

BACKGROUND
Cancer accounts for 23% of all deaths and is the leading cause of death for people under age 85. About 1.4 million individuals in the United States were diagnosed with cancer in 2006 and 560,000 died of the disease. The corresponding numbers for head and neck squamous cell carcinoma (HNSCC) are 11,200 and 40,000, respectively.1
Radiotherapy plays a crucial role in the treatment for HNSCC. However, the 5-year relapse-free survival rate for patients with locally advanced HNSCC is 30-40%, and most patients die from locoregional disease progression. Tumor repopulation, hypoxia, intrinsic radioresistance, and dose-limiting toxicities are among the chief causes of poor outcome. The efficacy of altered fractionation (AF) regimens and combinations of radiotherapy with chemotherapy has been intensively investigated. Recent meta-analyses showed that collectively AF and radiotherapy plus concurrent chemotherapy increase the 5-year survival rate by 3.4% and 8%, respectively.2, 3 Consequently, most centers have adopted AF and concurrent radiochemotherapy (mostly cisplatin) for the non-surgical treatment of intermediate stage and locally advanced HNSCC, respectively. Unfortunately, the systemic toxicities of chemotherapy and other side effects, particularly mucositis, of the combined therapy can be severe.
Increasing knowledge of molecular radiation biology spurred the development of rational strategies for combining radiotherapy with molecular therapeutics. Laboratory and clinical investigations on epidermal growth factor receptor (EGFR) revealed its role in carcinogenesis, tumor progression, and response to therapy. Collectively, this line of research validated the concept of selective modulation of tumor response to radiotherapy by targeting a specific growth factor signaling pathway and established a new treatment option for locally advanced HNSCC. This article summarizes relevant findings of laboratory and clinical investigations on EGFR put into historical perspective in order to assess the direction of future investigations in radiation oncology. The number of references is capped in compliance with the journal policy. A recent review by Nyati et al.4 is a complementary reference.
DISCOVERY AND STRUCTURE OF EPIDERMAL GROWTH FACTOR RECEPTOR
A protein promoting neurite outgrowth in a mouse tumor was characterized in 1965 as epidermal growth factor (EGF), as it stimulated epithelial cell proliferation.5 Its receptor was identified a decade later along with the demonstration that its tyrosine-specific phosphorylation activated intracellular signal transduction.6 The EGFR was later characterized as a 170-kDa transmembrane receptor tyrosine kinase (RTK) of the ERBB family, which is essential for normal development. Its cDNA sequence (3633 bp)7, chromosomal location (7p12-p22)8, genomic structure, and amino-acid sequence were subsequently identified.9
Structurally, the EGFR is composed of four extracellular domains, a hydrophobic transmembrane region, a juxta-membrane domain, an intracellular protein tyrosine kinase domain (PTK) containing the ATP-binding pocket, and a regulatory carboxyl terminal domain (Fig. 1). It is monomeric in the basal state, but binding of a ligand produces an extended and stabilized conformation, which promotes homo- and hetero-dimerization10 and activates signal transduction.
Figure 1. Schematic illustration of EGFR structure in its tethered (A) and untethered (B) form.
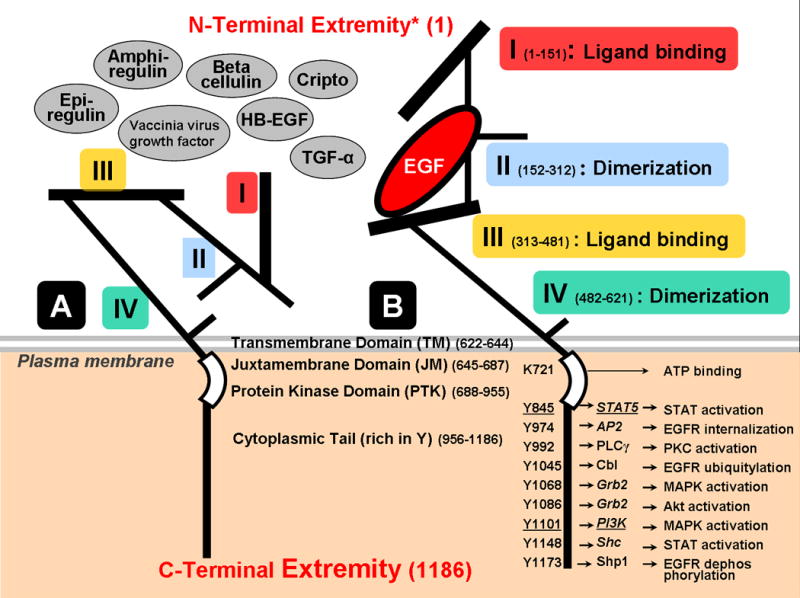
There are 4 extracellular domains, collectively called ectodomain, of which domains I and III (also referred to as L1 and L2) are involved in ligand binding and the cystein rich domains II and IV (CR1 and CR2) in dimerization. The remainder of the structure consists of a hydrophobic transmembrane domain (TM), a juxta-membrane domain (JM), an intracellular protein tyrosine kinase domain (PTK), including the ATP binding pocket with receptor kinase activity (RTK), and a regulatory carboxyl terminal domain (reviewed by Jorissen et al.73). The eight ligands (12 ligands for all ERBB receptors together) presently known are EGF, TGF-α, HB-EGF, amphiregulin, betacellulin, epiregulin, vaccinia virus growth factor and cripto. Binding of a ligand (e.g., EGF in Panel B) to Domains I and III alters and stabilizes the spatial configuration promoting homo- and hetero-dimerization10 and subsequent activation (also see Fig. 2).
Tyrosines of the EGFR cytoplasmic tail (e.g., Src activation sites underlined) represent docking sites for adaptors15 or for downstream proteins. Phosphorylation of the EGFR C-terminus, by autophosphorylation or transphosphorylation by other kinases such as Src16 and Jak-2, provides specific docking sites for specific interaction domains of intracellular signal transducers and adaptors, leading to their colocalization and to the assembly of multicomponent signaling “particles.” Signaling proteins that associate directly with some EGFR tyrosines are illustrated in panel B.
FUNCTION OF EPIDERMAL GROWTH FACTOR RECEPTOR
EGFR activation initiates multiple layers of signaling and amplification governing biological responses. Briefly, EGFR serves as a node in interacting networks, as its carboxyl terminal domain contains numerous distinct docking sites for various molecules in response to stimuli (Fig. 1).11 EGFR heterodimerizes with other growth factors12 such as other ERBB receptors and IGF-1R13 at the cell surface. Heterodimerization is facilitated by raft colocalization14 and result in trans-autophosphorylation through horizontal interactions.14, 15 In addition, non-RTKs (e.g., Src) can participate in EGFR-mediated transactivation.16 These dynamic interactions are regulated by site- and time-specific phosphorylations of tyrosine (Fig. 1), serine, and threonine residues. These events initiate the recruitment and phosphorylation of several intracellular substrates (signaling proteins and adaptors like Grb2 and Shc), leading to phosphorylation cascades. A specific ligand triggers preferential activation of downstream signaling pathways (Fig. 1, 2). For example, EGF might preferentially activate the PLC pathway17, whereas TGF-α might favor the Jak-STAT pathway.18
Figure 2. EGFR activation and downstream signaling.
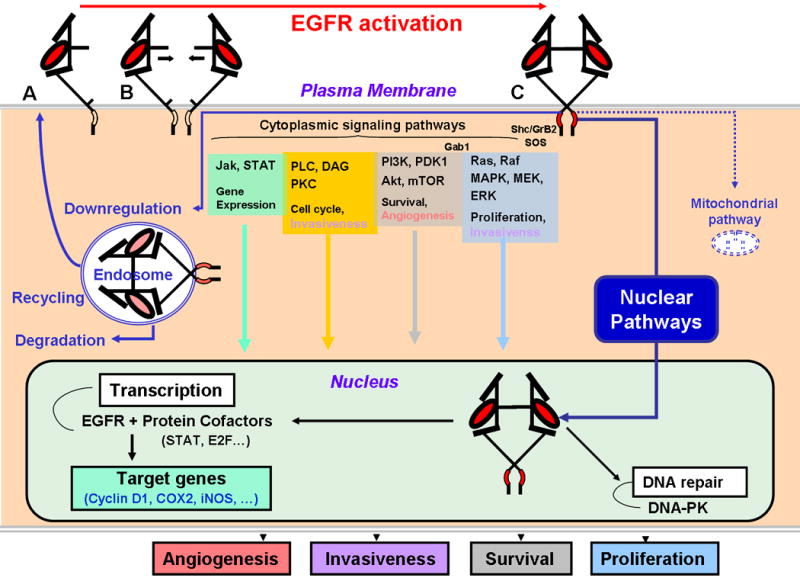
Upper panel illustrated ligand-induced conformational change followed by dimerization progressing from Panels A to C (see Jorissen et al. for complete review.73). This process triggers downstream signaling through four major cytoplasmic pathways depicted schematically. Nuclear influx of EGFR initiates interaction with transcription factors and proteins participating in DNA double-strand break repair. Endosomal degradation or recycling regulates EGFR signaling. A mitochondrial pathway has recently been described and its functions is under investigation.
Four major cytoplasmic downstream signaling routes of EGFR have been characterized (Fig. 2). The Ras-Raf-MAPK pathway leads to the activation of ERK1-2, which in turn regulates the transcription of molecules mediating cell proliferation, survival, and transformation. The PI3K and downstream serine/threonine kinase, Akt, govern cell growth, proliferation, survival, and motility. The Jak/STAT pathway translocates STAT molecules to the nucleus to induce gene transcription and mediate cell division, viability, motility, invasion, adhesion, and DNA repair. The PLC-DAG-calcium/calmoduline-PKC pathway also regulates cell cycle progression and cell motility.17
Nuclear EGFR pathways (Fig. 2), ligand-dependent19 and -independent20, 21, were recently identified. EGFR possesses nuclear localization sequence signals in its juxta-membrane domain (Fig. 1)22 for nuclear translocation as non-membrane-bound receptor through the nuclear pore complex, or through interaction with nuclear transport receptors such as importins α/β1 and exportins.21 Although EGFR lacks putative DNA binding domains, it has transactivation domains on its C-terminal extremity21 (Fig. 2) regulating synthesis of pro-mitogenic proteins.19 In addition, EGFR interacts with nuclear DNA-PK (Fig. 2) and promotes repair of radiation-induced DNA strand-breaks20 (discussed below in modulation of radiosensitivity). Mitochondrial pathway23 was recently described (Fig. 2).
Attenuation of EGFR signaling is through dephosphorylation of key residues and removal by endocytosis. Following clathrin-mediated endocytosis, EGFR is sorted into early endosomes and directed to multi-vesicular bodies and late endosomes for degradation or recycling.14, 24 Multiubiquitination of EGFR mediated by Cbl is essential for internalization and routing for lysosomal degradation.14 Deficiencies in this control mechanism can result in enhanced recycling and signal amplification.
EGFR IN CANCER
EGFR is highly expressed in most carcinomas. EGFR mRNA and protein are expressed abundantly in 90% of HNSCCs and less frequently in the adjacent dysplastic lesions or in histologically normal surrounding mucosa25, which imply that EGFR amplification plays a role in early carcinogenesis. Transcriptional targets of nuclear EGFR (Fig. 2)21 are involved in tumor progression.
The main mechanism of EGFR upregulation is transcriptional activation, secondary to autocrine production of TGF-α.26 TGF-α is closely related to EGF including binding to EGFR and thereby initiating signal transduction. It can be secreted by macrophages, T cells, and keratinocytes in response to tissue injury. High EGFR expression is often associated with poor prognosis and resistance to cytotoxic agents, including ionizing radiation (discussed below). High nuclear EGFR level has also been correlated with poor outcome in HNSCC.27
Gain of function may also occur through mutations. Activating mutations in the kinase domain found in non–small-cell lung cancer (NSCLC) appear to be rare in HNSCC. Deletion of exons 2-7 of the extracellular domain yields a constitutively active truncated EGFRvIII.28 It is prevalent in glioblastomas and to lesser extent in HNSCC.29 EGFRvIII and the kinase domain mutants activate survival pathways such as Akt.30 Cross-talk with other ERBB receptors can also lead to aberrant activation.
EGFR IN RADIOTHERAPY
A. Preclinical Studies
EGFR and tumor clonogen repopulation
Repopulation of tumor clonogens during treatment is one mechanism of resistance to radiotherapy31 (Fig. 3A). Schmidt-Ullrich et al. found that cancer cells surviving irradiation acquired a phenotype with upregulated EGFR and TGF-α.32 They further showed in vitro that therapeutic dose range of radiation increased EGFR tyrosine phosphorylation26, which was linked to critical components of mitogenic signaling pathways.33 This adaptive response produced radioresistance and was interpreted as an underlying mechanism for accelerated repopulation.
Figure 3. Integration of traditional and molecular radiology for the development of a novel combined therapy modality.
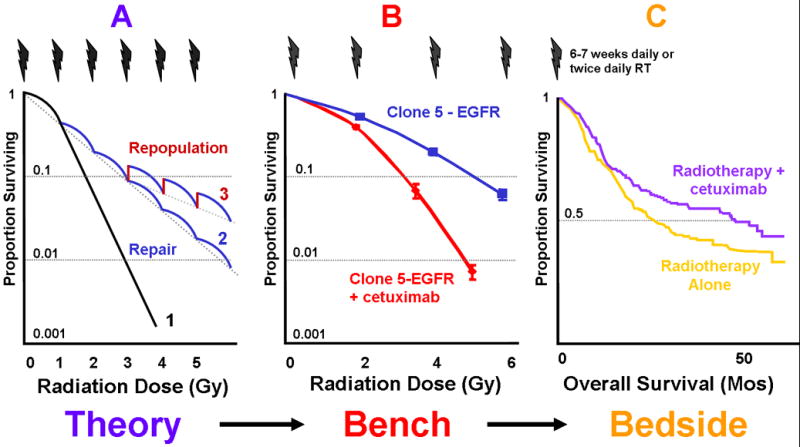
Panel A illustrates the survival curve of a single dose exposure along with the effects of sublethal damage repair (from curve 1 to 2) and clonogen repopulation (from curve 2 to 3) between fractions resulting in an increase in cell survival. Panel B shows that radiation resistance resulting from transduction of EGFR can be offset by blocking the EGFR by specific antibody.38 Panel C summarizes the results of a pivotal randomized clinical trial showing an improvement in overall survival, resulting from better local-regional control, by adding cetuximab to radiotherapy in patients with locally advanced HNSCC.49
Doses of 1-5 Gy induced a 2-to 5-fold increase in tyrosine phosphorylation within 5-10 min, as opposed to >5-fold rise induced by ligands in physiologic concentrations26, 33 This first phase of activation, falling to baseline within 10 min, was associated with stimulation of major signaling pathways with selective functional linkage to different ERBB receptors.33 MAPK, for example, peaked between 5-15 min and was linked to EGFR activation with additional contributions by Raf.26 The second phase starts after 30 min and triggers pro-proliferative responses and activation of transcription factors.34
Effect of EGFR on cellular radiation sensitivity
The first clue that EGFR expression might affect cellular radiation sensitivity in vivo emerged from a study on murine models by Akimoto and colleagues.35 They found that single-dose irradiation induced EGFR autophosphorylation and downstream signaling only in high EGFR-expressing tumors. This phenomenon was associated with relative radioresistance. Since clonogen repopulation plays no role in determining in vivo tumor response to single-dose irradiation36, these results suggest that EGFR contributes to determining intrinsic radiosensitivity. The data of a complementary correlative study37 using specimens of patients with HNSCC (see below) are consistent with this finding.
A follow-up study 38 revealed evidence for a causal relationship between EGFR expression and radioresistance. Transfection of a full-length human EGFR vector into a low EGFR-expressing murine carcinoma cell line resulted in an EGFR level-dependent increase in radioresistance measured by clonogenic cell survival assays. It also demonstrated that exposing EGFR transfectants to the anti-EGFR antibody cetuximab reduces the expression of EGFR and downstream Akt and MAPK and reverses the cellular radioresistance (Fig. 3B).
A subsequent experiment elucidated a mechanism by which EGFR affects radiation sensitivity.20 This thorough study showed that in contrast to cytoplasmic signaling induced by EGF, radiation triggeres nuclear EGFR translocation. This process is accompanied by a nuclear influx of proteins Ku70/80 and phosphatase 1, an increase in nuclear DNA-PK activity, and formation of DNA end-binding protein complexes containing DNA-PK, which plays a dominant role in repairing radiation-induced DNA double strand-breaks through a non-homologous end-joining mechanism. EGFR blockade by cetuximab abolishes nuclear EGFR import, diminishes radiation-induced activation of DNA-PK, inhibits DNA repair, and enhances cellular radiation sensitivity.20
EGFR inhibitors to enhance tumor response to radiotherapy
The concept that blockade of EGFR signaling might have antitumor activity was introduced in 1980s. Mendelsohn et al. provided the first pre-clinical demonstration of an antiproliferative effect of monoclonal antibodies (MAbs) directed at the EGF-binding site.39 Since then, numerous EGFR inhibitors were studied.
The potential modulation of radiation response with EGFR inhibitors attracted attention in the mid 1990s. Several investigators showed that EGFR antagonists inhibits radiation-induced EGFR phosphorylation and tumor cell proliferation.26, 40 Initial studies with cetuximab showed enhanced tumor response to single-dose and fractionated radiation in cell lines and in xenograft models using apoptosis41, 42, regrowth delay43 and tumor control44 as endpoints. Further studies showed that the magnitude of the radiation enhancement varies among EGFR antagonists.45 These findings sparked an interest in elucidating the mechanisms of radiation sensitization in vivo.
The available data suggest that cetuximab can potentially increase radiosensitivity through several processes. Briefly, these include (1) binding to domain III46 (Fig.1) and sterical blockade of domain I, thus preventing ligand-binding and ligand-independent (i.e. radiation-induced) activation34; (2) preventing EGFR from adopting the conformation needed for dimerization; (3) preventing EGFR from inducing autocrine ligand production; (4) inhibiting EGFR nuclear translocation and thus impairing EGFR-mediated DNA repair20; (5) inducing an antibody-dependent cellular toxicity 46; and (6) downregulating the expression of several pro-angiogenic factors, including VEGF, bFGF, IL8, and thereby promotes endothelial cell apoptosis and vasculature collapse.47 It should also be realized, however, that reassortment of cells in a relatively radioresistant G1 phase of the cell cycle might be a drawback associated with the use of EGFR antagonists for the treatment of slowly repopulation cells.
B. Correlative Biomarker Studies and Clinical Trials
A correlative biomarker analysis using specimens of patients with locally advanced HNSCC enrolled into a phase III trial showed that high EGFR expression was a strong, independent predictor of local-regional control (LRC) and overall survival after radiotherapy alone.37 Concurrently, Roberts et al. demonstrated in a pilot study48 the safety of combining cetuximab with radiotherapy and recommended a regimen consisting of one loading dose before radiotherapy, followed by seven weekly doses concurrent with radiotherapy.
Preclinical studies, biomarker analysis, and the pilot trial provided the impetus in completing a phase III study testing the efficacy of combining cetuximab with radiotherapy.49 This pivotal trial demonstrated that the addition of eight doses of cetuximab to radiotherapy improved LRC significantly (3-year rate: 47% vs 34%, p=0.005) without increasing radiation side effects, including mucositis, dysphagia, pain, etc. There were also significant increases in median survival time (from 29 to 49 months) and 3-year survival rate (from 45% to 55%), as shown in Fig. 3C. Consequently, cetuximab in combination with radiotherapy was approved as a frontline treatment for locally advanced HNSCC.
Collectively, coordinated studies established the proof-of-principle that modulating a perturbed signaling pathway can lead to a selective tumor radiosensitization and thereby truly improve the therapeutic index. Such clear improvement in the therapeutic index has not been accomplished by combining radiation with traditional chemotherapy.
Building on this success, rational combinations of radiotherapy, with or without chemotherapy, with other novel agents are being explored. In patients with locally advanced HNSCC, for example, the nonsurgical standard of care has changed from radiotherapy alone to concurrent radiochemotherapy after the phase III cetuximab trial had been launched. Therefore, trials have been commenced to assess the feasibility of combining cetuximab with radio chemotherapy. A phase II trial showed that the combination of radiotherapy-cetuximab with cisplatin resulted in 3-year overall survival of 76%, despite the occurrence of two early deaths (pneumonia and unknown cause).50 This regimen is now being evaluated in a phase III trial. The combination of radiotherapy-cetuximab with gemcitabine in HNSCC yielded a complete response rate of 77% and no major toxicities (Table 1).
Table 1.
Examples of ongoing clinical trials assessing combinations of radiotherapy with molecular therapeutics for the treatment of head-and-neck carcinomas. Preliminary results of some trials were presented at the 2006 Annual meeting of the American Society of Clinical Oncology (abstract number).
| Principal Investigator or Leader | N | Phase | Tumor Stage | RT combined with | Results or Endpoints |
|---|---|---|---|---|---|
| De La Garza, ASCO 2006 (#15502) | 20 | II | Locally advanced | Cetuximab + gemcitabine | CR 77%, 89% compliance, 42% G-3 mucositis, 21% G-3 rash |
| Langer (ECOG E3303) | 68 | II | Locally advanced | Cetuximab + cisplatin | PFS, LC, OS |
| Kataria (NCT00343083) | 60 | II | Locally advanced | Cetuximab + paclitaxel + carboplatin | LRC, PFS, pathologic RR, toxicities |
| Mesia (NCT00251381) | 90 | II | Locally advanced | Cetuximab | LRC, toxicity |
| Ang (RTOG 0522; NCT0026594) | 720 | II | Locally advanced | Cetuximab + cisplatin (accelerated fractionation) | DFS, OS, LRC, toxicity, QOL, and correlative study |
| Harari (RTOG 0234; NCT00084318) | 230 | II-R | Locally advanced | Cetuximab + cisplatin or docetaxel (adjuvant) | DFS, LRC,OS, toxicity, correlative study |
| Argiris (NCT00226239) | 40 | II | Locally advanced | Cetuximab + cisplatin | RR, toxicities, control and survival rates |
| Argiris (NCT00291707) | 40 | I | Locally advanced | Cetuximab + pemetrexed | MTD, DLT, Toxicity, RR |
| Cohen, ASCO 2006 (#5506) | 69 | II | Locally advanced | Gefitinib + 5FU + hydroxyurea | CR 89%, RR 98%, PFS 1-year 85% |
| Doss, ASCO 2006 (#5543) | 45 | II | Locally advanced | Gefitinib + paclitaxel + carboplatin | CR 32%, 1-year PFS 68% 1-year OS 86% 1 toxic death |
| Hainsworth (NCT00193284) | 50 | II | Locally advanced | Gefitinib +/- docetaxel after induction | RR + OS + TTP + toxicity |
| AstraZeneca (NCT00228488) | 60 | II | Locally advanced or first recurrence | Gefitinib | effect on gene expression profiles |
| Morris (NCT00083057) | 30 | I | Locally advanced or first recurrence | Gefitinib + paclitaxel | MTD, RR |
| AstraZeneca (NCT00233636) | 28 | II | Locally advanced | Gefitinib | RR, TTP, OS |
| Wadler (NCT00195078) | 29 | I-II | Locally advanced | Gefitinib + cisplatin | RR, DFS, duration of response, toxicity |
| Adelstein (NCT00352105) | 66 | I-II | Locally advanced | Gefitinib + cisplatin + FU | OS, DFS, toxicity |
| Raben (NCT00033449) | 30 | I | Locally advanced | Gefitinib + cisplatin + maintenance | MTD, toxicity, RR, PFS, OS |
| Le (NCT00185835) | 10 | I | First recurrence | Gefitinib + cisplatin (reirradiation) | Toxicity, DFS, OS, RR, duration of response, correlative study |
| Bensadoun (GORTEC 2004-02; NCT00169221) | 140 | II-R | Locally advanced | Cisplatin +/- gefitinib (post-operative adjuvant) | Safety, Efficacy |
| AstraZeneca (NCT00229723) | 224 | II | Locally advanced | Gefitinib + cisplatin | LCR, PFS, OS, tolerability, correlative study |
| Herchenhorn ASCO 2006 (abstract 5575) | I-II | Locally advanced | Erlotinib + cisplatin | Toxicity, RR | |
| Savvides ASCO 2006 (#5545) | 23 | I | Locally advanced | Erlotinib + docetaxel (+ maintenance) | CR: 83% |
| Brizel (NCT00140556) | 30 | I-II-R | Locally advanced | Erlotinib + bevacizumab + cisplatin (hyperfractionation) | RR, LRC, OS, Completion rate |
| Glisson (NCT00113347) | 24 | I | Locally advanced | Erlotinib + docetaxel | MTD, TTP, OS, swallowing function |
| Gillison (NCT00049166) | 48 | I | Locally advanced | Erlotinib +/- cisplatin (IMRT) | MTD, correlative study, PET |
| Klug (NCT00304278) | 20 | II | Locally advanced | Erlotinib intra-arterial + cisplatin | Safety, LRC |
| GSK Clinical trial (NCT00371566) | 90 | II-R | Locally advanced | Lapatinib | RR, toxicity |
| Harrington ASCO 2006 (#5553) | 17 | I | Locally advanced | Lapatinib + cisplatin | Toxicity: 2 DLT (Grade 3-4) |
| Seiwert ASCO 2006 (abstract 5530) | 43 | I | First recurrence | Bevacizumab + FU + hydroxyurea | 2-Year OS: 26 months |
| Savvides (NCT00281840) | 30 | II-R | Locally advanced | Bevacizumab + docetaxel | TTP, RR, duration of response, LRC, OS, toxicity |
| Prellop ASCO 2006 (#5582) | 28 | I-II | Locally advanced or first recurrence | Celecoxib + paclitaxel + carboplatin + maintenance | G3-4 febrile neutropenia: 24%, 2-Year OS: 65%, LC: 76% |
CR: complete response, DFS: disease-free survival; DLT: dose limiting toxicity; FU: fluorouracil; G: grade; IMRT: intensity modulated radiotherapy; LC: local control; LRC: local-regional control; MTD: maximally tolerated dose; OS: overall survival; PFS: progression free survival; QOL: quality of life; R: randomized; RR: response rate; TTP: time to tumor progression.
It is also important to realize that cetuximab does not benefit 85-90% of patients with locally advanced HNSCC. Over 50% of patients receiving the combination still developed local-regional relapse. It is thus critical to develop assays to identify such “resistant” tumors to personalize therapy. It is also vital to understand the biological basis of resistance to EGFR antagonists to develop alternative strategies.
PREDICTIVE BIOMARKERS FOR TUMOR RESPONSE TO ANTI-EGFR THERAPY
Emerging data indicate that EGFR expression, mainly measured by immunohistochemistry (IHC), is an independent predictor of HNSCC response to conventionally fractionated37 or accelerated51, 52 radiotherapy or radiotherapy plus chemotherapy. However, the predictive power varies among the series. The search for predictive biomarkers for response to EGFR antagonists has been disappointing. Counterintuitively, pre-treatment tumor EGFR expression was not found to predict response to EGFR antagonists.53, 54 Some patients with negative EGFR colorectal cancer even benefited from cetuximab.55
Noteworthy is that the assay methodology varied widely. A standard IHC assay generally deems a cell positive when >30,000 EGFR receptors are present. Unfortunately, the number and density of receptors required to mediate a given biologic effect is not known. This deficiency could partially account for the discrepancies between studies. In addition, quantitative-EGF binding experiments showed the presence of high- and low-affinity forms of EGFR on the cell surface56, whose proportions, roles, and dynamics are still largely unknown. Moreover, infiltrating inflammatory cells can also express EGFR and thereby further confounding the finding. So it is crucial to standardize the assays (fixative, storage time, scoring method) and perform better organized correlative analyses to resolve this important topic. Combining IHC assay with FISH might also yield better predictive power for the response to EGFR inhibitors, as shown in NSCLC.57
There appears to be a dose–response relationship between the incidence and severity of skin rash and a clinical benefit in some tumors53, 58 but data in HNSCC are contradictory.54, 59 Of note is that the recording and reporting of rash have not been standardized. Even if the association is further confirmed, rash might be useful only for titrating the dose of EGFR inhibitors in individual patients, e.g., escalating the dose until a rash appears, but not for identifying patients who might benefit most from the therapy.
OVERCOMING RESISTANCE TO EGFR INHIBITORS
Why EGFR antagonists do not affect the growth or radiation sensitivity of most HNSCC is unclear. A number of hypotheses and research directions for overcoming resistance are briefly summarized. First, cetuximab or TKIs given as single agents in the current dose regimens might not effectively suppress EGFR-mediated signaling. Therefore, the potential benefits of other dose regimens, other antibodies, alternative TKIs, antisense nucleotides, or various combinations of these agents have been investigated. A recent preclinical study showed that three additional doses of cetuximab given after concurrent radiation-cetuximab improved LRC compared to concurrent radiation-cetuximab alone.60 Other interesting antibodies are hR3 (longer half-life than cetuximab) and panitumumab (human MAb with higher affinity for EGFR). Radiotherapy plus hR3 yielded 3-year overall survival and 2-year disease-free survival rates of 67% and 65%, respectively, in patients with locally advanced HNSCC 61. A phase III trial showed a benefit of panitumumab compared to best supportive care in patients with metastatic colorectal cancers that have failed chemotherapy.58
Two types of TKIs are now available. Type 1, such as erlotinib and gefitinib, targets the kinase ATP binding site in its active conformation. The combination of erlotinib and gefitinib with various radiochemotherapy regimens are being evaluated (Table 1). CI-1033 is another TKI that binds irreversibly to all ERBB kinases (a pan-ERBB inhibitor), and EGFRvIII, resulting in a prolonged suppression of downstream signaling. In HNSCC cell lines, CI-1033 blocked cell growth, downregulated specific genes co-regulating in vivo neoplastic behavior, and sensitized cells to radiotherapy. Type II TKIs, such as lapatinib, have an additional binding site immediately adjacent to the ATP docking site. Its longer half-life correlates with a prolonged down-regulation of receptor tyrosine phosphorylation in tumor cells relative to erlotinib and gefitinib.62 It is being tested in phase II trials (Table 1).
Dual-agent targeting of the EGFR pathway (gefitinib or erlotinib plus cetuximab) or multi-target TKIs showed more pronounced inhibition of cell proliferation and tumor growth in preclinical models.45 Preliminary clinical studies (colorectal, HNSCC, and NSCLC) also showed that cetuximab plus gefitinib had a superior pharmacodynamic signal inhibition and greater clinical activity than either agent alone.
Second, EGFR mutations may result in aberrant signaling and poorer response to EGFR antagonists or radiotherapy. EGFRvIII was detected in a number of tumors and differed from wild-type EGFR (EGFRwt) in its preferential activation of downstream signaling pathways.30 In a series of 33 patients, EFGRvIII and EGFRwt were simultaneously expressed in 42% of HNSCCs.29 Transfection of EGFRvIII into HNSCC cell lines decreased cisplatin-induced apoptosis and cetuximab-induced growth inhibition. This observation formed the basis for investigating EGFRvIII-specific monoclonal antibody. Other somatic mutations of the kinase domain occur in 1% and 7% of Caucasian and Asian patients with HNSCC, respectively.63 Their biologic impact is largely unknown owing to the low incidence.
Third, constitutive activation of signaling pathways downstream of EGFR by upregulation of other ERBB receptors or RTKs can promote survival (see review by Kalyankrishna and Grandis18). For example, a high level of activated Akt can occur downstream of EGFR inhibition through alternative upstream-activated Src, Ras, mutated PTEN, or amplification of the PI3K catalytic subunit. The STAT3 and STAT5 pathways can be constitutively active in HNSCC. Overexpression of six major ERBB family ligands can activate ERBB receptors and IGF-1R, resulting in resistance to EGFR inhibition. Upregulation of IGF-1R, for instance, resulted in sustained signaling through the PI3K pathway, leading to antiapoptotic and proinvasion effects and resistance to a TKI in a glioblastoma model.13 Activation of EGFR-independent pathways, such as G protein-coupled receptors (GPCRs)64, may promote survival and resistance to EGFR inhibitors. Of note is that ionizing radiation can activate all ERBB receptors12, IGF-1R13, and some metalloproteases and integrins.18
Fourth, HNSCCs commonly express high level of VEGF, which supports tumor growth by stimulating angiogenesis. EGFR signaling also stimulates VEGF expression by tumor cells. One mechanism of acquired resistance to EGFR inhibitors is the selection of tumor cell subpopulations with increased angiogenic potential65, suggesting that VEGF might be upregulated by alternative pathways. Therefore, many preclinical studies address the efficacy of targeting EGFR and angiogenic pathways simultaneously. VEGF-A is a key regulator of tumor-induced endothelial cell proliferation and vascular permeability. Adding an anti-VEGFR antibody DC101 to cetuximab, for example, significantly reduced tumor vascularity, inhibited tumor growth, and increased apoptosis in both tumor and endothelial cells.66 ZD6474 (vandetanib) is a small molecule TKI with specificity towards VEGFR and EGFR. Preclinical studies of ZD6474 demonstrated radiation sensitization of various xenografts67 and reduction of microvascular density in tumors resistant to cetuximab or gefitinib. These results provided a rationale for the clinical evaluation of ZD6474 with taxanes or cetuximab. Preliminary data from a phase II trial testing sorafenib, a potent inhibitor of the Raf-1, B-Raf, VEGFR-2-3, and PDGFR-B pathways, in metastatic or recurrent HNSCC were recently reported.
COMBINING RADIOTHERAPY WITH OTHER MOLECULAR THERAPEUTICS
Numerous strategies are emerging based on better understanding of tumor biology. A comprehensive overview is beyond the scope of this article, but some strategies are briefly summarized to illustrate the need for extensive commitment and the scientific-practical obstacles to the development of novel therapeutic strategies.
Sensitizing tumors to radiotherapy by targeting the resistant hypoxic tumor cells has been attempted for many decades. The clinical results of oxygen mimetic agents (nitroimidazole compounds) have been disappointing with the exception of nimorazol in a Danish trial.68 The availability of tirapazamine (TPZ), a bioreductive cytotoxic agent that is toxic to hypoxic cells, has renewed interest in this field. Phase III trials testing its combination with radiation and cisplatin in the treatment of HNSCC were launched based on encouraging results of a phase II study.69 The first efficacy analysis showed no overall survival benefit in favor of TPZ. This finding along with the increased treatment-associated mortality observed in the experimental arm of the second trial led to early termination of this clinical development program in HNSCC (L. Peters, personal communication, 2006). A phase III trial assessing the efficacy of ARCON, which combines accelerated radiotherapy with carbogen (inhalation of hyperoxic gas) and nicotinamide (a vasoactive agent) to decrease chronic and perfusion-limited hypoxia, respectively, is approaching completion of patient accrual.
Cyclooxygenase-2 (COX-2), a key enzyme for the synthesis of prostaglandins (PGs), is another target. COX-2 prevents cell damage by ionizing radiation.70 COX-2 and PG overexpression are linked to carcinogenesis, tumor growth, facilitation of metastatic spread, and decreased immunosurveillance.70 In addition, macrophages and other inflammatory cells that infiltrate the tumor can produce COX-2 and thereby contributed to increased tumor radioresistance. PGs also enhance bFGF-induced angiogenesis through induction of VEGF.70 Celecoxib, a COX-2 inhibitor, enhances cellular sensitivity to radiation in vitro70 through inhibition of DNA repair processes and vasculature collapse. Unfortunately, the increased cardiovascular toxicity associated with long-term use of COX-2 inhibitors as chemoprevention in individuals with colorectal adenoma has prematurely dampened the interest, mostly from the industrial sector, in the drug’s development as a cancer therapy since 2005. It is worth stressing that the benefit-risk ratio might still be favorable in cancer patients, which is being addressed in an ongoing clinical trial (Table 1).
The concept of targeting angiogenesis in cancer has been pioneered by Folkman since 1970s. Agents targeting the VEGF-VEGFR signaling axis used to overcome tumor resistance to EGFR antagonists are presented above. In general, radiation oncologists have been skeptical about combining anti-angiogenic agents with radiotherapy because of concerns of inducing tumor hypoxia and thus diminish the response to radiotherapy. Recent preclinical data, however, suggest that some anti-angiogenic agents may induce a transient normalization window with increased blood flow and tumor oxygenation.71 In addition, emerging data show that bevacizumab (antibody against VEGF-A) added to chemotherapy increases the response rate and survival in a number of cancers. This feature made it interesting for combining bevacizumab with radiotherapy and chemotherapy for the treatment of cancers in which distant metastasis is the main pattern of relapse (e.g., nasopharyngeal carcinoma). Clinical trials testing the combination of bevacizumab with radiochemotherapy are ongoing in rectal cancers72 and HNSCC (Table 1). Agents targeting vascular endothelium, including combretastatin A-4, are also being tested.
Some ongoing trials combining radiotherapy with EGFR-inhibitors, anti-angiogenic agents, or other targeted therapies, as presented above, or at the 2006 annual ASCO meeting are summarized in Table 1.
CONCLUSION
Advances in the understanding of tumor biology have opened a new strategy for developing novel cancer therapy. Research on the EGFR signaling pathway exemplifies this quest. Discoveries of the contribution of the EGFR signaling pathways to several key cellular regulatory processes, their perturbation in epithelial neoplasms, and the improvement in tumor response to therapy led to the conception and completion of an integrated research program producing a new frontline therapy modality for locally advanced HNSCC. Although this pivotal phase III trial yielded the proof-of-principle that selective tumor sensitization to radiotherapy can be accomplished by modulating a perturbed signaling pathway, the clinical benefit resulting from this strategy was rather modest, and many questions remain to be addressed. For example, why the magnitude of EGFR expression does not correlate with tumor response to EGFR antagonists is poorly understood and even counter-intuitive. Further investigations are underway to identify biomarkers that predict the response to EGFR inhibitors and isolate the mechanisms that underlie the lack of cetuximab-mediated sensitization in the majority of HNSCC to radiotherapy. Lessons learned from the work on EGFR will contribute to the development of strategies to augment tumor response by modulating other signaling pathways individually or in combination, which will bring us closer to the implementation of personalized cancer therapy.
Acknowledgments
Supported by grants P01CA06294 and R01CA84415 awarded by the National Cancer Institute, Lavoisier awarded by the French Ministry of Foreign Affairs, and the Gilbert H. Fletcher Distinguished Chair. We thank fellows, visiting scientists, and the Head and Neck multidisciplinary team for their contributions to preclinical studies and clinical trials, and Ms. Cathy Ramirez for preparing this manuscript.
GLOSSARY
- Akt
also known as protein kinase B (PKB). There are three isoforms Akt1, Akt2, Akt3
- ATP
adenosine triphosphate
- bFGF
basic fibroblast growth factor
- B-Raf
v-raf murine sarcoma viral oncogene homolog B1
- Cbl
Casitas B-lineage lymphoma proto-oncogene (ubiquitin ligase; also called c-Cbl)
- DAG
diacylglycerol
- DNA-PK
DNA-dependent protein kinase
- EGFRvIII
truncated constitutively active variant of EGFR
- ERBB
family of receptor tyrosine kinase receptors including EGFR/HER1, HER2/ERBB2, HER3/ERBB3, HER4/ERBB4
- FISH
fluorescence in situ hybridization
- HB-EGF
heparin-binding EGF
- IGF-1R
Insulin Growth Factor 1 Receptor
- IL8
interleukin 8
- Jak
Janus kinase
- MAPK
mitogen-activated protein kinase
- PDGFR-B
platelet-derived growth factor B
- PI3K
phosphatidylinositol 3-serine/threonine kinase
- PKC
serine/threonine kinase protein kinase-C
- PLC
phospholipase c (PLC usually stands for PLC-gamma)
- PTEN
phosphatase and tensin homolog
- Raf
v-raf-1 murine leukemia viral oncogene homolog
- Raft
membrane microdomains rich in sphingolipids and cholesterol. Caveolae are sometimes considered as a caveolin-positive subset of lipid rafts
- Ras
rat sarcoma viral oncogene homolog
- SH2-domain
Src homology 2 protein domain
- Shc
SH2 containing transforming protein (protein adaptor)
- Src
sarcoma viral oncogene homolog (also called c-Src)
- STAT
signal transducer and activator of transcription
- TGF-α
transforming growth factor α
- TKI
low-molecular-weight ATP-competitive inhibitor of the receptor’s tyrosine kinase
- VEGF
vascular endothelial growth factor
- VEGFR
vascular endothelial growth factor receptor
- Y
tyrosine residue (amino acid)
Footnotes
Conflict of interest: none
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Jemal A, Siegel R, Ward E, et al. Cancer statistics, 2006. CA Cancer J Clin. 2006;56:106–130. doi: 10.3322/canjclin.56.2.106. [DOI] [PubMed] [Google Scholar]
- 2.Bourhis J, Armand CPignon JP. Update of MACH-NC (Meta-Analysis of Chemotherapy in Head & Neck Cancer) database focused on concomitant chemoradiotherapy. J Clin Oncol ASCO Annual Meeting Proceedings (Post-Meeting Edition) 2004;22:5505. [Google Scholar]
- 3.Bourhis J, Overgaard J, Audry H, et al. Hyperfractionated or accelerated radiotherapy in head and neck cancer: a meta-analysis. Lancet. 2006;368:843–854. doi: 10.1016/S0140-6736(06)69121-6. [DOI] [PubMed] [Google Scholar]
- 4.Nyati MK, Morgan MA, Feng FY, et al. Integration of EGFR inhibitors with radiochemotherapy. Nat Rev Cancer. 2006;6:876–885. doi: 10.1038/nrc1953. [DOI] [PubMed] [Google Scholar]
- 5.Cohen S. The stimulation of epidermal proliferation by a specific protein (EGF) Dev Biol. 1965;12:394–407. doi: 10.1016/0012-1606(65)90005-9. [DOI] [PubMed] [Google Scholar]
- 6.Cohen S, Carpenter G, King L., Jr Epidermal growth factor-receptor-protein kinase interactions. Co-purification of receptor and epidermal growth factor-enhanced phosphorylation activity. J Biol Chem. 1980;255:4834–4842. [PubMed] [Google Scholar]
- 7.Ullrich A, Coussens L, Hayflick JS, et al. Human epidermal growth factor receptor cDNA sequence and aberrant expression of the amplified gene in A431 epidermoid carcinoma cells. Nature. 1984;309:418–425. doi: 10.1038/309418a0. [DOI] [PubMed] [Google Scholar]
- 8.Davies RL, Grosse VA, Kucherlapati R, et al. Genetic analysis of epidermal growth factor action: assignment of human epidermal growth factor receptor gene to chromosome 7. Proc Natl Acad Sci U S A. 1980;77:4188–4192. doi: 10.1073/pnas.77.7.4188. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Haley J, Whittle N, Bennet P, et al. The human EGF receptor gene: structure of the 110 kb locus and identification of sequences regulating its transcription. Oncogene Res. 1987;1:375–396. [PubMed] [Google Scholar]
- 10.Burgess AW, Cho HS, Eigenbrot C, et al. An open-and-shut case? Recent insights into the activation of EGF/ErbB receptors. Mol Cell. 2003;12:541–552. doi: 10.1016/s1097-2765(03)00350-2. [DOI] [PubMed] [Google Scholar]
- 11.Schulze WX, Deng L, Mann M. Phosphotyrosine interactome of the ErbB-receptor kinase family. Mol Syst Biol. 2005;1:2005–0008. doi: 10.1038/msb4100012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Bowers G, Reardon D, Hewitt T, et al. The relative role of ErbB1-4 receptor tyrosine kinases in radiation signal transduction responses of human carcinoma cells. Oncogene. 2001;20:1388–1397. doi: 10.1038/sj.onc.1204255. [DOI] [PubMed] [Google Scholar]
- 13.Chakravarti A, Loeffler JS, Dyson NJ. Insulin-like Growth Factor Receptor I Mediates Resistance to Anti-Epidermal Growth Factor Receptor Therapy in Primary Human Glioblastoma Cells through Continued Activation of Phosphoinositide 3-Kinase Signaling. Cancer Res. 2002;62:200–207. [PubMed] [Google Scholar]
- 14.Warren CM, Landgraf R. Signaling through ERBB receptors: multiple layers of diversity and control. Cell Signal. 2006;18:923–933. doi: 10.1016/j.cellsig.2005.12.007. [DOI] [PubMed] [Google Scholar]
- 15.Schlessinger J. Cell signaling by receptor tyrosine kinases. Cell. 2000;103:211–225. doi: 10.1016/s0092-8674(00)00114-8. [DOI] [PubMed] [Google Scholar]
- 16.Kim YN, Dam P, Bertics PJ. Caveolin-1 phosphorylation in human squamous and epidermoid carcinoma cells: dependence on ErbB1 expression and Src activation. Exp Cell Res. 2002;280:134–147. doi: 10.1006/excr.2002.5623. [DOI] [PubMed] [Google Scholar]
- 17.Thomas SM, Coppelli FM, Wells A, et al. Epidermal growth factor receptor-stimulated activation of phospholipase Cgamma-1 promotes invasion of head and neck squamous cell carcinoma. Cancer Res. 2003;63:5629–5635. [PubMed] [Google Scholar]
- 18.Kalyankrishna S, Grandis JR. Epidermal growth factor receptor biology in head and neck cancer. J Clin Oncol. 2006;24:2666–2672. doi: 10.1200/JCO.2005.04.8306. [DOI] [PubMed] [Google Scholar]
- 19.Lin SY, Makino K, Xia W, et al. Nuclear localization of EGF receptor and its potential new role as a transcription factor. Nat Cell Biol. 2001;3:802–808. doi: 10.1038/ncb0901-802. [DOI] [PubMed] [Google Scholar]
- 20.Dittmann K, Mayer C, Fehrenbacher B, et al. Radiation-induced Epidermal Growth Factor Receptor Nuclear Import Is Linked to Activation of DNA-dependent Protein Kinase. J Biol Chem. 2005;280:31182–31189. doi: 10.1074/jbc.M506591200. [DOI] [PubMed] [Google Scholar]
- 21.Lo HW, Hung MC. Nuclear EGFR signalling network in cancers: linking EGFR pathway to cell cycle progression, nitric oxide pathway and patient survival. Br J Cancer. 2006;94:184–188. doi: 10.1038/sj.bjc.6602941. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Lo HW, Hsu SC, Ali-Seyed M, et al. Nuclear interaction of EGFR and STAT3 in the activation of the iNOS/NO pathway. Cancer Cell. 2005;7:575–589. doi: 10.1016/j.ccr.2005.05.007. [DOI] [PubMed] [Google Scholar]
- 23.Boerner JL, Biscardi JS, Silva CM, et al. Transactivating agonists of the EGF receptor require Tyr 845 phosphorylation for induction of DNA synthesis. Mol Carcinog. 2005;44:262–273. doi: 10.1002/mc.20138. [DOI] [PubMed] [Google Scholar]
- 24.Wang Y, Pennock S, Chen X, et al. Endosomal signaling of epidermal growth factor receptor stimulates signal transduction pathways leading to cell survival. Mol Cell Biol. 2002;22:7279–7290. doi: 10.1128/MCB.22.20.7279-7290.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Rubin Grandis J, Melhem MF, Barnes EL, et al. Quantitative immunohistochemical analysis of transforming growth factor-alpha and epidermal growth factor receptor in patients with squamous cell carcinoma of the head and neck. Cancer. 1996;78:1284–1292. doi: 10.1002/(SICI)1097-0142(19960915)78:6<1284::AID-CNCR17>3.0.CO;2-X. [DOI] [PubMed] [Google Scholar]
- 26.Schmidt-Ullrich RK, Mikkelsen RB, Dent P, et al. Radiation-induced proliferation of the human A431 squamous carcinoma cells is dependent on EGFR tyrosine phosphorylation. Oncogene. 1997;15:1191–1197. doi: 10.1038/sj.onc.1201275. [DOI] [PubMed] [Google Scholar]
- 27.Psyrri A, Yu Z, Weinberger PM, et al. Quantitative Determination of Nuclear and Cytoplasmic Epidermal Growth Factor Receptor Expression in Oropharyngeal Squamous Cell Cancer by Using Automated Quantitative Analysis. Clin Cancer Res. 2005;11:5856–5862. doi: 10.1158/1078-0432.CCR-05-0420. [DOI] [PubMed] [Google Scholar]
- 28.Wikstrand CJ, McLendon RE, Friedman AH, et al. Cell surface localization and density of the tumor-associated variant of the epidermal growth factor receptor, EGFRvIII. Cancer Res. 1997;57:4130–4140. [PubMed] [Google Scholar]
- 29.Sok JC, Coppelli FM, Thomas SM, et al. Mutant epidermal growth factor receptor (EGFRvIII) contributes to head and neck cancer growth and resistance to EGFR targeting. Clin Cancer Res. 2006;12:5064–5073. doi: 10.1158/1078-0432.CCR-06-0913. [DOI] [PubMed] [Google Scholar]
- 30.Lammering G, Hewit TH, Valerie K, et al. EGFRvIII-mediated radioresistance through a strong cytoprotective response. Oncogene. 2003;22:5545–5553. doi: 10.1038/sj.onc.1206788. [DOI] [PubMed] [Google Scholar]
- 31.Withers HR, Taylor JM, Maciejewski B. The hazard of accelerated tumor clonogen repopulation during radiotherapy. Acta Oncol. 1988;27:131–146. doi: 10.3109/02841868809090333. [DOI] [PubMed] [Google Scholar]
- 32.Schmidt-Ullrich RK, Valerie KC, Chan W, et al. Altered expression of epidermal growth factor receptor and estrogen receptor in MCF-7 cells after single and repeated radiation exposures. Int J Radiat Oncol Biol Phys. 1994;29:813–819. doi: 10.1016/0360-3016(94)90570-3. [DOI] [PubMed] [Google Scholar]
- 33.Schmidt-Ullrich RK, Contessa JN, Lammering G, et al. ERBB receptor tyrosine kinases and cellular radiation responses. Oncogene. 2003;22:5855–5865. doi: 10.1038/sj.onc.1206698. [DOI] [PubMed] [Google Scholar]
- 34.Amorino GP, Hamilton VM, Valerie K, et al. Epidermal growth factor receptor dependence of radiation-induced transcription factor activation in human breast carcinoma cells. Mol Biol Cell. 2002;13:2233–2244. doi: 10.1091/mbc.01-12-0572. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Akimoto T, Hunter NR, Buchmiller L, et al. Inverse relationship between epidermal growth factor receptor expression and radiocurability of murine carcinomas. Clin Cancer Res. 1999;5:2884–2890. [PubMed] [Google Scholar]
- 36.Milas L, Akimoto T, Hunter NR, et al. Relationship between cyclin D1 expression and poor radioresponse of murine carcinomas. Int J Radiat Oncol Biol Phys. 2002;52:514–521. doi: 10.1016/s0360-3016(01)02693-1. [DOI] [PubMed] [Google Scholar]
- 37.Ang KK, Berkey BA, Tu X, et al. Impact of epidermal growth factor receptor expression on survival and pattern of relapse in patients with advanced head and neck carcinoma. Cancer Research. 2002;62:7350–7356. [PubMed] [Google Scholar]
- 38.Liang K, Ang KK, Milas L, et al. The epidermal growth factor receptor mediates radioresistance. International Journal of Radiation Oncology Biology Physics. 2003;57:246–254. doi: 10.1016/s0360-3016(03)00511-x. [DOI] [PubMed] [Google Scholar]
- 39.Sato JD, Kawamoto T, Le AD, et al. Biological effects in vitro of monoclonal antibodies to human epidermal growth factor receptors. Molecular biology & medicine. 1983;1:511–529. [PubMed] [Google Scholar]
- 40.Balaban N, Moni J, Shannon M, et al. The effect of ionizing radiation on signal transduction: Antibodies to EGF receptor sensitize A431 cells to radiation. Biochimica et Biophysica Acta. 1996;1314:147–156. doi: 10.1016/s0167-4889(96)00068-7. [DOI] [PubMed] [Google Scholar]
- 41.Huang SM, Bock JM, Harari PM. Epidermal growth factor receptor blockade with C225 modulates proliferation, apoptosis, and radiosensitivity in squamous cell carcinomas of the head and neck. Cancer Res. 1999;59:1935–1940. [PubMed] [Google Scholar]
- 42.Saleh MN, Raisch KP, Stackhouse MA, et al. Combined modality therapy of A431 human epidermoid cancer using anti-EGFr antibody C225 and radiation. Cancer Biother Radiopharm. 1999;14:451–463. doi: 10.1089/cbr.1999.14.451. [DOI] [PubMed] [Google Scholar]
- 43.Milas L, Mason K, Hunter N, et al. In vivo enhancement of tumor radioresponse by C225 antiepidermal growth factor receptor antibody. Clin Cancer Res. 2000;6:701–708. [PubMed] [Google Scholar]
- 44.Nasu S, Ang KK, Fan Z, et al. C225 antiepidermal growth factor receptor antibody enhances tumor radiocurability. Int J Radiat Oncol Biol Phys. 2001;51:474–477. doi: 10.1016/s0360-3016(01)01671-6. [DOI] [PubMed] [Google Scholar]
- 45.Huang S, Armstrong EA, Benavente S, et al. Dual-agent molecular targeting of the epidermal growth factor receptor (EGFR): combining anti-EGFR antibody with tyrosine kinase inhibitor. Cancer Res. 2004;64:5355–5362. doi: 10.1158/0008-5472.CAN-04-0562. [DOI] [PubMed] [Google Scholar]
- 46.Li S, Schmitz KR, Jeffrey PD, et al. Structural basis for inhibition of the epidermal growth factor receptor by cetuximab. Cancer Cell. 2005;7:301–311. doi: 10.1016/j.ccr.2005.03.003. [DOI] [PubMed] [Google Scholar]
- 47.Huang SM, Harari PM. Modulation of radiation response after epidermal growth factor receptor blockade in squamous cell carcinomas: Inhibition of damage repair, cell cycle kinetics, and tumor angiogenesis. Clin Cancer Res. 2000;6:2166–2174. [PubMed] [Google Scholar]
- 48.Robert F, Ezekiel MP, Spencer SA, et al. Phase I study of anti--epidermal growth factor receptor antibody cetuximab in combination with radiation therapy in patients with advanced head and neck cancer. J Clin Oncol. 2001;19:3234–3243. doi: 10.1200/JCO.2001.19.13.3234. [DOI] [PubMed] [Google Scholar]
- 49.Bonner JA, Harari PM, Giralt J, et al. Radiotherapy plus Cetuximab for Squamous-Cell Carcinoma of the Head and Neck. N Engl J Med. 2006;354:567–578. doi: 10.1056/NEJMoa053422. [DOI] [PubMed] [Google Scholar]
- 50.Pfister DG, Su YB, Kraus DH, et al. Concurrent cetuximab, cisplatin, and concomitant boost radiotherapy for locoregionally advanced, squamous cell head and neck cancer: a pilot phase II study of a new combined-modality paradigm. J Clin Oncol. 2006;24:1072–1078. doi: 10.1200/JCO.2004.00.1792. [DOI] [PubMed] [Google Scholar]
- 51.Bentzen SM, Atasoy BM, Daley FM, et al. Epidermal Growth Factor Receptor Expression in Pretreatment Biopsies From Head and Neck Squamous Cell Carcinoma As a Predictive Factor for a Benefit From Accelerated Radiation Therapy in a Randomized Controlled Trial. J Clin Oncol. 2005;23:5560–5567. doi: 10.1200/JCO.2005.06.411. [DOI] [PubMed] [Google Scholar]
- 52.Eriksen JG, Steiniche T, Askaa J, et al. The prognostic value of epidermal growth factor receptor is related to tumor differentiation and the overall treatment time of radiotherapy in squamous cell carcinomas of the head and neck. Int J Radiat Oncol Biol Phys. 2004;58:561–566. doi: 10.1016/j.ijrobp.2003.09.043. [DOI] [PubMed] [Google Scholar]
- 53.Soulieres D, Senzer NN, Vokes EE, et al. Multicenter Phase II Study of Erlotinib, an Oral Epidermal Growth Factor Receptor Tyrosine Kinase Inhibitor, in Patients With Recurrent or Metastatic Squamous Cell Cancer of the Head and Neck. J Clin Oncol. 2004;22:77–85. doi: 10.1200/JCO.2004.06.075. [DOI] [PubMed] [Google Scholar]
- 54.Burtness B, Goldwasser MA, Flood W, et al. Phase III Randomized Trial of Cisplatin Plus Placebo Compared With Cisplatin Plus Cetuximab in Metastatic/Recurrent Head and Neck Cancer: An Eastern Cooperative Oncology Group Study. J Clin Oncol. 2005;23:8646–8654. doi: 10.1200/JCO.2005.02.4646. [DOI] [PubMed] [Google Scholar]
- 55.Chung KY, Shia J, Kemeny NE, et al. Cetuximab shows activity in colorectal cancer patients with tumors that do not express the epidermal growth factor receptor by immunohistochemistry. J Clin Oncol. 2005;23:1803–1810. doi: 10.1200/JCO.2005.08.037. [DOI] [PubMed] [Google Scholar]
- 56.Lax I, Bar-Eli M, Yarden Y, et al. Antibodies to two defined regions of the transforming protein pp60src interact specifically with the epidermal growth factor receptor kinase system. Proc Natl Acad Sci U S A. 1984;81:5911–5915. doi: 10.1073/pnas.81.19.5911. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Dziadziuszko R, Hirsch FR, Varella-Garcia M, et al. Selecting lung cancer patients for treatment with epidermal growth factor receptor tyrosine kinase inhibitors by immunohistochemistry and fluorescence in situ hybridization--why, when, and how? Clin Cancer Res. 2006;12:4409s–4415s. doi: 10.1158/1078-0432.CCR-06-0087. [DOI] [PubMed] [Google Scholar]
- 58.Gibson TB, Ranganathan A, Grothey A. Randomized phase III trial results of panitumumab, a fully human anti-epidermal growth factor receptor monoclonal antibody, in metastatic colorectal cancer. Clin Colorectal Cancer. 2006;6:29–31. doi: 10.3816/CCC.2006.n.01. [DOI] [PubMed] [Google Scholar]
- 59.Baselga J, Trigo JM, Bourhis J, et al. Phase II Multicenter Study of the Antiepidermal Growth Factor Receptor Monoclonal Antibody Cetuximab in Combination With Platinum-Based Chemotherapy in Patients With Platinum-Refractory Metastatic and/or Recurrent Squamous Cell Carcinoma of the Head and Neck. J Clin Oncol. 2005;23:5568–5577. doi: 10.1200/JCO.2005.07.119. [DOI] [PubMed] [Google Scholar]
- 60.Milas L, Fang F, Mason KA, et al. Importance of maintenance therapy in C225-induced enhancement of tumor control by fractionated radiation. Int J Radiat Oncol Biol Phys. 2007;67:568–572. doi: 10.1016/j.ijrobp.2006.09.044. [DOI] [PubMed] [Google Scholar]
- 61.Crombet T, Osorio M, Cruz T, et al. Use of the humanized anti-epidermal growth factor receptor monoclonal antibody h-R3 in combination with radiotherapy in the treatment of locally advanced head and neck cancer patients. J Clin Oncol. 2004;22:1646–1654. doi: 10.1200/JCO.2004.03.089. [DOI] [PubMed] [Google Scholar]
- 62.Wood ER, Truesdale AT, McDonald OB, et al. A unique structure for epidermal growth factor receptor bound to GW572016 (Lapatinib): relationships among protein conformation, inhibitor off-rate, and receptor activity in tumor cells. Cancer Res. 2004;64:6652–6659. doi: 10.1158/0008-5472.CAN-04-1168. [DOI] [PubMed] [Google Scholar]
- 63.Loeffler-Ragg J, Witsch-Baumgartner M, Tzankov A, et al. Low incidence of mutations in EGFR kinase domain in Caucasian patients with head and neck squamous cell carcinoma. Eur J Cancer. 2006;42:109–111. doi: 10.1016/j.ejca.2005.08.034. [DOI] [PubMed] [Google Scholar]
- 64.Daub H, Weiss FU, Wallasch C, et al. Role of transactivation of the EGF receptor in signalling by G-protein-coupled receptors. Nature. 1996;379:557–560. doi: 10.1038/379557a0. [DOI] [PubMed] [Google Scholar]
- 65.Viloria-Petit A, Crombet T, Jothy S, et al. Acquired Resistance to the Antitumor Effect of Epidermal Growth Factor Receptor-blocking Antibodies in Vivo: A Role for Altered Tumor Angiogenesis. Cancer Res. 2001;61:5090–5101. [PubMed] [Google Scholar]
- 66.Tonra JR, Deevi DS, Corcoran E, et al. Synergistic antitumor effects of combined epidermal growth factor receptor and vascular endothelial growth factor receptor-2 targeted therapy. Clin Cancer Res. 2006;12:2197–2207. doi: 10.1158/1078-0432.CCR-05-1682. [DOI] [PubMed] [Google Scholar]
- 67.Williams KJ, Telfer BA, Brave S, et al. ZD6474, a potent inhibitor of vascular endothelial growth factor signaling, combined with radiotherapy: schedule-dependent enhancement of antitumor activity. Clin Cancer Res. 2004;10:8587–8593. doi: 10.1158/1078-0432.CCR-04-1147. [DOI] [PubMed] [Google Scholar]
- 68.Overgaard J, Eriksen JG, Nordsmark M, et al. Plasma osteopontin, hypoxia, and response to the hypoxia sensitiser nimorazole in radiotherapy of head and neck cancer: results from the DAHANCA 5 randomised double-blind placebo-controlled trial. Lancet Oncol. 2005;6:757–764. doi: 10.1016/S1470-2045(05)70292-8. [DOI] [PubMed] [Google Scholar]
- 69.Rischin D, Peters L, Fisher R, et al. Tirapazamine, Cisplatin, and Radiation versus Fluorouracil, Cisplatin, and Radiation in patients with locally advanced head and neck cancer: a randomized phase II trial of the Trans-Tasman Radiation Oncology Group (TROG 98.02) J Clin Oncol. 2005;23:79–87. doi: 10.1200/JCO.2005.01.072. [DOI] [PubMed] [Google Scholar]
- 70.Milas L. Cyclooxygenase-2 (COX-2) enzyme inhibitors as potential enhancers of tumor radioresponse. Semin Radiat Oncol. 2001;11:290–299. doi: 10.1053/srao.2001.26018. [DOI] [PubMed] [Google Scholar]
- 71.Mauceri HJ, Hanna NN, Beckett MA, et al. Combined effects of angiostatin and ionizing radiation in antitumour therapy. Nature. 1998;394:287–291. doi: 10.1038/28412. [DOI] [PubMed] [Google Scholar]
- 72.Willett CG, Boucher Y, di Tomaso E, et al. Direct evidence that the VEGF-specific antibody bevacizumab has antivascular effects in human rectal cancer. Nat Med. 2004;10:145–147. doi: 10.1038/nm988. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Jorissen RN, Walker F, Pouliot N, et al. Epidermal growth factor receptor: mechanisms of activation and signalling. Exp Cell Res. 2003;284:31–53. doi: 10.1016/s0014-4827(02)00098-8. [DOI] [PubMed] [Google Scholar]