

Introduction
Anemia is a common problem in the fetal and neonatal period. The differential diagnosis of fetal and neonatal anemia is broad, and includes immune-mediated hemolysis, fetal hemorrhage, intrinsic erythrocyte disorders, and erythrocyte underproduction. This review will focus on genetic conditions leading to intrinsic disorders of the erythrocyte and erythrocyte underproduction. Intrinsic disorders of the erythrocyte, such as the hemoglobinopathies and glucose-6-phosphate dehydrogenase deficiency, are important causes of perinatal anemia and represent some of the most common inherited genetic diseases. Inherited abnormalities that lead to decreased erythrocyte production are much less common, but are important to recognize as they can cause severe anemia that is often associated with other congenital anomalies. Presentation of these disorders in the fetus and newborn is varied. Anemia may range from asymptomatic to life threatening and can be associated with other complications such as nonimmune hydrops fetalis (Table 1) and severe hyperbilirubinemia.
Table 1.
Intrinsic Disorders of the Erythrocyte Leading to Nonimmune Hydrops Fetalis
| Hemoglobin Disorders | Reference |
| α-Thalassemia | |
| Hemoglobin H Disease | 11-13 |
| Homozygous α-Thalassemia | 15 |
| Enzyme Deficiencies | |
| G6PD Deficiency | 47-49 |
| Pyruvate Kinase | 31-36 |
| Membrane Defects | |
| Hereditary Spherocytosis | 58 |
| Hereditary Elliptocytosis | 59-60 |
| Hereditary Stomatocytosis Syndromes | 65,66 |
| Decreased Erythrocyte Production | |
| Congenital Dyserythopeietic Anemias | 80 |
| Pearson Syndrome | 81 |
| Diamond-Blackfan Anemia | 72-73 |
The Neonatal Erythrocyte
Understanding the differences between neonatal and adult erythrocytes is critical in the evaluation of perinatal erythrocyte disorders. These differences include variation in erythrocyte size, shape, globin composition, and cellular metabolism.
Size and Shape
Fetal erythrocytes are larger and have more variation in shape than those of adults. They are largest early in gestation, with an MCV of approximately 150-180 fL.1 They slowly decrease in size reaching approximately 114 fL at term,2 and are similar to adult cells by 1 year of age. Peripheral blood smears of neonates show greater numbers of acanthocytes, target cells, stomatocytes, and immature erythrocytes than peripheral blood smears of adults.3 Neonatal erythrocytes also show decreased deformability and increased osmotic resistance during the first 4 to 6 weeks of life.4 When viewed under interference microscopy, one-half of erythrocytes in preterm infants and one-quarter of erythrocytes in term infants will have surface pits, compared to only 2.6% of erythrocytes in adults. These surface pits are thought to be a consequence of the poor splenic function of the neonate.5
Fetal hemoglobin
Fetal hemoglobin (Hb F) comprises 70-90% of the hemoglobin in the neonatal erythrocyte. In contrast to adult hemoglobin, fetal hemoglobin does not interact with 2,3-diphosphoglycerate (DPG), causing a shift of the hemoglobin dissociation curve to the left. This is crucial for the fetus, as the higher oxygen affinity of Hb F allows fetal erythrocytes to extract oxygen from maternal RBC. Error! Bookmark not defined.
In contrast to the adult RBC lifespan of 120 days, the lifespan of the neonatal erythrocyte is only 60 to 90 days with preterm infants having an even shorter life span of 35 to 50 days.6 Hb F has a tendency to denature and damage the membrane from within, contributing to the shortened the red cell lifespan. Fetal hemoglobin is also more soluble in strong phosphate buffers and is more resistant to acid denaturation than adult hemoglobin. Its resistance to acid denaturation is the basis of the kliehauerbetke test.7
Metabolism of the Neonatal Erythrocyte
Neonatal erythrocytes contain higher levels of ATP and consume more glucose than adult cells. They also have lower levels of glutathione peroxidase, methemoglobin reductase, and carbonic anydrase.3 The lower enzyme levels make neonatal erythrocytes more susceptible to oxidative damage, leading to the formation of methemoglobin and Heinz bodies,3,8 and likely contributing to the decreased life span of the neonatal erythorcyte.3
Hemoglobin Disorders
Developmental differences in globin chain synthesis are responsible for the different clinical manifestations of α-chain and β-chain defects in the perinatal period. The first α-like chain, ζ, is produced in significant amounts only in the first few months of gestation. By 9 weeks, α-globin is the major α-like globin in the human fetus, and embryonic hemoglobin, hemoglobin Portland (ζ2γ2) is found only in small amounts. In contrast, β-globin production begins late in gestation, and the switch from fetal hemoglobin (α2γ2) to adult hemoglobin (α2β2) is not complete until the end of the first year of life (Figure 1). Defects in α-globin synthesis therefore manifest in utero, while defects in β-chain synthesis may not become apparent until late infancy.1
Figure 1. Hemoglobin switching during embryonic, fetal, and adult development.
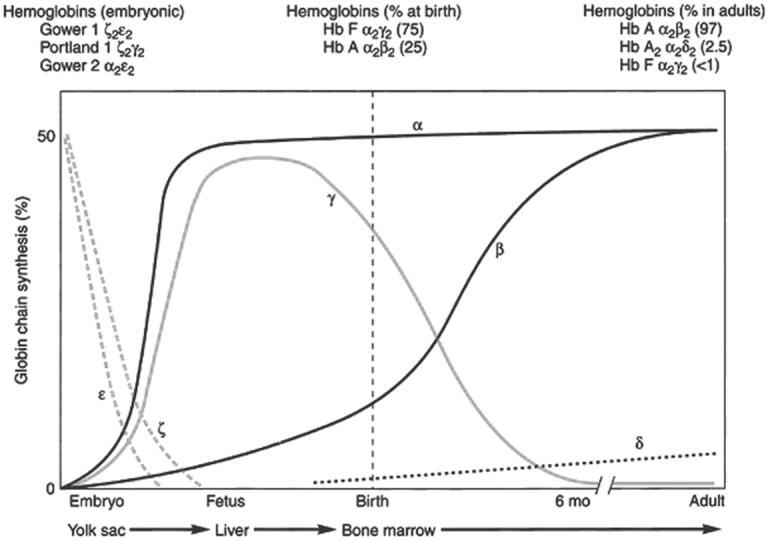
From: Steinberg MH, Benz Jr. EJ, Adewoye HA, and Ebert B. Pathobiology of the human erythrocyte and its hemoglobins. In Hoffman R, Benz EJ, Shattil, SJ: Hematology: Basic Principles and Practice, 4th ed., 2005. Churchill Livingston, Philadelphia. Page 442-454, with permission.
α-Thalassemia Syndromes
The α-thalassemias predominately affect patients of southeast asian, middle eastern, and mediterranean descent and carrier states are thought to provide protection from malaria.9 α-Thalassemia results from the deletion of one or more of the four α-globin genes which reside on the short arm of chromosome 16, and the clinical manifestations of α-thalassemia are directly related to the number of functional α-globin genes present.10 A single α-globin gene deletion results in an asymptomatic carrier state. Deletion of two genes results in α-thalassemia trait, which is characterized by microcytosis and mild anemia.
Hemoglobin H disease results from deletion of 3 α-globin genes. This leads to a significant imbalance in α– and β– like chain production and the formation of hemoglobin H (β4) and hemoglobin Barts (γ4). Patients with hemoglobin H disease are often born with a hypochromic, hemolytic anemia, and are at risk for significant neonatal hyperbilirubinemia. Hemoglobin H has been associated with hydrops fetalis.11-13 Children and adults with hemoglobin H disease may be transfusion dependent.14
Patients with deletion of all 4 α-globin genes, homozygous α-thalassemia, are severely affected in utero. The major hemoglobin is hemoglobin Barts (γ4), and survival of the fetus depends on the presence of embryonic hemoglobin Portland (ζ2γ2). Affected infants suffer from severe hemolytic anemia, high output cardiac failure, and hydrops fetalis.15 Maternal complications, such as preeclampia, preterm labor, and retained placenta are common in pregnancies affected by homozygous α-thalassemia.16-18 Without intervention, the majority of infants will die in utero or shortly after birth. The advent of intrauterine transfusions, long term transfusion and chelation therapy, and bone marrow transplantation has made it possible for some of these infants to survive into childhood.19
β-globin defects
Most β-globin chain defects, including sickle cell disease and β-thalassemia, do not present with anemia or hemolysis in the neonatal period, due to the presence of large amounts of fetal hemoglobin (α2γ2). One β-globin defect that does manifest in the perinatal period is γδβ-thalassemia. γδβ-Thalassemia is caused either by large deletions in the coding region of the β-globin cluster20-22 or in the β-globin promoter.23-24 Neonates with this disorder typically present with hypochromic, hemolytic anemia, and a prominent normoblastosis.25 Older children and adults with γδβ thalassemia have variable clinical manifestations similar to patients with β-thalassemia minor.26
Unstable Hemoglobins
Unstable hemoglobins typically exhibit lower solubility than normal hemoglobin leading to chronic nonspherocytic hemolytic anemia. They can affect both α- and β- chains and can cause severe hemolysis and hyperbilirubinemia in the newborn. The unstable hemoglobinopathies are sometimes called congenital Heinz bony anemias, because precipitation of the insoluble globin leads to the formation of Heinz bodies, which can be viewed on peripheral smear. The absence of Heinz bodies, particularly in the perinatal period, does not rule out the presence of unstable hemoglobinopathies, as they can be difficult to detect in young patients and in patients who have a functioning spleen. The best method for diagnosing unstable hemoglobin variants is demonstration of decreased solubility using the heat stability test and/or the isopropanol precipitation test. Diagnosis of unstable hemoglobinopathy can be made with hemoglobin electrophoresis, but roughly 30% of unstable hemoglobins are not detectable using this method.27
Two unstable hemoglobin variants are particularly interesting in the neonatal period. In hemoglobin F Poole, a glycine is substituted for a tryptophan at position 130 of the γ-globin chain, leading to fetal hemoglobin instability and hemolysis.27-28 Hb Hasharon results from a substitution of hisitidine for aspartate at position 47 of the α-globin gene.29 When paired with γ-globin, it has decreased stability, leading to neonatal hemolysis. When paired with the β-globin gene, Hb Hasharon is stable and the hemolysis subsides. In both Hb F Poole and Hb Hasharon, hemolysis subsides after the first few months of life as fetal hemoglobin (α2γ2) is replaced by adult hemoglobin (α2β2).27
Disorders of Erythrocyte Metabolism
Several enzymatic pathways are vital for the erythrocyte to function properly. The Embden-Meyerhof pathway is the major source of ATP in the erythrocyte and metabolizes approximately 90% of ATP in the erythrocyte. The energy generated by this process is stored as glutathione, pyridine nucleotides (NADH and NADPH), and ATP. This pathway is the major source of NADH in the erythrocyte, an essential co-factor for the enzyme NADH methemoglobin reducatase, which maintains hemoglobin in the reduced state, preventing methemoglobinemia. The hexose monophosphate shunt, also called the pentose phosphate pathway, generates the majority of NADPH in the erythrocyte through the metabolism of glucose-6-phosphate. NADPH is necessary to maintain adequate amounts of reduced glutathione, critical for protecting the erythrocyte from oxidative damage.
Pyruvate Kinase Deficiency
Pyruvate kinase (PK) deficiency is the most common inherited deficiency in the Embden-Meyerhof pathway. It is inherited in an autosomal recessive manner and is seen most often in patients of Northern European descent. Heterozygotes are generally normal or only mildly affected. Homozygotes or compound heterozygotes are affected with variable degrees of clinical severity.30 PK deficiency often presents in the neonatal period with anemia and jaundice, and patients can become transfusion dependent. More severe cases have been reported, including severe in utero anemia, hyperbilirubinemia requiring exchange transfusion, and hydrops.31-36
Diagnosis of PK deficiency may made by enzyme assay with erythrocytes exhibiting PK activity that is 5-40% of normal. Genetic studies can also reveal the diagnosis, and over 158 mutations have been described in the coding region of the PK gene.37 The majority of these mutations are private (i.e. not shared between families or ethnic groups). Missense mutations are most common, but point mutations, deletions, or insertions leading to alterations of the spicing site, frameshift, and early termination mutations have been reported.37-39 In addition to coding sequence mutations, disruption of the PK promoter can also lead to severe clinical manifestations.40 Treatment for PK deficiency is generally supportive, although some transfusion dependent patients have benefited from splenectomy and successful bone marrow transplantation has been reported.41 In the neonatal period, therapy is focused on the treatment of hyperbilirubinemia and anemia.
Glucose-6-Phosphate Dehydrogenase Deficiency
Glucose-6-phosphatase dehydrogenase (G6PD) deficiency affects nearly 400 million people.42 It is one of the most common genetic disorders, likely due to protection of carriers from Plasmodium falciparum infection.9,43 G6PD deficiency is the most common disorder of the hexose monophosphate shunt, the pathway responsible for generating NADPH, necessary for maintaining glutathione in its reduced state. Normal erythrocytes are able to significantly increase their production of NADPH in response to oxidative stress.44-45 Although generally asymptomatic in the absence of oxidative stress, affected patients are unable to increase production of NADPH when necessary,46 making their red cells vulnerable to oxidative damage and hemolysis. Chronic non-spherocytic hemolytic anemia occurs in a small subset of patients with such severe G6PD deficiency that they experience severe hemolysis even in the absence of oxidative stress.42
In the neonatal period, G6PD deficiency most commonly presents as jaundice, which can be severe enough to necessitate exchange transfusion. Rarely, severe intrauterine hemolysis and hydrops fetalis have been reported following maternal ingestion of oxidatitive agents47-49 and severe neonatal hemolysis has been reported in breast feeding infants following maternal ingestion of fava beans.50 There are several screening tests available for G6PD deficiency, including the dye discoloration test, the methemoglobin reduction test, and the fluorescence spot test.51 The reticulocytosis seen during hemolysis can make these screening tests inaccurate because reticulocytes have higher G-6-PD levels than mature cells, and can falsely elevate the amount of G6PD in a sample. Screening tests are also generally unable to detect female carriers.
More definitive diagnosis can be made through direct assay of enzyme activity in erythrocytes. Molecular diagnosis is also possible and over 100 mutations have been associated with G6PD deficiency. The majority of these mutations are missense mutations in the coding region of the gene. There have been no reports of large frameshift mutations or deletions, suggesting that total deficiency of G6PD is incompatible with life.42 Treatment for G6PD deficiency includes avoidance of oxiadative agents and supportive care during episodes of hemolysis.4 In the neonatal period, appropriate treatment of hyperbilirubinemia is important for the prevention of kernicterus and neonates with active hemolysis and hyperbilirubinemia have lower threshold levels for phototherapy and exchange transfusion than infants with non-hemolytic jaundice.52
Disorders of the Erythrocyte Membrane
The erythrocyte membrane is composed of lipids and proteins that interact to give the erythrocyte the deformability and flexibility required to endure circulatory stress. Quantitative or qualitative defects in membrane proteins lead to decreased membrane deformability, membrane instability, and subsequent hemolysis.
Hereditary spherocytosis
Hereditary spherocytosis (HS) is characterized by the presence of spherocytes on peripheral blood smear (Figure 2B). HS is the most common inherited anemia in people of northern European descent, occurring in 1/2500-5000 individuals.53-54 Deficiencies of the membrane proteins ankyrin, band 3, α-spectrin, β-spectrin and protein 4.2 have been described. The most common cause of typical dominant HS is ankyrin deficiency,55 followed by β-spectrin or band 3 deficiency, with nondominant cases caused by defects in protein 4.2 or α-spectrin.56-57 The membrane instability caused by these protein defects leads to a loss of membrane surface area, decreased deformability of cells, splenic entrapment, and hemolysis. Anemia is the most common manifestation of HS in neonates. Hyperbilirubinemia is also common and nearly half of neonates with HS become jaundiced. Cases of hydrops fetalis linked to membrane defects have been described.58
Figure 2. Wright stained peripheral blood smears.

(A) Normal. (B) Hereditary Spherocytosis. (C) Hereditary Elliptocytosis. (D) Hereditary Pyropoikilocytosis. Clinical and Molecular Aspects of Disorders of the Erythrocyte Membrane Skeleton, Gallagher PG, Tse WT, Forget BG in Seminars in Perinatology, 14:351-67, 1990, with permission
The presence of spherocytes on peripheral smear suggests the diagnosis of hereditary spherocytosis and diagnosis can be made by osmotic fragility testing. Standard osmotic fragility testing is normal in one-fourth of patients, making incubated osmotic fragility the test of choice. Osmotic fragility testing can also be used in neonates, but it is important to use neonatal osmotic fragility curves, as neonatal RBC have greater osmotic resistance than adult cells.4 Treatment of HS depends on severity, and splenectomy can reduce or eliminate hemolysis in severely affected patients.27
Hereditary Elliptocytosis
Hereditary Elliptocytosis is characterized by the presence of elliptical or oval cigar-shaped erythrocytes on peripheral blood smears of affected individuals (figure 2C). The erythrocyte life span is normal in the majority of patients with HE. Only 12% of patients with HE become symptomatic, and patients who have a shortened erythrocyte life span are the patients who tend to develop symptomatic anemia. HE is generally not symptomatic until 4-6 months of life, but neonatal jaundice, hemolysis, anemia, and hydrops fetalis have been reported.1, 59-60 Even in symptomatic infants, hemolysis usually abates by 1-2 years of age and the infant goes on to have mild typical HE.4
Hereditary Pyropoikilocytosis
Hereditary Pyropoikilocytosis (HPP) (Figure 2D) is characterized by the presence of pyknocytes on peripheral blood smear and increased erythrocyte thermal sensitivity.61 Erythrocyte morphology resembles that seen in patients with severe burns. It is most common in patients of African descent, and causes severe anemia and hemolysis in the neonatal period.1 The hemolysis tends to gradually improve through infancy and many older patients have clinical symptoms similar to those seen in HE.62 There is a strong relationship between HE and HPP, as many patients with HPP have a family history of typical HE.4
Diagnosis of HPP is generally made by reviewing the peripheral blood smear and family history. Increased incubated osmotic fragility and decreased MCV support the diagnosis. Mutations in α- and β- spectrin underlie HPP63 and more specialized tests, such as ektacytometry, spectrin dimmer self-association studies, and genomic DNA analysis are available, but generally not necessary for diagnosis. Treatment of HPP is generally supportive, but splenectomy has been used in severe cases. In the neonatal period, phototherapy or exchange transfusion may be needed to treat hyperbilirubinemia, and transfusion may be required for anemia.
Hereditary Stomatocytosis Syndromes
The hereditary stomatocytosis syndromes are disorders of erythrocyte hydration that are generally inherited in an autosomal dominant manner.64 Erythrocyte hydration is determined primarily by the intracellular concentration of sodium and potassium. Increased levels of intracellular sodium and potassium cause water to enter the cell, forming stomatocytes and resulting in overhydrated hereditary stomatocytosis (OHST). Decreased intracellular concentration of sodium and potassium results in cellular dehydration and the formation of xerocytes, leading to dehydrated hereditary stomatocytosis (DHST). Intermediate syndromes also exist.
OHST is characterized by stomatoctes on peripheral blood smear. The clinical presentation is variable, with some patients having pseudohyperkalemia, severe hemolysis, hyperbilirubinemia, and anemia, and other patients being asymptomatic. Hydrops fetalis has been reported.65 Diagnosis is based on peripheral blood smear, increased MCV, increased osmotic fragility, and increased concentrations of intracellular sodium.
DHST, also known as xerocytosis, is characterized by increased MCHC and decreased osmotic fragility. The MCV is spuriously elevated when measured by flow cytometry. Erythrocyte morphology is generally normal, although stomatocytes and target cells can be seen. Diagnosis is based on decreased intracellular concentration of sodium and a characteristic osmotic gradient ektacytometric curve. The neonatal manifestations of DHST are variable, and include hemolysis and pseudohyperkalemia. Prenatally, hydrops fetalis66and transient perinatal ascites have been reported.67-69 DHST has been mapped to 16q23-q24, but the precise molecular basis is not known.69 Treatment is supportive, although unlike the other disorders of the erythrocyte membrane, splenectomy is contraindicated, as splenectomized patients have a high risk of thrombosis.70
Decreased Erythrocyte Production
Decreased erythrocyte production is an uncommon, but important cause of neonatal anemia, and is often associated with other congenital anomalies. The differential diagnosis for decreased erythrocyte production includes genetic causes, infectious suppression, and bone marrow replacement syndromes. This review will focus genetic causes of erythrocyte underproduction.
Diamond-Blackfan Anemia
Diamond-Blackfan anemia (DBA) is a congenital pure red cell aplasia causing a moderate to severe anemia which often presents early in infancy.71 Hydrops fetalis is rare, but has been reported.72-73 The most common associated anomalies are thumb and radial abnormalities with others including microcephaly, high arched palate, hypertelorism, retrognathia, webbed neck, and low set ears. Diagnosis of DBA is made on the basis of bone marrow examination, which demonstrates an absence of erythrocyte precursors in an otherwise normocellular marrow. Elevated erythropoietin levels support the diagnosis of DBA.1 Treatment includes chronic transfusion therapy, steroids, and bone marrow transplantation.74
Schwachman-Diamond Syndrome
Schwachman-Diamond Syndrome (SDS) is a rare autosomal recessive disorder characterized by bone marrow failure, exocrine pancreatic failure, and skeletal abnormalities.75-77 Neonates with SDS can present with severe anemia, as well as overwhelming infection due to impaired immune function. Myelodysplastic syndrome develops in 10-44% of patients, and 5-24% progress to leukemia.77 Diagnosis of SDS is generally made clinically. SDS has been localized to the Schwachman-Bodian-Diamond Syndrome Gene on chromosome 7 and confirmatory genetic testing is now available.77 Therapy may include pancreatic enzyme replacement, G-CSF for febrile neutropenia,and monitoring for development of myelodysplastic syndrome.
Congenital Dyserythropoietic Anemia
Congenital Dyserythropoietic Anemia (CDA) is a rare group of disorders characterized by ineffective erythropoeisis, megaloblastic anemia, and secondary hemosiderosis. Three types of CDA have been described. Type I is characterized by autosomal recessive inheritance, megaloblastiod erythroid hyperplasia, and distinct nuclear chromatin bridges between cells. Type II, the most common variant, is characterized by multinuclear erythrocytes and a positive acidified serum test. Type III is characterized by multinuclear erythroblasts and macrocytosis. CDA frequently presents in the neonatal period and severe anemia,78 hyperbilirubinemia, and pulmonary hypertension have been reported.79 CDA can also present prenatally as hydrops fetalis.80 Therapy for CDA has included intrauterine transfusions and transplantation.
Pearson Syndrome
Pearson Syndrome is a rare, often fatal, mitochondrial disorder. It presents early in infancy and is characterized by refractory anemia, exocrine pancreatic insufficiency, and metabolic acidosis. Hepatic and renal manifestations are also common. One-fourth of patients present with anemia in the neonatal period, and hydrops fetails has been reported.81 Diagnosis is made based on clinical manifestations and bone marrow aspirate, which shows vaculated myeloid and erythroid precursors, decreased numbers of erythroblasts, and many sideroblasts. Pearson syndrome is caused by deletions in mitochondrial DNA and genetic diagnosis is possible.82-83
Aase Syndrome
Aase syndrome is a rare congenital, hypoplastic anemia associated with triphalangial thumbs.84 Aase syndrome can also be associated with boney abnormalities, growth failure, and unilateral cleft palate. The anemia in Aase syndrome is steroid responsive and generally improves with age.4
Fanconi's Anemia
Fanconi's anemia (FA) is a rare autosomal recessive disorder leading to bone marrow failure and increased susceptibility to leukemia and other cancers. Fanconi's anemia is often associated with other congenital abnormalities, such as café-au-lait spots, radial ray abnormalities, thumb abnormalities, short stature, and characteristic facies (broad nose, epicanthal folds, micrognathia.). Renal and genital anomalies are also common. Fanconi's may present in the neonatal period with cytopenias, congenital malformations, or both. Diagnosis is made on the basis of increased chromosomal breakage on exposure to alkalating agents,85 that becomes more pronounced with exposure to diepoxybutane86 or mitomycin C.87 Fanconi's anemia has been linked to mutations in FANCA, FANCC, FANCD1, FANCD2, FANCE, and FANCF, which are thought to form a complex important for DNA repair.88 Therapy for Fanconi's anemia includes androgens such as oral oxymethalone, which increase bone marrow cellularity. G-CSF and GM-CSF have also been used. Bone marrow transplantation89 or stem cell transplant with cord blood are current therapies.90
Acknowledgments
Supported in part by T32 HD07094, RO1 DK62039, and RO1 HL65448.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Handin RI, Lux SE, Stossel TP, editors. Principles and Practices of Hematology. Lippencott; Philadelphia, PA: 2003. Blood. [Google Scholar]
- 2.Forestier F, Daffos F, Catherine N, Renard M, Andreux JP. Developmental Hematopoiesis in normal Human Fetal Blood. Blood. 1991;77:2360–3. [PubMed] [Google Scholar]
- 3.Zipurksy A, Brown E, Palko J, Brown EJ. The erythrocyte differential in newborn infants. Amer J of Pediatr Hematol Oncol. 1983;5:45–51. [PubMed] [Google Scholar]
- 4.Christensen RD, editor. Hematologic problems of the neonate. WB Saunders; Philadelphia, PA: 2000. [Google Scholar]
- 5.Holroyde CP, Oski FA, Gardner FH. The “Pocked” erythrocyte. Red-cell surface alterations in reticuloendothelial immaturity of the neonate. N Engl J Med. 1969;281:516. doi: 10.1056/NEJM196909042811002. [DOI] [PubMed] [Google Scholar]
- 6.Pearson HA. Life-span of the fetal red blood cell. J Pediatr. 1967;70:166–71. doi: 10.1016/s0022-3476(67)80410-4. [DOI] [PubMed] [Google Scholar]
- 7.Kleihauer E, Braun H, Betke K. Demonstration of fetal hemoglobin in erythrocytes of a blood smear. Klin Wochenschr. 1957;35:637–8. doi: 10.1007/BF01481043. [DOI] [PubMed] [Google Scholar]
- 8.Gross RT, Bracci R, Rudolph N, Schroeder E, Kochen J. Hydrogen peroxide toxicity and detoxification in the erythrocytes of newborn infants. Blood. 1967;29:481–93. [PubMed] [Google Scholar]
- 9.Luzzatto L. Genetics of red cells and susceptibility to malaria. Blood. 1979;54:961–76. [PubMed] [Google Scholar]
- 10.Higgs DR, Vickers MA, Wilkie AO, Pretorius IM, Jarman AP, Weatherall DJ. A review of the molecular genetics of the human alpha-globin gene cluster. Blood. 1989;73:1081–1104. [PubMed] [Google Scholar]
- 11.Chan V, Chan TK, Liang ST, Ghosh A, Kan YW, Todd D. Hydrops fetalis due to an unusual form of Hb H disease. Blood. 1985;66:224–8. [PubMed] [Google Scholar]
- 12.Ko TM, Hseih FJ, Hsu PM, Lee TY. Molecular characterization of severe alpha-thalassemias causing hydrops fetalis in Taiwan. Am J Med Genet. 1991;39:317–20. doi: 10.1002/ajmg.1320390314. [DOI] [PubMed] [Google Scholar]
- 13.Chan V, Chan VW, Tang M, Lau K, Todd D, Chang TK. Molecular defects in Hb H hydrops fetalis. Br J Haematol. 1997;96:224–8. doi: 10.1046/j.1365-2141.1997.d01-2017.x. [DOI] [PubMed] [Google Scholar]
- 14.Ne-Win, Harano K, Harano T, et al. Common alpha-thalassemia deletions in transfusion-dependent thalassemia patients in the Southeast Asia region of Myanmar. Lab Hematol. 2006;12:139–42. doi: 10.1532/LH96.06017. [DOI] [PubMed] [Google Scholar]
- 15.Chui DH, Waye JS. Hydrops fetalis caused by alpha-thalassemia: an emerging healthcare problem. Blood. 1998;91:2213–22. [PubMed] [Google Scholar]
- 16.Hofstaetter C, Gonser M, Goelz R. Perinatal case report of unexpected thalassemia Hb Bart. Fetal Diagn Ther. 1993;8:418–22. doi: 10.1159/000263861. [DOI] [PubMed] [Google Scholar]
- 17.Guy G, Coady DJ, Jansen V, Snyder J, Zinberg S. alpha-Thalassemia hydrops fetalis: clinical and ultrasonographic considerations. Am J Obstet Gynecol. 1985;153:500–4. doi: 10.1016/0002-9378(85)90461-2. [DOI] [PubMed] [Google Scholar]
- 18.Liang ST, Wong VC, So WW, Ma HK, Chan V, Todd D. Homozygous alpha-thalassaemia: clinical presentation, diagnosis and management. A review of 46 cases. Br J Obstet Gynaecol. 1985;92:680–4. doi: 10.1111/j.1471-0528.1985.tb01447.x. [DOI] [PubMed] [Google Scholar]
- 19.Singer ST, Styles L, Bojanowski J, Quirolo K, Foote D, Vichinsky EP. Changing outcome of homozygous alpha-thalassemia: cautious optimism. J Pediatr Hematol Oncol. 2000;22:539–42. doi: 10.1097/00043426-200011000-00014. [DOI] [PubMed] [Google Scholar]
- 20.Pirastu M, Kan YW, Lin CC, Baine RM, Holbrook CT. Hemolytic disease of the newborn caused by a new deletion of the entire beta-globin cluster. J Clin Invest. 1983;72:602–9. doi: 10.1172/JCI111008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Orkin SH, Goff SC, Nathan DG. Heterogeneity of DNA deletion in gamma delta beta-Thalassemia. J Clin Invest. 1981;67:878–84. doi: 10.1172/JCI110105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Fortina P, Delgrosso K, Werner E, et al. A greater than 200 kb deletion removing the entire beta-like globin gene cluster in a family of Irish descent. Hemoglobin. 1991;15:23–41. doi: 10.3109/03630269109072482. [DOI] [PubMed] [Google Scholar]
- 23.Driscoll MC, Dobkin CS, Alter BP. Gamma delta beta-Thalassemia due to a de novo mutation deleting the 5′ beta-globin gene activation–region hypersensitive sites. Proc. Natl. Acad Sci. 1989;86:7470–7474. doi: 10.1073/pnas.86.19.7470. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Curtin PT, Kan YW. The inactive beta globin gene on a gamma delta beta thalassemia chromosome has a normal structure and functions normally in vitro. Blood. 1988;71:766–70. [PubMed] [Google Scholar]
- 25.Kan YW, Forget BG, Nathan DG. Gamma-beta Thalassemia: A cause of hemolytic disease of the newborn. New Engl J Med. 1972;286:129–34. doi: 10.1056/NEJM197201202860304. [DOI] [PubMed] [Google Scholar]
- 26.Bollekens JA, Forget BG. Delta beta thalassemia and hereditary persistence of fetal hemoglobin. Hematol Oncol Clin North Am. 1991;5:399–22. [PubMed] [Google Scholar]
- 27.Steinberg MH, Forget BG, Higgs DR, Nagel RL, editors. Genetics, Pathophysiology, and Clinical Management. Cambridge University Press; Cambridge, UK: 2001. Disorders of Hemoglobin. [Google Scholar]
- 28.Lee-Potter JP, Deacon-Smith RA, Simpkiss MJ, Kamuzora H, Lehmann H. A new cause of haemolytic anaemia in the newborn. A description of an unstable fetal haemoglobin: F Poole, alpha2-G-gamma2 130 trptophan yields glycine. J Clin Pathol. 1975;28:317–20. doi: 10.1136/jcp.28.4.317. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Charache S, Mondzac AM, Gessner U. Hemoglobin Hasharon (α247 his(CD5)β2): a hemoglobin found in low concentration. J Clin Invest. 1969;48:834–47. doi: 10.1172/JCI106041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Pissard S, Max-Audit I, Skopinski L, et al. Pyruvate Kinase deficiency in France: a 3-year study reveals 27 new mutations. Br J Haematol. 2006;133:683–9. doi: 10.1111/j.1365-2141.2006.06076.x. [DOI] [PubMed] [Google Scholar]
- 31.Gilsanz F, Vega MA, Gomez-Castillo E, Ruiz-Balda JA, Omenaca F. Fetal anemia due to pyruvate kinase deficiency. Arch Dis Child. 1993;69:523–4. doi: 10.1136/adc.69.5_spec_no.523. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Hennekam RC, Beemer FA, Cats BP, Jansen G, Staal GE. Hydrops Fetalis associated with red cell pyruvate kinase deficiency. Genet Couns. 1990;1:75–9. [PubMed] [Google Scholar]
- 33.Demina A, Varughese KI, Barbot J, Forman L, Beutler E. Six previously undescribed pyruvate kinase mutations causing enzyme deficiency. Blood. 1998;92:647–52. [PubMed] [Google Scholar]
- 34.Zanella A, Bianchi P. Red cell pyruvate kinase deficiency: from genetics to clinical manifestations. Baillieres Best Pract Res Clin Haematol. 2000;13:57–81. doi: 10.1053/beha.1999.0057. [DOI] [PubMed] [Google Scholar]
- 35.Diaz A, Gilsanz F, Martinez J, Perez-Benavente S, Meza NW, Bautista JM. Life-threatening nonspherocytic hemolytic anemia in a patient with a null mutation in the PKLR gene and no compensatory PKM gene expression. Blood. 2005;106:1851–6. doi: 10.1182/blood-2005-02-0555. [DOI] [PubMed] [Google Scholar]
- 36.Costa C, Albuisson J, Le TH, et al. Severe hemolytic anemia in a Vietnamese family associated with novel mutations in the gene encoding for pyruvate kinase. Haematologica. 2005;90:25–30. [PubMed] [Google Scholar]
- 37.Bianchi P, Zanella A. Hematologically important mutations: red cell pyruvate kinase. (Third Update) Blood Cells Mol Dis. 2000;26:47–53. doi: 10.1006/bcmd.2000.0276. [DOI] [PubMed] [Google Scholar]
- 38.Lezner C, Nurnberg P, Jacobasch G, Gerth C, Thiele BJ. Molecular analysis of 29 pyruvate kinase-deficient patients from central Europe with hereditary hemolytic anemia. Blood. 1997;89:1793–9. [PubMed] [Google Scholar]
- 39.Kanno H, Fuji H, Wei DC, et al. Frame shift mutation, exon skipping, and a two codon deletion caused by splice site mutations account for pyruvate kinase deficiency. Blood. 1997;89:4213–8. [PubMed] [Google Scholar]
- 40.van Wijk R, van Solinge WW, Nerlov C, et al. Disruption of a novel regulatory element in the erythroid-specific promoter of the human PKLR gene causes severe pyruvate kinase deficiency. Blood. 2003;101:1596–1602. doi: 10.1182/blood-2002-07-2321. [DOI] [PubMed] [Google Scholar]
- 41.Suvatte V, Tanphaichitr VS, Visuthisakchai S, et al. Bone marrow, peripheral blood and cord blood stem cell transplantation in children: ten years' experience at Siriraj Hospital. Int J Hematol. 1998;68:411–9. doi: 10.1016/s0925-5710(98)00083-8. [DOI] [PubMed] [Google Scholar]
- 42.Mehta A, Mason PJ, Vulliamy TJ. Glucose-6-Phosphate dehydrogenase deficiency. Baillieres Best Pract Res Clin Haematol. 2000;13:21–38. doi: 10.1053/beha.1999.0055. [DOI] [PubMed] [Google Scholar]
- 43.Ruwende C, Khoo SC, Snow RW, et al. Natural selection of hemi- and heterozygotes for G6PD deficiency in Africa by resistance to severe malaria. Nature. 1995;376:246–9. doi: 10.1038/376246a0. [DOI] [PubMed] [Google Scholar]
- 44.Ursini MV, Parrella A, Rosa G, Salzano S, Martini G. Enhanced expression of glucose-6-phosphate dehydrogenase in human cells sustaining oxidative stress. Biochem J . 1997;323:801–6. doi: 10.1042/bj3230801. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Salvemini F, Franze A, Iervolino A, Filosa S, Salzano S, Ursini MV. Enhanced glutathione levels and oxidoresistance mediated by increased glucose-6-phosphate dehydrogenase expression. J Biol Chem. 1999;274:2750–7. doi: 10.1074/jbc.274.5.2750. [DOI] [PubMed] [Google Scholar]
- 46.Gaetani GD, Parker JC, Kirkman HN. Intracellular restraint: a new basis for the limitation in response to oxidative stress in human erythrocytes containing low-activity variants of glucose-6-phosphate dehydrogenase. Proc Natl Acad Sci USA. 1974;71:3584–7. doi: 10.1073/pnas.71.9.3584. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Perkins RP. Hydrops fetalis and stillbirth in a male glucose-6-phosphate dehydrogenase-deficient fetus possibly due to maternal ingestion of sulfisoxazole; a case report. Am J Obstet Gynecol. 1971;111:379–81. doi: 10.1016/0002-9378(71)90781-2. [DOI] [PubMed] [Google Scholar]
- 48.Mentzer WC, Collier E. Hydrops fetalis associated with erythrocyte G-6-PD deficiency and maternal ingestion of fava beans and ascorbic acid. J Pediatr. 1975;86:565–7. doi: 10.1016/s0022-3476(75)80148-x. [DOI] [PubMed] [Google Scholar]
- 49.Corchia C, Balata A, Meloni GF, Meloni T. Favism in a female newborn infant whose mother ingested fava beans before delivery. J Pediatr. 1995;127:807–8. doi: 10.1016/s0022-3476(95)70178-8. [DOI] [PubMed] [Google Scholar]
- 50.Kaplan M, Vreman HJ, Hammerman C, et al. Favism by proxy in nursing glucose-6-phosphate dehydrogenase-deficient neonates. J Perinatol. 1998;18:477–9. [PubMed] [Google Scholar]
- 51.Kaplan M, Leiter C, Hammerman C, Rudensky B. Comparison of commercial screening tests for glucose-6-phosphate dehydrogenase deficiency in the neonatal period. Clin Chem. 1997;43:1236–7. [PubMed] [Google Scholar]
- 52.American Academy of Pediatrics Management of hyperbilirubinemia in the newborn infant 35 weeks or more of gestation. Pediatrics. 2004;114:297–316. doi: 10.1542/peds.114.1.297. [DOI] [PubMed] [Google Scholar]
- 53.Eber SW, Pekrun A, Neufeldt A, Schroter W. Prevalence of increased osmotic fragility of erythrocytes in German blood donors: screening using a modified glycerol lysis test. Ann Hematol. 1992;64:88–92. doi: 10.1007/BF01715351. [DOI] [PubMed] [Google Scholar]
- 54.Godal HC, Heisto H. High prevalence of increased osmotic fragility of red blood cells among Norwegian blood donors. Scand J Hematol. 1981;27:30–4. doi: 10.1111/j.1600-0609.1981.tb00448.x. [DOI] [PubMed] [Google Scholar]
- 55.Lux S, Tse W, Menninger J, et al. Hereditary spherocytosis associated with deletion of human erythrocyte ankyrin gene on chromosome 8. Nature. 1990;345:736–9. doi: 10.1038/345736a0. [DOI] [PubMed] [Google Scholar]
- 56.Gallagher PG. Update on the clinical spectrum and genetics of red blood cell membrane disorders. Curr Hematol Rep. 2004;3:85–91. [PubMed] [Google Scholar]
- 57.Gallagher PG. Red Cell Membrane Disorders. Hematology Am Soc Hematol Educ Program. 2005:13–8. doi: 10.1182/asheducation-2005.1.13. [DOI] [PubMed] [Google Scholar]
- 58.Whitfield CF, Follweiler JB, Lopresti-Morrow L, Miller BA. Deficiency of α-spectrin synthesis in burst-forming units-erythroid in lethal hereditary spherocytosis. Blood. 1991;78:3043–51. [PubMed] [Google Scholar]
- 59.Gallagher PG, Weed SA, Tse WT, et al. Recurrent fatal hydrops fetalis associated with a nucleotide substitution in the erythrocyte beta-spectrin gene. J Clin Invest. 1995;95:1174–82. doi: 10.1172/JCI117766. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Gallagher PG, Petruzzi MJ, Weed SA, et al. Mutation of a highly conserved residue of beta spectrin associated with fatal and near-fatal neonatal hemolytic anemia. J Clin Invest. 1997;99:267–77. doi: 10.1172/JCI119155. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Zarkowsky HS, Mohandas N, Speaker CB, Shohet SB. A congenital haemolytic anaemia with thermal sensitivity of the erythrocyte membrane. Br J Haematol. 1975;4:537–43. doi: 10.1111/j.1365-2141.1975.tb02740.x. [DOI] [PubMed] [Google Scholar]
- 62.MacDougall LG, Moodley G, Quirk M. The pyropoikilocytosis-elliptocytosis syndrome in a black South African infant: clinical and hematological features. Am J Pediatr Hematol Oncol. 1982;3:344–9. [PubMed] [Google Scholar]
- 63.Zhang Z, Weed SA, Gallagher PG, Morrow JS. Dynamic molecular modeling of pathogenic mutations in the spectrin self-association domain. Blood. 2001;98:1645–53. doi: 10.1182/blood.v98.6.1645. [DOI] [PubMed] [Google Scholar]
- 64.Delaunay J, Stewart G, Iolascon A. Hereditary dehydrated and overhydrated stomatocytosis: recent advances. Curr Opin Hematol. 1999;6:110–4. doi: 10.1097/00062752-199903000-00009. [DOI] [PubMed] [Google Scholar]
- 65.Grootenboer-Mignot S, Cretien A, Laurendeau I, et al. Sub-lethal hydrops as a manifestation of dehydrated hereditary stomatocytosis in two consecutive pregnancies. Prenat Diagn. 2003;23:380–4. doi: 10.1002/pd.598. [DOI] [PubMed] [Google Scholar]
- 66.Vicente-Gutierrez MP, Castello-Almazan I, Salvia-Roiges MD, et al. Nonimmune hydrops fetalis due to congenital xerocytosis. J Perinatol. 2005;25:63–5. doi: 10.1038/sj.jp.7211200. [DOI] [PubMed] [Google Scholar]
- 67.Entezami M, Becker R, Menssen HD, Marcinkowski M, Versmold HT. Xerocytosis with concomitant intrauterine ascites: first description and therapeutic approach. Blood. 1996;87:5392–3. [PubMed] [Google Scholar]
- 68.Sanchez M, Palacio M, Borrell A, Carmona F, Cobo T, Coll O, Cararach V. Prenatal diagnosis and management of fetal xerocytosis associated with ascites. Fetal Diagn Ther. 2005;20:402–5. doi: 10.1159/000086820. [DOI] [PubMed] [Google Scholar]
- 69.Grootenboer S, Schischmanoff PO, Laurendeau I, et al. Pleiotropic syndrome of dehydrated hereditary stomatocytosis, pseudohyperkalemia, and perinatal edema maps to 16q23-q24. Blood. 2000;96:2599–605. [PubMed] [Google Scholar]
- 70.Stewart GW, Amess JAL, Eber SW, Kingswood C, Lane PA, Smith B. Thrombo-embolic disease after splenectomy for hereditary stomatocytosis. Br J Haematol. 1996;93:303–10. doi: 10.1046/j.1365-2141.1996.4881033.x. [DOI] [PubMed] [Google Scholar]
- 71.Da Costa L, Willig TN, Fixler J, Mohandas N, Tchernia G. Diamond-Blackfan anemia. Curr Opin Pediatr. 2001;13:10–15. doi: 10.1097/00008480-200102000-00002. [DOI] [PubMed] [Google Scholar]
- 72.Rogers BB, Bloom SL, Buchanan GR. Autosomal dominantly inherited Diamond-Blackfan anemia resulting in nonimmune hydrops. Obstet Gynecol. 1997;89:805–7. doi: 10.1016/s0029-7844(97)00035-5. [DOI] [PubMed] [Google Scholar]
- 73.Saladi SM, Chattopadhyay T, Adiotomre PN. Nonimmune hydrops fetalis due to Diamond Blackfan anemia. Indian Pediatrics. 2004;41:187–8. [PubMed] [Google Scholar]
- 74.Roy V, Perez WS, Eapen M, et al. Non-Malignant Marrow Disorders Working Committee of the International Bone Marrow Transplant Registry. Bone Marrow Transplantation for Diamond-Blackfan Anemia. Biol Blood Marrow Transplant. 2005;11:600–8. doi: 10.1016/j.bbmt.2005.05.005. [DOI] [PubMed] [Google Scholar]
- 75.Makitie O, Ellis L, Durie PR, et al. Skeletal phenotype in patients with Shwachman-Diamond syndrome and mutations in SBDS. Clin Genet. 2004;65:101–12. doi: 10.1111/j.0009-9163.2004.00198.x. [DOI] [PubMed] [Google Scholar]
- 76.Shwachman H, Diamond LK, Oski FA, Khaw KT. The syndrome of pancreatic insufficiency and bone marrow dysfunction. J Pediatr. 1964;65:645–63. doi: 10.1016/s0022-3476(64)80150-5. [DOI] [PubMed] [Google Scholar]
- 77.Dror Y, Freedman MH. Swachman-diamond syndrome. Br J Haematol. 2002;118:701–13. doi: 10.1046/j.1365-2141.2002.03585.x. [DOI] [PubMed] [Google Scholar]
- 78.Kato K, Sugitani M, Kawataki M, et al. Congenital dyserythropoietic anemia type 1 with fetal onset of severe anemia. J Pediatr Hematol Oncol. 2001;23:63–6. doi: 10.1097/00043426-200101000-00016. [DOI] [PubMed] [Google Scholar]
- 79.Shalev H, Kapelushnik J, Moser A, Dgany O, Krasnov T, Tamary H. A comprehensive study of the neonatal manifestations of congenital dyserythropoietic anemia type I. J Pediatr Hematol Oncol. 2004;26:746–8. doi: 10.1097/00043426-200411000-00011. [DOI] [PubMed] [Google Scholar]
- 80.Remacha AF, Badell I, Pujol-Moix N, et al. Hydrops fetalis-associated dyserythropoietic anemia treated with intrauterine transfusions and bone marrow transplantation. Blood. 2002;100:356–8. doi: 10.1182/blood-2001-12-0351. [DOI] [PubMed] [Google Scholar]
- 81.Li CH, Lam CW, Lee CW, Kwong NS, Szeto SC. Pearson's syndrome: a rare cause of non-immune hydrops fetalis. Chin Med J (Engl) 2003;116:1952–4. [PubMed] [Google Scholar]
- 82.Jacobs LJ, Jongbloed RJ, Wijburg FA, et al. Pearson syndrome and the role of deletion dimmers and duplications in the mtDNA. J Inherit Metab Dis. 2004;27:47–55. doi: 10.1023/B:BOLI.0000016601.49372.18. [DOI] [PubMed] [Google Scholar]
- 83.Knerr I, Metzler M, Niemeyer CM, et al. Hematologic features and clinical course of an infant with Pearson syndrome caused by a novel deletion of mitochondrial DNA. J Pediatr Hematol Oncol. 2003;25:948–51. doi: 10.1097/00043426-200312000-00008. [DOI] [PubMed] [Google Scholar]
- 84.Muis N, Beemer FA, van Dijken P, Klep-de Pater JM. The Aase syndrome. Case report and review of the literature. Eur J Pediatr. 1986;145:153–7. doi: 10.1007/BF00441882. [DOI] [PubMed] [Google Scholar]
- 85.Auerbach AD, Wolman SR. Susceptibility of Fanconi's anaemia fibroblasts to chromosome damage by carcinogens. Nature. 1976;261:494–6. doi: 10.1038/261494a0. [DOI] [PubMed] [Google Scholar]
- 86.Auerbach AD. Fanconi anemia diagnosis and the diepoxybutane (DEB) test. Exp Hematol. 1993;21:731–3. [PubMed] [Google Scholar]
- 87.German J, Schonberg S, Caskie S, Warburton D, Falk C, Ray JH. A test for Fanconi's anemia. Blood. 1987;69:1637–41. [PubMed] [Google Scholar]
- 88.Taniguchi T, Garcia-Higuera I, Xu B, et al. Convergence of the Fanconi anemia and ataxia telangiectasia signaling pathways. Cell. 2002;109:459–72. doi: 10.1016/s0092-8674(02)00747-x. [DOI] [PubMed] [Google Scholar]
- 89.MacMillan ML, Auerbach AD, Davies SM, et al. Haematopoietic cell transplantation in patients with Fanconi anaemia using alternate donors: results of a total body irradiation dose escalation trial. Br J Haematol. 2000;109:121–9. doi: 10.1046/j.1365-2141.2000.01955.x. [DOI] [PubMed] [Google Scholar]
- 90.Grewal SS, Kahn JP, MacMillan ML, Ramsay NK, Wagner JE. Successful hematopoietic stem cell transplantation for Fanconi anemia from an unaffected HLA-genotype-identical sibling selected using preimplantation genetic diagnosis. Blood. 2004;103:1147–51. doi: 10.1182/blood-2003-02-0587. [DOI] [PubMed] [Google Scholar]