cycloheximide in papillary thyroid carcinoma cells. J Cell Biochem. 2004;92:1246–1256. doi: 10.1002/jcb.20157. [DOI] [PubMed] [Google Scholar]
- 19.Chen YH, Lin SJ, Lin MW, Tsai HL, Kuo SS, Chen JW, Charng MJ, Wu TC, Chen LC, Ding YA, Pan WH, Jou YS, Chau LY. Microsatellite polymorphism in promoter of heme oxygenase-1 gene is associated with susceptibility to coronary artery disease in type 2 diabetic patients. Human Genet. 2002;111:1–8. doi: 10.1007/s00439-002-0769-4. [DOI] [PubMed] [Google Scholar]
- 20.Cherrington JM, Strawn LM, Shawver LK. New paradigms for the treatment of cancer: the role of anti-angiogenesis agents. Adv Cancer Res. 2000;79:1–38. doi: 10.1016/s0065-230x(00)79001-4. [DOI] [PubMed] [Google Scholar]
- 21.Cisowski J, Loboda A, Jozkowicz A, Chen S, Agarwal A, Dulak J. Role of heme oxygenase-1 in hydrogen peroxide-induced VEGF synthesis: effect of HO-1 knockout. Biochem Biophys Res Commun. 2005;326:670–676. doi: 10.1016/j.bbrc.2004.11.083. [DOI] [PubMed] [Google Scholar]
- 22.Clark JE, Green CJ, Motterlini R. Involvement of the heme oxygenase-carbon monoxide pathway in keratinocyte proliferation. Biochem Biophys Res Commun. 1997;241:215–220. doi: 10.1006/bbrc.1997.7742. [DOI] [PubMed] [Google Scholar]
- 23.Coussens LM, Werb Z. Inflammation and cancer. Nature. 2002;420:860–867. doi: 10.1038/nature01322. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Cuenda A, Cohen P. Stress-activated protein kinase-2/p38 and a rapamycin-sensitive pathway are required for C2C12 myogenesis. J Biol Chem. 1999;274:4341–4346. doi: 10.1074/jbc.274.7.4341. [DOI] [PubMed] [Google Scholar]
- 25.De Matteis F, Dawson SJ, Pons N, Pipino S. Bilirubin and uroporphyrinogen oxidation by induced cytochrome P4501A and cytochrome P4502B: role of polyhalogenated biphenyls of different configuration. Biochem Pharmacol. 2002;63:615–624. doi: 10.1016/s0006-2952(01)00851-6. [DOI] [PubMed] [Google Scholar]
- 26.Deininger MH, Meyermann R, Trautmann K, Duffner F, Grote EH, Wickboldt J, Schluesener HJ. Heme oxygenase (HO)-1 expressing macrophages/microglial cells accumulate during oligodendroglioma progression. Brain Res. 2000;882:1–8. doi: 10.1016/s0006-8993(00)02594-4. [DOI] [PubMed] [Google Scholar]
- 27.Deshane J, Chen S, Caballero S, Grochot-Przeczek A, Was H, Lach R, Hill-Kapturczak N, Siegal GP, Dulak J, Jozkowicz A, Grant MB, Agarwal A. Stromal cell-derived factor-1 promotes angiogenesis via a heme oxygenase-1 dependent mechanism. J Exp Med. 2007;204:605–618. doi: 10.1084/jem.20061609. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Doi K, Akaike T, Fujii S, Tanaka S, Ikebe N, Beppu T, Shibahara S, Ogawa M, Maeda H. Induction of haem oxygenase-1, nitric oxide and ischaemia in experimental solid tumours and implications for tumour growth. Br J Cancer. 1999;80:1945–1954. doi: 10.1038/sj.bjc.6690624. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Duckers HJ, Boehm M, True AL, Yet SF, San H, Park JL, Clinton WR, Lee ME, Nabel GJ, Nabel EG. Heme oxygenase-1 protects against vascular constriction and proliferation. Nat Med. 2001;7:693–698. doi: 10.1038/89068. [DOI] [PubMed] [Google Scholar]
- 30.Dulak J, Jozkowicz A, Foresti R, Kasza A, Frick M, Huk I, Green CJ, Pachinger O, Weidinger F, Motterlini R. Heme oxygenase activity modulates vascular endothelial growth factor synthesis in vascular smooth muscle cells. Antioxid Redox Signal. 2002;4:229–240. doi: 10.1089/152308602753666280. [DOI] [PubMed] [Google Scholar]
- 31.Engelman JA, Berg AH, Lewis RY, Lin A, Lisanti MP, Scherer PE. Constitutively active mitogen-activated protein kinase kinase 6 (MKK6) or salicylate induces spontaneous 3T3-L1 adipogenesis. J Biol Chem. 1999;274:35630–35638. doi: 10.1074/jbc.274.50.35630. [DOI] [PubMed] [Google Scholar]
- 32.Engelman JA, Wykoff CC, Yasuhara S, Song KS, Okamoto T, Lisanti MP. Recombinant expression of caveolin-1 in oncogenically transformed cells abrogates anchorage-independent growth. J Biol Chem. 1997;272:16374–16381. doi: 10.1074/jbc.272.26.16374. [DOI] [PubMed] [Google Scholar]
- 33.Etoh T, Shibuta K, Barnard GF, Kitano S, Mori M. Angiogenin expression in human colorectal cancer: the role of focal macrophage infiltration. Clin Cancer Res. 2000;6:3545–3551. [PubMed] [Google Scholar]
- 34.Exner M, Schillinger M, Minar E, Mlekusch W, Schlerka G, Haumer M, Mannehalter C, Wagner O. Heme oxygenase-1 gene promoter microsatellite polymorphism is associated with restenosis after percutaneous transluminal angioplasty. J Endovasc Ther. 2001;8:433–440. doi: 10.1177/152660280100800501. [DOI] [PubMed] [Google Scholar]
- 35.Fang J, Sawa T, Akaike T, Akuta T, Sahoo SK, Khaled G, Hamada A, Maeda H. In vivo antitumor activity of pegylated zinc protoporphyrin: targeted inhibition of heme oxygenase in solid tumor. Cancer Res. 2003;63:3567–3574. [PubMed] [Google Scholar]
- 36.Fang J, Sawa T, Akaike T, Greish K, Maeda H. Enhancement of chemotherapeutic response of tumor cells by a heme oxygenase inhibitor, pegylated zinc protoporphyrin. Int J Cancer. 2004;109:1–8. doi: 10.1002/ijc.11644. [DOI] [PubMed] [Google Scholar]
- 37.Ferris CD, Jaffrey SR, Sawa A, Takahashi M, Brady SD, Barrow RK, Tysoe SA, Wolosker H, Baranano DE, Dore S, Poss KD, Snyder SH. Haem oxygenase-1 prevents cell death by regulating cellular iron. Nat Cell Biol. 1999;1:152–157. doi: 10.1038/11072. [DOI] [PubMed] [Google Scholar]
- 38.Foresti R, Clark JE, Green CJ, Motterlini R. Thiol compounds interact with nitric oxide in regulating heme oxygenase-1 induction in endothelial cells: involvement of superoxide and peroxynitrite anions. J Biol Chem. 1997;272:18411–18417. doi: 10.1074/jbc.272.29.18411. [DOI] [PubMed] [Google Scholar]
- 39.Franzmann EJ, Schroeder GL, Goodwin WJ, Weed DT, Fisher P, Lokeshwar VB. Expression of tumor markers hyaluronic acid and hyaluronidase (HYAL1) in head and neck tumors. Int J Cancer. 2003;106:438–445. doi: 10.1002/ijc.11252. [DOI] [PubMed] [Google Scholar]
- 40.Fujita H, Yamamoto M, Yamagami T, Hayashi N, Bishop TR, De Verneuil H, Yoshinaga T, Shibahara S, Morimoto R, Sassa S. Sequential activation of genes for heme pathway enzymes during erythroid differentiation of mouse Friend virus-transformed erythroleukemia cells. Biochim Biophys Acta. 1991;1090:311–316. doi: 10.1016/0167-4781(91)90195-r. [DOI] [PubMed] [Google Scholar]
- 41.Galbiati F, Volonte D, Liu J, Capozza F, Frank PG, Zhu L, Pestell RG, Lisanti MP. Caveolin-1 expression negatively regulates cell cycle progression by inducing G(0)/G(1) arrest via a p53/p21(WAF1/Cip1)-dependent mechanism. Mol Biol Cell. 2001;12:2229–2244. doi: 10.1091/mbc.12.8.2229. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Genter MB, Burman DM, Vijayakumar S, Ebert CL, Aronow BJ. Genomic analysis of alachlor-induced oncogenesis in rat olfactory mucosa. Physiol Genomics. 2002;12:35–45. doi: 10.1152/physiolgenomics.00120.2002. [DOI] [PubMed] [Google Scholar]
- 43.Ghattas MH, Chuang LT, Kappas A, Abraham NG. Protective effect of HO-1 against oxidative stress in human hepatoma cell line (HepG2) is independent of telomerase enzyme activity. Int J Biochem Cell Biol. 2002;34:1619–1628. doi: 10.1016/s1357-2725(02)00097-3. [DOI] [PubMed] [Google Scholar]
- 44.Glei M, Klenow S, Sauer J, Wegewitz U, Richter K, Pool-Zobel BL. Hemoglobin and hemin induce DNA damage in human colon tumor cells HT29 clone 19A and in primary human colonocytes. Mutat Res. 2006;594:162–171. doi: 10.1016/j.mrfmmm.2005.08.006. [DOI] [PubMed] [Google Scholar]
- 45.Gong P, Hu B, Cederbaum AI. Diallyl sulfide induces heme oxygenase-1 through MAPK pathway. Arch Biochem Biophys. 2004;432:252–260. doi: 10.1016/j.abb.2004.09.024. [DOI] [PubMed] [Google Scholar]
- 46.Gonzalez-Michaca L, Farrugia G, Croatt AJ, Alam J, Nath KA. Heme: a determinant of life and death in renal tubular epithelial cells. Am J Physiol Renal Physiol. 2004;286:F370–F377. doi: 10.1152/ajprenal.00300.2003. [DOI] [PubMed] [Google Scholar]
- 47.Goodman AI, Choudhury M, da Silva JL, Schwartzman ML, Abraham NG. Overexpression of the heme oxygenase gene in renal cell carcinoma. Proc Soc Exp Biol Med. 1997;214:54–61. doi: 10.3181/00379727-214-44069. [DOI] [PubMed] [Google Scholar]
- 48.Goon PK, Lip GY, Boos CJ, Stonelake PS, Blann AD. Circulating endothelial cells, endothelial progenitor cells, and endothelial microparticles in cancer. Neoplasia. 2006;8:79–88. doi: 10.1593/neo.05592. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Goswami A, Ranganathan P, Rangnekar VM. The phosphoinositide 3-kinase/Akt1/Par-4 axis: a cancer-selective therapeutic target. Cancer Res. 2006;66:2889–2892. doi: 10.1158/0008-5472.CAN-05-4458. [DOI] [PubMed] [Google Scholar]
- 50.Graham BL, Mink JT, Cotton DJ. Dynamic measurements of CO diffusing capacity using discrete samples of alveolar gas. J Appl Physiol. 1983;54:73–79. doi: 10.1152/jappl.1983.54.1.73. [DOI] [PubMed] [Google Scholar]
- 51.Hanselmann C, Mauch C, Werner S. Haem oxygenase-1: a novel player in cutaneous wound repair and psoriasis? Biochem J. 2001;353:459–466. doi: 10.1042/0264-6021:3530459. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Hara E, Takahashi K, Tominaga T, Kumabe T, Kayama T, Suzuki H, Fujita H, Yoshimoto T, Shirato K, Shibahara S. Expression of heme oxygenase and inducible nitric oxide synthase mRNA in human brain tumors. Biochem Biophys Res Commun. 1996;224:153–158. doi: 10.1006/bbrc.1996.0999. [DOI] [PubMed] [Google Scholar]
- 53.Hayashi S, Omata Y, Sakamoto H, Higashimoto Y, Hara T, Sagara Y, Noguchi M. Characterization of rat heme oxygenase-3 gene: implication of processed pseudogenes derived from heme oxygenase-2 gene. Gene. 2004;336:241–250. doi: 10.1016/j.gene.2004.04.002. [DOI] [PubMed] [Google Scholar]
- 54.Hayashi S, Takamiya R, Yamaguchi T, Matsumoto K, Tojo SJ, Tamatani T, Kitajima M, Makino N, Ishimura Y, Suematsu M. Induction of heme oxygenase-1 suppresses venular leukocyte adhesion elicited by oxidative stress: role of bilirubin generated by the enzyme. Circ Res. 1999;85:663–671. doi: 10.1161/01.res.85.8.663. [DOI] [PubMed] [Google Scholar]
- 55.Hayen W, Goebeler M, Kumar S, Riessen R, Nehls V. Hyaluronan stimulates tumor cell migration by modulating the fibrin fiber architecture. J Cell Sci. 1999;112:2241–2251. doi: 10.1242/jcs.112.13.2241. [DOI] [PubMed] [Google Scholar]
- 56.Henry F, Bretaudeau L, Barbieux I, Meflah K, Gregoire M. Induction of antigen presentation by macrophages after phagocytosis of tumour apoptotic cells. Res Immunol. 1998;149:673–679. doi: 10.1016/s0923-2494(99)80037-6. [DOI] [PubMed] [Google Scholar]
- 57.Hill M, Pereira V, Chauveau C, Zagani R, Remy S, Tesson L, Mazal D, Ubillos L, Brion R, Asghar K, Mashreghi MF, Kotsch K, Moffett J, Doebis C, Seifert M, Boczkowski J, Osinaga E, Anegon I. Heme oxygenase-1 inhibits rat and human breast cancer cell proliferation: mutual cross inhibition with indoleamine 2,3-dioxygenase. FASEB J. 2005;19:1957–1968. doi: 10.1096/fj.05-3875com. [DOI] [PubMed] [Google Scholar]
- 58.Hirai H, Kubo H, Yamaya M, Nakayama K, Numasaki M, Kobayashi S, Suzuki S, Shibahara S, Sasaki H. Microsatellite polymorphism in heme oxygenase-1 gene promoter is associated with susceptibility to oxidant-induced apoptosis in lymphoblastoid cell lines. Blood. 2003;102:1619–1621. doi: 10.1182/blood-2002-12-3733. [DOI] [PubMed] [Google Scholar]
- 59.Hirai K, Sasahira T, Ohmori H, Fujii K, Kuniyasu H. Inhibition of heme oxygenase-1 by zinc protoporphyrin IX reduces tumor growth of LL/2 lung cancer in C57BL mice. Int J Cancer. 2007;120:500–505. doi: 10.1002/ijc.22287. [DOI] [PubMed] [Google Scholar]
- 60.Ishikawa K, Navab M, Leitinger N, Fogelman AM, Lusis AJ. Induction of heme oxygenase-1 inhibits the monocyte transmigration induced by mildly oxidized LDL. J Clin Invest. 1997;100:1209–1216. doi: 10.1172/JCI119634. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Ishikawa T, Yoshida N, Higashihara H, Inoue M, Uchiyama K, Takagi T, Handa O, Kokura S, Naito Y, Okanoue T, Yoshikawa T. Different effects of constitutive nitric oxide synthase and heme oxygenase on pulmonary or liver metastasis of colon cancer in mice. Clin Exp Metastasis. 2003;20:445–450. doi: 10.1023/a:1025448403124. [DOI] [PubMed] [Google Scholar]
- 62.Iwasa H, Han J, Ishikawa F. Mitogen-activated protein kinase p38 defines the common senescence-signalling pathway. Genes Cells. 2003;8:131–144. doi: 10.1046/j.1365-2443.2003.00620.x. [DOI] [PubMed] [Google Scholar]
- 63.Jazwa A, Loboda A, Golda S, Cisowski J, Szelag M, Zagorska A, Sroczynska P, Drukala J, Jozkowicz A, Dulak J. Effect of heme and heme oxygenase-1 on vascular endothelial growth factor synthesis and angiogenic potency of human keratinocytes. Free Radic Biol Med. 2006;40:1250–1263. doi: 10.1016/j.freeradbiomed.2005.11.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Ji P, Diederichs S, Wang W, Boing S, Metzger R, Schneider PM, Tidow N, Brandt B, Buerger H, Bulk E, Thomas M, Berdel WE, Serve H, Muller-Tidow C. MALAT-1, a novel noncoding RNA, and thymosin beta4 predict metastasis and survival in earlystage non-small cell lung cancer. Oncogene. 2003;22:8031–8041. doi: 10.1038/sj.onc.1206928. [DOI] [PubMed] [Google Scholar]
- 65.Johnson TM, Yu ZX, Ferrans VJ, Lowenstein RA, Finkel T. Reactive oxygen species are downstream mediators of p53-dependent apoptosis. Proc Natl Acad Sci U S A. 1996;93:11848–11852. doi: 10.1073/pnas.93.21.11848. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Jozkowicz A, Dulak J. Effects of protoporphyrins on production of nitric oxide and expression of vascular endothelial growth factor in vascular smooth muscle cells and macrophages. Acta Biochim Pol. 2003;50:69–79. [PubMed] [Google Scholar]
- 67.Jozkowicz A, Huk I, Nigisch A, Weigel G, Dietrich W, Motterlini R, Dulak J. Heme oxygenase and angiogenic activity of endothelial cells: stimulation by carbon monoxide and inhibition by tin protoporphyrin-IX. Antioxid Redox Signal. 2003;5:155–162. doi: 10.1089/152308603764816514. [DOI] [PubMed] [Google Scholar]
- 68.Jozkowicz A, Huk I, Nigisch A, Weigel G, Weidinger F, Dulak J. Effect of prostaglandin-J(2) on VEGF synthesis depends on the induction of heme oxygenase-1. Antioxid Redox Signal. 2002;4:577–585. doi: 10.1089/15230860260220076. [DOI] [PubMed] [Google Scholar]
- 69.Kapturczak MH, Wasserfall C, Brusko T, Campbell-Thompson M, Ellis TM, Atkinson MA, Agarwal A. Heme oxygenase-1 modulates early inflammatory responses: evidence from the heme oxygenase-1-deficient mouse. Am J Pathol. 2004;165:1045–1053. doi: 10.1016/S0002-9440(10)63365-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Karjalainen JM, Eskelinen MJ, Kellokoski JK, Reinikainen M, Alhava EM, Kosma VM. p21(WAF1/CIP1) expression in stage I cutaneous malignant melanoma: its relationship with p53, cell proliferation and survival. Br J Cancer. 1999;79:895–902. doi: 10.1038/sj.bjc.6690143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Keyse SM, Tyrrell RM. Heme oxygenase is the major 32-kDa stress protein induced in human skin fibroblasts by UVA radiation, hydrogen peroxide, and sodium arsenite. Proc Natl Acad Sci U S A. 1989;86:99–103. doi: 10.1073/pnas.86.1.99. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Khelifi AF, Prise VE, Tozer GM. Effects of tin-protoporphyrin IX on blood flow in a rat tumor model. Exp Biol Med. 2003;228:481–485. doi: 10.1177/15353702-0322805-10. [DOI] [PubMed] [Google Scholar]
- 73.Kikuchi A, Yamaya M, Suzuki S, Yasuda H, Kubo H, Nakayama K, Handa M, Sasaki T, Shibahara S, Sekizawa K, Sasaki H. Association of susceptibility to the development of lung adenocarcinoma with the heme oxygenase-1 gene promoter polymorphism. Hum Genet. 2005;116:354–360. doi: 10.1007/s00439-004-1162-2. [DOI] [PubMed] [Google Scholar]
- 74.Kim HP, Wang X, Galbiati F, Ryter SW, Choi AM. Caveolae compartmentalization of heme oxygenase-1 in endothelial cells. FASEB J. 2004;18:1080–1089. doi: 10.1096/fj.03-1391com. [DOI] [PubMed] [Google Scholar]
- 75.Kim HP, Wang X, Nakao A, Kim SI, Murase N, Choi ME, Ryter SW, Choi AM. Caveolin-1 expression by means of p38beta mitogen-activated protein kinase mediates the antiproliferative effect of carbon monoxide. Proc Natl Acad Sci U S A. 2005;102:11319–11324. doi: 10.1073/pnas.0501345102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Kimpara T, Takeda A, Watanabe K, Itoyama Y, Ikawa S, Watanabe M, Arai H, Sasaki H, Higuchi S, Okita N, Takase S, Saito H, Takahashi K, Shibahara S. Microsatellite polymorphism in the human heme oxygenase-1 gene promoter and its application in association studies with Alzheimer and Parkinson disease. Hum Genet. 1997;100:145–147. doi: 10.1007/s004390050480. [DOI] [PubMed] [Google Scholar]
- 77.Kobayashi T, Okada F, Fujii N, Tomita N, Ito S, Tazawa H, Aoyama T, Choi SK, Shibata T, Fujita H, Hosokawa M. Thymosin-beta4 regulates motility and metastasis of malignant mouse fibrosarcoma cells. Am J Pathol. 2002;160:869–882. doi: 10.1016/s0002-9440(10)64910-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Kocanova S, Buytaert E, Matroule JY, Piette J, Golab J, de Witte P, Agostinis P. Induction of heme-oxygenase 1 requires the p38(MAPK) and PI3K pathways and suppresses apoptotic cell death following hypericin-mediated photodynamic therapy. Apoptosis. 2007;12:731–741. doi: 10.1007/s10495-006-0016-x. [DOI] [PubMed] [Google Scholar]
- 79.Koiso Y, Nakajima O, Matsumura D, Fujimoto Y, Hashimoto Y. Chemical control of cell differentiation of human myeloleukemia K562 cell line. Yakugaku Zasshi. 2000;120:104–112. doi: 10.1248/yakushi1947.120.1_104. [DOI] [PubMed] [Google Scholar]
- 80.Kweon MH, Adhami VM, Lee JS, Mukhtar H. Constitutive overexpression of Nrf2-dependent heme oxygenase-1 in A549 cells contributes to resistance to apoptosis induced by epigallocatechin 3-gallate. J Biol Chem. 2006;281:33761–33772. doi: 10.1074/jbc.M604748200. [DOI] [PubMed] [Google Scholar]
- 81.Lavrovsky Y, Schwartzman ML, Levere RD, Kappas A, Abraham NG. Identification of binding sites for transcription factors NF-kappa B and AP-2 in the promoter region of the human heme oxygenase-1 gene. Proc Natl Acad Sci U S A. 1994;91:5987–5991. doi: 10.1073/pnas.91.13.5987. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Li W, Tanaka K, Marioka K, Uesaka T, Yamada N, Takamori A, Handa M, Tanabe S, Idaya A. Thymidine phosphorylase gene transfer inhibits vascular smooth muscle cell proliferation by upregulating heme oxygenase-1. Artherioscler Thromb Vasc Biol. 2005;25:1370–1375. doi: 10.1161/01.ATV.0000168914.85107.64. [DOI] [PubMed] [Google Scholar]
- 83.Lee PJ, Alam J, Wiegand GW, Choi AM. Overexpression of heme oxygenase-1 in human pulmonary epithelial cells results in cell growth arrest and increased resistance to hyperoxia. Proc Natl Acad Sci U S A. 1996;93:10393–10398. doi: 10.1073/pnas.93.19.10393. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Lee TS, Chau LY. Heme oxygenase-1 mediates the anti-inflammatory effect of interleukin-10 in mice. Nat Med. 2002;8:240–246. doi: 10.1038/nm0302-240. [DOI] [PubMed] [Google Scholar]
- 85.Levenson AS, Thurn KE, Simons LA, Veliceasa D, Jarrett J, Osipo C, Jordan VC, Volpert OV, Satcher RL, Jr, Gartenhaus RB. MCT-1 oncogene contributes to increased in vivo tumorigenicity of MCF7 cells by promotion of angiogenesis and inhibition of apoptosis. Cancer Res. 2005;65:10651–10656. doi: 10.1158/0008-5472.CAN-05-0845. [DOI] [PubMed] [Google Scholar]
- 86.Li Volti G, Sacerdoti D, Sangras B, Vanella A, Mezentsev A, Scapagnini G, Falck JR, Abraham NG. Carbon monoxide signaling in promoting angiogenesis in human microvessel endothelial cells. Antioxid Redox Signal. 2005;7:704–710. doi: 10.1089/ars.2005.7.704. [DOI] [PubMed] [Google Scholar]
- 87.Lim IK. TIS21 (/BTG2/PC3) as a link between ageing and cancer: cell cycle regulator and endogenous cell death molecule. J Cancer Res Clin Oncol. 2006;132:417–426. doi: 10.1007/s00432-006-0080-1. [DOI] [PubMed] [Google Scholar]
- 88.Lin EY, Pollard JW. Role of infiltrated leucocytes in tumour growth and spread. Br J Cancer. 2004;90:2053–2058. doi: 10.1038/sj.bjc.6601705. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Lin F, Girotti AW. Hyperresistance of leukemia cells to photodynamic inactivation after long-term exposure to hemin. Cancer Res. 1996;56:4636–4643. [PubMed] [Google Scholar]
- 90.Lin SC, Liu CJ, Yeh WI, Lui MT, Chang KW, Chang CS. Functional polymorphism in NFKB1 promoter is related to the risks of oral squamous cell carcinoma occurring on older male areca (betel) chewers. Cancer Lett. 2006;243:47–54. doi: 10.1016/j.canlet.2005.11.019. [DOI] [PubMed] [Google Scholar]
- 91.Liu XM, Chapman GB, Wang H, Durante W. Adenovirus-mediated heme oxygenase-1 gene expression stimulates apoptosis in vascular smooth muscle cells. Circulation. 2002;105:79–84. doi: 10.1161/hc0102.101369. [DOI] [PubMed] [Google Scholar]
- 92.Liu ZM, Chen GG, Ng EK, Leung WK, Sung JJ, Chung SC. Upregulation of heme oxygenase-1 and p21 confers resistance to apoptosis in human gastric cancer cells. Oncogene. 2004;23:503–513. doi: 10.1038/sj.onc.1207173. [DOI] [PubMed] [Google Scholar]
- 93.Loboda A, Jazwa A, Wegiel B, Jozkowicz A, Dulak J. Heme oxygenase-1-dependent and -independent regulation of angiogenic gene expression: effect of cobalt protoporphyrin and cobalt chloride on VEGF and IL-8 synthesis in human microvascular endothelial cells. Cell Mol Biol. 2005;51:347–355. [PMC free article] [PubMed] [Google Scholar]
- 94.Lokeshwar VB, Cerwinka WH, Lokeshwar BL. HYAL1 hyaluronidase: a molecular determinant of bladder tumor growth and invasion. Cancer Res. 2005;65:2243–2250. doi: 10.1158/0008-5472.CAN-04-2805. [DOI] [PubMed] [Google Scholar]
- 95.Lokeshwar VB, Obek C, Pham HT, Wei D, Young MJ, Duncan RC, Soloway MS, Block NL. Urinary hyaluronic acid and hyaluronidase: markers for bladder cancer detection and evaluation of grade. J Urol. 2000;163:348–356. doi: 10.1016/s0022-5347(05)68050-0. [DOI] [PubMed] [Google Scholar]
- 96.Lokeshwar VB, Obek C, Soloway MS, Block NL. Tumor-associated hyaluronic acid: a new sensitive and specific urine marker for bladder cancer. Cancer Res. 1997;57:773–777. [PubMed] [Google Scholar]
- 97.Lokeshwar VB, Rubinowicz D, Schroeder GL, Forgacs E, Minna JD, Block NL, Nadji M, Lokeshwar BL. Stromal and epithelial expression of tumor markers hyaluronic acid and HYAL1 hyaluronidase in prostate cancer. J Biol Chem. 2001;276:11922–11932. doi: 10.1074/jbc.M008432200. [DOI] [PubMed] [Google Scholar]
- 98.Lokeshwar VB, Young MJ, Goudarzi G, Iida N, Yudin AI, Cherr GN, Selzer MG. Identification of bladder tumor-derived hyaluronidase: its similarity to HYAL1. Cancer Res. 1999;59:4464–4470. [PubMed] [Google Scholar]
- 99.Lutton JD, Griffin MO, Nishimura M, Levere RD, Kappas A, Abraham NG, Shibahara S. Coexpression of erythropoietin and heme oxygenase genes in Hep3B cells. Hepatology. 1993;17:861–868. [PubMed] [Google Scholar]
- 100.Madan AK, Yu K, Dhurandhar N, Cullinane C, Pang Y, Beech DJ. Association of hyaluronidase and breast adenocarcinoma invasiveness. Oncol Rep. 1999;6:607–609. doi: 10.3892/or.6.3.607. [DOI] [PubMed] [Google Scholar]
- 101.Maines MD, Abrahamsson PA. Expression of heme oxygenase-1 (HSP32) in human prostate: normal, hyperplastic, and tumor tissue distribution. Urology. 1996;47:727–733. doi: 10.1016/s0090-4295(96)00010-6. [DOI] [PubMed] [Google Scholar]
- 102.Maines MD. Heme oxygenase: function, multiplicity, regulatory mechanisms, and clinical applications. FASEB J. 1988;2:2557–2568. [PubMed] [Google Scholar]
- 103.Maines MD. The heme oxygenase system: a regulator of second messenger gases. Annu Rev Pharmacol Toxicol. 1997;37:517–554. doi: 10.1146/annurev.pharmtox.37.1.517. [DOI] [PubMed] [Google Scholar]
- 104.Malinda KM, Goldstein AL, Kleinman HK. Thymosin beta 4 stimulates directional migration of human umbilical vein endothelial cells. FASEB J. 1997;11:474–481. doi: 10.1096/fasebj.11.6.9194528. [DOI] [PubMed] [Google Scholar]
- 105.Marinissen MJ, Tanos T, Bolos M, de Sagarra MR, Coso OA, Cuadrado A. Inhibition of heme oxygenase-1 interferes with the transforming activity of the Kaposi sarcoma herpesvirus encoded G protein-coupled receptor. J Biol Chem. 2006;281:11332–11346. doi: 10.1074/jbc.M512199200. [DOI] [PubMed] [Google Scholar]
- 106.Matsumoto A, Hanayama R, Nakamura M, Suzuki K, Fujii J, Tatsumi H, Taniguchi N. A high expression of heme oxygenase-1 in the liver of LEC rats at the stage of hepatoma: the possible implication of induction in uninvolved tissue. Free Radic Res. 1998;28:383–391. doi: 10.3109/10715769809070807. [DOI] [PubMed] [Google Scholar]
- 107.Mayerhofer M, Florian S, Krauth MT, Aichberger KJ, Bilban M, Marculescu R, Printz D, Fritsch G, Wagner O, Selzer E, Sperr WR, Valent P, Sillaber C. Identification of heme oxygenase-1 as a novel BCR/ABL-dependent survival factor in chronic myeloid leukemia. Cancer Res. 2004;64:3148–3154. doi: 10.1158/0008-5472.can-03-1200. [DOI] [PubMed] [Google Scholar]
- 108.McAllister SC, Hansen SG, Ruhl RA, Raggo CM, DeFilippis VR, Greenspan D, Fruh K, Moses AV. Kaposi sarcoma-associated herpesvirus (KSHV) induces heme oxygenase-1 expression and activity in KSHV-infected endothelial cells. Blood. 2004;103:3465–3473. doi: 10.1182/blood-2003-08-2781. [DOI] [PubMed] [Google Scholar]
- 109.McCoubrey WK, Huang TJ, Maines MD. Isolation and characterization of a cDNA from the rat brain that encodes hemoprotein heme oxygenase-3. Eur J Biochem. 1997;247:725–732. doi: 10.1111/j.1432-1033.1997.00725.x. [DOI] [PubMed] [Google Scholar]
- 110.McDaid J, Yamashita K, Chora A, Ollinger R, Strom TB, Li XC, Bach FH, Soares MP. Heme oxygenase-1 modulates the alloimmune response by promoting activation-induced cell death of T cells. FASEB J. 2005;19:458–460. doi: 10.1096/fj.04-2217fje. [DOI] [PubMed] [Google Scholar]
- 111.Morooka T, Nishida E. Requirement of p38 mitogen-activated protein kinase for neuronal differentiation in PC12 cells. J Biol Chem. 1998;273:24285–24288. doi: 10.1074/jbc.273.38.24285. [DOI] [PubMed] [Google Scholar]
- 112.Morse D, Pischke SE, Zhou Z, Davis RJ, Flavell RA, Loop T, Otterbein SL, Otterbein LE, Choi AM. Suppression of inflammatory cytokine production by carbon monoxide involves the JNK pathway and AP-1. J Biol Chem. 2003;278:36993–36998. doi: 10.1074/jbc.M302942200. [DOI] [PubMed] [Google Scholar]
- 113.Motterlini R, Foresti R, Bassi R, Calabrese V, Clark JE, Green CJ. Endothelial heme oxygenase-1 induction by hypoxia: modulation by inducible nitric-oxide synthase and S-nitrosothiols. J Biol Chem. 2000;275:13613–13620. doi: 10.1074/jbc.275.18.13613. [DOI] [PubMed] [Google Scholar]
- 114.Nagata Y, Takahashi N, Davis RJ, Todokoro K. Activation of p38 MAP kinase and JNK but not ERK is required for erythropoietin-induced erythroid differentiation. Blood. 1998;92:1859–1869. [PubMed] [Google Scholar]
- 115.Nakasa K, Kitayama M, Fukuda H, Kimura K, Yanagawa T, Ishii T, Nakashima K, Yamada K. Oxidative stress-related proteins A170 and heme oxygense-1 are differently induced in the rat cerebellum under kainate-mediated excitotoxicity. Neurosci Lett. 2000;282:57–60. doi: 10.1016/s0304-3940(00)00836-3. [DOI] [PubMed] [Google Scholar]
- 116.Nihal M, Ahmad N, Mukhtar H, Wood GS. Anti-proliferative and proapoptotic effects of (-)-epigallocatechin-3-gallate on human melanoma: possible implications for the chemoprevention of melanoma. Int J Cancer. 2005;20:513–521. doi: 10.1002/ijc.20785. [DOI] [PubMed] [Google Scholar]
- 117.Nishie A, Ono M, Shono T, Fukushi J, Otsubo M, Onoue H, Ito Y, Inamura T, Ikezaki K, Fukui M, Iwaki T, Kuwano M. Macrophage infiltration and heme oxygenase-1 expression correlate with angiogenesis in human gliomas. Clin Cancer Res. 1999;5:1107–1113. [PubMed] [Google Scholar]
- 118.Nishiya T, Kataoka H, Mori K, Goto M, Sugawara T, Furumara K. Tienilic acid enhances hyperbilirubinemia in Eisai hyperbilirubinuria rats through hepatic multidrug resistance-associated protein 3 and heme oxygenase-1 induction. Toxicol Sci. 2006;91:651–659. doi: 10.1093/toxsci/kfj162. [DOI] [PubMed] [Google Scholar]
- 119.Noël A, Gilles C, Bajou K, Devy L, Kebers F, Lewalle JM, Maquoi E, Munaut C, Remacle A, Foidart JM. Emerging roles for proteinases in cancer. Invasion Metastasis. 1997;17:221–239. [PubMed] [Google Scholar]
- 120.Nowis D, Legat M, Grzela T, Niderla J, Wilczek E, Wilczynski GM, Glodkowska E, Mrowka P, Issat T, Dulak J, Jozkowicz A, Was H, Adamek M, Wrzosek A, Nazarewski S, Makowski M, Stoklosa T, Jakobisiak M, Golab J. Heme oxygenase-1 protects tumor cells against photodynamic therapy-mediated cytotoxicity. Oncogene. 2006;25:3365–3374. doi: 10.1038/sj.onc.1209378. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121.Okamoto I, Krogler J, Endler G, Kaufmann S, Mustafa S, Exner M, Mannhalter C, Wagner O, Pehamberger H. A microsatellite polymorphism in the heme oxygenase-1 gene promoter is associated with risk for melanoma. Int J Cancer. 2006;119:1312–1315. doi: 10.1002/ijc.21937. [DOI] [PubMed] [Google Scholar]
- 122.Ollinger R, Bilban M, Erat A, Froio A, McDaid J, Tyagi S, Csizmadia E, Graca-Souza AV, Liloia A, Soares MP, Otterbein LE, Usheva A, Yamashita K, Bach FH. Bilirubin: a natural inhibitor of vascular smooth muscle cell proliferation. Circulation. 2005;112:1030–1039. doi: 10.1161/CIRCULATIONAHA.104.528802. [DOI] [PubMed] [Google Scholar]
- 123.Otterbein LE, Bach FH, Alam J, Soares M, Tao LH, Wysk M, Davis RJ, Flavell RA, Choi AM. Carbon monoxide has anti-inflammatory effects involving the mitogen-activated protein kinase pathway. Nat Med. 2000;6:422–428. doi: 10.1038/74680. [DOI] [PubMed] [Google Scholar]
- 124.Pae HO, Oh GS, Choi BM, Chae SC, Chung HT. Differential expression of heme oxygenase-1 in CD25- and CD25 + subsets of human CD4 + T cells. Biochem Biophys Res Commun. 2003;306:701–705. doi: 10.1016/s0006-291x(03)01037-4. [DOI] [PubMed] [Google Scholar]
- 125.Patel S, Turner PR, Stubberfield C, Barry E, Rohlff CR, Stamys A, McKenzie E, Young K, Tyson K, Terrett J, Box G, Eccles S, Page MJ. Hyaluronidase gene profiling and role of hyal-1 overexpression in an orthotopic model of prostate cancer. Int J Cancer. 2002;97:416–424. doi: 10.1002/ijc.1638. [DOI] [PubMed] [Google Scholar]
- 126.Petrache I, Otterbein LE, Alam J, Wiegand GW, Choi AM. Heme oxygenase-1 inhibits TNF-alpha-induced apoptosis in cultured fibroblasts. Am J Physiol Lung Cell Mol Physiol. 2000;278:L312–L319. doi: 10.1152/ajplung.2000.278.2.L312. [DOI] [PubMed] [Google Scholar]
- 127.Peyton KJ, Reyna SV, Chapman GB, Ensenat D, Liu XM, Wang H, Schafer AI, Durante W. Heme oxygenase-1-derived carbon monoxide is an autocrine inhibitor of vascular smooth muscle cell growth. Blood. 2002;99:4443–4448. doi: 10.1182/blood.v99.12.4443. [DOI] [PubMed] [Google Scholar]
- 128.Pierre F, Tache S, Petit CR, Van der Meer R, Corpet DE. Meat and cancer: haemoglobin and haemin in a low-calcium diet promote colorectal carcinogenesis at the aberrant crypt stage in rats. Carcinogenesis. 2003;24:1683–1690. doi: 10.1093/carcin/bgg130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129.Pinlaor S, Hiraku Y, Ma N, Yongvanit P, Semba R, Oikawa S, Murata M, Sripa B, Sithithaworn P, Kawanishi S. Mechanism of NO-mediated oxidative and nitrative DNA damage in hamsters infected with Opisthorchis viverrini: a model of inflammation-mediated carcinogenesis. Nitric Oxide. 2004;11:175–183. doi: 10.1016/j.niox.2004.08.004. [DOI] [PubMed] [Google Scholar]
- 130.Poss KD, Tonegawa S. Reduced stress defense in heme oxygenase 1-deficient cells. Proc Natl Acad Sci U S A. 1997;94:10925–10930. doi: 10.1073/pnas.94.20.10925. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 131.Price JT, Thompson EW. Mechanisms of tumour invasion and metastasis: emerging targets for therapy. Expert Opin Ther Targets. 2006;6:217–233. doi: 10.1517/14728222.6.2.217. [DOI] [PubMed] [Google Scholar]
- 132.Sacca P, Caballero F, Batlle A, Vazquez E. Cell cycle arrest and modulation of HO-1 expression induced by acetyl salicylic acid in hepatocarcinogenesis. Int J Biochem Cell Biol. 2004;36:1945–1953. doi: 10.1016/j.biocel.2004.01.029. [DOI] [PubMed] [Google Scholar]
- 133.Sahoo SK, Sawa T, Fang J, Tanaka S, Miyamoto Y, Akaike T, Maeda H. Pegylated zinc protoporphyrin: a water-soluble heme oxygenase inhibitor with tumor-targeting capacity. Bioconjug Chem. 2002;13:1031–1038. doi: 10.1021/bc020010k. [DOI] [PubMed] [Google Scholar]
- 134.Sawa T, Wu J, Akaike T, Maeda H. Tumor-targeting chemotherapy by a xanthine oxidase-polymer conjugate that generates oxygen-free radicals in tumor tissue. Cancer Res. 2000;60:666–671. [PubMed] [Google Scholar]
- 135.Sawle P, Foresti R, Mann BE, Johnson TR, Green CJ, Motterlini R. Carbon monoxide-releasing molecules (CO-RMs) attenuate the inflammatory response elicited by lipopolysaccharide in RAW264.7 murine macrophages. Br J Pharmacol. 2005;145:800–810. doi: 10.1038/sj.bjp.0706241. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 136.Schacter BA, Kurz P. Alterations in hepatic and splenic microsomal electron transport system components, drug metabolism, heme oxygenase activity, and cytochrome P-450 turnover in Murphy-Sturm lymphosarcoma-bearing rats. Cancer Res. 1982;42:3557–3564. [PubMed] [Google Scholar]
- 137.Schillinger M, Exner M, Minar E, Mlekusch W, Mullner M, Mannhalter C, Bach FH, Wagner O. Heme oxygenase-1 genotype and restenosis after balloon angioplasty: a novel vascular protective factor. J Am Coll Cardiol. 2004;43:950–957. doi: 10.1016/j.jacc.2003.09.058. [DOI] [PubMed] [Google Scholar]
- 138.Schillinger M, Exner M, Mlekusch W, Domanovits H, Huber K, Mannhalter C, Wagner O, Minar E. Heme oxygenase-1 gene promoter polymorphism is associated with abdominal aneurysm. Thromb Res. 2002;106:131–136. doi: 10.1016/s0049-3848(02)00100-7. [DOI] [PubMed] [Google Scholar]
- 139.Schlegel A, Volonte D, Engelman JA, Galbiati F, Mehta P, Zhang XL, Scherer PE, Lisanti MP. Crowded little caves: structure and function of caveolae. Cell Signal. 1998;10:457–463. doi: 10.1016/s0898-6568(98)00007-2. [DOI] [PubMed] [Google Scholar]
- 140.Schwartsburd PM. Chronic inflammation as inductor of pro-cancer microenvironment: pathogenesis of dysregulated feedback control. Cancer Metastasis Rev. 2003;22:95–102. doi: 10.1023/a:1022220219975. [DOI] [PubMed] [Google Scholar]
- 141.Shibahara S, Nakayama M, Kitamuro T, Udono-Fujimori R, Takahashi T. Repression of heme oxygenase-1 expression as a defense strategy in humans. Exp Biol Med. 2003;228:472–473. doi: 10.1177/15353702-0322805-08. [DOI] [PubMed] [Google Scholar]
- 142.Silver BJ, Hamilton BD, Toossi Z. Suppression of TNF-alpha gene expression by hemin: implications for the role of iron homeostasis in host inflammatory responses. J Leukoc Biol. 1997;62:547–552. doi: 10.1002/jlb.62.4.547. [DOI] [PubMed] [Google Scholar]
- 143.Simpson MA. Concurrent expression of hyaluronan biosynthetic and processing enzymes promotes growth and vascularization of prostate tumors in mice. Am J Pathol. 2006;169:247–257. doi: 10.2353/ajpath.2006.060032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 144.Snyder SH, Baranano DE. Heme oxygenase: a font with multiple messengers. Neuropsychopharmacology. 2001;25:294–298. doi: 10.1016/S0893-133X(01)00275-5. [DOI] [PubMed] [Google Scholar]
- 145.Soares MP, Lin Y, Anrather J, Csizmadia E, Takigami K, Sato K, Grey ST, Colvin RB, Choi AM, Poss KD, Bach FH. Expression of heme oxygenase-1 can determine cardiac xenograft survival. Nat Med. 1998;4:1073–1077. doi: 10.1038/2063. [DOI] [PubMed] [Google Scholar]
- 146.Song R, Mahidhara RS, Zhou Z, Hoffman RA, Seol DW, Flavell RA, Biliar TR, Otterbein LE, Choi AM. Carbon monoxide inhibits T lymphocyte proliferation via caspase-dependent pathway. J Immunol. 2004;172:1220–1226. doi: 10.4049/jimmunol.172.2.1220. [DOI] [PubMed] [Google Scholar]
- 147.Song W, Su H, Song S, Paudel HK, Schipper HM. Over-expression of heme oxygenase-1 promotes oxidative mitochondrial damage in rat astroglia. J Cell Physiol. 2006;206:655–663. doi: 10.1002/jcp.20509. [DOI] [PubMed] [Google Scholar]
- 148.Sosne G, Chan CC, Thai K, Kennedy M, Szliter EA, Hazlett LD, Kleinman HK. Thymosin beta 4 promotes corneal wound healing and modulates inflammatory mediators in vivo. Exp Eye Res. 2001;72:605–608. doi: 10.1006/exer.2000.0985. [DOI] [PubMed] [Google Scholar]
- 149.Straume O, Akslen LA. Importance of vascular phenotype by basic fibroblast growth factor, and influence of the angiogenic factors basic fibroblast growth factor/fibroblast growth factor receptor-1 and ephrin-A1/EphA2 on melanoma progression. Am J Pathol. 2002;160:1009–1019. doi: 10.1016/S0002-9440(10)64922-X. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 150.Struckmann K, Schraml P, Simon R, Elmenhorst K, Mirlacher M, Kononen J, Moch H. Impaired expression of the cell cycle regulator BTG2 is common in clear cell renal cell carcinoma. Cancer Res. 2004;64:1632–1638. doi: 10.1158/0008-5472.can-03-1687. [DOI] [PubMed] [Google Scholar]
- 151.Stuhlmeier KM. Activation and regulation of Hsp32 and Hsp70. Eur J Biochem. 2000;267:1161–1167. doi: 10.1046/j.1432-1327.2000.01112.x. [DOI] [PubMed] [Google Scholar]
- 152.Sunamura M, Duda DG, Ghattas MH, Lozonschi L, Motoi F, Yamauchi J, Matsuno S, Shibahara S, Abraham NG. Heme oxygenase-1 accelerates tumor angiogenesis of human pancreatic cancer. Angiogenesis. 2003;6:15–24. doi: 10.1023/a:1025803600840. [DOI] [PubMed] [Google Scholar]
- 153.Suzuki M, Iso-o N, Takeshita S, Tsukamoto K, Mori I, Sato T, Ohno M, Nagai R, Ishizaka N. Facilitated angiogenesis induced by heme oxygenase-1 gene transfer in a rat model of hindlimb ischemia. Biochem Biophys Res Commun. 2003;302:138–143. doi: 10.1016/s0006-291x(03)00114-1. [DOI] [PubMed] [Google Scholar]
- 154.Taille C, Almolki A, Benhamed M, Zedda C, Megret J, Berger P, Leseche G, Fadel E, Yamaguchi T, Marthan R, Aubier M, Boczkowski J. Heme oxygenase inhibits human airway smooth muscle proliferation via a bilirubin-dependent modulation of ERK1/2 phosphorylation. J Biol Chem. 2003;278:27160–27168. doi: 10.1074/jbc.M300364200. [DOI] [PubMed] [Google Scholar]
- 155.Tanaka S, Akaike T, Fang J, Beppu T, Ogawa M, Tamura F, Miyamoto Y, Maeda H. Antiapoptotic effect of haem oxygenase-1 induced by nitric oxide in experimental solid tumour. Br J Cancer. 2003;88:902–909. doi: 10.1038/sj.bjc.6600830. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 156.Tenhunen R, Marver HS, Schmid R. The enzymatic conversion of heme to bilirubin by microsomal heme oxygenase. Proc Natl Acad Sci U S A. 1968;61:748–755. doi: 10.1073/pnas.61.2.748. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 157.Terry CM, Clikeman JA, Hoidal JR, Callahan KS. Effect of tumor necrosis factor-alpha and interleukin-1 alpha on heme oxygenase-1 expression in human endothelial cells. Am J Physiol. 1998;274:H883–H891. doi: 10.1152/ajpheart.1998.274.3.H883. [DOI] [PubMed] [Google Scholar]
- 158.Torisu-Itakura H, Furue M, Kuwano M, Ono M. Co-expression of thymidine phosphorylase and heme oxygenase-1 in macrophages in human malignant vertical growth melanomas. Jpn J Cancer Res. 2000;91:906–910. doi: 10.1111/j.1349-7006.2000.tb01033.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 159.Torti FM, Torti SV. Regulation of ferritin genes and protein. Blood. 2002;99:3505–3516. doi: 10.1182/blood.v99.10.3505. [DOI] [PubMed] [Google Scholar]
- 160.Tozer GM, Prise VE, Motterlini R, Poole BA, Wilson J, Chaplin DJ. The comparative effects of the NOS inhibitor, N-omeganitro-L-arginine, and the haemoxygenase inhibitor, zinc protoporphyrin IX, on tumour blood flow. Int J Radiat Oncol Biol Phys. 1998;42:849–853. doi: 10.1016/s0360-3016(98)00303-4. [DOI] [PubMed] [Google Scholar]
- 161.Tsui TY, Siu YT, Schlitt HJ, Fan ST. Heme oxygenase-1-derived carbon monoxide stimulates adenosine triphosphate generation in human hepatocyte. Biochem Biophys Res Commun. 2005;336:898–902. doi: 10.1016/j.bbrc.2005.08.187. [DOI] [PubMed] [Google Scholar]
- 162.Tsuji MH, Yanagawa T, Iwasa S, Tabuchi K, Onizawa K, Bannai S, Toyooka H, Yoshida H. Heme oxygenase-1 expression in oral squamous cell carcinoma as involved in lymph node metastasis. Cancer Lett. 1999;138:53–59. doi: 10.1016/s0304-3835(98)00372-3. [DOI] [PubMed] [Google Scholar]
- 163.van Horssen R, Ten Hagen TL, Eggermont AM. TNF-alpha in cancer treatment: molecular insights, antitumor effects, and clinical utility. Oncologist. 2006;11:397–408. doi: 10.1634/theoncologist.11-4-397. [DOI] [PubMed] [Google Scholar]
- 164.Vigneswaran N, Wu J, Sacks P, Gilcrease M, Zacharias W. Microarray gene expression profiling of cell lines from primary and metastatic tongue squamous cell carcinoma: possible insights from emerging technology. J Oral Pathol Med. 2005;34:77–86. doi: 10.1111/j.1600-0714.2004.00258.x. [DOI] [PubMed] [Google Scholar]
- 165.Volonte D, Zhang K, Lisanti MP, Galbiati F. Expression of caveolin-1 induces premature cellular senescence in primary cultures of murine fibroblasts. Mol Biol Cell. 2002;13:2502–2517. doi: 10.1091/mbc.01-11-0529. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 166.Wang WS, Chen PM, Hsiao HL, Wang HS, Liang WY, Su Y. Overexpression of the thymosin beta-4 gene is associated with increased invasion of SW480 colon carcinoma cells and the distant metastasis of human colorectal carcinoma. Oncogene. 2004;23:6666–6671. doi: 10.1038/sj.onc.1207888. [DOI] [PubMed] [Google Scholar]
- 167.Was H, Cichon T, Smolarczyk R, Rudnicka D, Stopa M, Chevalier C, Leger JJ, Lackowska B, Grochot A, Bojkowska K, Ratajska A, Kieda C, Szala S, Dulak J, Jozkowicz A. Overexpression of heme oxygenase-1 in murine melanoma: increased proliferation and viability of tumor cells, decreased survival of mice. Am J Pathol. 2006;169:2181–2198. doi: 10.2353/ajpath.2006.051365. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 168.Watanabe N, Niitsu Y, Umeno H, Kuriyama H, Neda H, Yamauchi N, Maeda M, Urushizaki I. Toxic effect of tumor necrosis factor on tumor vasculature in mice. Cancer Res. 1988;48:2179–2183. [PubMed] [Google Scholar]
- 169.Weiss G, Lutton JD, Fuchs D, Werner-Felmayer G, Bock G, Abraham NG, Kappas A, Levere RD, Wachter H. Comparative effects of heme and metalloporphyrins on interferon-gamma-mediated pathways in monocytic cells (THP-1) Proc Soc Exp Biol Med. 1993;202:470–475. doi: 10.3181/00379727-202-43561. [DOI] [PubMed] [Google Scholar]
- 170.Willis D, Moore AR, Frederick R, Willoughby DA. Heme oxygenase: a novel target for the modulation of the inflammatory response. Nat Med. 1996;2:87–90. doi: 10.1038/nm0196-87. [DOI] [PubMed] [Google Scholar]
- 171.Yan Y, Higashi K, Yamamura K, Fukamachi Y, Abe T, Gotoh S, Sugiura T, Hirano T, Higashi T, Ichiba M. Different responses other than the formation of DNA-adducts between the livers of carcinogen-resistant rats (DRH) and carcinogen-sensitive rats (Donryu) to 3′-methyl-4-dimethylaminoazobenzene administration. Jpn J Cancer Res. 1998;89:806–813. doi: 10.1111/j.1349-7006.1998.tb00632.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 172.Yanagawa T, Omura K, Harada H, Nakaso K, Iwasa S, Koyama Y, Onizawa K, Yusa H, Yoshida H. Heme oxygenase-1 expression predicts cervical lymph node metastasis of tongue squamous cell carcinomas. Oral Oncol. 2004;40:21–27. doi: 10.1016/s1368-8375(03)00128-3. [DOI] [PubMed] [Google Scholar]
- 173.Yokoyama S, Mita S, Okabe A, Abe M, Ogawa M. Prediction of radiosensitivity in human esophageal squamous cell carcinomas with heme oxygenase-1: a clinicopathological and immunohistochemical study. Oncol Rep. 2001;8:355–358. doi: 10.3892/or.8.2.355. [DOI] [PubMed] [Google Scholar]
- 174.Zhang WL, Tsuneishi S, Nakamura H. Induction of heat shock proteins and its effects on glial differentiation in rat C6 glioblastoma cells. Kobe J Med Sci. 2001;47:77–95. [PubMed] [Google Scholar]
- 175.Zhao M, Liu Y, Bao M, Kato Y, Han J, Eaton JW. Vascular smooth muscle cells proliferation requires both p38 and BMK1 MAP kinases. Arch Biochem Biophys. 2002;400:199–207. doi: 10.1016/S0003-9861(02)00028-0. [DOI] [PubMed] [Google Scholar]
- 176.Zwerina J, Tzima S, Hayer S, Redlich K, Hoffman O, Hanslik-Schnabel B, Smolen JS, Kolias G, Schett G. Heme oxygenase 1 (HO-1) regulates osteoclastsogenesis and bone resorption. FASEB J. 2005;19:2011–2013. doi: 10.1096/fj.05-4278fje. [DOI] [PubMed] [Google Scholar]