

Abstract
The structural organization in a peptide/membrane supramolecular complex is best described by knowledge of the peptide orientation plus its time-dependent and spatial fluctuations. The static orientation, defined by the peptide tilt and a rotation about its molecular axis, is accessible through a number of spectroscopic methods. However, peptide dynamics, although relevant to understand the functionality of these systems, remains largely unexplored. Here, we describe the orientation and dynamics of Trp-flanked and Lys-flanked hydrophobic peptides in a lipid bilayer from molecular dynamics simulations. A novel view is revealed, where collective nontrivial distributions of time-evolving and ensemble peptide orientations closely represent the systems as studied experimentally. Such global distributions are broad and unveil the existence of orientational states, which depend on the anchoring mode of interfacial residues. We show that this dynamics modulates 2H quadrupolar splittings and introduces ambiguity in the analysis of NMR data. These findings demonstrate that structural descriptions of peptide/membrane complexes are incomplete, and in cases even imprecise, without knowledge of dynamics.
INTRODUCTION
Lipid membranes, in their physiologically relevant liquid-crystal phase, are characterized by the partial order and high fluidity of constituent amphiphilic rodlike molecules, defining a lamellar arrangement with a centered hydrophobic layer, flanked by hydrated polar regions (1). This complex organization conditions the structure, orientation, and dynamics of polypeptides there embedded, which are restrained with respect to the membrane plane while still being free to diffuse within the lipid bilayer (2).
From a practical point of view, a restricted environment reduces the number of degrees of freedom and simplifies the structure problem in protein-membrane complexes. Thus, despite the difficulties to obtain high resolution information from these systems with classical techniques (x-ray crystallography and high resolution NMR), alternative spectroscopic methods have yielded new insights into their molecular organization in the form of orientation parameters (3–8). These methods are applied to complexes of α-helical peptides with membranes, and give mainly the tilt of the peptide molecule and the rotation about its principal axis. The peptide tilt is profusely measured, as it relates directly to the mechanisms of membrane-active peptides, like those forming pores or promoting membrane fusion. Additionally, the tilt is also important to define the response of the peptide to interactions with the membrane, according to ideas illustrated by the mattress model and explained under the concept of hydrophobic mismatch (9,10). On the other hand, the peptide rotation, despite being more scarcely studied (8,11–14), is relevant to fix the position of anchoring residues with respect to the membrane interface and to display the helix face that can be used for possible intermolecular interactions.
Notwithstanding the importance of the above static structural descriptions, a complete understanding of the molecular organization of peptide membrane complexes necessitates knowledge about their dynamics. Recent relaxation studies by solid-state NMR experiments show transmembrane polypeptides undergoing both axial diffusion and small amplitude off-axis reorientation, in the nanosecond and microsecond, respectively, time regimes (15,16). Including the later movements in the models used to interpret raw spectroscopic data can be expected to improve the end orientation results. However, this would require a detailed understanding of the mechanisms of subjacent dynamic processes. Molecular dynamics (MD) simulations have the potential to provide such a description (17). While still limited by the quality of the underlying models and accessible time- and length-scales, MD methods are increasingly applied to biomembrane systems (18–29). Thus, pure lipid bilayers and protein-membrane complexes can now be studied at atomic detail for up to hundreds of nanoseconds, reproducing critical physicochemical properties when compared with experiments. This allows the investigation of relevant phenomena, like self-assembly of lipid aggregates (19–21), phase transitions (22,23), formation and closure of pores (24), insertion and folding of membrane peptides (25,26,29), or hydrophobic-mismatch adaptations (27,28).
In this work, we study the orientation and dynamics of hydrophobic peptides in a dimyristoylphosphatidylcholine (DMPC) bilayer by means of MD simulations. The peptides belong to series of well-characterized transmembrane systems, introduced by Davis and co-workers (30), and profusely studied by the group of Killian (4,7,11,31–35); namely, we use acetyl-GW2L17W2A-ethanolamine (WLP23) and acetyl-GK2L17K2A-ethanolamine (KLP23) (33). The investigation of multiple replica simulations allows accounting explicitly for time-evolving and ensemble orientation distributions of the peptides. This provides a comprehensive description of dynamic peptide-membrane complexes, consisting of fluctuating orientational states. Finally, we show that the use of these explicit orientation distributions yields a coherent interpretation of the 2H-NMR data available for the same systems.
METHODS
Simulations
Software and general simulation conditions
We use the GROMACS package for all our simulations (36). The united-atom lipid parameters were adapted from the work of Berger and co-workers (37) and the peptides used the GROMOS force field. A time step of 4 fs was used (38). To ensure that the lipids were in the fluid phase, the temperature was set to 308 K, coupled to a Berendsen thermostat (39) with a coupling constant of 0.1 ps. The pressure was coupled semiisotropically to a Berendsen barostat (39), with a coupling constant of 1.0 ps. Both, the short-range electrostatic and van der Waals interactions were calculated using a cutoff of 1.0 nm, while for long-range electrostatics we used a PME algorithm (40).
Setup protocol
A total of 10 simulations were performed: one for a purely lipid system, conducted up to 50 ns; five for a WLP23-membrane complex, run for 200 ns each; and four for KLP23-membrane complex, up to 300-ns each. In all cases the lipid bilayer consisted of 128 DMPC lipids (64 per leaflet), in presence of 3655 water molecules. Chloride ions were included in simulations with the KLP23 peptide to ensure a electrically neutral simulation cell. The starting DMPC bilayer was downloaded from http://moose.bio.ucalgary.ca/downloads.
Peptides were generated as ideal α-helices using the software Swiss PDB viewer (41) (http://www.expasy.org/spdbv), with the N-termini acetylated and the C-termini amidated. They were solvated with SPC water molecules in a cubic box of 5.0 nm, energy minimized and simulated during 5 ns with position restraints on the peptide backbone. A single relaxed peptide was inserted symmetrically across the bilayer, with its long axis aligned with the membrane normal and the system was solvated in a bath of water. After energy minimization, preequilibrium simulations were performed for 2 ns, with position restraints on the peptide backbone atoms, before the production runs.
Data analysis
Definition and calculation of membrane-peptide orientation
The static orientation of a rodlike α-helical peptide in a lipid membrane is defined by the tilt (τ) of the helix long-axis (H) with respect to the membrane normal (N, here corresponding to the z axis), and the polarity or azimuthal rotation (ρ) of the helix about its molecular long-axis. The azimuthal rotation ρ is found in the plane perpendicular to the helix axis, as the angle between a vector t pointing in the direction of the tilt and a reference vector r pointing into the Cα of an arbitrary residue, here Gly1 (Fig. 1).
FIGURE 1.

Static orientation of a helical peptide, bound to a lipid bilayer. A pair of angles, tilt (τ) and rotation (ρ), is sufficient to define the peptide orientation. The value τ is the angle formed between the molecular long axis (H) of the helix and the membrane normal (N). The value ρ is the angle between the direction of the peptide tilt (t) and a vector r perpendicular to H, pointing to the Cα carbon of a reference residue, here Gly1.
The helix axis was calculated for each frame of the simulations by using the backbone atoms of the 17 central Leu residues. The tilt vector is calculated as t = h × (h × n), with h and n being unit vectors parallel to the helix axis (H) and the membrane normal (N), respectively (Fig. 1). In practice, throughout the simulations the peptide structure diverged slightly from ideal. This was found not to affect significantly the calculated helix axis and the values of τ, but it can affect ρ, as it may change the relative angular position of the reference Gly1. Thus, representative values of ρ were calculated as averages of rotations referred to residues in the center of the peptide (Leu11-Leu14) and translated into the virtual ideal position of Gly1, using a pitch of 100° between contiguous residues.
Experiment-like orientations
Reported experimental τ- and ρ-values for WLP23 and KLP23 (33) have been determined from 2H-NMR quadrupole splittings (Δν) of the −CβD3 group in deuterated Ala, substituting, one by one, four central Leu residues (11–14) of the helix. To make a fair comparison of these experimental values with our simulations, we calculated virtual (experiment-like) 2H splittings that would correspond to the orientation of Cα-Cβ bonds in residues 11–14, from which we obtained experiment-like τ- and ρ-angles by using a fitting procedure similar to the GALA (geometric analysis of labeled alanines) (11). Briefly, assuming that the membrane normal is aligned with the magnetic field, instantaneous (Δνins) static splittings for each residue in a particular frame of the trajectory are first calculated as
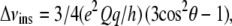 |
(1) |
where (e2Qq/h) is the nuclear quadrupole coupling constant, taken as 168.0 kHz for a C-D group (7,11), θ is the orientation of the Cα–Cβ bond (virtually the Cα–CβD3 vector) with respect to the membrane normal. Δνins values are multiplied by one-third to account for fast motional averaging about the Cα–CβD3 bond in virtual methylene deuteron splittings. These are then averaged over the full trajectory, for each individual simulation, or over a complete set of trajectories, for a global ensemble corresponding to each peptide, and the absolute value of the average (Δνaver) is considered for further treatments. Finally, experiment-like orientations {τ, ρ} are obtained for each set of four experiment-like splittings from a fit of the theoretical equation (11,33),
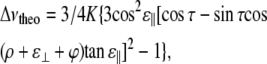 |
(2) |
where 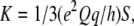 with the factor 1/3 associated with fast rotation about the Cα–CβD3 bond and S being an order parameter accounting for peptide dynamics. The angle ɛ‖ is that defined between the Cα–Cβ bond vector of the residue under consideration and the helix axis, ɛ⊥ is an angle between the Cα–Cβ bond and a vector from the helix axis to the Cα, both projected onto a plane perpendicular to the helix axis, and ϕ is the rotation pitch angle between the Cα of the reference Gly1 and the Cα of the residue considered for each particular Δνaver splitting. As in the experimental studies we want to compare with Özdirekcan et al. (33), we use S = 0.875, giving K = 49 kHz, a pitch of 100° between contiguous residues, corresponding to an ideal α-helix, and a constant estimated value of −43.3° for the angle ɛ⊥. Then, values of τ, ρ, and ɛ‖ are optimized during the fitting procedure, while minimizing root mean-squared deviations between Δνaver and Δνtheo.
with the factor 1/3 associated with fast rotation about the Cα–CβD3 bond and S being an order parameter accounting for peptide dynamics. The angle ɛ‖ is that defined between the Cα–Cβ bond vector of the residue under consideration and the helix axis, ɛ⊥ is an angle between the Cα–Cβ bond and a vector from the helix axis to the Cα, both projected onto a plane perpendicular to the helix axis, and ϕ is the rotation pitch angle between the Cα of the reference Gly1 and the Cα of the residue considered for each particular Δνaver splitting. As in the experimental studies we want to compare with Özdirekcan et al. (33), we use S = 0.875, giving K = 49 kHz, a pitch of 100° between contiguous residues, corresponding to an ideal α-helix, and a constant estimated value of −43.3° for the angle ɛ⊥. Then, values of τ, ρ, and ɛ‖ are optimized during the fitting procedure, while minimizing root mean-squared deviations between Δνaver and Δνtheo.
Lipid order parameters
The order of hydrocarbon chains in lipid bilayers is characterized by the deuterium order parameter SCD measured through 2H-NMR experiments. If θi is the angle between a C-D bond of a methylene group, i, and the bilayer normal, aligned with the direction of the applied magnetic field, the order parameter of that particular group is defined as
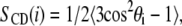 |
(3) |
where the brackets denote time and ensemble average. The absolute value of the order parameters of the methylene segments is reported. Since we employed a united-atom force field, the order parameters were calculated from the positions of the carbon atoms along the chain (42).
Peptide structure analysis
Profiles of secondary structure of peptides along the simulation trajectories were obtained with the help of standard GROMACS analysis tools, which use the DSSP algorithm (43).
RESULTS AND DISCUSSION
Orientation and dynamics of the membrane peptides
In the axially ordered lamellar phase of a lipid membrane system, the orientation of an embedded helical-peptide is well described through the pair of angles {τ, ρ}, giving, respectively, the helix tilt with respect to the membrane normal and the helix rotation about its molecular axis (see Methods and Fig. 1). However, membrane peptides are far from being rigid rodlike molecules at a unique and fixed orientation. In the fluidlike membrane, peptides experience complex whole-body movements, varying their tilt or their rotation and introducing time- and ensemble-dependent orientational diversity. Additionally, internal peptide dynamics and distortions from often assumed ideal α-helices, may affect the direction of the molecular axis and the reference for determining the ρ-angle.
In all our simulations (see a list in Table 1), the peptides were found to maintain an α-helical structure along the full trajectories (shown in Fig. 1 of the Supplementary Material). Thus, for the analysis and discussions below we will focus mainly on whole-body time-dependent fluctuations and variability of orientation states within a molecular ensemble.
TABLE 1.
Summary of simulations of peptide/membrane complexes with parameters defining peptide orientation
| Orientational parameters*
|
||||||||
|---|---|---|---|---|---|---|---|---|
| Simulation number | Total simulation time (ns) | Direct (averaged angles)
|
Indirect (from calculated 2H splittings)
|
|||||
| Peptide | τ (°)† | ρ (°)† | τ (°) | ρ (°) | ɛ‖ (°) | RMSD‡ (KHz) | ||
| WLP23 | 1 | 200 | 40 ± 5 | 171 ± 18 | 37 | 169 | 55.3 | 1.10 |
| 2 | 200 | 16 ± 10 | 115 ± 86 | 10 | 108 | 56.0 | 0.35 | |
| 3 | 200 | 39 ± 5 | 177 ± 14 | 40 | 181 | 55.9 | 1.70 | |
| 4 | 200 | 36 ± 5 | 122 ± 17 | 38 | 124 | 57.7 | 0.76 | |
| 5 | 200 | 24 ± 6 | 235 ± 29 | 25 | 245 | 56.6 | 0.20 | |
| Global | 1000 | 31 ± 12 | 173 ± 58 | 19 | 167 | 56.1 | 0.50 | |
| Experiment¶ | — | — | — | 8 | 176 | 58.7 | 0.40 | |
| KLP23 | 1 | 300 | 35 ± 7 | 347 ± 22 | 36 | 347 | 56.4 | 1.11 |
| 2 | 300 | 12 ± 7 | 204 ± 107 | 4 | 281 | 58.2 | 0.48 | |
| 3 | 300 | 15 ± 6 | 200 ± 63 | 11 | 221 | 57.6 | 1.48 | |
| 4 | 300 | 20 ± 7 | 188 ± 48 | 16 | 199 | 57.6 | 0.22 | |
| Global | 1200 | 20 ± 11 | 206 ± 104 | 6 | 260 | 58.7 | 1.52 | |
| Experiment¶ | — | — | — | 8 | 265 | 59.3 | 0.70 | |
Peptide tilt
After a quick displacement away from the starting orientation, where the helix axis was aligned with the membrane normal, τ oscillates with variable amplitude and frequency throughout the complete simulation. Some representative cases are shown in Fig. 2, graphs A and B. Due to the slow relaxation of peptide reorientation (15,16) we do not expect reaching equilibrium within the simulated times. Thus, we will use here the term “stabilization time” as the approximate time needed for the system to evolve outside the starting state, into a relatively stable characteristic orientation, for which we will perform our analysis. This process and the characteristic tilts are rather heterogeneous among the two peptides and corresponding replicas. For WLP23 (Fig. 2 A), simulations 1, 3, and 4 appear to stabilize slowly (within ∼50 ns) and passing through successive stages of small to intermediate tilts maintained for a few nanoseconds, before reaching relatively large (∼40°) and stable tilts (Fig. 2 A, black line). Simulations 2 and 5, however, evolve more rapidly outside the initial state (within ∼10 ns), remaining stable at smaller tilt angles (∼10° and ∼20°, respectively). After 150 ns, and for the rest of the explored time, the peptide tilt of simulation 2 increases up to ∼30° (Fig. 2 A, red line). For KLP23 (Fig. 2 B), stabilization occurs within the first 10 ns with characteristic tilts generally smaller than in the case of WLP23, except for simulation 1, which oscillates within the first ∼50 ns and stabilizes at values close to 35° (Fig. 2 B, black line). The mean tilt angles for each simulation were calculated after discarding the first 50 ns (longest stabilization time), and they are given in Table 1.
FIGURE 2.

Time evolution of peptide orientation. The graphs represent instantaneous values of the angles τ (A and B) and ρ (C and D) along the time coordinate for representative case examples of WLP23 (A and C) and KLP23 (B and D). In panels A and C, data from simulations 1, 2, and 5 are represented with colors black, red, and orange, respectively. In panel B, data from simulations 1, 2, 3, and 4 are drawn black, red, green, and blue, and in panel D data from simulations 2, 3, and 4 are red, green, and blue, respectively.
Peptide rotation
The time evolution of the instantaneous rotation angle is shown in Fig. 2, C and D, for representative simulations. Because, by definition (Fig. 1), ρ depends on the direction of the tilt, and the peptide can be assumed to incline initially at random, the azimuthal rotation may start at any value. For the same reason, a small and fluctuating τ, as occurring often for short simulation times, may easily originate abrupt fluctuations of ρ. After the consensus 50-ns stabilization time (defined above from the evolution of τ), the helix rotation tends to equilibrate, although with persisting fluctuations (Fig. 2, C and D). These are of small amplitude (±20°) for τ > 20°, or large jumplike transitions, for τ < 20°, the latter case being most typical for KLP23. As for the peptide tilt, the mean azimuthal angle for each replica is given in Table 1.
As observed before in other MD studies (25,28), the average tilts of the simulations are systematically larger than values derived experimentally from 2H-NMR (33). With respect to the azimuthal rotations, although they tend to approach the experimental angles, there are also some notable discrepancies (see Table 1). However, as we shall see, the explicit dynamics of the system, in the form of a large spread of angular values and distinguishable orientational states, has an impact on the interpretation of experimental data.
Distributions of orientations
Time-dependent distributions
The diversity and relative weight of peptide orientations is better represented by frequency distributions of tilt and rotation angles, as depicted in Fig. 3. In general, the distributions of τ for individual simulations approximate to unimodal, Gaussian-like probability density functions, with characteristic standard deviations of ∼10°, similar to other reported distributions from MD simulations (25). Exceptions are simulation 2 of WLP23 (Fig. 3 A), which shows a principal τ-mode at ∼12°, and a second less populated mode at ∼32°, and simulation 2 of KLP23 (Fig. 3 B), with an asymmetric peak of maximum frequency at ∼7° and a tail toward long τ-values.
FIGURE 3.

Frequency distributions of orientational parameters. Shown are probability densities of τ (A and B) and ρ (C and D) from peptides WLP23 (A and C) and KLP23 (B and D). The graphs show data from simulations 1 (black, continuous line), 2 (black, dashed line), 3 (black, dashed-dotted line), 4 (shaded, dashed line), and 5 (shaded, dashed-dotted line). Global distributions of orientations found in the complete set of simulations, for each peptide, are drawn as a shaded continuous line.
The individual distributions of ρ are more broad, with standard deviations larger than 20°. Simulations of WLP23 give a set of partially overlapping and fairly symmetric, single bell-shaped peaks between ∼100° and 260°, although runs 2 and 5 show significant intensity outside this interval (Fig. 3 C). In contrast, for KLP23 (Fig. 3 D), only simulation 1 is unimodal and centered around ∼350°, while ρ distributions from runs 3 and 4 are overlaps of partially resolved peaks around ∼195° and simulation 2 shows a very large spread of values, with resolved broad peaks in both the 350° and 195° regions.
Although all simulations display considerable orientational diversity, a comparison among them suggests that each one represents only part of the total dynamics of the system. One may think of increasing the available configurational space by increasing the simulation time, although this does not appear feasible within a reasonable limit (note that our simulations are 200–300-ns long). We may also enlarge this space through a global treatment of a set of replicas, as if they represented a small molecular ensemble. As we show below, such a global analysis suggests a complex orientational landscape, with a broad structural diversity and the emergence of different states.
Global distributions
For each of the two peptides the collective distribution of τ gives an asymmetric profile with a large spread of angles. In the case of WLP23, this shows a preference for large tilts, with a most prominent peak at ∼38° and a mean at 31° ± 12° (Fig. 3 A). However, for KLP23, the principal peak seats at relatively small tilts (around 15°), although coexisting with a resolved peak at larger tilts (at ∼37°), all together averaging at 20° ± 11° (Fig. 3 B).
The two peptides show also distinguishable global ρ distributions (Fig. 3, C and D; violet lines). In this case, due to their complexity, the global analysis is more clearly made from representations of tilt vectors (see Fig. 1), which can be plotted directly over an Edmundson's helical-wheel of the peptides (Fig. 4, A and B). Making the modulus of each tilt vector proportional to the occurrence of its corresponding ρ-angle, the average ρ is determined from the direction of the resultant vector. For WLP23, the tilt vectors group densely in a sector between 100° and 260°, with their sum forming an angle ρ = 173° with respect to the reference vector (Fig. 4 A). In contrast, the complex global distribution for KLP23 (Fig. 3 D, shaded continuous) corresponds to tilt vectors populating two sectors (Fig. 4 D), with a global average defining a ρ = 206°. These global ensemble averages of ρ approximate to values determined from experiments (33) with differences within the standard deviation. However, the global tilt angles are considerably larger in the simulations (Table 1). The origin of these discrepancies is discussed later with more detail.
FIGURE 4.

Vectorial representation of peptide rotations. Tips of tilt vectors, depicted as open circles, are drawn over an Edmundson helical wheel of the peptides WLP23 (A) and KLP23 (B). Only most important residues are represented: reference Gly1 and anchoring Trp (A) or Lys (B), with solid representation for residues at the N-terminus and shaded representation for residues at the C-terminus. The ρ-angles are defined with respect to the reference vector pointing into Gly1, fixed at the horizontal axis (see Fig. 1 for detailed definitions). Being the modulus of each tilt vector proportional to the occurrence of the corresponding rotation, the resultant tilt vector (arrow) marks the average ρ.
Orientational states
The global distributions appear generally asymmetric and/or multimodal (Fig. 3). Although this could be due to incomplete sampling of possible orientations, it suggests the existence of characteristic populations or states within the ensemble. A plot of {τ, ρ} pairs against their frequency (Fig. 5) permits defining these possible states. In the case of WLP23 there is a most probable state with τ around 38° and well-defined values of ρ close to 180°. Additionally, there are much less probable orientations for smaller tilts, which for τ < 20° correspond to a very wide distribution of ρ-values (Fig. 5 A). Thus, a representative structure of this peptide in DMPC is that defined by the pair of angles {τ = 38°, ρ = 173°}, depicted in Fig. 6 A. On the other hand, KLP23 is clearly more dynamic (Fig. 5 B). For this peptide the small tilts again correspond to a broad peak in the ρ-dimension, which is most intense at ∼195°, while the large tilts correspond to a well-resolved ρ-peak around 350°. We can then define two representative states for KLP23, with pairs of most probable angles {τ = 18°, ρ = 195°}, depicted in Fig. 6 B, and {τ = 37°, ρ = 350°}, depicted in Fig. 6 C.
FIGURE 5.

Global distributions of pairs of peptide orientational angles. (A) WLP23. (B) KLP23.
FIGURE 6.

Characteristic structures of peptide-membrane complexes. The models were chosen out of the complete set of trajectories to match the most populated {τ, ρ} pairs (Fig. 5). (A) WLP23 at τ = 38°, ρ = 173°. (B) KLP23 at τ = 18°, ρ = 195°. (C) KLP23 at τ = 37°, ρ = 350°. The lipid acyl tails are depicted in light gray and headgroup atoms are shown in red. The peptides are drawn as light blue ribbons, showing only side chains of anchoring residues, Trp (A) and Lys (B, C), in yellow and dark blue, respectively. The Cα of Gly1, marking the reference for the rotation angle, is shown as a green sphere.
The just-described large orientational variability, including the existence of alternative states, stable within at least a few hundreds of nanoseconds, contrasts with views of a static and well-defined membrane peptide orientation. Membrane peptides require relatively long simulation times to equilibrate (28) which, for whole-body orientational properties, due to their long correlation times (15,16), might be up to a few microseconds. Here, simulations needed up to several tens of nanoseconds before acquiring characteristic orientations, which, despite still outside equilibrium, may be close to possible stable states. Note that, although each simulation samples principally one state (Fig. 3), the large overlap among the individual distributions shows some level of transition between states within the 200–300-ns time range. Moreover, for small tilt values, transitions between ρ-states are clearly facilitated (as in simulations 2 of both peptides).
Implications for the analysis of experimental data
The systems under study are among the best-characterized peptide-lipid complexes. In particular, it has been reported from solid-state 2H-NMR that peptides of the WLP/WALP and KLP/KALP families possess well-defined pairs of τ and ρ in DMPC lipid bilayers (11,33). Special attention has been paid to the tilt angles, since the values determined through 2H-NMR methods are in general surprisingly small, even under positive hydrophobic mismatch, and compared with other experimental tilts from transmembrane peptides (31,44–46). Although dimerization has been claimed among the possible causes for the small tilts, no proof for oligomers at relevant conditions has been documented (34). With respect to the rotational angles, the data are more scarce and in general show that ρ adopts characteristic values that depend on the type of interfacial residues (33).
Influence of rotational dynamics on 2H NMR observables
We have seen above that simulated KLP23 and WLP23 peptides in membranes mainly present tilt angles larger than derived from 2H-NMR experiments, while the helix azimuthal rotations are closer to the reported experimental data, specially for the global ensemble averages (Table 1). We should note, however, that experimentally determined orientations are, as well, calculated values, obtained on the basis of an assumed model where the unknown peptide dynamics is largely simplified. For instance, internal motions, wobbling and fluctuations of the helix rotation are, at most, collectively imputed as an order parameter factor. To test the impact of explicit peptide dynamics in the structural interpretation of 2H-NMR data, we calculate orientation angles from the simulations by applying the same model and mathematical framework as it was used for the experiments (7,11,33). Thus, static 2H quadrupolar splittings corresponding to the four residues labeled for the NMR experiments (residues 11–14) were first back-calculated from each frame of the simulations, using Eq. 1, and subsequently averaged over a full trajectory. The tilt and rotation angles were then obtained by fitting a theoretical curve given by Eq. 2 (called quadrupolar wave (11)) to a set of experiment-like splittings. For most simulations of WLP23 and for simulation 1 of KLP23, the τ- and ρ-angles so obtained are very similar to the values determined directly from the corresponding time averages. However, for run 2 of WLP23 and runs 2–4 of KLP23, the experiment-like tilts are smaller than the values calculated directly (Table 1). Simulations of this second group are characterized by broad distributions of ρ (Fig. 4), which effectively reduce the calculated quadrupolar splittings (see Table 1 of Supplementary Material). Interestingly, this affects mainly the fitted tilts, which get close to the experimental values (Table 1). Thus, it appears that a large spreading of the ρ-angle leads to a reduction of the tilt angles obtained by this method.
As we would expect from a representative ensemble distribution of orientation states, the collective treatment of inferred quadrupolar splittings for each peptide yields experiment-like splittings which generally compare closely with values determined from solid-state 2H-NMR (Table 1 of the Supplementary Material). This shows that our limited sets of simulations are sufficient to reproduce an important part of the peptide orientational landscape. Consequently, the experiment-like {τ, ρ} pairs are in good agreement with the corresponding solid-state 2H-NMR values (Table 1). It is important to notice that although for some of the individual simulations the calculated splittings (and corresponding fitted angles) approach the experimental ones, the best results for each peptide were obtained when considering globally all the corresponding simulations. Thus, the dynamics exhibited by a limited number of replicas, within 200–300 ns time windows, appears a fair representation of the dynamics of the peptide/membrane complex. Moreover, the latter results stress that the rotational diversity defining these systems is contributed by the different orientational states, each of them exhibiting time-dependent fluctuations.
The findings described above solve a recurrent paradox about the orientation of WLP and KLP peptides studied by 2H-NMR, and offer an explanation (most likely extensible to WALP and KALP peptides) for the small tilts, barely reacting to mismatch, generally obtained in these cases (7,11,13,32,33). While a uniform rotation about the peptide long axis is to be discarded from the inequality of the splittings for different residues around the helix (7,11,33), such inequality is compatible with nonuniform broad fluctuations of ρ, as demonstrated here. This rotational dynamics, in a nanosecond timescale, can correspond to the axial diffusion with a 10−8–10−7 s correlation time reported from solid-state NMR relaxation experiments for peptides very similar to KLP23 (15,16). The same relaxation studies report small amplitude off-axis reorientations in the 10−6–10−5 s time regime, which may correspond to the dynamics of the tilt, including slow exchange between the tilt states inferred from the simulations.
Reinterpretation of experiments including dynamics
For real case experimental studies, in the absence of an explicit description of the underlying rotation dynamics, ideal distributions of ρ could be evaluated. Using a Gaussian distribution reduces the calculated quadrupolar splittings with only small variations of the phase of theoretical quadrupolar waves. Thus, the extracted value of τ will be smaller, with a very similar ρ-angle (the mean of the distribution). However, this allows multiple fittings of similar quality for a broad range of tilts, depending on an arbitrary choice of the standard deviation of the distribution. For example, the four experimental splittings measured for WLP23 in DMPC (33) can be fitted for τ-values of 8°, 20°, and 30°, using distributions with standard deviations of 0°, 71°, and 87° (Fig. 7). Although one might consider the system to be underdetermined with only four data points, all them in a helix turn, the use of eight experimental 2H splittings well distributed along the helix, as measured for WALP23 in DMPC (11), does not lift the ambiguity (see Fig. 2 of the Supplementary Material). Despite the unresolved τ, the ρ-angle is better determined, with values 176°, 185°, and 186°, respectively, for the three alternative fittings considered in Fig. 7. It should be realized, however, that the ambiguity will also affect ρ in cases with multimodal distributions of this latter parameter, like in the Lys-flanked peptide (see below). A detailed systematic evaluation of the expected influence of orientational dynamics on 2H NMR data will be published elsewhere.
FIGURE 7.

Best fit of quadrupolar splitting waves. Theoretical waves calculated by Eq. 2 are fitted to experimental values for WLP23 in DMPC (solid circles) (33). (Solid line, τ = 8°, ρ = 176°, SD = 0.0°, error = 0.4 kHz, and ɛ‖ = 58.7°. Dashed line, τ = 20°, ρ = 185°, SD = 71°, error = 0.25 kHz, and ɛ‖ = 58.7°. Shaded line, τ = 30°, ρ = 186°, SD = 87°, error = 0.17 kHz, and ɛ‖ = 59.7°.)
At this point, the question arises about possible consequences of extensive molecular motions on other experimental NMR observables, typically used to obtain orientational information, like 15N chemical shifts and 15N-1H dipolar couplings. Both parameters are usually measured in two-dimensional correlation spectra, known as polarization inversion spin exchange at the magic angle (PISEMA) (47), from which characteristic patterns, or polarization index of the slant angle (PISA) wheels, are obtained and assigned to well-defined peptide orientations (48,49). Unfortunately, there are no PISEMA spectra published so far for WLP/WALP or KLP/KALP model peptides, which would provide an independent experimental measurement of orientation for those cases and allow comparison with our simulation results. However, because both the 15N chemical shift anisotropy and 15N-1H dipolar interaction tensors align almost parallel to the axis of rotation (helix long axis), we expect this type of measurement to be much less influenced by rotational dynamics than the 2H NMR splittings. Supporting this idea, a recent hydrophobic mismatch study of cell-signaling peptides in bilayers of different thickness, using both MD simulations and PISEMA experiments, found a very good agreement in the tilts obtained from the two methods (50). In fact, simulated PISEMA spectra, considering motions around a central ρ-value, predict only small effects on the corresponding PISA wheels (51). However, the same study also shows a significant influence of librational motions of the peptide planes and wobble motions about a central tilt. This may cause ambiguous results which can be alleviated by increasing the number of spectral assignments. Clearly, more extensive investigations, combining NMR experiments, MD simulations, and including dynamics for the interpretation of spectra, are needed to completely solve this issue.
What determines peptide orientation and dynamics?
The orientation of membrane peptides is an important structural quality to define the molecular organization in peptide/membrane complexes. This is usually rationalized within the mattress model (9), which basically explains peptide-membrane interaction as a mutual adaptation dominated by rules of the hydrophobic mismatch (10), defined in turn as the difference between the hydrophobic length of the peptide and the hydrophobic width of the lipid bilayer. However, other intrinsic characteristics of the peptide and the bilayer lipids, like the nature of interfacial peptide residues and the lipid headgroup, may interplay together with hydrophobic matching to condition the type and properties of peptide-lipid interactions. Additionally, the dynamics of the membrane allows orientational fluctuations of embedded peptides, which may originate alternative peptide-membrane binding states and facilitate transitions between them. We will discuss now briefly how the dynamic peptide orientation is achieved through mutual adaptations of the peptide and the membrane.
Peptide adaptation to membrane binding
The apolar core of a DMPC bilayer is ∼23.0 Å thick (52). In turn, the hydrophobic length of the two peptides used in this study can in principle be approximated to 25.5 Å, considering the central 17 Leu residues and a 1.5 Å rise per residue for an ideal α-helix. Therefore, we are under conditions of positive hydrophobic mismatch and the peptides are expected to tilt to alleviate unfavorable contacts. However, the response of the lipid bilayer and uncertainty, or variability, of anchoring interactions between residues at the peptide ends and groups in the membrane interface, make mismatch, a priori, difficult to determine. For instance, although according to the simple calculation above, WLP23 and KLP23 would exhibit the same hydrophobic length, the first one shows clearly larger tilts, both for individual simulations and global averages (Table 1). This demonstrates a different effect of the Trp and Lys residues on the effective peptide orientation, either directly, by a different contribution to the total hydrophobic length, or indirectly, through distinct localized interactions of each type of residue at the membrane interface. In practice, both effects can be observed. The large rings of Trp are embedded at the level of the glycerol group, partially penetrating the hydrophobic core of the membrane (Fig. 6 A). A similar localization has been found for tryptophan and tryptophan analogs by NMR and MD simulations (53–55). This relatively deep interfacial position is in agreement with the large free energies of transfer of Trp, both to the interface (ΔGwif = −1.8 kcal/mol) and to n-octanol (ΔGwoct = −2.1 kcal/mol) in whole-residue hydrophobicity scales, with a difference favorable for insertion in the membrane core (ΔGwoct-ΔGwif = −0.3 kcal/mol) (56). Thus, the total effective hydrophobic length for WLP23 should account, at least partially, for the contribution of the flanking Trp residues, implying larger values of positive mismatch and peptide tilt. On the other hand, the positively charged Lys in KLP23 is more promiscuous, leading to alternative binding states. It shows a preference to interact with the polar phosphoryl groups of the phospholipids, which, in principle, restrains the peptide to relatively smaller tilt angles (Fig. 6 B). However, the long aliphatic chain with large conformational flexibility of Lys allows also a deeper binding of this residue, while its charged amino group can still reach the polar region via snorkeling. This latter possibility corresponds to a larger effective peptide hydrophobic-length and a larger tilt (Fig. 6 C).
With respect to the rotational angle, it is thought to be determined mainly by the position and type of the N- and C-terminal peptide-anchoring residues. For instance, ρ-values differing by ∼100° have been found experimentally for the WLP23 and KLP23 peptides (33). As the Trp and Lys interfacial residues occupy equivalent positions around the helix, the observed rotation angles were ascribed to differences in their mode of interaction with the bilayer headgroup region. From global average treatments of the simulations we arrive to ρ-values similar to the experimental ones (Table 1). However, the details of the distributions show that KLP23 is best described by two orientational states (Fig. 3, B and D, and Fig. 4 B): one associated with an average ρ at ∼195°, similar to the characteristic rotation of WLP23 (173°), and a second with an average ρ at ∼350°. Therefore, there seems to be a preferred azimuthal rotation characteristic of both peptides and determined by the position of the anchoring residues about the helix, where the tilt vector points between the two N-terminal Trp or Lys residues (Figs. 4 and 6, A and B). Additionally, the type of interfacial residue also matters. Thus, different from Trp, the large and flexible chain of Lys is involved in stabilizing alternative orientations with a tilt vector pointing toward Gly1 (Fig. 4 and Fig. 6 C).
Membrane response to a TM peptide
Peptide binding across a membrane affects its structure and dynamics. Typical consequences are appreciable changes in the electron density profiles, measured by x-ray diffraction (57), and variations in the order parameters of lipid tails, determined by 2H-NMR (31), both indicating changes in the membrane thickness. Molecular dynamics simulations can assess these membrane properties directly, allowing a useful comparison with experimental studies and a detailed evaluation of underlying biophysical phenomena.
The order parameters of lipid tails from simulations of a pure DMPC bilayer are in good agreement with those from 2H-NMR experiments (58). If represented against the aliphatic carbon position (Fig. 8), the shape of the curve is well reproduced by simulations, although the absolute values are slightly smaller for some methylene groups. In presence of WLP23 or KLP23, we observed an increase of the order parameters, averaged over the total set of simulations, for the 30 lipids nearest to the peptide (Fig. 8). Although there are no experimental order parameters available for membrane complexes with WLP23 or KLP23, a similar increase of order parameters has been reported in the presence of WALP19 in DMPC, at a peptide/lipid molar ratio of 1:30 (31). Nevertheless, we noticed that the increase of the bilayer width accompanying the changes in the order parameters is only marginal, as reported from experiments (57), and peptide tilting appears to be the principal adaptation upon peptide/membrane interaction.
FIGURE 8.

Lipid order parameters. Represented are absolute values of S for methylene groups of the sn-2 acyl chain of DMPC. Shaded lines correspond to pure lipid bilayers, analyzed by 2H-NMR experiments (58) (stars, continuous line) or MD simulations (diamonds, dashed line). The solid lines are order parameters in the presence of peptides, determined from experiments (WALP19 (31), triangles, dashed-dotted line) or simulations (WLP23, circles, continuous line, and KLP23, squares, dashed line). Note that the experimental data correspond to carbons C3 to C14, while simulated data extend from C2 to C13.
CONCLUSIONS
Despite the limited time window and small number of replicas, conforming to a reduced molecular ensemble, our observations can be considered an explicit representation of the complex orientational dynamics of trans-membrane peptides. The existence of fluctuating alternative {τ, ρ} states, arrestable within at least a few hundreds of nanoseconds, is the most relevant property. Such a view contrasts with often envisioned static, well-defined membrane peptide orientations and stresses the importance of dynamics to describe the molecular organization of these systems. Dynamic models should thus ideally accompany structural definitions in peptide/membrane complexes as a necessary ingredient to understand their functionality, especially in cases of intrinsically dynamic processes like membrane insertion, pore formation, membrane fusion, and intermolecular peptide association.
We have demonstrated as well the importance of properly considering peptide dynamics to determine orientational parameters from spectroscopic 2H-NMR data, giving an explanation to counterintuitive small tilts in model WLP/WALP and KLP/KALP peptides. Although general implicit distributions are not easy to find, explicit dynamics representations obtained from a limited number of MD simulations can be used to guide the calculation and interpretation of 2H NMR splittings from experimental data. Such rotational dynamics of the peptide is expected to have a more limited influence on 15N chemical shift and 15N-1H dipolar coupling NMR observables.
Finally, the fact that the orientation of the peptides, and particularly the rotational angles, are well reproduced by the MD simulations, implies that the principal features of the peptide-membrane interaction are well captured. This opens the possibility to analyze such interactions from atomic-detailed dynamic models, with particular emphasis in the membrane interface.
SUPPLEMENTARY MATERIAL
To view all of the supplemental files associated with this article, visit www.biophysj.org.
Supplementary Material
Acknowledgments
Note added in proof: While this article was under review, an independent MD study on the model peptide WALP23 has been completed (59). Similar to our work, this latter investigation also shows that the large fluctuation of the peptide rotational angle reduces back-calculated 2H-NMR splittings, which can be the origin of abnormally small tilts when determined from experiments.
Erik Strandberg and Siewert Jan Marrink are gratefully acknowledged for critical reading of the manuscript.
This work has been supported by grants from the Spanish Ministerio de Educación y Ciencia (CTQ2004-03444 and FPU fellowship (S.E.)).
Editor: Anthony Watts.
References
- 1.Nagle, J. F., and S. Tristram-Nagle. 2000. Structure of lipid bilayers. Biochim. Biophys. Acta. 1469:159–195. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Singer, S. J., and G. L. Nicolson. 1972. The fluid mosaic model of the structure of cell membranes. Science. 175:720–731. [DOI] [PubMed] [Google Scholar]
- 3.Arkin, I. T. 2006. Isotope-edited IR spectroscopy for the study of membrane proteins. Curr. Opin. Chem. Biol. 10:394–401. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.de Planque, M. R., E. Goormaghtigh, D. V. Greathouse, R. E. Koeppe 2nd, J. A. Kruijtzer, R. M. Liskamp, B. de Kruijff, and J. A. Killian. 2001. Sensitivity of single membrane-spanning α-helical peptides to hydrophobic mismatch with a lipid bilayer: effects on backbone structure, orientation, and extent of membrane incorporation. Biochemistry. 40:5000–5010. [DOI] [PubMed] [Google Scholar]
- 5.Marassi, F. M., and S. J. Opella. 2000. A solid-state NMR index of helical membrane protein structure and topology. J. Magn. Reson. 144:150–155. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Salgado, J., S. L. Grage, L. H. Kondejewski, R. S. Hodges, R. N. McElhaney, and A. S. Ulrich. 2001. Membrane-bound structure and alignment of the antimicrobial β-sheet peptide gramicidin S derived from angular and distance constraints by solid state 19F-NMR. J. Biomol. NMR. 21:191–208. [DOI] [PubMed] [Google Scholar]
- 7.van der Wel, P. C., E. Strandberg, J. A. Killian, and R. E. Koeppe 2nd. 2002. Geometry and intrinsic tilt of a tryptophan-anchored transmembrane α-helix determined by 2H NMR. Biophys. J. 83:1479–1488. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Bechinger, B., C. Aisenbrey, and P. Bertani. 2004. The alignment, structure and dynamics of membrane-associated polypeptides by solid-state NMR spectroscopy. Biochim. Biophys. Acta. 1666:190–204. [DOI] [PubMed] [Google Scholar]
- 9.Mouritsen, O. G., and M. Bloom. 1984. Mattress model of lipid-protein interactions in membranes. Biophys. J. 46:141–153. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Owicki, J. C., and H. M. McConnell. 1979. Theory of protein-lipid and protein-protein interactions in bilayer membranes. Proc. Natl. Acad. Sci. USA. 76:4750–4754. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Strandberg, E., S. Özdirekcan, D. T. Rijkers, P. C. van der Wel, R. E. Koeppe 2nd, R. M. Liskamp, and J. A. Killian. 2004. Tilt angles of transmembrane model peptides in oriented and non-oriented lipid bilayers as determined by 2H solid-state NMR. Biophys. J. 86:3709–3721. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Arkin, I. T., K. R. MacKenzie, and A. T. Brunger. 1997. Site-directed dichroism as a method for obtaining rotational and orientational constraints for oriented polymers. J. Am. Chem. Soc. 119:8973–8980. [Google Scholar]
- 13.Strandberg, E., P. Wadhwani, P. Tremouilhac, U. H. Durr, and A. S. Ulrich. 2006. Solid-state NMR analysis of the PGLa peptide orientation in DMPC bilayers: structural fidelity of 2H-labels versus high sensitivity of 19F-NMR. Biophys. J. 90:1676–1686. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Goodyear, D. J., S. Sharpe, C. W. Grant, and M. R. Morrow. 2005. Molecular dynamics simulation of transmembrane polypeptide orientational fluctuations. Biophys. J. 88:105–117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Fares, C., J. Qian, and J. H. Davis. 2005. Magic angle spinning and static oriented sample NMR studies of the relaxation in the rotating frame of membrane peptides. J. Chem. Phys. 122: Art. No 194908.
- 16.Davis, J. H., M. Auger, and R. S. Hodges. 1995. High resolution 1H nuclear magnetic resonance of a transmembrane peptide. Biophys. J. 69:1917–1932. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Hansson, T., C. Oostenbrink, and W. F. van Gunsteren. 2002. Molecular dynamics simulations. Curr. Opin. Struct. Biol. 12:190–196. [DOI] [PubMed] [Google Scholar]
- 18.Saiz, L., and M. L. Klein. 2002. Computer simulation studies of model biological membranes. Acc. Chem. Res. 35:482–489. [DOI] [PubMed] [Google Scholar]
- 19.Marrink, S. J., E. Lindahl, O. Edholm, and A. E. Mark. 2001. Simulation of the spontaneous aggregation of phospholipids into bilayers. J. Am. Chem. Soc. 123:8638–8639. [DOI] [PubMed] [Google Scholar]
- 20.Bond, P. J., J. M. Cuthbertson, S. S. Deol, and M. S. Sansom. 2004. MD simulations of spontaneous membrane protein/detergent micelle formation. J. Am. Chem. Soc. 126:15948–15949. [DOI] [PubMed] [Google Scholar]
- 21.Esteban-Martin, S., and J. Salgado. 2007. Self-assembling of peptide/membrane complexes by atomistic molecular dynamics simulations. Biophys. J. 92:903–912. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Marrink, S. J., and A. E. Mark. 2004. Molecular view of hexagonal phase formation in phospholipid membranes. Biophys. J. 87:3894–3900. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.de Vries, A. H., S. Yefimov, A. E. Mark, and S. J. Marrink. 2005. Molecular structure of the lecithin ripple phase. Proc. Natl. Acad. Sci. USA. 102:5392–5396. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Leontiadou, H., A. E. Mark, and S. J. Marrink. 2004. Molecular dynamics simulations of hydrophilic pores in lipid bilayers. Biophys. J. 86:2156–2164. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Im, W. 2005. Interfacial folding and membrane insertion of designed peptides studied by molecular dynamics simulations. Proc. Natl. Acad. Sci. USA. 102:6771–6776. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Nymeyer, H., T. B. Woolf, and A. E. Garcia. 2005. Folding is not required for bilayer insertion: replica exchange simulations of an α-helical peptide with an explicit lipid bilayer. Proteins. 59:783–790. [DOI] [PubMed] [Google Scholar]
- 27.Venturoli, M., B. Smit, and M. M. Sperotto. 2005. Simulation studies of protein-induced bilayer deformations, and lipid-induced protein tilting, on a mesoscopic model for lipid bilayers with embedded proteins. Biophys. J. 88:1778–1798. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Kandasamy, S. K., and R. G. Larson. 2006. Molecular dynamics simulations of model trans-membrane peptides in lipid bilayers: a systematic investigation of hydrophobic mismatch. Biophys. J. 90:2326–2343. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Bond, P. J., and M. S. Sansom. 2006. Insertion and assembly of membrane proteins via simulation. J. Am. Chem. Soc. 128:2697–2704. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Davis, J. H., D. M. Clare, R. S. Hodges, and M. Bloom. 1983. Interaction of a synthetic amphiphilic polypeptide and lipids in a bilayer structure. Biochemistry. 22:5298–5305. [Google Scholar]
- 31.de Planque, M. R., D. V. Greathouse, R. E. Koeppe 2nd, H. Schafer, D. Marsh, and J. A. Killian. 1998. Influence of lipid/peptide hydrophobic mismatch on the thickness of diacylphosphatidylcholine bilayers. A 2H NMR and ESR study using designed transmembrane α-helical peptides and gramicidin A. Biochemistry. 37:9333–9345. [DOI] [PubMed] [Google Scholar]
- 32.de Planque, M. R., J. A. Kruijtzer, R. M. Liskamp, D. Marsh, D. V. Greathouse, R. E. Koeppe 2nd, B. de Kruijff, and J. A. Killian. 1999. Different membrane anchoring positions of tryptophan and lysine in synthetic transmembrane α-helical peptides. J. Biol. Chem. 274:20839–20846. [DOI] [PubMed] [Google Scholar]
- 33.Özdirekcan, S., D. T. Rijkers, R. M. Liskamp, and J. A. Killian. 2005. Influence of flanking residues on tilt and rotation angles of transmembrane peptides in lipid bilayers. A solid-state 2H NMR study. Biochemistry. 44:1004–1012. [DOI] [PubMed] [Google Scholar]
- 34.Sparr, E., W. L. Ash, P. V. Nazarov, D. T. Rijkers, M. A. Hemminga, D. P. Tieleman, and J. A. Killian. 2005. Self-association of transmembrane α-helices in model membranes: importance of helix orientation and role of hydrophobic mismatch. J. Biol. Chem. 280:39324–39331. [DOI] [PubMed] [Google Scholar]
- 35.Killian, J. A., and T. K. Nyholm. 2006. Peptides in lipid bilayers: the power of simple models. Curr. Opin. Struct. Biol. 16:473–479. [DOI] [PubMed] [Google Scholar]
- 36.Lindahl, E., B. Hess, and D. van der Spoel. 2001. GROMACS 3.0: a package for molecular simulation and trajectory analysis. J. Mol. Model. 7:306–317. [Google Scholar]
- 37.Berger, O., O. Edholm, and F. Jahnig. 1997. Molecular dynamics simulations of a fluid bilayer of dipalmitoylphosphatidylcholine at full hydration, constant pressure, and constant temperature. Biophys. J. 72:2002–2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Feenstra, K. A., B. Hess, and H. J. C. Berendsen. 1999. Improving efficiency of large time-scale molecular dynamics simulations of hydrogen-rich systems. J. Comput. Chem. 20:786–798. [DOI] [PubMed] [Google Scholar]
- 39.Berendsen, H. J. C., J. P. M. Postma, W. F. van Gunsteren, A. DiNola, and J. R. Haak. 1984. Molecular-dynamics with coupling to an external bath. J. Chem. Phys. 81:3684–3690. [Google Scholar]
- 40.Essmann, U., L. Perera, M. L. Berkowitz, T. Darden, H. Lee, and L. G. Pedersen. 1995. A smooth particle mesh Ewald method. J. Chem. Phys. 103:8577–8593. [Google Scholar]
- 41.Schwede, T., J. Kopp, N. Guex, and M. C. Peitsch. 2003. SWISS-MODEL: an automated protein homology-modeling server. Nucleic Acids Res. 31:3381–3385. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Egberts, E., S. J. Marrink, and H. J. C. Berendsen. 1994. Molecular dynamics simulation of a phospholipid membrane. Eur. Biophys. J. 22:423–436. [DOI] [PubMed] [Google Scholar]
- 43.Kabsch, W., and C. Sander. 1983. Dictionary of protein secondary structure: pattern recognition of hydrogen-bonded and geometrical features. Biopolymers. 22:2577–2637. [DOI] [PubMed] [Google Scholar]
- 44.Park, S. H., and S. J. Opella. 2005. Tilt angle of a trans-membrane helix is determined by hydrophobic mismatch. J. Mol. Biol. 350:310–318. [DOI] [PubMed] [Google Scholar]
- 45.Koehorst, R. B., R. B. Spruijt, F. J. Vergeldt, and M. A. Hemminga. 2004. Lipid bilayer topology of the transmembrane α-helix of M13 major coat protein and bilayer polarity profile by site-directed fluorescence spectroscopy. Biophys. J. 87:1445–1455. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Duong-Ly, K. C., V. Nanda, W. F. Degrado, and K. P. Howard. 2005. The conformation of the pore region of the M2 proton channel depends on lipid bilayer environment. Protein Sci. 14:856–861. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Wu, C. H., A. Ramamoorthy, and S. J. Opella. 1994. High resolution heteronuclear dipolar solid-state NMR spectroscopy. J. Magn. Reson. 109:270–272. [Google Scholar]
- 48.Marassi, F. M., and S. J. Opella. 2000. A solid-state NMR index of helical membrane protein structure and topology. J. Magn. Res. 144:150–155. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Wang, J., J. Denny, C. Tian, S. Kim, Y. Mo, F. Kovacs, Z. Song, K. Nishimura, Z. Gan, R. Fu, J. R. Quine, and T. A. Cross. 2000. Imaging membrane protein helical wheels. J. Magn. Reson. 144:162–167. [DOI] [PubMed] [Google Scholar]
- 50.Ramamoorthy, A., S. K. Kandasamy, D. K. Lee, S. Kidambi, and R. G. Larson. 2007. Structure, topology, and tilt of cell-signaling peptides containing nuclear localization sequences in membrane bilayers determined by solid-state NMR and molecular dynamics simulation studies. Biochemistry. 30:965–975. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Straus, S. K., W. R. Scott, and A. Watts. 2003. Assessing the effects of time and spatial averaging in 15N chemical shift/15N–1H dipolar correlation solid state NMR experiments. J. Biomol. NMR. 26:283–295. [DOI] [PubMed] [Google Scholar]
- 52.de Planque, M. R., and J. A. Killian. 2003. Protein-lipid interactions studied with designed transmembrane peptides: role of hydrophobic matching and interfacial anchoring. Mol. Membr. Biol. 20:271–284. [DOI] [PubMed] [Google Scholar]
- 53.Norman, K. E., and H. Nymeyer. 2006. Indole localization in lipid membranes revealed by molecular simulation. Biophys. J. 91:2046–2054. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Gaede, H. C., W. M. Yau, and K. Gawrisch. 2005. Electrostatic contributions to indole-lipid interactions. J. Phys. Chem. B. 109:13014–13023. [DOI] [PubMed] [Google Scholar]
- 55.Persson, S., J. A. Killian, and G. Lindblom. 1998. Molecular ordering of interfacially localized tryptophan analogs in ester- and ether-lipid bilayers studied by 2H-NMR. Biophys. J. 75:1365–1371. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.White, S. H., and W. C. Wimley. 1998. Hydrophobic interactions of peptides with membrane interfaces. Biochim. Biophys. Acta. 1376:339–352. [DOI] [PubMed] [Google Scholar]
- 57.Weiss, T. M., P. C. van der Wel, J. A. Killian, R. E. Koeppe 2nd, and H. W. Huang. 2003. Hydrophobic mismatch between helices and lipid bilayers. Biophys. J. 84:379–385. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Petrache, H. I., S. W. Dodd, and M. F. Brown. 2000. Area per lipid and acyl length distributions in fluid phosphatidylcholines determined by 2H NMR spectroscopy. Biophys. J. 79:3172–3192. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Suat Özdirekcan, Catherine Etchebest, J. Antoinette Killian, and Patrick F. J. Fuchs. On the orientation of a designed transmembrane peptide: towards the right tilt angle? J. Am. Chem. Soc. In press. [DOI] [PubMed]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.