

Abstract
AtT-20 cells expressing the wild-type kappa opioid receptor (KOR) increased phospho-p38 MAPK following treatment with the kappa agonist U50,488. The increase was blocked by the kappa antagonist norbinaltorphimine and not evident in untransfected cells. In contrast, U50,488 treatment of AtT-20 cells expressing KOR having alanine substituted for serine-369 (KSA) did not increase phospho-p38. Phosphorylation of serine 369 in the KOR carboxyl terminus by G-protein receptor kinase 3 (GRK3) was previously shown to be required for receptor desensitization, and the results suggest that p38 MAPK activation by KOR may require arrestin recruitment. This hypothesis was tested by transfecting arrestin3-(R170E), a dominant positive form of arrestin that does not require receptor phosphorylation for activation. AtT-20 cells expressing both KSA and arrestin3-(R170E) responded to U50,488 treatment with an increase in phospho-p38 consistent with the hypothesis. Primary cultured astrocytes (glial fibrillary acidic protein-positive) and neurons (γ-aminobutyric acid-positive) isolated from mouse striata also responded to U50,488 by increasing phospho-p38 immunolabeling. p38 activation was not evident in either striatal astrocytes or neurons isolated from KOR knock-out mice or GRK3 knock-out mice. Astrocytes pretreated with small interfering RNA for arrestin3 were also unable to activate p38 in response to U50,488 treatment. Furthermore, in striatal neurons, the kappa-mediated phospho-p38 labeling was colocalized with arrestin3. These findings suggest that KOR may activate p38 MAPK in brain by a GRK3 and arrestin-dependent mechanism.
Kappa opioid receptors are G-protein-coupled receptors (GPCRs)2 that are widely expressed throughout the brain and are activated by the endogenous opioid peptides derived from prodynorphin (1, 2). Several reports have characterized the signal transduction events initiated by KOR activation. By coupling to the G-protein Gαi/o, KOR inhibits adenylate cyclase, increases potassium conductance, decreases calcium conductance, and mobilizes intracellular calcium (3). More recently, KOR has been recognized to activate the extracellular signal-regulated kinase (ERK 1/2) (4, 5). This activation has been demonstrated to persist for several hours following agonist treatment, suggesting a role for KOR in long term growth and gene regulation. Other studies have demonstrated that KOR can activate c-Jun amino-terminal kinase (6). These studies suggest that KOR can activate multiple signaling pathways that result in the immediate and long term cellular effects of kappa opioids.
Sustained agonist exposure causes GPCR phosphorylation and desensitization (7). For the kappa opioid receptor, G-protein receptor kinase 3 (GRK3) phosphorylation of serine 369 in the carboxyl-terminal domain of KOR initiates arrestin-dependent receptor desensitization and internalization(8,9). Recently, new evidence suggests that the arrestin-bound GPCRis not inactive and instead can recruit MAPK signaling modules (10). For example, for the chemokine receptor (CXCR4), β-arrestin2 (also called arrestin3) is involved in receptor-mediated p38 MAPK activation (11). Activation of the p38 MAPK pathways has been demonstrated to play a role in the stress response, activation of apoptotic pathways, and induction of cell proliferation.
To assess whether GRK-dependent arrestin association is required for KOR activation of MAPK, we compared the KOR-GFP and the GRK-resistant KOR(S369A)-GFP (KSA) expressed in AtT-20 cells. In addition, using cultured primary astrocytes and neurons from wild-type mice and mice lacking GRK3 (GRK3−/−), we assessed the role of GRK in mediating KOR activation of p38 in native cells. The results suggest that phosphorylation of KOR serine 369 by GRK and subsequent arrestin binding leads to activation of the p38 stress kinase.
EXPERIMENTAL PROCEDURES
Chemicals
Norbinaltorphimine HCl and (−)U50,488 were obtained from Tocris (Ellisville, MO). Dynorphin B was from Bachem AG (Belmont, CA). Salvinorin A was a gift of Daniel Seibert. All other drugs were purchased from Calbiochem. Drugs were dissolved in water or ethanol (salvinorin A) unless otherwise indicated.
Cell Culture and Transfection of AtT-20 Cells
AtT-20 cells expressing rat KOR-GFP and rat KOR(S369A)-GFP (KSA) were generated as described previously (8) where ≥90% of cells express the GFP-tagged receptor. KOR-GFP and KSA-GFP AtT-20 cells were grown in Dulbecco’s modified medium with 10% horse serum (Sigma). For experiments using the dominant positive arrestin3(R170E), KOR-GFP or KSA-GFP AtT-20 cells at ≥70% confluence were transfected with 5 μg of cDNA coding for the dominant positive arrestin3(R170E)-YFP (12, 13) for 3–4 h using the SuperFect reagent (Qiagen) according to the manufacturer’s protocol. cDNA for the dominant positive point mutant arrestin3-(R170E) was provided by Dr. Vsevolod Gurevich (Vanderbilt, TN). BglII and KpnI restriction sites were added to the 5′ and 3′ ends of the cDNA through polymerase chain reaction using the respective oligonucleotides: 5′-Arr3 BglI(5′-ATGCATAGATCTATGGGGGAGAAACCC-3′) and 3′-Arr3 KpnI (5′-GGCCCGCGGTACCTAGCAGAACTGGTC-3′). The resulting product was purified, digested, and then inserted into the multiple cloning site of the pEYFP-C1 vector (Clontech). The 5′ oligonucleotide used to insert the BglII restriction site ensured that the coding sequence for arrestin3 remained in-frame with the EYFP sequence; thus, the resulting construct codes for a fusion protein with an EYFP tag added to the amino terminus of the arrestin.
Radioligand Binding of KOR and KSA-GFP AtT-20 Cells
KOR-GFP and KSA-GFP-expressing AtT-20 cells were grown as described above. For each membrane preparation, three 100-mm dishes with cells at 80% confluence were used. Dishes were washed twice with PBS and then scraped with a rubber policeman in 2 ml of 50 mM Tris buffer (pH 7.4) on ice. The contents of each plate were combined and homogenized using a Dounce homogenizer. Samples were centrifuged at 30,000 × g for 20 min, supernatant was removed, and pellet was resuspended in Tris buffer, washed, and rehomogenized. Following centrifugation, supernatant was removed, and pellet was stored at −80 °C. For saturation binding experiments membrane pellets were resuspended and homogenized in Tris buffer. Total binding of [3H]diprenorphine (PerkinElmer Life Sciences) (50 Ci/mmol) was determined using tubes containing 50 μg of membrane protein from KOR-GFP or KSA-GFP AtT-20 cells, Tris buffer, and [3H]diprenorphine ranging in concentration from 6 pM to 20 nM. A parallel set of tubes also contained 10 μM naloxone to define nonspecific binding. Tubes were incubated for 30 min in a 37 °C shaking water bath, and samples were filtered through a multiscreen GF/B plate (Millipore, Billerica, MA). Filter plates were washed and filtered three times with ice-cold Tris buffer. Radioactivity retained on filters was counted by liquid scintillation. Protein concentrations were determined using the bicinchoninic assay method (Pierce) as described in the manufacturer’s protocol. Receptor levels and binding affinity (Bmax, Kd) for the KOR-GFP and KSA-GFP were calculated using a non-linear least squares curve fitting program (GraphPad Prism 4.0, San Diego, CA). Receptor expression levels were determined to be similar in both the KOR-GFP (Bmax 2.7 ± 0.169 pmol/mg of protein, Kd 0.40 ± 0.08 nM) and KSA-GFP (Bmax 3.3 ± 0.11 pmol/mg of protein, Kd 0.37 ± 0.11 nM) AtT-20 cell lines.
Primary Cultures of Astrocytes
This protocol has been described previously (14) and was adapted for these studies. In this protocol, >95% of cells are glial fibrillary acidic protein (GFAP)-positive, indicating that the culture is primarily comprised of astrocytes. Neonatal mice (postnatal days, 1–3) were decapitated, and striata were dissected and placed in 2 ml of culture medium at 4 °C containing NeurobasalA (Invitrogen) supplemented with GlutaMAX, 2% B27, 50 units of penicillin, and 50 μg/ml streptomycin (Invitrogen). The striatal tissue was coarsely cut and transferred to sterile culture medium containing 40 units/ml active papain (Sigma). The cell-papain solution was incubated at 30 °C for 30 min on a platform rotating at 120 rpm. The solution was then replaced with 2 ml of culture media and triturated 15–20 times. Following settling (about 2 min), the supernatant was passed through a 70-μm nylon cell strainer (BD Biosciences). This process of settling and straining was repeated twice more. Cells were then plated in 0.5 ml of culture medium at a density of 2.5 × 105 cells/ml on 6-well plates or 12-mm glass coverslips (PGC Scientifics, Gaithersburg, MD) placed in 24-well plates (BD Biosciences). Prior to use, coverslips were acid-washed, soaked in ethanol, and rinsed in sterile water before overnight treatment with 50 mg/ml poly-D-lysine (135 kDa, Sigma). 1 h after plating and incubation (5% CO2, 95% air, 37 °C), unattached cells were aspirated, and the culture medium was replaced. Following 3–4 days in culture, plating media was replaced with complete medium containing 1% fetal bovine serum (Gemini Bio-Products, Woodland, CA). Cover-slips with astrocytes were used for immunocytochemistry 1–2 weeks after plating. 6-well plates were used for immunoblotting experiments about 21 days after plating.
Striatal Neuronal Cultures
This procedure was described previously (15) and adapted for these studies. In this procedure, >90% of the neuronal cell culture stained positive for γ-aminobutyric acid (GABA), consistent with the endogenous neuronal populations in the striatum. The striatal region was dissected from 1–3-day-old C57BL/6 mice, incubated in media containing 20 units/ml papain and 0.5 M kynurenic acid for 45 min at 37 °C, and triturated using fire-polished Pasteur pipettes as described previously (15). Cells were plated on poly-D-lysine-and collagen-treated glass coverslips at a density of 75,000 cells per coverslip. Neuronal medium containing 50% minimal essential medium (MEM), 39% Ham’s F12 medium, 10% horse serum, 1% fetal bovine serum, 0.45% glucose, 0.1 mg/ml apotransferrin, 0.5 mM kynurenic acid, 100 units/ml penicillin, and 100 μg/ml streptomycin was added 1 h after initial plating. Cells were grown in a humidified 5% CO2 incubator at 37 °C and used after 21 days in culture. Cultures of striatal astrocytes and neurons were made from neonatal KOR−/− and GRK3−/− mice on a C57Bl/6 background (Charles River Laboratories), genotyped and characterized as described previously (16).
Immunocytochemistry and Confocal Microscopy
KOR-GFP and KOR(S369A)-GFP AtT-20 cells, primary cultured astrocytes and striatal neurons were grown on poly-D-lysine coverslips in 24-well plates and placed in a 37 °C (5% CO2) incubator. After drug treatment, cells were washed three times with PBS and then fixed in 4% paraformaldehyde for 20 min, washed in PBS 3×, blocked for 2 h in 0.3% gelatin, 0.025% Triton X-100 in PBS (blocking buffer) at room temperature, and then incubated overnight with primary antibody in blocking buffer. The following primary antibody concentrations were used: goat-anti-rabbit phospho-p38 (Thr-180/Tyr-182) MAPK antibody (Cell Signaling, Beverly, MA) 1:500, guinea pig anti-GFAP (Chemicon, Temecula, CA) 1:1000. Following primary antibody exposure and 3× wash in PBS, coverslips were exposed to secondary antibody. Secondary antibody for AtT-20 cells was the goat-anti-rabbit Alexa Fluor 555 (Molecular Probes, Eugene, OR). For the anti-GFAP and anti-phospho-p38, donkey anti-guinea pig IgG rhodamine conjugate (1:300, Jackson ImmunoResearch, West Grove, PA) and goat-anti-rabbit IgG 488, Alexa Fluor conjugate (1:400, Molecular Probes) were diluted in blocking buffer and incubated with coverslips for 1 h at room temp. AtT-20 cells were incubated with Topro3 stain (Topro3, Molecular Probes, 1:1000) for 15 min following secondary antibody incubation and then washed two times in PBS prior to mounting on slides to visualize nuclei. Striatal neurons were stained with the following antibodies. Mouse anti-phospho-p38 (Thr-180/Tyr-182) primary-antibody (Cell Signaling) was used at a 1:250 dilution in PBS, guinea pig anti-GABA was used at a dilution of 1:1000 in PBS, rabbit anti-arrestin3 (Santa Cruz Biotechnology, Santa Cruz, CA) antibody was used at a dilution of 1:300 in PBS, and the blocking buffer was composed of 5% goat-serum, 3% bovine serum albumin, and 0.5% TX-100. The secondary antibodies used for the neuronal staining were goat-anti-mouse IgG 488, goat-anti-rabbit IgG 555, and goat-anti-guinea pig IgG 633 (1:500, Molecular Probes). Coverslips were mounted using VECTASHIELD (Vector Laboratories, Burlington, CA) and sealed with nail polish. Fluorescent GFP signals were excited at 488 nm, Alexa Fluor 555 fluorescent signals were excited at 543 nm, and fluorescent YFP signals (excited at 440 nm–470 nm) and fluorescent Topro3 signals (excited at 633 nm) were detected and merged as appropriate. All imaging was performed at the University of Washington W. M. Keck Imaging Center for Advanced Studies in Neural Signaling.
Transfection of Arrestin3 siRNA
The chemically synthesized siRNA with 19-nucleotide duplex RNA and 2-nucleotide 3′ terminal dTdT overhangs were purchased from (Invitrogen). The siRNA sequence targeting mouse arrestin3 (NM_145429) was 5′-GGACCGGAAAGUGUUUGUG-3′. Lipofectamine 2000 (Invitrogen) was added to OptiMEM (Invitrogen) according to the manufacturer’s instructions, whereas RNA mixtures at a final concentration of 25 nM were prepared in MEM. RNA mixtures were added dropwise to the Lipofectamine 2000 mixture and incubated at room temperature for 30 min. After the incubation, the total mixture was added to cells in a 6-well tissue culture plate containing astrocytes for 4 h. Some wells received Lipofectamine 2000 only as a negative control. Additional MEM was added to each well or plate 24 h after the addition of the RNA mixtures. 24 h later, the siRNA/MEM mixture was replaced with fresh medium. Cells were harvested on the third day following transfection for quantification of phospho-p38, arrestin3, and actin immunoreactivity as described.
Immunoblotting
KOR-GFP and KSA-GFP-expressing AtT-20 cells, primary striatal astrocytes, and primary striatal neurons were cultured as described above. All cells were serum-starved 24 h prior to drug treatment. Cells were treated with the appropriate ligands and time points in cell culture medium and then lysed in 350 ml of lysis buffer containing 50 mM Tris-HCl, 300 mM NaCl, 1 mM EDTA, 1 mM Na3VO4, 1 mM NaF, 10% glycerol, 1% Nonidet P-40, 1:100 of phosphatase inhibitor mixture set 1 (Calbiochem), and 1:100 of protease inhibitor mixture set 1 (Calbiochem). Lysates were sonicated for 20 s and then centrifuged for 15 min (14000 × g, 4 °C), pellet was discarded, and sample supernatants were stored at −20 °C. Protein concentration was determined by Pierce bicinchoninic assay with bovine serum albumin as the standard before loading 20 μg onto non-denaturing 10% bisacrylamide precast gels (Invitrogen) and running at 150 V for 1.5 h. For determination of molecular weights, Benchmark prestained standards (Invitrogen) were loaded along with protein samples. Blots were transferred to nitrocellulose (Whatman, Middlesex, UK) for 1.5 h at 30 mV, blocked in TBS/5% bovine serum albumin for 1 h, incubated overnight at 4 °C with a 1:1000 dilution of goat-anti-rabbit phospho-p38 MAPK antibody or goat-anti-rabbit phospho-ERK 1/2 (Thr-202/Tyr-204) antibody (Cell Signaling) or mouse anti-arrestin3 (1:300, sc-13140, Santa Cruz Biotechnology). Following overnight incubation, membranes were washed 4 × 15 min in TBST (Tris-buffered saline, 1% Tween 20) and then incubated with the IRDye™ 800-conjugated affinity-purified anti-rabbit or anti-mouse IgG at a dilution of 1:10,000 in a 1:1 mixture of 5% milk/TBS and Li-Cor blocking buffer (Li-Cor Biosciences, Lincoln, NE) for 1 h at room temperature. Membranes were then washed 4 × 15 min in TBST, 1 × 10 min in TBS to remove Tween 20 (which can cause high background fluorescence on the Odyssey imaging system) and analyzed as described below. Membranes were reprobed with rabbit anti-β-actin (3 h at room temperature, 5% milk/TBS) and secondary antibody (as above) to confirm equal protein loading.
Data Analysis
Immunoblots were scanned using the Odyssey infrared imaging system (Li-Cor Biosciences). Band intensity was measured using the Odyssey software, which subtracts background and calculates band density in pixels. Data were normalized to a percentage of control sample band intensity (basal, 100%) and plotted using GraphPad (GraphPad Prism 4.0) software. Concentration-response data were fit using non-linear regression (Prism 4.0). Statistical significance was taken as p < 0.05 or p < 0.01 as determined by the Student’s t test or analysis of variance followed by Dunnett’s post hoc test where appropriate.
RESULTS
Kappa Agonist-induced Phosphorylation of p38 MAPK Is Dependent on KOR Phosphorylation and Arrestin Activation
Treatment of AtT-20 cells expressing KOR-GFP with the selective KOR agonist, 10μM U50,488, for 15 min at 37 °C resulted in an increase in phospho-p38 MAPK and resulted in internalization of KOR-GFP receptors as shown by the punctate green fluorescence clustered in the cytosol (Fig. 1, A and B). The activation of p38 was blocked by pretreatment with the selective KOR antagonist 1 μM norbinaltorphimine (norBNI) (Fig. 1B, inset) and was not evident in untransfected AtT-20 cells (data not shown). AtT-20 cells expressing the mutant KOR, having a serine-to-alanine substitution at residue 369 (KSA-GFP), did not demonstrate an increase in phospho-p38 immunoreactivity and did not show KOR internalization after treatment with 10 μM U50,488 for 15 min at 37 °C (Fig. 1, C and D). The differences in response to agonist were not due to differences in receptor expression or receptor localization; KOR-GFP and KSA-GFP were expressed at equivalent levels as measured by [3H]diprenorphine saturation binding (see “Experimental Procedures”). These results are consistent with our previous report showing that KSA-GFP receptors can still activate G-protein-gated inwardly rectifying potassium (KIR3) currents in this cell type but do not show rapid desensitization (9).
FIGURE 1. KOR activation of p38 MAPK is phosphorylation- and arrestin-dependent in AtT-20 cells.

KOR-GFP- and KSA-GFP-expressing AtT-20 cells were grown on cover-slips as described under “Experimental Procedures.” Cells were then fixed and labeled with anti-phospho-p38 MAPK (red), and green fluorescence is the GFP signal corresponding to the KOR or KSA receptor. Blue fluorescence corresponds to the Topro3 nuclear staining. KOR-GFP receptor was visualized (A) in the absence of U50,488 treatment. Following 10 μM U50,488 treatment for 15 min at 37 °C (B), KOR-GFP was internalized and phospho-p38 MAPK staining was increased. The inset in B shows KOR-GFP cells pretreated with 1 μM norBNI, prior to U50,488 treatment, and shows blockade of KOR-mediated phospho-p38 staining. KSA-GFP receptors distribute in a similar manner to the KOR in the absence of agonist (C ); however, they did not internalize or increase phospho-p38 MAPK upon the addition of 10 μM U50,488 for 15 min (D). Following transfection of the dominant positive arrestin R170E, the KSA-GFP receptor is able to respond to U50,488 treatment as demonstrated by its internalization (F ) and phospho-p38 MAPK (F, inset) activation; white arrows point to phospho-p38 KSA-GFP overlay. All experiments were performed on 2– 4 independent experiments.
Dominant Positive Arrestin3-R170E Rescues KSA-mediated p38 Activation
To assess whether KSA-GFP failed to induce p38 phosphorylation because agonist-bound KSA-GFP was unable to recruit arrestin and internalize as effectively as KOR-GFP (8, 9), we transfected the dominant positive arrestin3-(R170E)-YFP into KSA-GFP AtT-20 cells. This form of arrestin has been previously demonstrated to be constitutively active and can bind to the agonist-bound receptor independently of receptor phosphorylation (12, 13). In KSA-GFP-expressing AtT-20 cells transfected with arrestin3-(R170E)-YFP and then stimulated with 10 μM U50,488, the KSA-GFP receptor was able to internalize and cause activation of p38 (Fig. 1F). Successful transfection of arrestin3-(R170E)-YFP was confirmed by increased cytosolic YFP labeling in KSA-GFP cells (10–15% of total cell population). Transfection of the pEYFP-C1 vector alone in KSA-GFP cells failed to induce a significant increase in phospho-p38 MAPK following U50,488 stimulation (data not shown).
The effect of the arrestin3-(R170E)-YFP transfection on KSA-GFP mediated p38 activation was also quantified using immunoblotting. We found that transfection of the dominant positive arrestin3 resulted in an increase in KSA-GFP-mediated p38 activation following treatment with 10 μM U50,488 for 15 or 30 min (Fig. 2, A and B). In addition, KOR-GFP cells transfected with arrestin3-(R170E)-YFP and stimulated with 10 μM U50,488 caused activation of p38 at an equal magnitude to the KOR-GFP cells without the dominant positive arrestin (data not shown), suggesting that the arrestin3-(R170E)-YFP transfection, by itself, does not increase p38 activation following agonist stimulation. These results suggest that serine 369 phosphorylation and the subsequent recruitment of arrestin were required for maximal KOR-GFP activation of p38.
FIGURE 2. Dominant positive arrestin restores KSA-GFP mediated p38 MAPK activation.

KSA-GFP-expressing AtT-20 cells were transiently transfected with the dominant positive arrestin (R170E) and then exposed to U50,488 treatment. A, mean band intensities expressed as a percentage of basal-untreated control ± S.E. of KSA-GFP-expressing AtT-20 cells. Following 10 μM U50,488 treatment for 15 and 30 min (KSA U50 15, KSA U50 30), phospho-p38 levels were not significantly different from untreated controls. In KSA-GFP/R170E cells, 10 μM U50,488 treatment for 15 or 30 min (R170E U50 15, R170E U50 30) facilitated agonist-dependent p38 MAPK activation. n = 3, from separate transfections and experiments. *, significantly different from basal, p < 0.05, using the student’s t test. P-p38, phospho-p38 MAPK. B, representative Western blot data for phospho-p38 MAPK (P-p38) in KSA-GFP expressing A + T-20 cells.
KOR-induced Phosphorylation of ERK 1/2 Was Independent of GRK/Arrestin
Stimulation of KOR-GFP with 10 μM U50,488 induced phosphorylation of p38 MAPK maximally at 15 min (Fig. 3, A and C). Concentration-response curves for U50,488 and dynorphin-B for activating p38 at the 15-min time point were also generated. U50,488 and the KOR endogenous peptide agonist, dynorphin-B (prodynorphin 228–240), had similar potencies (EC50) for activation of p38 (200 ± 43 nM and 60 ± 24 nM, respectively, n = 3) in KOR-GFP cells (Fig. 5A). Activation of KSA-GFP by U50,488 was unable to cause significant phosphorylation of p38 (Fig. 3, A–C). In contrast, equivalent U50,488-stimulated ERK 1/2 phosphorylation was evident for both the KOR-GFP and the KSA-GFP receptor-expressing AtT-20 cells (Fig. 3). These data suggest that KOR activates p38 and ERK 1/2 with different kinetics and via different signal transduction mechanisms.
FIGURE 3. KOR-GFP but not KSA-GFP mediates p38 phosphorylation following agonist treatment.
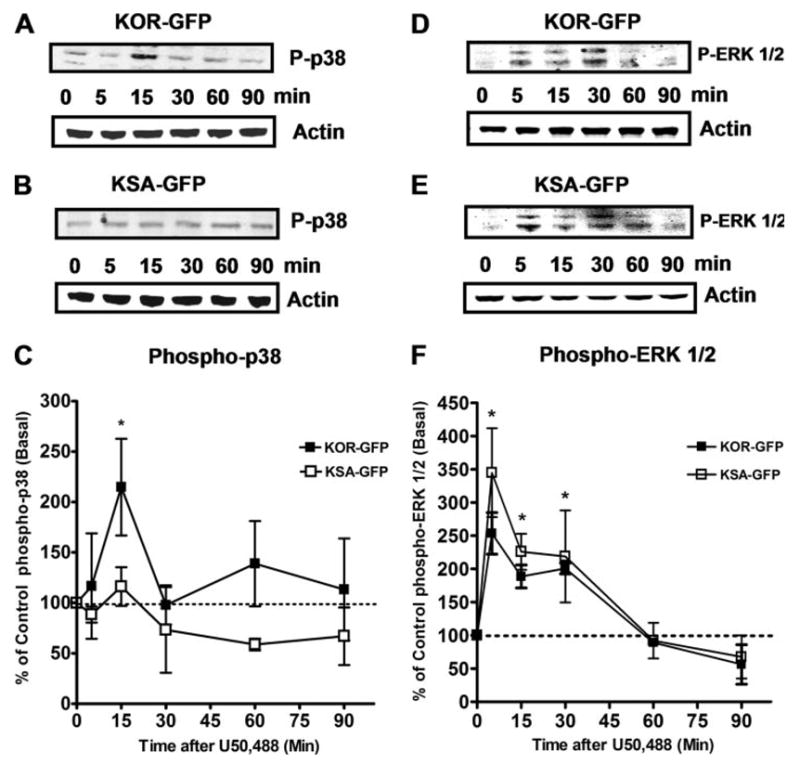
A–C, AtT-20 cells expressing either KOR-GFP or KSA-GFP were treated with 10 μM U50,488 for different times at 37 °C and then resolved by Western blot. A, representative Western blot data for phospho-p38 MAPK (P-p38) and β-actin protein loading control in KOR-GFP-expressing AtT-20 cells. B, representative Western blot data for phospho-p38 MAPK and β-actin protein loading in KSA-GFP AtT-20 cells. C, ■ are the mean band intensities expressed as a percentage of basal-untreated control ± S.E. of KOR-GFP mediated p38 MAPK phosphorylation; □ represent mean band intensities expressed as a percentage of basal-untreated control ± S.E. from AtT-20 cells expressing KSA-GFP. Phospho-p38 levels were only significantly (*) increased at the 15-min post-U50,488 time point for wild-type KOR-GFP. n = 3– 4, where each n represents an independent experiment. *, significantly different from basal, p < 0.05 using the student’s t test. D, representative Western blot data for phospho-ERK 1/2 (P-ERK 1/2) and β-actin protein loading control in KOR-GFP-expressing AtT-20 cells. E, representative Western blot data for phospho-ERK 1/2 MAPK and β-actin protein loading in KSA-GFP AtT-20 cells. F, ■ are the mean band intensities expressed as a percentage of basal-untreated control ± S.E. of KOR-GFP mediated ERK 1/2 phosphorylation; □ represent mean band intensities expressed as a percentage of basal-untreated control ± S.E. from AtT-20 cells expressing KSA-GFP and show a similar increase in the relative U50,488-stimulated ERK 1/2 activation. n = 3– 4, where each n represents an independent experiment. *, significantly different from basal for both KOR-GFP and KSA-GFP groups, p < 0.05 using the student’s t test.
FIGURE 5. KOR-mediated p38 activation in AtT-20 cells and Striatal Astrocytes is concentration-dependent.
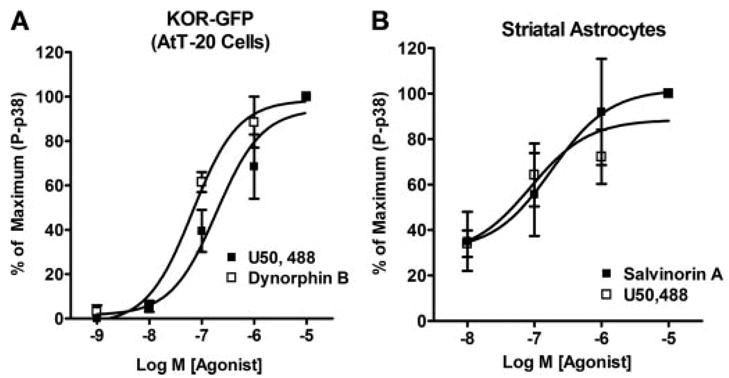
A, phospho-p38 MAPK (P-p38) concentration-response curves for the KOR agonists U50,488 and the endogenous KOR agonist-peptide dynorphin B in AtT-20 cells expressing KOR-GFP. B, phospho-p38 concentration-response curves for the KOR agonists U50,488 and salvinorin A in primary striatal astrocytes. All agonist treatments were performed at the 15-min time point. n = 3– 4, with each n taken from a separate cell culture and experiment.
KOR in Striatal Astrocytes Activates p38 MAPK in a GRK3-dependent Manner
Since transfected cell systems may have limited physiological relevance, we next used primary cultures of astrocytes derived from mouse striata to determine the signaling properties of endogenously expressed KOR. It has been previously determined that mu and kappa opioid receptors are endogenously expressed by astrocytes and that these receptors can activate ERK 1/2 (4, 5). However, KOR activation of p38 MAPK in primary striatal astrocytes has not been shown previously. U50,488 treatment caused an increase in the phospho-p38 staining in glial fibrillary acidic protein (GFAP)-positive cells (Fig. 4D), suggesting that KOR activates p38 in astrocytes. The U50,488-induced increase in phospho-p38 labeling was blocked by 1 μM norBNI (data not shown). In addition, concentration-response curves for activation of p38 in wild-type astrocytes were determined for the KOR-selective agonists (17, 18) U50,488 and salvinorin A by Western blotting (Fig. 5B). U50,488 and salvinorin A activated p38 with similar potency (U50,488 EC50 = 501 ± 348 nM; salvinorin A EC50 = 316 nM± 304 nM) and efficacy (2-fold over basal). In wild-type astrocytes, 10 μM U50,488 caused a 2-fold increase over basal in phospho-p38 (Fig. 6C) that was blocked by pretreatment with 1 μM norBNI (Fig. 6, A and C). In astrocyte cultures derived from striata of KOR−/− mice, 10 μM U50,488 had no significant effect on phospho-p38 levels (Fig. 6, A and C). The p38 activator anisomycin (50 μM, 15 min) caused a robust increase in phospho-p38 (650 ± 102%, n = 5) in KOR−/− cultures demonstrating that the p38 machinery is intact in these cells (Fig. 6A). Taken together, these results demonstrate that the U50,488-mediated increase in phospho-p38 in primary striatal astrocytes was KOR-dependent.
FIGURE 4. Primary cultures of striatal astrocytes show increased phospho-p38 MAPK (P-p38) activation in response to KOR stimulation.

Primary cultures of striatal astrocytes were grown on coverslips as described under “Experimental Procedures.” Cultures were treated with either vehicle or 10 μM U50,488 for 15 min. A–C show vehicle (basal) phospho-p38 (A, fluorescein isothiocyanate, green) and GFAP (B, rhodamine, red) merged (C, yellow) and demonstrate minimal phospho-p38 staining in the absence of agonist. However, D–F show increased 10 μM U50,488-stimulated phospho-p38 (D, green). This increase in phospho-p38 is colocalized with the astrocyte marker, GFAP (E, red) colocalization (F, yellow). The white arrows point to cells with particularly bright overlay following U50,488 treatment. Images shown are representative of at least 4 separate experiments from individual primary cultures.
FIGURE 6. U50,488-stimulated p38-MAPK activation in primary striatal astrocytes and neurons is KOR-mediated and GRK3-dependent.
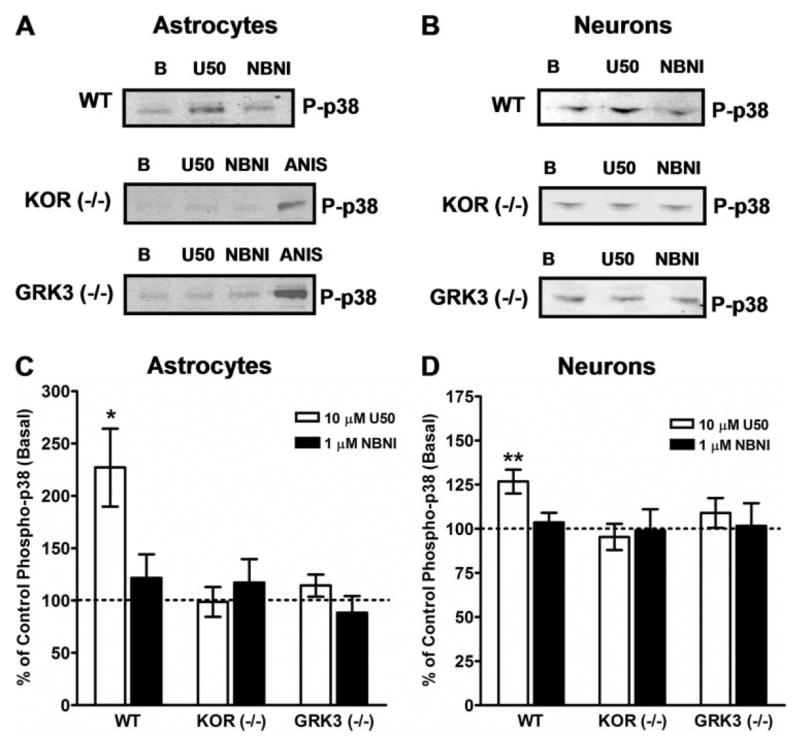
A and B, wild type (W T), kappa opioid receptor knock-out (KOR−/−), and G-protein-coupled receptor kinase knock-out (GRK3−/−) primary striatal astrocytes were cultured as described under “Experimental Procedures.” Astrocyte cultures were treated with vehicle (B), 10 μM U50,488 (U50), or U50,488 in the presence of 1 μM norbinaltorphimine (NBNI, 1-h pretreatment) for 15 min at 37 °C. KOR activation resulted in phosphorylation of p38 MAPK (p-p38) that was blocked by NBNI. U50,488-stimulated p38 activation was not evident in astrocytes from KOR−/−mice. In addition, U50,488 treatment did not induce p38 activation in astrocytes from GRK3−/− mice. In KOR−/− or GRK3−/− mice, anisomycin (ANIS), an efficacious activator of p38 MAPK (50 μM, 15 min, 37 °C), effectively stimulated p38 severalfold over basal, demonstrating an intact p38 MAPK system in these cultures. A, representative Western blots for phospho-p38 from W T, KOR−/−, and GRK3−/−primary striatal astrocytes. B, representative Western blots for phospho-p38 from W T, KO−/−, and GRK3−/−primary striatal neurons. C, data are the mean ± S.E. of U50,488-stimulated phospho-p38 in astrocytes from W T, KOR−/−, and GRK3−/−mice as a percentage of basal (dashed line). n = 7– 8, with each n taken from separate primary cell cultures and experiments. *, p < 0.05 for W T astrocytes plus U50,488 versus basal using the student’s t test. C and D, W T kappa opioid receptor knock-out (KOR−/−) and G-protein-coupled receptor kinase knock-out (GRK3−/−) primary striatal neurons were grown in culture as described under “Experimental Procedures.” KOR activation resulted in phosphorylation of p38 MAPK (p-p38) that was blocked by NBNI. U50,488-stimulated p38 activation was not evident in neurons from either KOR−/− mice or GRK3−/− mice. D, data are the mean ± S.E. of U50,488-stimulated phospho-p38 in neurons from W T, KOR−/−, and GRK3−/− mice as a percentage of basal (dashed line). n = 8 –10, with each n taken from separate primary cell cultures and experiments. **, p < 0.01 for W T astrocytes plus U50,488 versus basal using analysis of variance and Dunnett’s post hoc test.
To determine whether GRK3-mediated KOR phosphorylation was involved in p38 activation, we used primary cultures from GRK3−/− mice. In these cultures, 10 μM U50,488 had no effect on phospho-p38 (Fig. 6, A and C). These data suggest that in primary striatal astrocytes, KOR activates p38 MAPK by a GRK3-dependent mechanism.
KOR in Striatal Neurons Activate p38 MAPK in a GRK3-dependent Manner
KOR is known to function in the striatum and has also been implicated in stress-induced potentiation of cocaine-conditioned place preference (16, 19). Therefore, we also cultured primary striatal neurons to determine whether KOR activated p38 in these cells. Using Western blotting techniques in striatal neuronal cultures, treatment with 10 μM U50,488 for 15 min caused a significant (30%) increase in phospho-p38 that was blocked by 1 μM norBNI pretreatment (Fig. 6, B and D). In KOR−/− primary striatal neurons, 10 μM U50,488 treatment for 15 min did not cause a significant increase in phospho-p38 (Fig. 6, B and D), whereas anisomycin (10 min, 50 μM) caused a robust increase in phospho-38, demonstrating a functional p38 signaling pathway in these cultures (data not shown). Finally, we examined whether the KOR-mediated p38 phosphorylation in striatal neurons was dependent on receptor phosphorylation by GRK3. In striatal neuronal cultures from GRK3−/− mice, treatment with 10 μM U50,488 for 15 min did not increase phospho-p38 (Fig. 6D). These data are consistent with the hypothesis that KOR activation caused phosphorylation of p38 in primary striatal cultures and that this process was GRK3-dependent.
Confocal microscopy was used to confirm and visualize staining for phospho-p38 following U50,488 treatment. A majority of the striatal neurons (>90%) were GABAergic. In these studies, we found that 25–30% of the GABA-positive neurons displayed an increase in phosho-p38 following U50,488 treatment (which is consistent with the fraction of cells expressing KOR, data not shown). This stimulation was also blocked by norBNI (data not shown). Following U50,488 treatment, the phospho-p38 labeling was clustered as compared with the untreated striatal neurons (Fig. 7, A and E). In addition, U50,488 treatment caused the phospho-p38 labeling to be colocalized with arrestin3 immunoreactivity in striatal neurons (Fig. 7G). These data further support an interaction between KOR, arrestin3, and phospho-p38.
FIGURE 7. Primary cultures of striatal neurons showincreasedphospho-p38MAPKco-localized with arrestin3 in response to KOR stimulation.
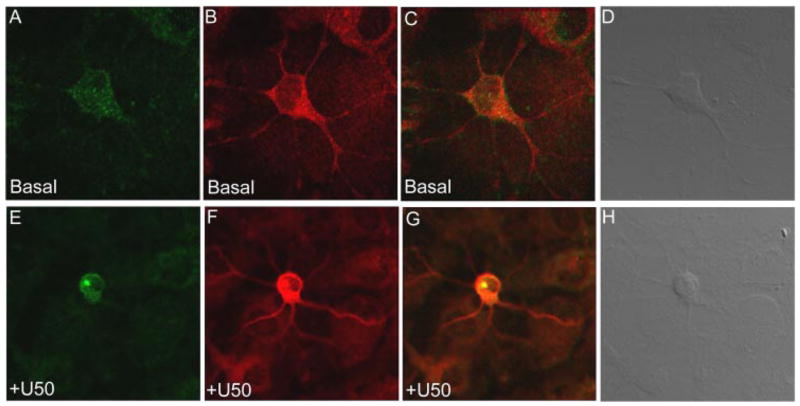
Primary striatal neurons were grown on coverslips as described under “Experimental Procedures.” Cultures were treated with either vehicle (basal) or 10 μM U50,488 for 15 min. A–C, show vehicle (basal) phospho-p38 (A, Alexa Fluor 488, green) and arrestin3 (B, Alexa Fluor 555, red) merged (C ) and demonstrate diffuse phospho-p38 labeling in the absence of agonist. However, E–G show a clustered, punctate, and increased staining for 10 μM U50,488-stimulated phospho-p38 (E) colocalized with arrestin3 (G, yellow). Images shown are representative of at least 4 separate experiments from individual primary cultures.
KOR-mediated p38 Activation Is Arrestin3-dependent in Striatal Astrocytes
To assess the role of arrestin3 in the activation of p38, we used siRNA to “knock down” the expression in cultured astrocytes. Treatment with siRNA for mouse arrestin3 effectively decreased arrestin3 expression as shown by immunoblotting for the 55-kDa arrestin3 protein (Fig. 8A). The siRNA treatment decreased arrestin3 expression in striatal astrocytes by 55 ± 6% S.E. (n = 6). Control astrocytes treated with similar transfection reagents, in the absence of siRNA, showed no differences in arrestin expression (data not shown). Furthermore, the siRNA knock down of arrestin3 resulted in a (62 ± 2% S.E., n = 6) decrease in U50,488-stimulated p38 activation (Fig. 8B). These results suggest that arrestin3 is required for KOR-mediated p38 activation in striatal astrocytes.
FIGURE 8. U50,488-stimulated phospho-p38 is decreased in arrestin3 siRNA-treated astrocytes.

Wild-type primary striatal astrocytes were grown in culture as described. Individual astrocyte cultures were treated with siRNA for arrestin3 as outlined under “Experimental Procedures.” A, representative Western blot for the 55-kDa protein arrestin3 (Arr3) in primary striatal astrocytes in the presence and absence (+,−) of siRNA for arrestin3; the β-actin control for protein loading and similar protein levels following siRNA treatment is also shown. B, data are the mean ± S.E. of U50,488-mediated phospho-p38 in striatal astrocytes in the absence (Control (+U50)) and presence of siRNA for arrestin3 (siRNA (+U50)). n = 6, with each n taken from separate primary cultures and siRNA treatment experiments. *, p < 0.05 using the students t test.
Additionally, we used inhibitors for signaling intermediates that have been previously described to be involved in p38 activation in other systems to identify the signal transduction pathway involved in KOR-mediated p38 activation (20, 21). Since MAPK activation by GPCRs has been previously demonstrated to involve Src, c-Raf, and phospholipase C (PLC) (4, 22, 23), we treated astrocytes with inhibitors for these enzymes. The Src tyrosine kinase inhibitor, PP2 (24) (1 μM), had no significant effect on U50,488-mediated p38 activation (n = 6). In addition, pretreatment with the c-Raf inhibitor GW5037 (30 nM), which displays 100-fold selectivity (25) for c-Raf (over Cdk1, c-Src, ERK2, MEK, and p38), did not significantly decrease KOR-mediated p38 activation (n = 6). PLC has also been implicated in GPCR-mediated MAPK activation in astrocytes, but we found that a 1-h pretreatment with the PLC inhibitor U-73122 (1 μM) did not significantly decrease the U50,488-mediated phospho-p38 (n = 5). The U-73122 structurally related PLC inhibitor negative isoform, U-73343, also had no effect on U50,488-mediated p38 activation. When striatal astrocytes were treated with PP2 or GW5037 in the absence of U50,488, the levels of phospho-p38 were not significantly different from vehicle control. The PI-3 kinase inhibitor, LY294002 was also used in these studies; however, due to its insolubility in water, we used Me2SO as a vehicle. The vehicle (0.1% Me2SO final) caused a significant increase (3-fold) in phospho-p38 in these astrocytes, precluding the determination of the role of the PI-3 kinase pathway in the KOR-mediated p38 activation. Taken together, these data suggest that the classical Src/Raf or the PLC pathway does not mediate KOR activation of p38. However, prior treatment with non-selective tyrosine kinase inhibitor genistein (10 μM) did significantly attenuate U50,488-mediated phospho-p38 (*, p < 0.05 students t test, n = 6), suggesting that these signaling events are mediated by activation of tyrosine kinase.
DISCUSSION
The principal finding of this study was that activation of KOR induced phosphorylation of p38 MAPK in AtT-20 cells, striatal astrocytes, and striatal neurons. KOR activation of p38 was blocked by a receptor mutation that blocked GRK/arrestin-dependent desensitization. Similarly, in both striatal astrocytes and neurons, p38 activation was blocked by GRK3 gene knock-out, and in astrocytes, p38 activation was blocked by arrestin3 suppression. These findings suggest that KOR activation of p38 may require GRK3 phosphorylation of the ser-369 in the carboxyl-terminal domain of KOR and subsequent association of the scaffolding protein arrestin. This hypothesis is consistent with prior reports suggesting that arrestin binding to GPCR may enable MAPK activation (7, 10).
Activation of MAPK by GPCRs has been demonstrated for several GPCR receptor classes in a variety of cell lines and systems (26); however, the underlying mechanisms vary depending on receptor type, cell type, and stimulus. KOR has been demonstrated to activate the ERK1/2 MAPK pathway in immortalized rat cortical astrocytes and C6 glioma cells (4, 5). KOR signaling to ERK1/2 in this cell type was mediated by the PI-3 kinase pathway, protein kinase C ζ and calcium (4). In addition, KOR has been shown through Src and focal adhesion kinase (FAK) to activate the c-Jun amino-terminal kinase (JNK), another member of the MAPK family (6). Thus, KOR-dependent activation of ERK1/2 and JNK has been found to be mediated by overlapping yet distinct signaling partners.
The role of arrestins in regulating opioid receptor desensitization, phosphorylation, and internalization has been extensively studied. The recent discovery that arrestins can also act as scaffolding proteins suggests that arrestin association with GPCRs may not stop signaling. Arrestin association may instead act as a switch that shifts the coupling of KOR from the acute regulation of ion channel conductance to a different mode of signaling involving p38 MAPK activation. Initially, it was demonstrated that arrestins can bind to Src family kinases and recruit them to the agonist-bound GPCR (27). It was later reported for some receptor types that arrestin recruitment is involved in GPCR-mediated ERK 1/2 and JNK3 activation (28, 29). These signaling mechanisms have also been examined using in vivo models in which arrestin scaffolding to MAPK has been implicated in behavioral consequences (30). There are fewer reports that have examined the role of arrestin in p38 regulation. For example, for the CXCR4 receptor and angiotensin 1A receptor, activation of p38 MAPK and chemotaxis was blocked when arrestin expression was suppressed (11, 31).
In our experiments, we found that KOR stimulated both p38 MAPK and ERK 1/2 activation in AtT-20 cells but that only the p38 MAPK activation was sensitive to the deletion of receptor phosphorylation site serine 369, in the mutant KSA-GFP-expressing cells. Our hypothesis is that KOR activation of PI-3 kinase and subsequent phosphorylation of ERK1/2 may be independent of GRK and arrestin; however, this concept requires further validation.
The KOR-mediated increase in phospho-p38 was found to be substantially larger in cultured astrocytes than in neurons. The basis for this difference may be due to the heterogeneity of the neuronal population. We found that only 30% of the neurons expressed KOR, and a similar fraction of neurons showed an increase in phospho-p38 staining following U50,488 treatment. In contrast the cultured astrocytes were more homogeneous, and a larger fraction expressed KOR. However, alternative explanations were not excluded.
The dominant positive R170E arrestin has previously been reported to bind to agonist-occupied receptors and mediate desensitization independently of receptor phosphorylation (12, 13). In these reports, the kinetics of desensitization using the arrestin3(R170E) mutant and a phosphorylation-insensitive delta opioid receptor were similar to the wild type (13), making this recombinant protein a useful tool. Using KSA-GFP-expressing AtT-20 cells cotransfected with arrestin3(R170E), we found that U50,488 was able to stimulate KSA-GFP and cause activation of p38. These data support the hypothesis that KOR-mediated p38 activation requires arrestin association. Interestingly, the KSA-GFP/R170E transfected cells had a more sustained activation of p38 than the wild-type KOR-GFP phospho-p38 response. The basis for the brief activation of p38 by wild-type KOR in the absence of arrestin3(R170E) is not clear from these studies but may depend on the duration of arrestin-KOR association.
In siRNA knockdown studies for arrestin3, the KOR-mediated increase in phoshpo-p38 was decreased, further supporting a role for arrestin in this signaling process. Consistent with this observation, the striatal neuron imaging studies showed that phospho-p38 colocalized with arrestin3 following U50,488 treatment. Nevertheless, a role for arrestin2 was not excluded. Arrestin3 antibody may cross-react with arrestin2, and the arrestin3 siRNA may have also affected arrestin2 expression.
GPCR-mediated ERK1/2 signaling has been demonstrated to vary depending on receptor type and signaling environment (26). However, the mechanisms by which GPCRs couple to the p38 MAPK are less well characterized. It has been suggested that p38 MAPK activation by GPCRs is regulated by Src, Raf, and PLC (26, 32, 33). At Src and Raf kinase-selective concentrations, the Src inhibitor PP2 and the Raf tyrosine inhibitor, GW5037, were both unable to significantly block KOR-mediated p38 activation in primary striatal astrocytes. A PLC inhibitor was unable to significantly decrease kappa stimulation of p38 phosphorylation. PI-3 kinase has been reported to mediate GPCR activation of p38 (34), but the PI-3 kinase inhibitor LY294002 vehicle control produced nonspecific p38 activation. Future studies using other approaches such as siRNA or antisense oligonucleotides may better resolve the possible role of PI-3 kinase in KOR-mediated p38 activation. Nevertheless, the tyrosine kinase inhibitor genistein did block, and these results suggest that KOR may activate p38 MAPK through a tyrosine kinase pathway. The present results suggest that KOR-mediated p38 activation follows a different signal transduction pathway than the Src, PLC, and PI-3 kinase pathways used by KOR to activate ERK 1/2-mediated signaling (4, 5).
The p38 MAPK pathway has been demonstrated to play a major role in environmental stress and inflammatory signals, including cytokine activation (35, 36). In epithelial cell types, p38 activation has been shown to be involved in chemotaxis and proliferative responses (9, 31). The activation of the p38 MAPK pathway in astrocytes results in the production of interleukin-1β, interleukin-6, and tumor necrosis factor-α and enzymes including COX-2 and inducible nitric oxide synthase (37). The activation of p38 by KOR in astrocytes may represent a means for opioid regulation of astrocyte proliferation and the production of inflammatory mediators. These effects on astrocytes may mediate the actions of endogenous dynorphins released within the spinal cord following sciatic nerve ligation (38).
KOR activation of p38 in striatal astrocytes may also serve as a protective role for neuronal stability. Since astrocytes have been recently implicated in synapse number and networking (39, 40), KOR activation on astrocytes may also serve to enhance neuronal transmission. The activation of p38 MAPK in neurons has been demonstrated to serve multiple functions. One such function is the modulation of synaptic plasticity by affecting long term depression (41, 42). Since KOR has previously been demonstrated to regulate tonic dopamine release in striatum (43), the activation of p38 by KOR in striatum may represent a mechanism by which opioids regulate synaptic transmission over time.
The proposed model of KOR activation of p38 is likely to have important in vivo implications; however, conclusions based on transfected cells or cells derived from knock-out animals must be interpreted conservatively. Overexpression of genes may enable signal transduction coupling events that are not evident under more physiological conditions. Gene deletion by knock-out strategies or suppression of gene expression by siRNA approaches may simultaneously alter compensatory gene expression. The results presented in the present study are internally consistent, and the concept that KOR signaling may couple to p38 via an arrestin scaffolding process is intriguing. Nevertheless, further validation and behavioral analysis of the implications of this hypothesis will be required.
Acknowledgments
We thank John Pintar for the KOR−/− mice; Marc Caron and Robert Lefkowitz for the GRK3−/− mice; and Vsevold Gurevich for the bovine arrestin3 R170E mutant. We also thank Andrea Francois, Michael Soskis, and Greg Martin for technical assistance.
Footnotes
This work was supported by U. S. Public Health Service Grant P01-DA15916 (to C. C.) and by National Institute on Drug Abuse Grant F32-DA20430 (to M. R. B. ).
The abbreviations used are: GPCR, G-protein-coupled receptor; ERK, extracellular signal-regulated kinase; JNK, c-Jun amino-terminal kinase; MAPK, mitogen-activated protein kinase; KOR, kappa opioid receptor; KSA, kappa opioid receptor serine 369 to alanine mutant; GRK3, G-protein-coupled receptor kinase 3; MEM, minimal essential medium; PBS, phosphate-buffered saline; MAPK; R170E, dominant positive arrestin3; RT, reverse transcriptase; siRNA, small inhibitory ribonucleic acid; TBS, Tris-buffered saline.
References
- 1.Chavkin C, James JF, Goldstein A. Science. 1982;215:413–415. doi: 10.1126/science.6120570. [DOI] [PubMed] [Google Scholar]
- 2.Dhawan BN, Cesselin R, Raghubir T, Reisine PB, Portoghese PS, Hamon M. Pharmcol Rev. 1996;48:568–586. [PubMed] [Google Scholar]
- 3.Piros ET, Hales TG, Evans CJ. Neurochem Res. 1996;21:1277–1285. doi: 10.1007/BF02532368. [DOI] [PubMed] [Google Scholar]
- 4.Belcheva MM, Clark AL, Haas PD, Serna JS, Hahn JW, Kiss A, Coscia C. J Biol Chem. 2005;280:27662–27669. doi: 10.1074/jbc.M502593200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Bohn LM, Belcheva MM, Coscia C. J Neurochem. 2000;74:564–573. doi: 10.1046/j.1471-4159.2000.740564.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Kam AY, Chan AS, Wong AH. J Pharmacol Exp Ther. 2004;310:301–310. doi: 10.1124/jpet.104.065078. [DOI] [PubMed] [Google Scholar]
- 7.Peirce KL, Lefkowitz RJ. Nat Rev Neurosci. 2001;2:727–733. doi: 10.1038/35094577. [DOI] [PubMed] [Google Scholar]
- 8.McLaughlin JP, Xu M, Mackie K, Chavkin C. J Biol Chem. 2003;278:34631–34640. doi: 10.1074/jbc.M304022200. [DOI] [PubMed] [Google Scholar]
- 9.Appleyard SM, Celver J, Pineda V, Kovoor A, Wayman GA, Chavkin C. J Biol Chem. 1999;274:23802–23807. doi: 10.1074/jbc.274.34.23802. [DOI] [PubMed] [Google Scholar]
- 10.Lefkowitz RJ, Shenoy SK. Science. 2005;308:512–517. doi: 10.1126/science.1109237. [DOI] [PubMed] [Google Scholar]
- 11.Sun Y, Cheng Z, Pei G. J Biol Chem. 2002;277:49212–49221. doi: 10.1074/jbc.M207294200. [DOI] [PubMed] [Google Scholar]
- 12.Kovoor A, Celver J, Abdryashitov RI, Chavkin C, Gurevich VV. J Biol Chem. 1999;274:6831–6834. doi: 10.1074/jbc.274.11.6831. [DOI] [PubMed] [Google Scholar]
- 13.Celver J, Vishnivetskiy S, Chavkin C, Gurevich VV. J Biol Chem. 2002;277:9043–9048. doi: 10.1074/jbc.M107400200. [DOI] [PubMed] [Google Scholar]
- 14.Brewer GJ. J Neurosci Methods. 1997;71:143–145. doi: 10.1016/s0165-0270(96)00136-7. [DOI] [PubMed] [Google Scholar]
- 15.Macey TA, Gurevich VV, Neve KA. Mol Pharmacol. 2004;66:1635–1642. doi: 10.1124/mol.104.001495. [DOI] [PubMed] [Google Scholar]
- 16.McLaughlin JP, Marton-Popovici M, Chavkin C. J Neurosci. 2003;23:5674–5683. doi: 10.1523/JNEUROSCI.23-13-05674.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Chavkin C, Sud S, Jin W, Stewart J, Zjawiony JK, Seibert DJ, Toth BA, Hufeisen SJ, Roth BL. J Pharmacol Exp Ther. 2004;308:1197–1203. doi: 10.1124/jpet.103.059394. [DOI] [PubMed] [Google Scholar]
- 18.Wang Y, Tang K, Inan S, Siebert D, Holzgrabe U, Lee DYW, Huang P, Li JG, Cowan A, Liu-Chen LC. J Pharmacol Exp Ther. 2005;312:220–230. doi: 10.1124/jpet.104.073668. [DOI] [PubMed] [Google Scholar]
- 19.Thompson AC, Zapata A, Justice JB, Jr, Vaughan RA, Sharpe LG, Shippenberg TS. J Neurosci. 2000;20:9333–9340. doi: 10.1523/JNEUROSCI.20-24-09333.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Mocsai A, Jakus Z, Vantus T, Berton G, Lowell CA, Ligeti E. J Immunol. 2000;164:4321–4331. doi: 10.4049/jimmunol.164.8.4321. [DOI] [PubMed] [Google Scholar]
- 21.Nagao M, Yamauchi J, Kaziro Y, Itoh H. J Biol Chem. 1998;273:22892–22898. doi: 10.1074/jbc.273.36.22892. [DOI] [PubMed] [Google Scholar]
- 22.Chan AS, Yeung WW, Wong YH. J Neurochem. 2005;94:1457–1470. doi: 10.1111/j.1471-4159.2005.03304.x. [DOI] [PubMed] [Google Scholar]
- 23.Della Rocca GJ, Maudsley S, Daaka Y, Lefkowitz RJ, Luttrell LM. J Biol Chem. 1999;274:13978–13984. doi: 10.1074/jbc.274.20.13978. [DOI] [PubMed] [Google Scholar]
- 24.Hanke JH, Gardner JP, Dow RL, Chnagelian PS, Brissette WH, Weringer EJ, Pollock BA, Connelly PA. J Biol Chem. 1996;271:695–701. doi: 10.1074/jbc.271.2.695. [DOI] [PubMed] [Google Scholar]
- 25.Lackey K, Cory M, Davis R, Frye SV, Harris PA, Hunter RN, Jung DK, McDonald OB, McNutt RW, Peel MR, Rukowske RD, Veal JM, Wood ER. Bioorg Med Chem Lett. 2000;10:223–226. doi: 10.1016/s0960-894x(99)00668-x. [DOI] [PubMed] [Google Scholar]
- 26.Marinissen MJ, Gutkind JS. Trends Pharmacol Sci. 2001;22:368–376. doi: 10.1016/s0165-6147(00)01678-3. [DOI] [PubMed] [Google Scholar]
- 27.Lutrell LM, Ferguson SSG, Daaka Y, Miller WE, Maudsley S, Della Rocca GJ, Lin FT, Kawakatsu H, Owada K, Luttrell DK. Science. 1999;283:655–661. doi: 10.1126/science.283.5402.655. [DOI] [PubMed] [Google Scholar]
- 28.Lutrell LM, Roudabush FL, Choy EW, Miller WE, Field ME, Pierce KL, Lefkowitz RJ. Proc Nat Acad Sci U S A. 2001;98:2449–2454. doi: 10.1073/pnas.041604898. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.McDonald PH, Chow CW, Miller WE, LaPorte SA, Field ME, Lin FT, Davis RJ, Lefkowitz RJ. Science. 2000;290:1574–1577. doi: 10.1126/science.290.5496.1574. [DOI] [PubMed] [Google Scholar]
- 30.Beaulieu JM, Sotnikova TD, Marion S, Lefkowitz RJ, Gainetdinov RR, Caron MG. Cell. 2005;122:261–273. doi: 10.1016/j.cell.2005.05.012. [DOI] [PubMed] [Google Scholar]
- 31.Hunton DL, Barnes WG, Kim J, Ren XR, Violin JD, Reiter E, Milligan G, Dhavalkumar DP, Lefkowitz RJ. Mol Pharmacol. 2005;67:1229–1236. doi: 10.1124/mol.104.006270. [DOI] [PubMed] [Google Scholar]
- 32.Lowes VL, Ip NY, Wong YH. Neurosignals. 2002;11:5–19. doi: 10.1159/000057317. [DOI] [PubMed] [Google Scholar]
- 33.Gutkind JS. Sci STKE. 2000;40:REI. doi: 10.1126/stke.2000.40.re1. [DOI] [PubMed] [Google Scholar]
- 34.Lopez-Ilasaca M, Crespo P, Pellici PG, Gutkind JS, Wetzker R. Science. 1997;275:394–397. doi: 10.1126/science.275.5298.394. [DOI] [PubMed] [Google Scholar]
- 35.Tibbles LA, Woodgett JR. CMLS Cell Mol Life Sci. 1999;55:1230–1254. doi: 10.1007/s000180050369. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Kumar S, Boehm J, Lee JC. Nat Rev Drug Discov. 2003;2:717–726. doi: 10.1038/nrd1177. [DOI] [PubMed] [Google Scholar]
- 37.Deleo JA, Yezierski RP. Pain. 2001;90:1–6. doi: 10.1016/s0304-3959(00)00490-5. [DOI] [PubMed] [Google Scholar]
- 38.Xu M, Petraschka M, McLaughlin JP, Westenbroek RE, Caron MG, Lefkowitz RJ, Czyzyk TA, Pintar JE, Terman GW, Chavkin C. J Neurosci. 2004;24:4576–4584. doi: 10.1523/JNEUROSCI.5552-03.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Ullian EM, Sapperstein SK, Christopherson KS, Barres BA. Science. 2001;291:657–661. doi: 10.1126/science.291.5504.657. [DOI] [PubMed] [Google Scholar]
- 40.Pascual O, Casper KB, Kubera C, Zhang J, Revilla-Sanchez R, Sul JY, Takano H, Moss S, McCarthy K, Haydon P. Science. 2005;310:113–116. doi: 10.1126/science.1116916. [DOI] [PubMed] [Google Scholar]
- 41.Bolshakov VY, Carboni L, Cobb MH, Siegelbaum SA, Belardetti F. Nature. 2000;11:1107–1112. doi: 10.1038/80624. [DOI] [PubMed] [Google Scholar]
- 42.Thomas GM, Huganir RL. Nat Rev Neurosci. 2004;5:173–183. doi: 10.1038/nrn1346. [DOI] [PubMed] [Google Scholar]
- 43.Ronken E, Mulder AH, Schoffelmeer AN. J Neurochem. 1993;61:1634–1639. doi: 10.1111/j.1471-4159.1993.tb09797.x. [DOI] [PubMed] [Google Scholar]