

Abstract
Summary
Intracellular nicotinamide phosphoribosyltransferase (iNampt) is an essential enzyme in the NAD biosynthetic pathway. An extracellular form of this protein (eNampt) has been reported to act as a cytokine named PBEF or an insulin-mimetic hormone named visfatin, but its physiological relevance remains controversial. Here we show that eNampt does not exert insulin-mimetic effects in vitro or in vivo but rather exhibits robust NAD biosynthetic activity. Haplodeficiency and chemical inhibition of Nampt cause defects in NAD biosynthesis and glucose-stimulated insulin secretion in pancreatic islets in vivo and in vitro. These defects are corrected by the administration of nicotinamide mononucleotide (NMN), a product of the Nampt reaction. A high concentration of NMN is present in mouse plasma, and plasma eNampt and NMN levels are reduced in Nampt heterozygous females. Our results demonstrate that Nampt-mediated systemic NAD biosynthesis is critical for β cell function, suggesting a vital framework for the regulation of glucose homeostasis.
Introduction
Nicotinamide adenine dinucleotide (NAD) and its derivative compounds are essential coenzymes in cellular redox reactions in all living organisms. NAD also participates in a number of important signaling pathways in mammalian cells, including mono- and poly-ADP-ribosylation and synthesis of cyclic ADP-ribose and nicotinate adenine dinucleotide phosphate (Belenky et al., 2007; Rongvaux et al., 2003). Recently, it has also been demonstrated that NAD and its derivatives play an important role in transcriptional regulation (Denu, 2003; Lin and Guarente, 2003). In particular, the discovery that the evolutionarily conserved Sir2 family catalyzes protein deacetylation in an NAD-dependent manner (Imai et al., 2000; Landry et al., 2000; Smith et al., 2000) has drawn much attention to this novel role for NAD. Despite the importance of NAD in these biological events, the regulation of NAD biosynthesis in vertebrates remains poorly understood.
In prokaryotes and lower eukaryotes, such as yeast, NAD is synthesized by the de novo pathway via quinolinic acid and by the salvage pathway via nicotinic acid (Belenky et al., 2007; Revollo et al., 2007; Rongvaux et al., 2003, see Supplementary Figure 1A). In vertebrates, NAD biosynthesis is markedly different from that of yeast and invertebrates. Mammals predominantly use nicotinamide rather than nicotinic acid as a precursor for NAD biosynthesis (Magni et al., 1999). Instead of the deamidation to nicotinic acid, nicotinamide is directly converted to nicotinamide mononucleotide (NMN) by nicotinamide phosphoribosyltransferase (Nampt) (Supplementary Figures 1B and 1C). NMN is then converted to NAD by nicotinamide/nicotinic acid mononucleotide adenylyltransferase (Nmnat).
Nampt has very ancient origins as an NAD biosynthetic enzyme. Originally, Nampt was identified as the product of the NadV gene in Haemophilus ducreyi (Martin et al., 2001) and has been found even in some bacteriophages (Miller et al., 2003). The homology of Nampt proteins between bacteriophage, bacteria, and vertebrates is unusually high (Revollo et al., 2004; Rongvaux et al., 2003). We and other groups characterized the biochemical nature of mammalian Nampt (Revollo et al., 2004; Rongvaux et al., 2002; van der Veer et al., 2005). We demonstrated that Nampt is the rate-limiting component in the NAD biosynthetic pathway from nicotinamide and regulates the activity of the NAD-dependent deacetylase Sirt1 in mammalian cells (Revollo et al., 2004). Furthermore, we and other groups have recently determined crystal structures of the Nampt apoenzyme, the Nampt-NMN complex, and the complex of Nampt and its potent chemical inhibitor FK866 (Khan et al., 2006; Kim et al., 2006; Wang et al., 2006), demonstrating that this enzyme is a dimeric type II phosphoribosyltransferase. These studies have firmly established the biochemical and structural basis of Nampt as an NAD biosynthetic enzyme.
Interestingly, the gene encoding human Nampt was originally isolated as a presumptive cytokine named pre-B cell colony-enhancing factor (PBEF) that enhances the maturation of B cell precursors in the presence of interleukin (IL)-7 and stem cell factor (Samal et al., 1994). Although this function of PBEF has not been reconfirmed to date, several groups have since reported a cytokine-like function of PBEF (Jia et al., 2004; Moschen et al., 2007; Ognjanovic and Bryant-Greenwood, 2002; Ye et al., 2005). Additionally, Nampt/PBEF has recently been re-identified as a “new visceral fat-derived hormone” named visfatin (Fukuhara et al., 2005). Strikingly, visfatin was reported to exert insulin-mimetic effects in cultured cells and to lower plasma glucose levels in mice by binding to and activating the insulin receptor. However, the physiological relevance of visfatin remains controversial (Arner, 2006; Revollo et al., 2007; Sethi, 2007; Stephens and Vidal-Puig, 2006). Subsequent studies have brought conflicting results regarding a possible connection of visfatin to obesity, type 2 diabetes, and other metabolic complications (see reviews, Sethi, 2007; Stephens and Vidal-Puig, 2006). Additionally, because this protein lacks a signal sequence for secretion, it has been suggested that the presence of Nampt/PBEF/visfatin in extracellular compartments might be simply due to either cell lysis or cell death (Hug and Lodish, 2005; Stephens and Vidal-Puig, 2006).
Here we show that Nampt/PBEF/visfatin functions as an intra- and extracellular NAD biosynthetic enzyme. Therefore, we will mainly use Nampt as the name of this protein through the rest of this paper. The extracellular form of Nampt (eNampt), which is positively secreted through a non-classical secretory pathway, does not show insulin-mimetic effects in vitro or in vivo but rather exhibits robust, even higher NAD biosynthetic activity compared to its intracellular form (iNampt). Nampt-deficient heterozygous (Nampt+/−) female mice show moderately impaired glucose tolerance and reduced glucose-stimulated insulin secretion. Consistent with this in vivo phenotype, primary islets isolated from Nampt+/− mice show significant decreases in NAD biosynthesis and insulin secretion in response to glucose. A similar phenotype is also observed in wild-type primary islets treated with FK866. These defects in NAD biosynthesis and glucose-stimulated insulin secretion can be corrected by the administration of the Nampt reaction product, nicotinamide mononucleotide (NMN), in vivo and in vitro, demonstrating that the observed defects are due to the lack of the NAD biosynthetic activity of Nampt. Strikingly, a high concentration of NMN is found in mouse plasma, and plasma eNampt and NMN levels are reduced in Nampt+/− females. These findings strongly suggest that Nampt-mediated systemic NAD biosynthesis plays a critical role in the regulation of β cell function, providing important insights to develop preventive/therapeutic interventions against metabolic complications, such as type 2 diabetes.
Results
The Nampt protein is highly expressed in brown adipose tissue, liver, kidney, and heart in mice
It has been shown that Nampt has both intra- and extracellular forms in mammals, but their physiological functions have been the matter of debate (Revollo et al., 2007). To elucidate the physiological role of Nampt, we first surveyed the tissue distribution of the intracellular Nampt protein (iNampt) in mice (Figure 1A). Among the tissues examined, brown adipose tissue (BAT), liver, and kidney showed the highest levels of iNampt protein expression, and heart showed an intermediate level of iNampt. White adipose tissue (WAT), lung, spleen, testis, and muscle showed very low levels of iNampt, and levels of iNampt in the brain and pancreas were undetectable. These findings suggest that the requirement of Nampt-mediated NAD biosynthesis might vary widely among different tissues.
Figure 1.

Tissue distribution of intracellular Nampt in mice and production of intra- and extracellular Nampt during adipocyte differentiation. A) Distribution of intracellular Nampt (iNampt) in mouse tissues. 22.5 μg of each tissue extract from a C57BL/6 mouse was analyzed by Western blotting with Nampt- and actin-specific antibodies. 5 μg of cell extract from a Nampt-overexpressing NIH3T3 cell line (Nampt1) was loaded as a reference. WAT, white adipose tissue; BAT, brown adipose tissue. B) Production of intra- and extracellular Nampt (iNampt and eNampt) during differentiation of HIB-1B brown preadipocytes. Upper panel: Confluent cultures of HIB-1B cells were differentiated, and cell extracts were prepared at the indicated days. 45 μg of each cell extract was analyzed. Lower panel: 20 μl of each culture supernatant collected at the indicated days was analyzed. C) Production of iNampt and eNampt during differentiation of 3T3-L1 white preadipocytes. Upper panel: 45 μg of each cell extract collected at the indicated days was analyzed. Middle panel: 20 μl of culture supernatant was analyzed. Lower panel: Concentrated culture supernatant was analyzed at day 8 to detect eNampt. 10 μg of the cell extract from Nampt-overexpressing fibroblasts (Nampt1) was loaded as a reference in each experiment. D) Production of iNampt and eNampt during differentiation of human SGBS white preadipocytes. Left and middle panels: 13 μg of each cell extract and 25 μl of each culture supernatant (10-fold concentrated) collected at the indicated points were analyzed. Adiponectin production is also shown as a positive control for adipocyte differentiation. Pre, undifferentiated preadipocytes; d4, differentiating adipocytes at day 4; d8, mature adipocytes; CM, control medium; SN, supernatant. Right panel: iNampt protein expression was analyzed in tissue extracts from human subcutaneous (sc) and visceral (visc) white adipose tissues (WAT).
Differentiated adipocytes produce extracellular Nampt (eNampt)
Because Nampt has been reported to act as an adipokine (Fukuhara et al., 2005), we examined the production of intra- and extracellular Nampt by brown and white adipocytes. We compared iNampt and eNampt levels between the mouse preadipocyte cell lines, HIB-1B and 3T3-L1, that can differentiate into brown and white adipocytes, respectively. iNampt was highly induced during the differentiation of HIB-1B cells (Figure 1B, upper panel). In parallel with this induction, we also detected increasing amounts of eNampt from day 2 to 8 in HIB-1B conditioned media (Figure 1B, lower panel). This extracellular, immunodetectable Nampt was purified from HIB-1B conditioned media, and its physical identity was confirmed by tandem mass spectrometric peptide analyses (data not shown). We estimated that the concentration of eNampt in HIB-1B conditioned media at day 8 was ∼100 ng/ml (data not shown), a concentration comparable to that of adiponectin in 24h culture supernatants of differentiated human adipocytes (Wang et al., 2005). During the differentiation of 3T3-L1 white preadipocytes, a similar induction of iNampt was detected, although the final induction level of iNampt in differentiated 3T3-L1 cells was significantly lower than that in differentiated HIB-1B cells (Figure 1C, upper panel). A very low level of eNampt was detectable in 3T3-L1 conditioned media at day 8 after concentration (Figure 1C, middle and lower panels). iNampt protein levels also mildly increased during the differentiation of human SGBS white preadipocytes (Figure 1D, left panel). eNampt was detectable in concentrated SGBS conditioned media at day 8 when SGBS cells fully differentiated and started secreting adiponectin (Figure 1D, middle panel). The iNampt protein expression was also confirmed in human subcutaneous and visceral white adipose tissue extracts (Figure 1D, right panel).
We further examined whether other cell types are capable of producing eNampt. We examined NIH3T3 (a mouse fibroblast cell line), HEK293 (a human embryonic kidney cell line), Hepa1-6 (a mouse hepatocyte cell line), COS-7 (a SV40-transformed monkey kidney cell line), and Chinese hamster ovary (CHO) cells. None of these cell lines naturally produced eNampt. However, detectable amounts of eNampt were observed only in conditioned media from COS-7 and CHO cells when Nampt is highly overexpressed (data not shown). Thus, eNampt is produced in a cell type-dependent manner, suggesting that eNampt secretion is a highly regulated process. In this regard, HIB-1B cells, which can produce high levels of both iNampt and eNampt, provide a useful, robust culture system to further investigate the production and function of eNampt.
eNampt is positively secreted through a non-classical secretory pathway
It has been speculated that the apparent “secretion” of eNampt might be simply due to either cell lysis or cell death (Hug and Lodish, 2005; Stephens and Vidal-Puig, 2006). We therefore determined whether the production of eNampt is due to positive secretion or to cell lysis/death. We constructed C-terminally FLAG-tagged versions of Nampt (Nampt-FLAG) as well as dihydrofolate reductase (Dhfr) and preprolactin (Ppl) as controls for intracellular and extracellular proteins, respectively (Dhfr-FLAG and Ppl-FLAG). HIB-1B cell lines that stably expressed these FLAG-tagged proteins were established and differentiated. Dhfr-FLAG was detected exclusively in cell extracts (Supplementary Figure 2A), whereas prolactin-FLAG (Prl-FLAG) was detected predominantly in supernatants (Supplementary Figure 2B). Significant levels of Nampt-FLAG were detected in both cell extracts and supernatants in these cell lines, demonstrating that the presence of eNampt in supernatants is not due to cell lysis or cell death. We also treated these cell lines with brefeldin A (BFA), an inhibitor of protein secretion through the Golgi-ER system. While eNampt-FLAG levels in the supernatant were not affected by BFA, the secretion of Prl-FLAG was significantly inhibited by BFA (Supplementary Figure 2C). This finding suggests that eNampt is not secreted through a classical Golgi-ER system but rather through a non-classical secretory pathway. We also confirmed similar results by transfecting CHO cells with each of the FLAG-tagged constructs (data not shown). These results demonstrate that specific cell types (adipocytes and CHO cells) positively secrete eNampt through a non-classical secretory pathway.
eNampt produced by differentiated adipocytes is enzymatically active
Using differentiated HIB-1B brown adipocytes as a model system, we next analyzed the enzymatic activities of iNampt and eNampt. Interestingly, we noticed that untagged eNampt co-immunoprecipitated with eNampt-FLAG (Figure 2A), indicating that eNampt forms a dimer, as observed in the crystal structure (Wang et al., 2006). The dimer formation of Nampt was also confirmed as a ~100 kDa peak in size exclusion chromatography (Supplementary Figure 3). To examine if dimerization is necessary for Nampt enzymatic activity, we mutated Ser199 and Ser200, both of which play key roles in forming a symmetrical dimer interface (Wang et al., 2006, see Supplementary Figure 3A), to aspartic acid. The S199D and S200D Nampt mutants do not dimerize properly and show decreased enzymatic activity (Supplementary Figure 3B and Supplementary Table 1), suggesting that dimerization is important for Nampt enzymatic activity.
Figure 2.
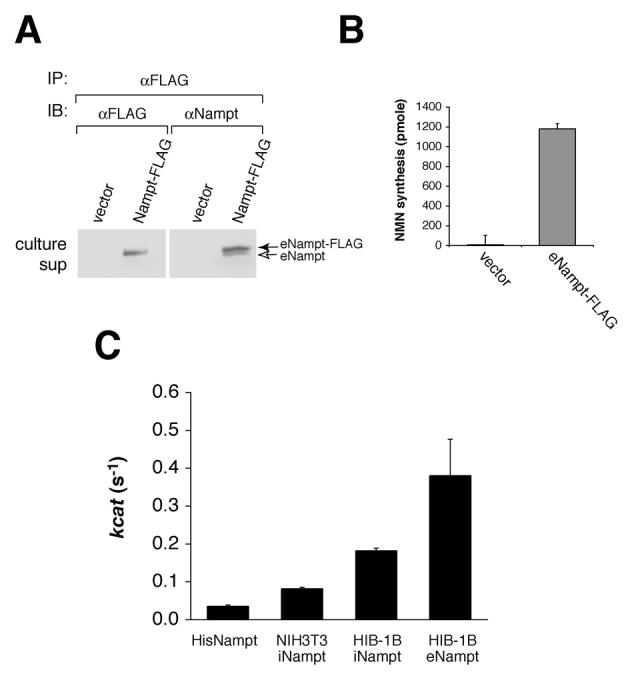
eNampt produced by differentiated HIB-1B brown adipocytes has high Nampt enzymatic activity. A) eNampt-FLAG co-immunoprecipitates with untagged eNampt from culture supernatants. The eNampt-FLAG protein was immunoprecipitated with an anti-FLAG antibody from 8 ml of each culture supernatant and blotted with either the same antibody or an anti-Nampt antibody. The culture supernatants of the vector-transfected cells were used as a control. B) The eNampt-FLAG protein immunoprecipitated from culture supernatants of differentiated Nampt-FLAG HIB-1B cells has Nampt enzymatic activity. Results are presented as mean ± SE (n=3). C) The kcat values of the bacterially produced His-tagged recombinant Nampt and intra- and extracellular Nampt-FLAG from NIH3T3 and differentiated HIB-1B cells were calculated by measuring NMN synthesis and quantifying the amount of Nampt by Western blotting (data not shown). Results are presented as mean ± SEM (n=7 for His-tagged Nampt, 3 for iNampt from NIH3T3, 6 for iNampt from HIB-1B, and 4 for eNampt from HIB-1B), and all differences in pair-wise comparisons are statistically significant with the Student's t test (p < 0.05).
The immunoprecipitates from the HIB-1B culture supernatants were subjected to enzyme-coupled fluorometric assays (Revollo et al., 2004) to compare the enzymatic activities of iNampt and eNampt. The immunoprecipitated eNampt-FLAG protein exhibited robust activity, while immunoprecipitates from culture supernatants of the backbone vector-transfected HIB-1B cells showed no activity (Figure 2B). We also measured the Nampt enzymatic activity of the bacterially produced recombinant Nampt and the iNampt-FLAG immunoprecipitated from HIB-1B and NIH-3T3 cellular extracts and calculated their kcat values. Surprisingly, the eNampt-FLAG secreted from differentiated HIB-1B cells showed a significantly higher kcat value (0.380/s) than recombinant Nampt (0.035/s, ∼11-fold), iNampt-FLAG from NIH3T3 cells (0.082/s, ∼5-fold), and iNampt-FLAG from differentiated HIB-1B cells (0.182/s, ∼2-fold) (Figure 2C). Taken together, our findings provide strong evidence that Nampt functions both intra- and extracellularly as a robust, dimeric NAD biosynthetic enzyme.
eNampt does not exert insulin-mimetic effects in vitro and in vivo
Fukuhara et al. (2005) reported that the same protein, re-named as visfatin, exerted insulin-mimetic effects on cultured cells by binding to and activating the insulin receptor. Therefore, we examined whether eNampt/visfatin indeed exerts insulin-like effects.
We first investigated the potency of exogenously administered Nampt/visfatin (eNampt/visfatin) to induce adipocyte differentiation in the human SGBS preadipocyte cell line. While SGBS preadipocytes fully differentiated into mature adipocytes in the presence of insulin (20 nM), we did not detect differentiation into mature adipocytes at all with equimolar concentrations of eNampt/visfatin (Figure 3A). Additionally, both eNampt/visfatin proteins produced in bacteria and mammalian cells failed to mimic the effect of insulin to induce expression of adipocyte differentiation markers, such as adiponectin and PPARγ (Figure 3B). We also examined whether eNampt/visfatin could enhance glucose uptake in differentiated human SGBS and mouse 3T3-L1 adipocytes. While insulin shows the dose-dependent stimulation of glucose uptake, 100 nM of eNampt/visfatin did not show any effect on glucose uptake in those differentiated adipocytes (Figure 3C).
Figure 3.
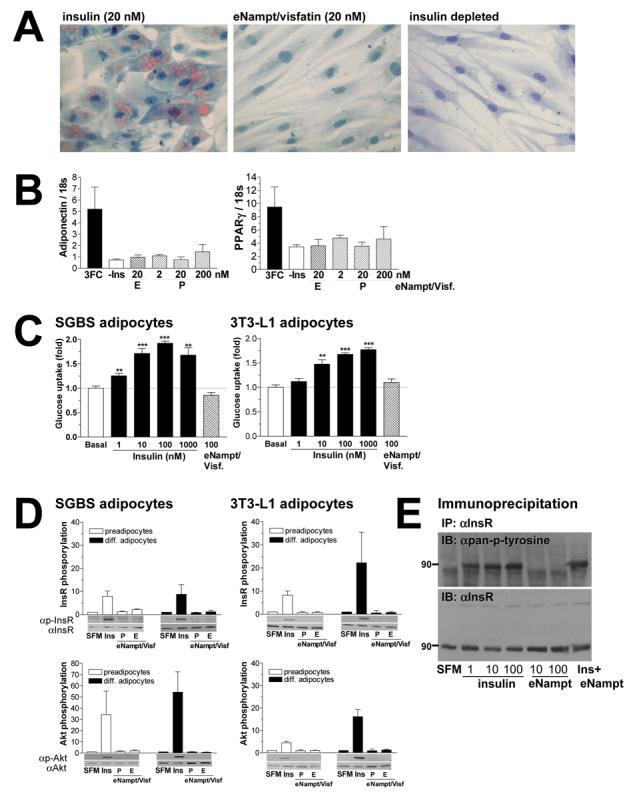
eNampt does not exert insulin-mimetic effects on adipogenesis, glucose uptake, and insulin signaling in cultured cells. A) Differentiation of human SGBS preadipocytes was induced over a period of 8 days in standard induction medium containing 20 nM insulin or 20 nM eNampt/visfatin. For negative control, cells were treated in induction medium without insulin. Mature adipocytes were stained with Sudan III to visualize lipid accumulation at day 8. B) mRNA expression levels of two adipocyte differentiation markers, adiponectin and PPARγ, were analyzed with real-time quantitative RT-PCR. SGBS preadipocytes were differentiated in standard induction medium with 20nM insulin (3FC) or eNampt/visfatin proteins produced in bacteria (P) or in mammalian cells (E) at concentrations indicated. C) Differentiated SGBS and 3T3-L1 adipocytes were incubated with increasing concentrations of insulin or 100 nM eNampt/visfatin produced in mammalian cells. Glucose uptake was determined using [14C]-2-deoxyglucose at 0.5 μCi/ml for 5 min. Experiments were performed in quadriplicates, and glucose uptake was normalized to the amount of protein. Basal glucose uptake in non-stimulated cells is assigned as 1. D) The effect of eNampt/visfatin on phosphorylation of the insulin receptor (InsR, upper panels) and Akt/PKB kinase (Akt, lower panels) was examined in undifferentiated and differentiated human SGBS and mouse 3T3-L1 cells. Cells were starved overnight and then exposed to serum free medium (SFM), 10 nM insulin (Ins), or 10 nM eNampt/visfatin produced in bacteria (P) or in mammalian cells (E). Signals of phosphorylated proteins are normalized to those of non-phosphorylated proteins, and values are shown relative to the signal in serum free medium (n ≥ 3). E) Insulin receptor phosphorylation was examined in R-IR cells treated with different concentrations of insulin and eNampt/visfatin. Cells were treated with the indicated concentrations of insulin, eNampt/visfatin produced in mammalian cells or both (10 nM each). The insulin receptor protein (InsR) was immunoprecipitated with an anti-InsR antibody 29B4 and blotted with an anti-pan-phosphotyrosine antibody (upper panel). The membrane was re-probed with another anti-InsR antibody C19 (lower panel).
All results are expressed as mean ± SEM.
Because we were unable to reproduce the reported insulin-mimetic activity of visfatin for adipogenesis and glucose uptake, we next examined whether eNampt/visfatin could induce insulin signaling. In this experiment, we employed several different cell models: human SGBS cell line, mouse 3T3-L1 cell line that was also used in the original report of visfatin (Fukuhara et al., 2005), and R- and R-IR cells, mouse fibroblast cell lines in which the endogenous insulin-like growth factor I (IGF-I) receptor is disrupted (R-), but the human insulin receptor is stably overexpressed (R-IR) (Miura et al., 1995). Although insulin (10 nM) induced significant phosphorylation of the insulin receptor and a downstream signaling kinase Akt in each of these cell lines, equimolar concentrations of eNampt/visfatin failed to induce phosphorylation of either insulin receptor or Akt (Figure 3D and Supplementary Figure 4A). The dose-dependent phosphorylation of the insulin receptor was also observed in R-IR cells with 1-100 nM of insulin, but 10 and 100 nM of eNampt/visfatin were unable to induce phosphorylation in these cells (Figure 3E). Additionally, conditioned media prepared from either control or Nampt-overexpressing COS-7 cells did not induce phosphorylation of the insulin receptor or Akt (Supplementary Figure 4B).
We also examined whether eNampt/visfatin reduces blood glucose levels in vivo. Consistent with our results from cell culture experiments, we were unable to see any immediate decrease in blood glucose levels even when very high doses (i.e. 20-fold higher than previously reported) of the recombinant Nampt were injected (Supplementary Figure 4C). Thus, in contrast to the original report of visfatin, we were unable to reproduce the insulin-mimetic function of visfatin for adipogenesis, glucose uptake, insulin signaling, or blood glucose reduction.
Nampt-deficient heterozygous (Nampt+/−) mice show impaired glucose tolerance and reduced glucose-stimulated insulin secretion
To further elucidate the physiological role of Nampt, we generated Nampt-deficient mice using a 129/Ola ES cell line created and designated RRT307 by the BayGenomics consortium (baygenomics.ucsf.edu). In the RRT307 ES cell line, the Nampt gene locus is disrupted by the insertion of a β-galactosidase-neomycin (β-geo) exon-trap vector between exon 8 and exon 9. As a result of this insertion, the C-terminal 128 amino acids are truncated so that a ∼190 kDa fusion protein of Nampt and β-geo is produced. Because the deleted residues include critical amino acids that contribute to the Nampt catalytic site, such as Gly384 and Arg392 (Wang et al., 2006), this fusion protein is expected to be enzymatically deficient. Consistent with this notion, mice homozygous for the Nampt mutant allele died prior to embryonic day 10.5 (data not shown). Nampt heterozygous (Nampt+/−) mice were overtly normal, but they showed significant decreases in iNampt levels in all tissues examined (heart, liver, kidney, and BAT) and expressed the Nampt-β-geo fusion protein (Supplementary Figures 5A-D). We also measured total NAD levels in BAT and liver from Nampt+/− and control mice (Supplementary Figure 5E). While NAD levels were significantly decreased ∼66% of control in Nampt+/− BAT, no significant difference was observed in NAD levels between Nampt+/− and control livers. This presumably reflects the strong contribution of the tryptophan-kynurenine pathway for de novo NAD biosynthesis in the liver (Magni et al., 2004). Interestingly, female Nampt+/− mice showed a significant decrease in plasma eNampt levels (∼50% of control eNampt levels), consistent with a decrease in tissue iNampt levels, while a similar decrease was not observed in Nampt+/− males (Supplementary Figure 5F).
Both male and female Nampt+/− mice showed normal body weights and mostly normal fed and fasted glucose levels (Figures 4A and 4B). Nampt+/− and control mice did not differ significantly in the percent islet area and islet morphology (Supplementary Figures 6A and 6B). However, Nampt+/− female mice showed moderately impaired glucose tolerance during intraperitoneal glucose tolerance tests (IPGTTs) as compared to control mice (Figure 4C), while males did not show this phenotype (Supplementary Figures 6C and 6D). We also measured plasma insulin levels in female Nampt+/− and control mice during IPGTTs. Nampt+/− mice showed significant decreases in insulin secretion at 15 and 30 min after glucose injection compared to control littermates, consistent with the IPGTT results (Figure 4D). We also conducted insulin tolerance tests on these female mice, but no difference was detected between Nampt+/− and control mice (Figure 4E). These results suggest that Nampt haplodeficiency significantly affects glucose-stimulated insulin secretion in pancreatic β cells and causes impaired glucose tolerance in mice.
Figure 4.
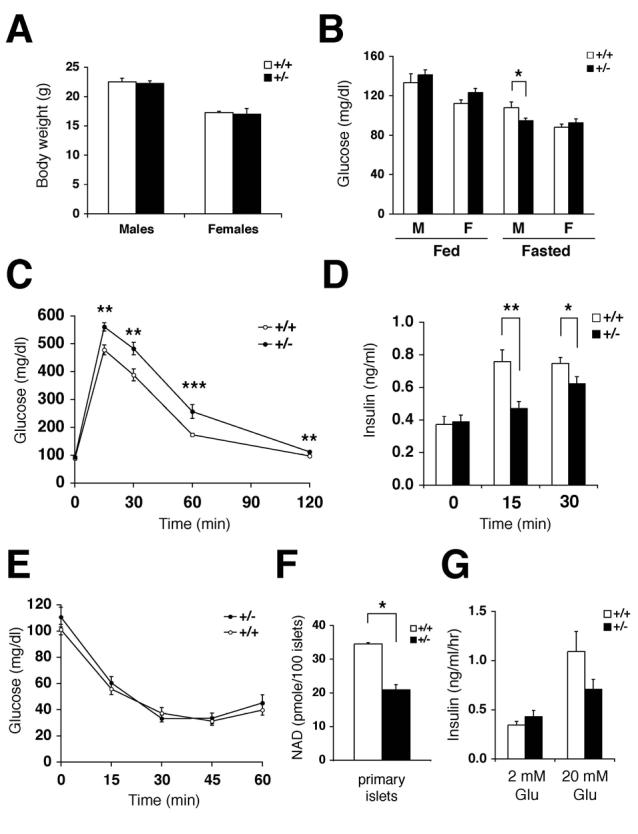
Nampt+/− mice show moderately impaired glucose tolerance and reduced glucose-stimulated insulin secretion. A) Body weights of Nampt+/− and wild-type littermates at 8 weeks of age (n=7-15). B) Fed and fasted glucose levels in males (M) and females (F) of Nampt+/− and control mice (n=10-18). C) Intraperitoneal glucose tolerance tests (IPGTTs). Nampt+/− (n=13) and control (n=18) females were injected with PBS and fasted for 12-14 hrs. Dextrose (3 g/kg body weight) was injected intraperitoneally, and blood glucose levels were measured. D) Plasma insulin levels in Nampt+/− and control female littermates at 0, 15, and 30 min time points in IPGTTs. Nampt+/− mice (n=14), control mice (n=17) for 0 and 30 min time points; Nampt+/− mice (n=7), control mice (n=11) for 15 min time point. E) Insulin tolerance tests (ITTs). Nampt+/− (n=9) and control (n=13) females were injected with human insulin (0.75 U/kg body weight) after fasting for 4 hrs, and blood glucose levels were measured. F) NAD levels (pmole) in primary islets isolated from Nampt+/− and control female mice. NAD levels were measured by HPLC in duplicates of primary islets pooled from three individual mice of each genotype. G) Insulin secreted (ng/ml/hr) from Nampt+/− and control islets at the indicated glucose concentrations (n=4 mice for each genotype). Isolated primary islets were cultured overnight in the RPMI media containing 1 μM nicotinamide prior to insulin secretion experiments.
All results are expressed as mean ± SEM. *p ≤ 0.05, ** p ≤ 0.01, *** p ≤ 0.001.
To confirm these in vivo results, we also examined glucose-stimulated insulin secretion in isolated primary islets. Wild-type primary islets had a low level of iNampt protein expression (∼10-fold lower than liver), and Nampt+/− islets exhibited a significant decrease in iNampt expression (Supplementary Figures 6E and 6F). Because Nampt has the KM of 0.92 μM for nicotinamide (Revollo et al., 2004), which falls within the physiological range of plasma nicotinamide concentrations in mammals (Revollo et al., 2007), we cultured isolated primary islets overnight in RPMI media containing 1 μM nicotinamide prior to insulin secretion experiments. Under this experimental condition, primary islets isolated from Nampt+/− female mice showed a ∼40% decrease in NAD levels and a comparable decrease in glucose-stimulated insulin secretion compared to those from wild-type littermates (Figures 4F and 4G). Therefore, consistent with the in vivo results, these findings support the notion that Nampt haplodeficiency causes defects in NAD biosynthesis and glucose-stimulated insulin secretion in pancreatic β cells.
The defects in Nampt+/− mice and islets can be corrected by administering NMN
If the glucose intolerance observed in Nampt+/− female mice is indeed due to the lack of the NAD biosynthetic activity of Nampt, the administration of NMN, a product of the Nampt enzymatic reaction, might be able to correct their defects. To test this possibility, we used the same Nampt+/− and control female cohorts used for the previous IPGTTs (see legend for Figure 4C) and injected them intraperitoneally with NMN (500 mg/kg body weight) ∼14 hrs prior to the IPGTTs. Interestingly, after NMN administration, there was no difference in blood glucose levels in IPGTTs between Nampt+/− and control female mice (Figure 5A). In addition, NMN-treated Nampt+/− and control mice also had similar plasma insulin levels at each time point (Figure 5B). These data clearly indicate that NMN administration corrects the defect in glucose-stimulated insulin secretion observed in Nampt+/− mice. We also examined the effect of NMN on glucose-stimulated insulin secretion in primary islets isolated from Nampt+/− and control female mice that were cultured overnight in the RPMI media containing 1 μM nicotinamide and 50 μM NMN. The addition of NMN significantly augmented insulin secretion in response to 20 mM glucose in both Nampt+/− and control islets, and after the NMN treatment, glucose-stimulated insulin secretion did not differ between Nampt+/− and control islets (Figure 5C). These results strongly indicate that the defects observed in Nampt+/− mice and islets are not due to the lack of the insulin-mimetic activity of visfatin but are rather due to the lack of the NAD biosynthetic activity of Nampt.
Figure 5.

NMN administration ameliorates the defects in Nampt+/− mice and islets. A) IPGTTs after NMN administration. The same Nampt+/− (n=18) and control (n=19) cohorts that were used for the IPGTTs shown in Figure 4C were injected with NMN (500 mg/kg body weight) ∼14 hrs prior to IPGTTs. B) Plasma insulin levels in Nampt+/− and control female littermates at 0, 15, and 30 min time points in IPGTTs. Nampt+/− mice (n=16), control mice (n=17) for 0 and 30 min time points; Nampt+/− mice (n=10), control mice (n=8) for 15 min time point. C) Insulin secreted (ng/ml/hr) from NMN-treated Nampt+/− and control islets at the indicated glucose concentrations (n=4 mice for each genotype). Isolated primary islets were cultured overnight in the RPMI media containing 1 μM nicotinamide plus 50 μM NMN prior to insulin secretion experiments. D) NAD levels (pmole) in wild-type primary islets treated overnight with NMN, FK866, or the combination of these compounds at the indicated concentrations. NAD levels were measured by HPLC in triplicates of primary islets pooled from four individual wild-type mice. E) Insulin secreted (ng/ml/hr) at the indicated glucose concentrations from control, FK866-treated, and FK866 plus NMN-treated wild-type primary islets. The experiments were conducted in triplicates of primary islets pooled from four individual mice cultured for 48h in the RPMI media containing 1 μM nicotinamide and indicated compounds (10 nM for FK866 and 100 μM for NMN) prior to insulin secretion experiments.
All results are expressed as mean ± SEM. *p ≤ 0.05, ** p ≤ 0.01, *** p ≤ 0.001.
To further confirm the effect of NMN on NAD biosynthesis and insulin secretion, we measured NAD levels in primary islets cultured overnight with NMN and/or FK866, a potent chemical inhibitor of Nampt (Hasmann and Schemainda, 2003). While 50 μM NMN enhanced NAD biosynthesis by ∼30%, 10 nM FK866 inhibited it by ∼80% in primary islets (Figure 5D), suggesting that Nampt-mediated NAD biosynthesis is critical for primary islets. Interestingly, in the presence of FK866, the administration of 50-100 μM NMN restored NAD levels in a dose-dependent manner (Figure 5D). Therefore, primary islets are clearly able to use exogenous NMN to maintain normal levels of NAD. Glucose-stimulated insulin secretion was suppressed by ∼60% in primary islets after 48 hrs of FK866 treatment, and 100 μM NMN increased insulin secretion in the FK866-treated primary islets above control islets (Figure 5E). Taken together, these findings demonstrate that administration of NMN ameliorates the defects in NAD biosynthesis and glucose-stimulated insulin secretion caused by Nampt haplodeficiency or the Nampt inhibitor FK866 in vivo and in vitro.
NMN circulates systemically in blood
eNampt has been reported to exist in human and mouse plasma (Sethi, 2007; Stephens and Vidal-Puig, 2006), and we also confirmed the observation by Western blotting (Supplementary Figure 5F). The high NAD biosynthetic activity of eNampt suggests that it might synthesize NMN in blood circulation. Therefore, we tested whether we could detect NMN in mouse plasma by HPLC. Interestingly, mouse plasma extracts showed a sharp peak at the same elution time as that of standard NMN (Figure 6A). To confirm its identity, we spiked mouse plasma extracts with 100 pmole of NMN and found that the peak of interest was significantly increased (Figure 6B). We also injected NMN (500 mg/kg body weight) into mice and found that the peak of interest increased significantly 15 min after NMN injection (Figure 6C), indicating that this particular peak represents NMN. To further examine the nature of this peak, we collected fractions that contained this peak and analyzed them by ion trap tandem mass spectrometry. Standard NMN showed the signature m/z of 335 and 123 (Figure 6D, upper panel). The same signature spectra of NMN were detected in the peak fractions from mouse plasma extracts (Figure 6D, lower panel), demonstrating that this peak is indeed NMN.
Figure 6.

NMN circulates systemically in mouse blood. A) HPLC chromatograms of mouse plasma extracts and standard NMN. The extract that corresponded to 5 μl of plasma and standard NMN were run in an isocratic condition at a flow rate of 1 ml/min. The peak indicated with an arrow showed the same elution time as that of standard NMN. B) HPLC chromatograms of mouse plasma extracts with or without a spike of 100 pmole of NMN. Extracts that corresponded to 2.5 μl of plasma were run at a flow rate of 0.7 ml/min. The peak indicated with arrows showed a significant increase after spiking NMN. C) HPLC chromatograms of mouse plasma extracts at 0 and 15 min time points after NMN injection (500 mg/kg body weight). The peaks indicated by arrows showed an increase at 15 min time point. D) Ion trap tandem mass spectrometry analysis for the fractions that contain the peaks indicated by arrows in HPLC chromatograms. Upper panel, standard NMN; lower panel, the peak fraction of a mouse plasma extract. The signature m/z of 123 (a) and 335 (b) for NMN were detected in the peak fraction. E) Plasma NMN concentrations in control and Nampt+/− male and female mice. The extracts corresponding to 2.5 μl of plasma were analyzed by HPLC, and the NMN peaks were quantitated for control (n=3) and Nampt+/− (n=4) mice at ∼8 months of age. Results are expressed as mean ± SEM.
We measured plasma NMN levels by HPLC and calculated its concentrations in control and Nampt+/− male and female mice. Wild-type males and females had similar plasma NMN concentrations (80-90 μM) (Figure 6E). Interestingly, Nampt+/− females showed a ∼35% decrease in plasma NMN concentrations compared to wild-type littermates, whereas Nampt+/− males had similar values to controls (Figure 6E). These sex-related differences are consistent with the differences in plasma eNampt levels between Nampt+/− males and females (Supplementary Figure 5F). This disparity in plasma eNampt and NMN levels between genders is likely the cause of the IPGTT defects observed only in Nampt+/− females. Taken together, these results clearly demonstrate that NMN, the product of the Nampt enzymatic reaction, circulates systemically in mammalian blood.
Discussion
Nampt/PBEF/visfatin as an intra- and extracellular NAD biosynthetic enzyme that regulates pancreatic β cell function
In this study, we have provided several lines of evidence demonstrating that Nampt functions as an intra- and extracellular NAD biosynthetic enzyme and plays an important role in the regulation of glucose-stimulated insulin secretion in pancreatic β cells. First, fully differentiated mouse and human adipocytes produce eNampt. eNampt is positively secreted through a non-classical secretory pathway, and the presence of eNampt is not due to cell lysis or cell death. Second, eNampt produced by differentiated adipocytes exhibits robust NAD biosynthetic activity that is even higher than iNampt, indicating that Nampt can function as an extracellular enzyme. Third, eNampt does not have any insulin-mimetic activity in vitro and in vivo. Fourth, Nampt+/− female mice show impaired glucose tolerance and significantly reduced insulin secretion in IPGTTs. Primary islets isolated from Nampt+/− mice or treated with FK866 also show significant decreases in NAD biosynthesis and insulin secretion in response to glucose. Fifth, these defects in NAD biosynthesis and glucose-stimulated insulin secretion can be corrected by the exogenous administration of NMN in vivo and in vitro. Finally, we have demonstrated that a high concentration of NMN circulates systemically in mouse plasma. These results clearly reveal an important functional mode of Nampt as an extracellular NAD biosynthetic enzyme and strongly support the physiological significance of Nampt-mediated systemic NAD biosynthesis in the regulation of β cell function.
The physiologically relevant function of secreted eNampt
While the function of iNampt is well established as an essential NAD biosynthetic enzyme (Revollo et al., 2004; Rongvaux et al., 2002; van der Veer et al., 2005), the physiological role of eNampt remains controversial. In this study, we attempted to reproduce the reported insulin-mimetic effects of eNampt/visfatin on adipogenesis, glucose uptake, cellular insulin signaling, and blood glucose levels in mice. Surprisingly, we were unable to obtain evidence supporting the insulin-mimetic activity of eNampt/visfatin. Furthermore, we found that Nampt+/− mice and islets have defects in NMN/NAD biosynthesis and glucose-stimulated insulin secretion. These defects are ameliorated by NMN administration in Nampt+/− mice and islets. Thus, these findings strongly indicate that the defects observed in Nampt+/− mice and islets are not due to the lack of the proposed insulin-mimetic activity of visfatin but are due to the lack of Nampt-catalyzed NMN production. This conclusion is also supported by the findings that the chemical inhibition of Nampt by FK866 also causes defects in NAD biosynthesis and glucose-stimulated insulin secretion in primary islets and that these defects are ameliorated by exogenous NMN administration. Therefore, we conclude that it is the NAD biosynthetic activity of Nampt, not the insulin-mimetic activity of visfatin, that is physiologically important for the regulation of glucose metabolism.
The cell type-dependent regulation of eNampt secretion
The fact that eNampt is positively secreted through a non-classical secretory pathway in a cell type-dependent manner indicates that the secretion of eNampt is a highly regulated process. Because Nampt-FLAG had different enzymatic activities dependent on the cell type and the compartment, it is very likely that a post-translational modification is responsible for both or either the altered enzymatic activity and/or the secretion of eNampt. The treatment of eNampt-FLAG from HIB-1B conditioned media with calf intestinal phosphatase and N-acetylglucosaminidase did not change its enzymatic activity (data not shown), and an extensive mass spectrometric search for the modification has so far been unsuccessful. Further investigation will be necessary to identify the presumed modification of eNampt.
Interestingly, adipocytes apparently acquire the ability to modify and secrete Nampt during their differentiation process. Among several cell types examined, only fully differentiated adipocytes naturally produce eNampt. Thus, it is conceivable that adipose tissue might be one of the major sites for the production of eNampt in vivo. Under normal physiological conditions, white adipose tissue does not appear to produce high levels of iNampt and eNampt. However, in certain pathophysiological conditions that cause significant changes in adipose tissue mass, structure, or function, such as obesity or type 2 diabetes, white adipose tissue might secrete more eNampt. Indeed, it has been reported that circulating levels of eNampt are increased by hyperglycemia (Haider et al., 2006b). Additionally, there are several clinical studies reporting that higher plasma eNampt levels are associated with higher BMI and percent body fat (Berndt et al., 2005), type 2 diabetes (Chen et al., 2006), and obesity in children (Haider et al., 2006a). Therefore, it will be of great importance to identify the stimuli that cause increased production of eNampt in white adipocytes in these pathophysiological conditions.
Nampt-mediated systemic NAD biosynthesis and its importance in the regulation of glucose metabolism
The findings that eNampt possesses robust NAD biosynthetic activity and that 80-90 μM of NMN circulates in mouse plasma are striking, as it has been thought that NAD biosynthesis is an entirely intracellular process (Magni et al., 1999; Rongvaux et al., 2003). Interestingly, only Nampt+/− females, but not males, have reduced levels of plasma eNampt and NMN as well as defects in glucose metabolism. Although it is currently unclear why and how Nampt+/− males maintain their plasma eNampt and NMN levels, these results suggest that the maintenance of high NMN levels by eNampt in blood circulation is critical for normal β cell function, probably because pancreatic islets have very low levels of iNampt compared to other tissues. Furthermore, NMN concentrations in wild-type mouse plasma are within the range that can enhance and restore NAD biosynthesis and glucose-stimulated insulin secretion in normal and FK866-treated primary islets in vitro, respectively. Based on these findings, we propose that NMN functions as an essential plasma metabolite that can modulate pancreatic β cell function (Figure 7). Given that fully differentiated adipocytes are a natural producer of eNampt, adipose tissue may regulate β cell function through secretion of eNampt and extracellular biosynthesis of NMN (Figure 7). Intriguingly, individuals homozygous for either of two SNP variants in the Nampt gene promoter region have lower fasting plasma insulin levels (Bailey et al., 2006). Therefore, circulating NMN levels, maintained by plasma eNampt, may play an important role in regulating β cell function in physiological and pathophysiological conditions. It will be of great interest to measure eNampt activity and NMN levels in plasma samples from patients with obesity and/or type 2 diabetes by developing high-throughput detection methods.
Figure 7.

A model for the regulation of insulin secretion by Nampt-mediated systemic NAD biosynthesis in pancreatic β cells. See texts for details. eNampt, extracellular Nampt; iNampt, intracellular Nampt; NMN, nicotinamide mononucleotide; Nmnat, NMN adenylyltransferase.
Although the molecular mechanism underlying attenuated glucose-stimulated insulin secretion in Nampt+/− islets is still unclear, alterations in NAD levels could alter activities of important enzymes in metabolic pathways such as glycolysis or fatty acid oxidation in pancreatic β cells (Figure 7). These changes in NAD would also affect other NAD-dependent enzymes, such as NAD-dependent deacetylase Sirt1 or poly-ADP-ribose polymerase PARP (Figure 7). Sirt1 is particularly interesting in this regard because we previously demonstrated that it promotes glucose-stimulated insulin secretion in β cells using pancreatic β cell-specific Sirt1-overexpressing (BESTO) mice (Moynihan et al., 2005). Interestingly, the phenotypes observed in BESTO mice are the opposite of those observed in Nampt+/− mice. Furthermore, NMN administration can also augment glucose-stimulated insulin secretion in aged BESTO mice (Mills and Imai, unpublished results), suggesting that Sirt1 is a downstream regulator in pancreatic β cells that responds to alterations in systemic NAD biosynthesis. Although further investigation will be necessary to test the model, the discovery that Nampt-mediated systemic NAD biosynthesis is a critical contributor to β cell function opens up important avenues for the development of therapeutics to treat metabolic diseases, such as type 2 diabetes.
Experimental Procedures
All experimental procedures are provided in Supplementary Materials.
Supplementary Material
Acknowledgments
We thank Perry Bickel for 3T3-L1 and HIB-1B cells, Wayne Yokoyama and Christopher Nicchitta for Dhfr and Ppl cDNAs, respectively, George Gokel for a fluorometer, Gene Wade Sherrow for insulin measurements, Marjorie Case for mass spec analysis, and Roy Tauscher and Antje Berthold for their technical assistance. We also thank Nada Abumrad, Irving Boime, Jean Shaffer, and Tom Baranski for their critical comments and suggestions. We also thank the members of the Imai and the Kiess labs for their help and encouragement. J.R.R. is a fellow supported by the Lucille P. Markey Special Emphasis Pathway in Human Pathology. A.K. and W.K. are supported by grants from and the Deutsche Forschungsgemeinschaft (DFG) KFO152: “Atherobesity”, projects BE 1264/10-1 (to W.K.) and KO 3512/1-1 (to A.K), the Translational Centre for Regenerative Medicine (project 1082MN), and the German Diabetes Association (to A.K.) J.M. is supported by grants from the Muscular Dystrophy Association and NIH (NS36358). R.R.T. is supported in part by the National Centers of Research Resources (P41RR00945) and NIDDK (P30 DK52574). S.I. is an Ellison Medical Foundation Scholar in Aging and also supported by grants from NIA (AG024150), American Diabetes Association, Juvenile Diabetes Research Foundation, and the Washington University Clinical Nutrition Research Unit (DK56341). J.M. and C.W are members of the scientific advisory board of Sirtris Pharmaceuticals, Inc. S.I. and J.R.R. are the inventor and the co-inventor, respectively, of the intellectual properties regarding the uses of Nampt and NMN, one of which is licensed to Sirtris Pharmaceuticals, Inc.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Arner P. Visfatin--a true or false trail to type 2 diabetes mellitus. J Clin Endocrinol Metab. 2006;91:28–30. doi: 10.1210/jc.2005-2391. [DOI] [PubMed] [Google Scholar]
- Bailey SD, Loredo-Osti JC, Lepage P, Faith J, Fontaine J, Desbiens KM, Hudson TJ, Bouchard C, Gaudet D, Perusse L, et al. Common Polymorphisms in the Promoter of the Visfatin Gene (PBEF1) Influence Plasma Insulin Levels in a French-Canadian Population. Diabetes. 2006;55:2896–2902. doi: 10.2337/db06-0189. [DOI] [PubMed] [Google Scholar]
- Belenky P, Bogan KL, Brenner C. NAD+ metabolism in health and disease. Trends Biochem Sci. 2007;32:12–19. doi: 10.1016/j.tibs.2006.11.006. [DOI] [PubMed] [Google Scholar]
- Berndt J, Kloting N, Kralisch S, Kovacs P, Fasshauer M, Schon MR, Stumvoll M, Bluher M. Plasma visfatin concentrations and fat depot-specific mRNA expression in humans. Diabetes. 2005;54:2911–2916. doi: 10.2337/diabetes.54.10.2911. [DOI] [PubMed] [Google Scholar]
- Chen MP, Chung FM, Chang DM, Tsai JC, Huang HF, Shin SJ, Lee YJ. Elevated plasma level of visfatin/pre-B cell colony-enhancing factor in patients with type 2 diabetes mellitus. J Clin Endocrinol Metab. 2006;91:295–299. doi: 10.1210/jc.2005-1475. [DOI] [PubMed] [Google Scholar]
- Denu JM. Linking chromatin function with metabolic networks: Sir2 family of NAD(+)-dependent deacetylases. Trends Biochem Sci. 2003;28:41–48. doi: 10.1016/s0968-0004(02)00005-1. [DOI] [PubMed] [Google Scholar]
- Fukuhara A, Matsuda M, Nishizawa M, Segawa K, Tanaka M, Kishimoto K, Matsuki Y, Murakami M, Ichisaka T, Murakami H, et al. Visfatin: a protein secreted by visceral fat that mimics the effects of insulin. Science. 2005;307:426–430. doi: 10.1126/science.1097243. [DOI] [PubMed] [Google Scholar]
- Haider DG, Holzer G, Schaller G, Weghuber D, Widhalm K, Wagner O, Kapiotis S, Wolzt M. The adipokine visfatin is markedly elevated in obese children. J Pediatr Gastroenterol Nutr. 2006a;43:548–549. doi: 10.1097/01.mpg.0000235749.50820.b3. [DOI] [PubMed] [Google Scholar]
- Haider DG, Schaller G, Kapiotis S, Maier C, Luger A, Wolzt M. The release of the adipocytokine visfatin is regulated by glucose and insulin. Diabetologia. 2006b;49:1909–1914. doi: 10.1007/s00125-006-0303-7. [DOI] [PubMed] [Google Scholar]
- Hasmann M, Schemainda I. FK866, a highly specific noncompetitive inhibitor of nicotinamide phosphoribosyltransferase, represents a novel mechanism for induction of tumor cell apoptosis. Cancer Res. 2003;63:7436–7442. [PubMed] [Google Scholar]
- Hug C, Lodish HF. Medicine. Visfatin: a new adipokine. Science. 2005;307:366–367. doi: 10.1126/science.1106933. [DOI] [PubMed] [Google Scholar]
- Imai S, Armstrong CM, Kaeberlein M, Guarente L. Transcriptional silencing and longevity protein Sir2 is an NAD-dependent histone deacetylase. Nature. 2000;403:795–800. doi: 10.1038/35001622. [DOI] [PubMed] [Google Scholar]
- Jia SH, Li Y, Parodo J, Kapus A, Fan L, Rotstein OD, Marshall JC. Pre-B cell colony-enhancing factor inhibits neutrophil apoptosis in experimental inflammation and clinical sepsis. J Clin Invest. 2004;113:1318–1327. doi: 10.1172/JCI19930. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Khan JA, Tao X, Tong L. Molecular basis for the inhibition of human NMPRTase, a novel target for anticancer agents. Nat Struct Mol Biol. 2006;13:582–588. doi: 10.1038/nsmb1105. [DOI] [PubMed] [Google Scholar]
- Kim MK, Lee JH, Kim H, Park SJ, Kim SH, Kang GB, Lee YS, Kim JB, Kim KK, Suh SW, Eom SH. Crystal Structure of Visfatin/Pre-B Cell Colony-enhancing Factor 1/Nicotinamide Phosphoribosyltransferase, Free and in Complex with the Anti-cancer Agent FK-866. J Mol Biol. 2006;362:66–77. doi: 10.1016/j.jmb.2006.06.082. [DOI] [PubMed] [Google Scholar]
- Landry J, Slama JT, Sternglanz R. Role of NAD+ in the deacetylase activity of the SIR2-like proteins. Biochem Biophys Res Commun. 2000;278:685–690. doi: 10.1006/bbrc.2000.3854. [DOI] [PubMed] [Google Scholar]
- Lin S-J, Guarente L. Nicotinamide adenine dinucleotide, a metabolic regulator of transcription, longevity and disease. Curr Opin Cell Biol. 2003;15:241–246. doi: 10.1016/s0955-0674(03)00006-1. [DOI] [PubMed] [Google Scholar]
- Magni G, Amici A, Emanuelli M, Orsomando G, Raffaelli N, Ruggieri S. Enzymology of NAD+ homeostasis in man. Cell Mol Life Sci. 2004;61:19–34. doi: 10.1007/s00018-003-3161-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Magni G, Amici A, Emanuelli M, Raffaelli N, Ruggieri S. Enzymology of NAD+ synthesis. Adv Enzymol Relat Areas Mol Biol. 1999;73:135–182. doi: 10.1002/9780470123195.ch5. [DOI] [PubMed] [Google Scholar]
- Martin P, Shea R, Mulks M. Identification of a plasmid-encoded gene from Haemophilus ducreyi which confers NAD independence. J Bacteriol. 2001;183:1168–1174. doi: 10.1128/JB.183.4.1168-1174.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miller ES, Heidelberg JF, Eisen JA, Nelson WC, Durkin AS, Ciecko A, Feldblyum TV, White O, Paulsen IT, Nierman WC, et al. Complete genome sequence of the broad-host-range vibriophage KVP40: comparative genomics of a T4-related bacteriophage. J Bacteriol. 2003;185:5220–5233. doi: 10.1128/JB.185.17.5220-5233.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miura M, Surmacz E, Burgaud JL, Baserga R. Different effects on mitogenesis and transformation of a mutation at tyrosine 1251 of the insulin-like growth factor I receptor. J Biol Chem. 1995;270:22639–22644. doi: 10.1074/jbc.270.38.22639. [DOI] [PubMed] [Google Scholar]
- Moschen AR, Kaser A, Enrich B, Mosheimer B, Theurl M, Niederegger H, Tilg H. Visfatin, an adipocytokine with proinflammatory and immunomodulating properties. J Immunol. 2007;178:1748–1758. doi: 10.4049/jimmunol.178.3.1748. [DOI] [PubMed] [Google Scholar]
- Moynihan KA, Grimm AA, Plueger MM, Bernal-Mizrachi E, Ford E, Cras-Meneur C, Permutt MA, Imai S. Increased dosage of mammalian Sir2 in pancreatic β cells enhances glucose-stimulated insulin secretion in mice. Cell Metab. 2005;2:105–117. doi: 10.1016/j.cmet.2005.07.001. [DOI] [PubMed] [Google Scholar]
- Ognjanovic S, Bryant-Greenwood GD. Pre-B-cell colony-enhancing factor, a novel cytokine of human fetal membranes. Am J Obstet Gynecol. 2002;187:1051–1058. doi: 10.1067/mob.2002.126295. [DOI] [PubMed] [Google Scholar]
- Revollo JR, Grimm AA, Imai S. The NAD biosynthesis pathway mediated by nicotinamide phosphoribosyltransferase regulates Sir2 activity in mammalian cells. J Biol Chem. 2004;279:50754–50763. doi: 10.1074/jbc.M408388200. [DOI] [PubMed] [Google Scholar]
- Revollo JR, Grimm AA, Imai S. The regulation of nicotinamide adenine dinucleotide biosynthesis by Nampt/PBEF/visfatin in mammals. Curr Opin Gastroenterol. 2007;23:164–170. doi: 10.1097/MOG.0b013e32801b3c8f. [DOI] [PubMed] [Google Scholar]
- Rongvaux A, Andris F, Van Gool F, Leo O. Reconstructing eukaryotic NAD metabolism. Bioessays. 2003;25:683–690. doi: 10.1002/bies.10297. [DOI] [PubMed] [Google Scholar]
- Rongvaux A, Shea RJ, Mulks MH, Gigot D, Urbain J, Leo O, Andris F. Pre-B-cell colony-enhancing factor, whose expression is up-regulated in activated lymphocytes, is a nicotinamide phosphoribosyltransferase, a cytosolic enzyme involved in NAD biosynthesis. Eur J Immunol. 2002;32:3225–3234. doi: 10.1002/1521-4141(200211)32:11<3225::AID-IMMU3225>3.0.CO;2-L. [DOI] [PubMed] [Google Scholar]
- Samal B, Sun Y, Stearns G, Xie C, Suggs S, McNiece I. Cloning and characterization of the cDNA encoding a novel human pre-B-cell colony-enhancing factor. Mol Cell Biol. 1994;14:1431–1437. doi: 10.1128/mcb.14.2.1431. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sethi JK. Is PBEF/visfatin/Nampt an authentic adipokine relevant to the metabolic syndrome? Curr Hypertens Rep. 2007;9:33–38. doi: 10.1007/s11906-007-0007-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smith JS, Brachmann CB, Celic I, Kenna MA, Muhammad S, Starai VJ, Avalos JL, Escalante-Semerena JC, Grubmeyer C, Wolberger C, Boeke JD. A phylogenetically conserved NAD+-dependent protein deacetylase activity in the Sir2 protein family. Proc Natl Acad Sci USA. 2000;97:6658–6663. doi: 10.1073/pnas.97.12.6658. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stephens JM, Vidal-Puig AJ. An update on visfatin/pre-B cell colony-enhancing factor, an ubiquitously expressed, illusive cytokine that is regulated in obesity. Curr Opin Lipidol. 2006;17:128–131. doi: 10.1097/01.mol.0000217893.77746.4b. [DOI] [PubMed] [Google Scholar]
- van der Veer E, Nong Z, O'Neil C, Urquhart B, Freeman D, Pickering JG. Pre-B-cell colony-enhancing factor regulates NAD+-dependent protein deacetylase activity and promotes vascular smooth muscle cell maturation. Circ Res. 2005;97:25–34. doi: 10.1161/01.RES.0000173298.38808.27. [DOI] [PubMed] [Google Scholar]
- Wang B, Jenkins JR, Trayhurn P. Expression and secretion of inflammation-related adipokines by human adipocytes differentiated in culture: integrated response to TNF-alpha. Am J Physiol Endocrinol Metab. 2005;288:E731–740. doi: 10.1152/ajpendo.00475.2004. [DOI] [PubMed] [Google Scholar]
- Wang T, Zhang X, Bheda P, Revollo JR, Imai S, Wolberger C. Structure of Nampt/PBEF/visfatin, a mammalian NAD(+) biosynthetic enzyme. Nat Struct Mol Biol. 2006;13:661–662. doi: 10.1038/nsmb1114. [DOI] [PubMed] [Google Scholar]
- Ye SQ, Simon BA, Maloney JP, Zambelli-Weiner A, Gao L, Grant A, Easley RB, McVerry BJ, Tuder RM, Standiford T, et al. Pre-B-cell colony-enhancing factor as a potential novel biomarker in acute lung injury. Am J Respir Crit Care Med. 2005;171:361–370. doi: 10.1164/rccm.200404-563OC. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.