

Abstract
It has been shown that the propensity of a protein to form amyloid-like fibrils can be predicted with high accuracy from the knowledge of its amino acid sequence. It has also been suggested, however, that some regions of the sequences are more important than others in determining the aggregation process. Here, we have addressed this issue by constructing a set of “sequence scrambled” variants of the first 29 residues of horse heart apomyoglobin (apoMb1-29), in which the sequence was modified while maintaining the same amino acid composition. The clustering of the most amyloidogenic residues in one region of the sequence was found to cause a marked increase of the elongation rate (kagg) and a remarkable shortening of the lag phase (tlag) of the fibril growth, as determined by far-UV circular dichroism and thioflavin T fluorescence. We also show that taking explicitly into consideration the presence of aggregation-promoting regions in the predictive methods results in a quantitative agreement between the theoretical and observed kagg and tlag values of the apoMb1-29 variants. These results, together with a comparison between homologous segments from the family of globins, indicate the existence of a negative selection against the clustering of highly amyloidogenic residues in one or few regions of polypeptide sequences.
INTRODUCTION
It has been suggested that amyloid formation is a fundamental characteristic of the chemistry of polypeptide chains (1,2). Most proteins, if not all, can form amyloid-like fibrils of particularly high thermodynamic stability under appropriate conditions (1). It is also increasingly recognized that, from bacteria to humans, some proteins can form amyloid-like fibrils in vivo that fulfill a variety of biological functions (2,3). Examples are the sequestration of melanin pigments in mammals (3–5), the transfer of inheritable information in fungal prion proteins (6), and the modulation of the activity of a bactericide peptide from Klebsiella pneumoniae by its own aggregation (7). Other proteins, which by contrast are designed by evolution to remain soluble, form extracellular amyloid fibrils or intracellular inclusions with amyloid-like characteristics following a failure to remain in such a soluble state, and give rise to well-described pathological states (2). These range from neurodegenerative conditions, such as Alzheimer's disease, Parkinson's disease, and spongiform encephalopathies, to systemic amyloidoses including light chain amyloidosis or dialysis-related amyloidosis (2,8). The considerable tendency of proteins to form amyloid-like fibrils, as well as the unique molecular organization of these well-ordered and self-organized structures, has also led many investigators to explore the biotechnological applications of these fibrillar aggregates as nanomolecular materials (9,10).
The generic ability of polypeptide chains to aggregate into morphologically similar amyloid-like fibrils, regardless of the structures and sequences of the precursor proteins, has suggested that the underlying physicochemical principles behind this process may be rationalized on relatively simple, universally valid grounds. It was first shown that the effect of a mutation on the aggregation rate of a peptide or protein in its unfolded state is determined by the effect that such mutation has on simple characteristics of the primary sequence, like charge, hydrophobicity or propensity to form β-sheet structure (11). Using these or related factors, predictive algorithms were developed that are able to determine the change in the rate or propensity of aggregation of an unstructured polypeptide chain following mutation (11–15), the absolute aggregation rate (16,17), or the most aggregation-prone regions within a sequence (12,15,17–22). These algorithms can identify “amyloidogenic” or “aggregation-promoting” regions of the sequence that overlap with satisfactory accuracy with those sequence portions known to form, from experimental results, the β-core of the fibrils in a number of test cases. These studies clearly show (and even provide a rational explanation) that a limited portion of the sequence plays a key role in the aggregation process and participates in the formation of the β-core of the mature fibrils (23–25). On the other hand, to a first approximation, the aggregation rate appears to correlate to the average aggregation propensity of the polypeptide chain, with no distinction between amyloidogenic and nonamyloidogenic regions (13,16,17).
To investigate the importance of the regions of the sequence with a high aggregation propensity, as opposed to the entire sequence, in determining the aggregation behavior of an unstructured polypeptide, we have used a peptide corresponding to the first 29 residues of horse heart apomyoglobin (apoMb1-29). Full-length apoMb is a well-described system that has been extensively used for folding as well as aggregation studies (26–29); apoMb1-29 is an unstructured peptide that is soluble at neutral pH, but self-assembles at pH 2.0 into amyloid-like fibrils that are morphologically, structurally, and tinctorially indistinguishable from those formed by naturally amyloidogenic proteins (30). The core of the fibrils is formed by the region of the sequence spanning from residue 7 to residue 16 or 18, as probed by proteolysis studies (30).
We have designed four scrambled sequence variants of apoMb1-29. These variants feature the same length and amino acid composition as the wild-type peptide, but a scrambled sequence resulting in a modified aggregation propensity profile along the sequence relative to the wild-type, as predicted by the algorithms mentioned above. We shall describe that the clustering of the most amyloidogenic residues in one region of the sequence, resulting into a wide and high peak in the aggregation propensity profile, leads to a marked change in the kinetics of aggregation, with a reduction of the lag phase by over 5000-fold and a 40-fold enhancement of the fibril elongation rate. We will also show that aggregation propensity profiles of this type have clearly been negatively selected by evolution in the structural family of globins, emphasizing that prevention of aggregation has been an important driving force in the evolution of natural amino acid sequences.
MATERIALS AND METHODS
Calculation of the intrinsic aggregation propensity profile of a sequence
Except for the aggregation propensity profiles reported in Fig. 1, B–D, all the aggregation propensity profiles and the derived parameters were calculated according to Pawar et al. (18). In brief, the intrinsic aggregation propensities (pagg) of the 20 naturally occurring amino acid residues were taken from the aforementioned work at pH 2.0 (also listed in the Supplementary Material, Table S1). The pagg value of an individual amino acid along a sequence was increased by αpat Ipat if it belonged to a five-residue or longer sequence stretch having a pattern of alternating polar and nonpolar residues (see Table S1 for viewing which residues are considered polar and nonpolar, respectively). As in the previous work αpat and Ipat values were set equal to 0.39 and 1, respectively (18). We then considered:
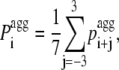 |
(1) |
where 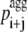 is the pagg value of the individual amino acid at position i + j (with j varying from −3 to +3). With this definition
is the pagg value of the individual amino acid at position i + j (with j varying from −3 to +3). With this definition 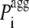 is the intrinsic aggregation propensity of a seven-residue window centered at position i and resulting from the average of the pagg values of its seven residues. The
is the intrinsic aggregation propensity of a seven-residue window centered at position i and resulting from the average of the pagg values of its seven residues. The 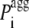 values were not calculated for the first three residues at the N-terminus and the last three residues at the C-terminus, due to the chosen window of seven residues.
values were not calculated for the first three residues at the N-terminus and the last three residues at the C-terminus, due to the chosen window of seven residues.
FIGURE 1.

Aggregation propensity profiles of wild-type and scrambled sequence variants of apoMb1-29. The aggregation propensity profiles for panels A–D are calculated according to Pawar et al. (18), Sanchez de Groot et al. (19), Fernandez-Escamilla et al. (12), and Tartaglia et al. (13), respectively. The profiles correspond to wild-type (black), P1 (orange), P2 (green), P3 (red), and P4 (blue) apoMb1-29 variants. In panel C the P1 and P2 variants have aggregation propensity of 0 throughout their sequences.
To define a profile of intrinsic aggregation propensity along the sequence we generated randomly Ns sequences of length N using the natural occurrences of the 20 amino acids (reported in Table S1) as their actual probabilities in the random procedure. The average (μagg) and standard deviation (σagg) of the 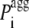 values within the Ns sequences were defined as:
values within the Ns sequences were defined as:
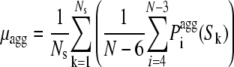 |
(2) |
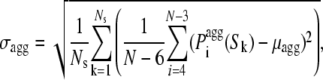 |
(3) |
where Sk is the sequence No. k (with k varying from 1 to Ns), N is the number of residues in each sequence and Ns is the number of sequences. N and Ns were set equal to 29 and 10,000 in this study, respectively. We then calculated the intrinsic Z-score of aggregation of a seven-residue window centered at position i (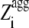 ) using:
) using:
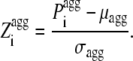 |
(4) |
The plot of 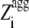 versus residue number (from residue 4 to residue N − 3) represents the intrinsic aggregation propensity profile (
versus residue number (from residue 4 to residue N − 3) represents the intrinsic aggregation propensity profile (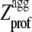 ), as reported (18).
), as reported (18).
The aggregation propensity profiles in Fig. 1, B–D, were calculated according to Sanchez de Groot et al. (19), Fernandez-Escamilla et al. (12), and Tartaglia et al. (13), respectively. For the analysis in Fig. 1 C the following parameters were used: pH = 2.0; T = 310 K; ionic strength = 0.01 M; trifluoroethanol concentration = 0%; protein stability = −3 kcal/mol (12). The horizontal line corresponds to the threshold of significance as defined by the authors. For the analysis in Fig. 1 D, the relative change of aggregation rate considering a single-point mutation F→X was calculated for each residue i of the apoMb1-29 variants. To allow the comparison between all plots reported in Fig. 1, A–D, a sliding window of seven residues was used in all cases.
Calculation of the aggregation parameters of a polypeptide chain (Pagg, Zagg, Sagg, Z0agg)
The aggregation propensity (Pagg) and aggregation Z-score (Zagg) of a sequence of N residues were calculated as:
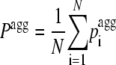 |
(5) |
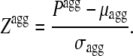 |
(6) |
We define Sagg as the total surface of the aggregation propensity profile (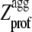 ) that lies above a
) that lies above a 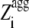 threshold of 1. This was achieved using (G.-G. Tartaglia and M. Vendruscolo, unpublished results):
threshold of 1. This was achieved using (G.-G. Tartaglia and M. Vendruscolo, unpublished results):
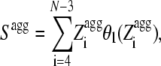 |
(7) |
where θ1(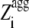 ) is 1 for
) is 1 for 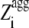 ≥ 1 and 0 for
≥ 1 and 0 for 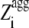 < 1. Since Sagg appears to be an important determinant of aggregation rate, the Zagg values of a set of intrinsically disordered proteins were recalculated (
< 1. Since Sagg appears to be an important determinant of aggregation rate, the Zagg values of a set of intrinsically disordered proteins were recalculated (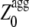 ) using:
) using:
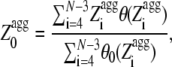 |
(8) |
where θ0(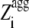 ) is 1 for
) is 1 for 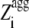 ≥ 0 and 0 for
≥ 0 and 0 for 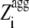 < 0.
< 0. 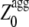 represents the aggregation Z-score of a sequence resulting from the regions of the sequence with a higher aggregation propensity. The
represents the aggregation Z-score of a sequence resulting from the regions of the sequence with a higher aggregation propensity. The 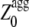 values were analyzed with the experimental aggregation rates in a multiple regression fitting to determine newly optimized pagg as described (16). Finally the
values were analyzed with the experimental aggregation rates in a multiple regression fitting to determine newly optimized pagg as described (16). Finally the 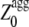 values of the apoMb1-29 variants were determined using Eqs. 5, 6, and 8.
values of the apoMb1-29 variants were determined using Eqs. 5, 6, and 8.
Fourier transform infrared spectroscopy
All the apoMb1-29 peptides were purchased from GenScript (Piscataway, NJ). Each of the apoMb1-29 lyophilized peptides was dissolved in 50 μL of a solution containing 10 mM HCl, pH 2.0, at a peptide concentration equal or higher than 5 mg/ml, and incubated 1 week at room temperature. Spectra were collected at room temperature using a Jasco FTIR-4200 spectrometer (Tokyo, Japan). Sample aliquots were placed between CaF2 windows, separated by a 6-μM-thick spacer. The sample and detector compartments were thoroughly purged with N2. Spectra represent averages of 200 scans recorded between 4000 and 400 cm−1 at a resolution of 4 cm−1. All spectra were baseline corrected, blank subtracted, and smoothed using a means-movement function.
Aggregation kinetics probed by far-ultraviolet circular dichroism
Aggregation of wild-type and scrambled sequence apoMb1-29 variants was initiated as previously described for the wild-type peptide (30). Briefly, the lyophilized peptides were dissolved in 10 mM Tris-HCl, pH 8.3, centrifuged for 5 min at 16,000 rpm, and filtered using 0.02 μM Anatop filters (Whatman, Brentford, UK). The samples were then diluted to reach a peptide concentration of 0.2 mg/ml, led to pH 2.0 adding a minimal amount of 2 M HCl, and incubated at 37°C. Far-UV CD spectra were acquired at regular time intervals at 37°C using a 1-mm pathlength cuvette and a Jasco J-810 spectropolarimeter. This was equipped with a thermostated cell holder connected to a circulating Thermo Haake C25P water bath (Karlsruhe, Germany). Each spectrum, recorded as the average of five scans, was blank subtracted and smoothed using a fast Fourier transform filter. The residual ellipticity at 216 nm was plotted versus time. For the P4 apoMb1-29 variant, the resulting plot was fitted to the following single exponential function:
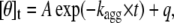 |
(9) |
where A is the total change of mean residue ellipticity from time 0 to ∞, kagg is the apparent aggregation rate constant, and q is the final mean residue ellipticity at time ∞. For the wild-type, P1, P2, and P3 apoMb1-29 variants, the plots were fitted to the following empirical sigmoid function (31):
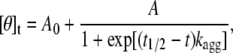 |
(10) |
where A and kagg have the same meaning as for Eq. 9, A0 is mean residue ellipticity at time 0, and t1/2 is the midpoint of aggregation. The lag time of aggregation, tlag, was determined from kagg and t1/2 as follows (31):
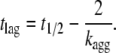 |
(11) |
Aggregation kinetics probed by thioflavin T fluorescence
The apoMb1-29 peptides were prepared and incubated as described above for the CD measurements. At different incubation times aliquots of 60 μl were mixed with 440 μl of 10 mM phosphate buffer, pH 6.0, containing 25 μM ThT (Sigma-Aldrich, St. Louis, MO). The steady-state fluorescence of the resulting samples was measured at 25°C using a 2 × 10-mm pathlength cuvette and a Perkin-Elmer LS-55 fluorimeter (Wellesley, MA) equipped with a thermostated cell compartment attached to a Haake F8 water bath. The excitation and emission wavelengths were 440 and 485 nm, respectively. All measured fluorescence values were blank subtracted and normalized to the percentage of the maximum value. Plots of fluorescence values versus time were analyzed with a procedure of best fit, using a single exponential (Eq. 9) or a sigmoid (Eq. 10) function, as described above.
Construction of the peptide database from the globin family
All sequences contained in the globin structural family of the Prosite database (code PS01033) were aligned. Alignment was achieved using structural criteria, which has been proposed to be a preferred method due to the extremely low sequence identity between some of the globin family members (32). In practice, a multiple sequence alignment against a secondary structure pattern extracted from the three-dimensional structure of horse heart myoglobin was performed using the ClustalW software. Only the sequence fragments starting and ending at positions corresponding to the 1st and 29th residues of apoMb1-29, respectively, and with a length comprised between 27 and 31 amino acids, were considered. The parameters Pagg, Zagg, and Sagg were then calculated for all sequences from this data set as described above (Eqs. 5–7).
RESULTS
Design of “scrambled sequence” variants of apoMb1-29
As detailed in the Introduction section, several computational methods have been developed to identify the regions of the sequence, within an unstructured peptide or protein, with a high potential to promote amyloid aggregation and form the β-core of the resulting fibrils. Using four of these programs, the most amyloidogenic region of wild-type apoMb1-29 is consistently predicted to be in all cases the central region of the sequence, encompassing approximately residues 9–14 (Fig. 1). This prediction is in agreement with the experimental data obtained with proteolysis showing that the region spanning approximately from residue 7 to residue 16 or 18 forms the β-core of the fibrils (30).
To assess the role of this central amyloidogenic region of the sequence relative to the overall amino acid composition of apoMb1-29, we designed four diverse variants of apoMb1-29 in which the sequence was scrambled with respect to the wild-type sequence, i.e., it was changed while maintaining the same length and amino acid composition. This reorganization of the sequence was achieved by introducing minimal changes, typically shifting some residues from the edge regions of the sequence to the central portion. The sequences of the designed “scrambled sequence” variants, named P1, P2, P3, and P4 apoMb1-29, are reported in Table 1. Using the terminology and parameter definition described in Materials and Methods and previously reported (18), wild-type apoMb1-29 and the four designed variants have the same values of overall aggregation score (termed Zagg, Table 1).
TABLE 1.
Predicted parameters of apoMb1-29 aggregation
| Variant | Sequence | Zagg* | Sagg† |
 ‡ ‡
|
|---|---|---|---|---|
| WT | GLSDGEWQQVLNVWGKVEADIAGHGQEVL | 0.37 | 0.59 | 0.625 |
| P1 | GLSDGEWQQVENVWGKVEADIAGHGQLVL | 0.37 | 0.00 | 0.624 |
| P2 | GLSDGEWQQVLENVWGKVEADIAGHGQVL | 0.37 | 0.32 | 0.607 |
| P3 | GLSDGEQQVLWINVWGKVEADAGHGQEVL | 0.37 | 4.15 | 0.683 |
| P4 | GLSDGEQQVLVWIVNVWGKEADAGHGQEL | 0.37 | 7.74 | 0.941 |
Aggregation Z-score of the entire sequence, calculated as described in Materials and Methods, and previously (18).
Prominence of the aggregation promoting region, calculated as described in Materials and Methods.
New aggregation Z-score, calculated as described in Materials and Methods.
Despite their identical Zagg values, the apoMb1-29 variants have different aggregation propensity profiles relative to the wild-type peptide (Fig. 1). The prominence of the central aggregation-promoting region above the critical threshold of normalized aggregation propensity (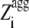 = 1; see Materials and Methods) is very different in the various cases, with the P4 variant having the highest and largest peak (Fig. 1 A). To have a quantitative measure of the prominence of the aggregation-prone region, we introduced the Sagg parameter. Sagg represents the total surface of the aggregation propensity profile that lies above the threshold
= 1; see Materials and Methods) is very different in the various cases, with the P4 variant having the highest and largest peak (Fig. 1 A). To have a quantitative measure of the prominence of the aggregation-prone region, we introduced the Sagg parameter. Sagg represents the total surface of the aggregation propensity profile that lies above the threshold 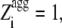 and correlates to both the height and width of the central aggregation-promoting region (see Materials and Methods for details). The Sagg parameter spans from a value of 0 for P1 to a value of 7.74 for P4, the wild-type having an intermediate value of 0.59 (Table 1). Remarkably, the trend of prominence of the aggregation-prone region predicted by Pawar et al. (18) for the designed apoMb1-29 variants was confirmed by three other prediction methods, lending further support to the robustness of the design (Fig. 1, B–D).
and correlates to both the height and width of the central aggregation-promoting region (see Materials and Methods for details). The Sagg parameter spans from a value of 0 for P1 to a value of 7.74 for P4, the wild-type having an intermediate value of 0.59 (Table 1). Remarkably, the trend of prominence of the aggregation-prone region predicted by Pawar et al. (18) for the designed apoMb1-29 variants was confirmed by three other prediction methods, lending further support to the robustness of the design (Fig. 1, B–D).
All apoMb1-29 variants form amyloid-like fibrils at low pH
As a first step we established that the designed scrambled sequence mutants do form amyloid-like fibrils in vitro, as it has been previously demonstrated for the wild-type peptide (30). At pH 8.3 wild-type apoMb1-29 shows a far-UV CD spectrum typical of largely unfolded polypeptide chains, with a minimum at ≈200 nm (Fig. 2 A, dashed line). Similar far-UV CD spectra were obtained at pH 8.3 for the scrambled sequence variants analyzed here (data not shown). Following acidification at pH 2.0, the far-UV CD spectra were initially typical of an unfolded peptide (Fig. 2 A, dotted line). However, after 4 days at pH 2.0, all the apoMb1-29 variants had spectra with a minimum mean residue ellipticity between 214 and 218 nm and a maximum between 195 and 199 nm, indicating a high content of β-sheet structure (Fig. 2 A, solid lines).
FIGURE 2.

Far-UV CD and FTIR spectra of apoMb1-29 variants. (A) Far-UV CD spectra. (B) FTIR spectra. The spectra refer to wild-type at pH 8.3 immediately after dissolution (black, dashed line), wild-type at pH 2.0 immediately after pH decrease (black, dotted line), wild-type at pH 2.0, and t = 4 days (black, solid line), P1 at pH 2.0 and t = 4 days (orange), P2 at pH 2.0 and t = 4 days (green), P3 at pH 2.0 and t = 4 days (red), and P4 at pH 2.0 and t = 4 days (blue).
When working with protein aggregates the obtained far-UV CD spectra are likely to be affected by light scattering phenomena, due to the large size of the aggregates and short wavelength in the far-UV region of light. We therefore analyzed the apoMb1-29 variants using FTIR spectroscopy, which is not significantly affected by light scattering due to the long wavelength that is used in this spectroscopic technique. The FTIR spectra were recorded for three representative apoMb1-29 variants incubated at pH 2.0 for a few days, namely the wild-type, P1 and P4. The FTIR spectra presented an amide I band with a maximum in the region 1620–1635 cm−1, further confirming the presence of a largely β-sheet structure content (Fig. 2 B). The differences in the far-UV CD and FTIR spectra between the apoMb1-29 variants probably reflect the differences of sequence between them and consequently between the structures of the resulting aggregates.
The addition of each aggregated peptide to a ThT solution resulted in a significant enhancement of the fluorescence at 485 nm, with a fluorescence signal at the equilibrium at least 40 times higher than the one at t = 0 (Fig. 3 A). All these spectroscopic properties obtained with far-UV CD, FTIR, and ThT fluorescence spectroscopies were shown to be associated with the formation of amyloid-like fibrils for wild-type apoMb1-29 (30). Therefore, we concluded that the four scrambled sequence variants formed amyloid-like fibrils under the conditions used here.
FIGURE 3.

Kinetics of aggregation for apoMb1-29 variants. (A) Aggregation was followed by ThT fluorescence (excitation 440 nm, emission 485 nm). (B) Aggregation was followed by far-UV CD at 216 nm. (C) Comparison of ThT and far-UV CD kinetics of aggregation for three representative variants. In all panels open symbols and solid lines refer to ThT fluorescence data; solid symbols and dashed lines refer to CD data; the kinetic traces correspond to wild-type (circles), P1 (inverted triangles), P2 (triangles), P3 (diamonds), and P4 (squares) apoMb1-29 variants. In all cases the insets show the P4 traces on an extended timescale. In all panels the lines through the data points of the P4 variant represent the best fits to a single exponential function (Eq. 9); the lines through the data points of the wild-type, P1, P2, and P3 variants represent the best fits to a sigmoid function (Eq. 10). The parameters calculated for each variant from these analyses (kagg and tlag) are reported in Table 2.
Differences in the aggregation kinetics of the apoMb1-29 variants are determined by differences in the Sagg values
We then followed the kinetics of aggregation of the apoMb1-29 variants by ThT fluorescence and far-UV CD (Fig. 3, A–B). The kinetic traces of P4 apoMb1-29 did not present any detectable lag phase, but only a rapid exponential phase (Fig. 3, A–B, insets). These traces were analyzed with a procedure of best fit using a single exponential function (Eq. 9). By contrast, the kinetic traces of wild-type and P1, P2, and P3 apoMb1-29 presented an important lag phase followed by an exponential growth phase (Fig. 3, A–B). The data were analyzed using a sigmoid function (Eq. 10). The rate constants for the exponential phase (kagg) and the lengths of the lag phase (tlag) obtained for all the variants are reported in Table 2. The kinetic traces obtained for wild-type apoMb1-29, as well as the resulting kagg and tlag values, are consistent with those previously reported (30). For each variant, the kinetic traces and the resulting kagg and tlag values obtained in independent experiments and with the two different techniques (CD and ThT fluorescence) were highly coherent (Table 2; Fig. 3 C).
TABLE 2.
Measured parameters of apoMb1-29 aggregation
| Variant |
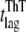 (h)* (h)*
|
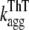 (h−1)* (h−1)*
|
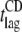 (h)* (h)*
|
 (h−1)* (h−1)*
|
|---|---|---|---|---|
| WT | 15 ± 2 | 0.24 ± 0.02 | 16.4 ± 0.3 | 0.20 ± 0.04 |
| P1 | 29 ± 6 | 0.20 ± 0.01 | 24 ± 6 | 0.28 ± 0.01 |
| P2 | 28.1 ± 0.3 | 0.5 ± 0.1 | 28.0 ± 0.4 | 0.5 ± 0.2 |
| P3 | 20.7 ± 0.2 | 0.64 ± 0.07 | 21.3 ± 0.3 | 1.2 ± 0.4 |
| P4 | 0† | 10 ± 1 | 0† | 9 ± 5 |
Obtained from the analyses of the kinetic plots reported in Fig. 3, A and B. The entries give the mean ± SE in two or three independent experiments, or the value ± SE in the curve fit (case of the ThT values of P3 and P4, and of the CD values of WT and P4).
Lower than the experimental dead time (10 s).
These results show that the various mutants have very different values of tlag and kagg (Table 2) despite their identical amino acid compositions and Zagg values (Table 1). To elucidate the determinants of the observed differences in aggregation time course and resulting kagg and tlag parameters, we assessed whether the kinetic parameters of aggregation correlated with the prominence of the aggregation-promoting region (Sagg). A significant linear correlation was found between kagg and Sagg (Fig. 4 A; r = 0.96; p = 0.01). Thus, the differences in the Sagg values between the apoMb1-29 variants seem to account for the differences between their aggregation rates.
FIGURE 4.

Dependence of the kinetic parameters of aggregation on the Sagg parameter. (A) Correlation between the natural logarithm of the rate constant for the exponential phase (kagg) and Sagg. (B) Correlation between the lag time of aggregation (tlag) and Sagg. Both panels report the mean ± SE on both kagg and tlag, obtained after averaging the ThT fluorescence and CD data reported in Table 2.
Sagg also correlates linearly to tlag, although the p-value lies slightly above the statistical significance threshold of 0.05 in this case (Fig. 4 B; r = 0.84; p = 0.07). This correlation is nevertheless satisfying, considering that the individual pagg used to calculate Sagg were optimized on experimental kagg data rather than tlag values (18). Moreover, it has been demonstrated that the values of kagg and tlag are strictly correlated for a number of variants of insulin, glucagon, and the 40-residue form of the amyloid-β peptide (33), explaining why both kagg and tlag correlate with Sagg. None of the other parameters tested, namely the width of the main aggregation-promoting region on the profile, its height, shape (width/height), number of peaks, or Sagg normalized by the number of peaks, gave any better correlation for either tlag or kagg (results not shown).
Definition of an accurate predictor of aggregation propensity for unstructured polypeptides
Having realized that the most amyloidogenic regions of the sequence, which correspond to the highest values of the 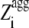 in the profile, mostly determine the kinetic parameters of aggregation of an unstructured protein or peptide. a new Z-score for aggregation was defined, termed
in the profile, mostly determine the kinetic parameters of aggregation of an unstructured protein or peptide. a new Z-score for aggregation was defined, termed 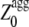 (G.-G. Tartaglia and M. Vendruscolo, unpublished results). The
(G.-G. Tartaglia and M. Vendruscolo, unpublished results). The 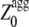 value of an unstructured polypeptide chain is calculated from its sequence by considering only the
value of an unstructured polypeptide chain is calculated from its sequence by considering only the 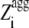 values that appear to be sufficiently high in the profile, after a new optimization of the individual pagg (see Materials and Methods for details). The
values that appear to be sufficiently high in the profile, after a new optimization of the individual pagg (see Materials and Methods for details). The 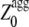 values for all the apoMb1-29 variants studied here are reported in Table 1. Unlike the Zagg values, the
values for all the apoMb1-29 variants studied here are reported in Table 1. Unlike the Zagg values, the 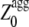 values are different among the apoMb1-29 variants, with the P4 mutant featuring the highest value. Both the experimentally determined kagg and tlag values correlate with
values are different among the apoMb1-29 variants, with the P4 mutant featuring the highest value. Both the experimentally determined kagg and tlag values correlate with 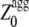 providing statistically significant r- and p-values (r = 0.96, p = 0.01, and r = 0.91, p = 0.03, respectively).
providing statistically significant r- and p-values (r = 0.96, p = 0.01, and r = 0.91, p = 0.03, respectively).
Comparison with other peptides from the structural family of globins
To evaluate the biological significance of the observed aggregation behavior of the apoMb1-29 variants analyzed here, we compared their sequences and aggregation propensity profiles to those of the corresponding peptides from the structural family of globins. All known sequences of globins from different organisms, including those of evolutionary distant phyla such as eubacteria, were aligned. Following alignment, the 745 sequence fragments corresponding to the sequence of apoMb1-29 studied here were considered (see Materials and Methods for details). The distributions of the Zagg and Sagg values calculated over this database are shown in Fig. 5. The apoMb1-29 variants studied in this work have Zagg values inside the interval mean ± 2 SD obtained from the overall database (Fig. 5, inset). Wild-type, P1, and P2 apoMb1-29 also have “ordinary” Sagg values, i.e., in the interval mean ± 2 SD (Fig. 5). On the contrary, both P3 and P4, particularly the latter, have Sagg values significantly higher than the mean value of the dataset (higher than mean ± 2 SD; Fig. 5). The P3 and P4 variants were designed without adding additional residues with a high aggregation propensity, but simply by scrambling the existing sequence of apoMb1-29. Thus it appears that the clustering of amino acids with high individual propensities to aggregate in the center of the sequence, and their resulting high kagg and low tlag values, represent an unusual situation within the structural family of globins.
FIGURE 5.

Distribution of Sagg (main panel) and Zagg (inset) among the structural family of globins. In both the major and inset figures the vertical solid and dashed lines indicate the mean and mean ± 2 SD values, respectively. The Sagg values for all the apoMb1-29 variants studied here are shown with dotted vertical lines. The Zagg value for all apoMb1-29 variants is 0.37 and falls within the mean ± 2 SD interval.
DISCUSSION
The clustering of residues with high amyloidogenic propensity promotes aggregation
Methods developed to predict the absolute aggregation rate of a polypeptide chain and the change of rate following mutation often consider the aggregation propensity of each residue to be independent of its position along the sequence. As a result, scrambled sequences are predicted to have similar propensities and rates of aggregation as their wild-type counterpart. However, aggregation appears to be driven by intermolecular interactions that take place between a limited set of sequence regions (34–37). The question therefore arises as to the importance of such regions in the determination of the aggregation rate, and whether or not the residues involved should be considered as partially dependent on the neighboring residues.
In this work we have shown that five initially unstructured peptides, sharing the same length and amino acid composition but with scrambled sequences, present very different behaviors in terms of amyloid fibril formation kinetics. The lag times, corresponding to the rates of formation of oligomeric nuclei during the lag phase, span from <10 s (the dead time in recording the aggregation trace of P4 apoMb1-29) to 30 h. Moreover, the rate constants of fibril elongation differ by almost 2 orders of magnitude. In particular, the clustering of the most amyloidogenic residues within one region of the sequence increases by ∼40-fold the fibril elongation rate, and shortens the lag time below detectable levels (Table 2). Scrambled variants of a sequence corresponding to the N-terminal and middle regions of the Sup-35 prion were also found to aggregate in vitro with different kinetics (38). In a recent development of their algorithm that predicts amyloid aggregation propensity, Ventura and colleagues defined a parameter that for each peak of the profile measures the area lying above a certain threshold (15). They noticed that half of the sequence segments experimentally known to be involved in aggregation correspond to the peaks of major area within the various analyzed sequences (15). Simulation studies reinforced all these experimental results. A lattice-based computer simulation showed an increasing propensity to aggregate as the hydrophobic residues became concentrated in fewer continuous subsequences (39). Using a toy model to simulate the aggregation of unfolded polypeptides, the dispersion of critical residues was identified as a determinant of the aggregation rate (40). Hence, the distribution of residues with high amyloidogenic propensities within the sequence appears to be an important determinant of aggregation kinetics, both kagg and tlag.
How can we explain the increased aggregation rate constants and decreased lag times observed when the most amyloidogenic residues are gathered within the sequence? The correlations that we observe between Sagg and both kagg and tlag suggest a possible rationalization at the molecular level. During the lag phase, a series of thermodynamically unfavorable oligomeric assemblies of increasing size are formed. These oligomers become stable when they exceed the size of the “critical nucleus”, which depends on the specific sequence and the conditions of the solution. Further addition of monomers to the critical nucleus is then thermodynamically favorable (41). The clustering of the amino acids that are more prone to aggregate, and thus that are thought to establish the intermolecular interactions that drive aggregation, may create the possibility of realizing critical nuclei of small size by increasing the strength of intermolecular interactions. It could also increase the probability of productive collisions of monomers (lag phase) or of monomers or oligomers with a growing fibril (elongation phase), thus contributing to accelerate both the lag and elongation phases, respectively (42).
The clustering of residues with high amyloidogenic propensities is negatively selected
A number of proteins are intrinsically disordered, i.e., they normally exist in an unstructured state unless they are bound to their substrates or macromolecular targets (43). Even globular proteins, forming a compact folded state during most of their lifetimes, can transiently adopt an unfolded state, particularly during biosynthesis, translocation, or stress conditions. Thus, avoiding aggregation from an unstructured state is generally an issue for natural proteins. If the clustering of residues with high aggregation propensities is so deleterious, one would expect it to be negatively selected by evolution. Our analysis of the globin family clearly indicates that unbalanced distributions of amyloidogenic residues, with a resulting clustering of the most highly amyloidogenic ones within one or few portions of the sequence (such as in P3 and P4 apoMb1-29), has been selected against by natural evolution in the N-terminal trait of this structural family (Fig. 5). P3 and P4 apoMb1-29 variants are artificially designed sequences, which are probably incompatible with the native fold of the whole apoMb protein. However, aggregation profiles with shape and Sagg values comparable to those of P3 and P4 can be obtained with one or two single-point mutations in the central region of the apoMb1-29 sequence, a situation that is more realistic from an evolutionary point of view (data not shown). The fact remains that sequences with profiles and Sagg values similar to those of the P3 and P4 variants are very rare, if not at all absent, indicating the existence of a negative selection against such an unfavorable situation.
Hints of negative selection against long stretches of hydrophobic residues (44–46), patterns of alternating polar and nonpolar residues (47), or stretches with very high aggregation propensity, calculated according to the TANGO algorithm (48), have been observed on more extended databases. All these results are highly consistent with our findings, as hydrophobicity and patterns of alternating polar and nonpolar residues contribute to the amyloidogenic propensity of each amino acid (18). By constructing an artificial system in which the only changed parameter was the dispersion of the amino acid residues within a sequence, we provided an experimental confirmation of what was suggested from the database analysis: the deleterious effect of, and negative selection against, long stretches of highly amyloidogenic residues. This adds to the accumulating evidence that protein sequences have evolved sequence and structural adaptations to prevent aggregation (reviewed in Monsellier and Chiti (49)).
Existence of a “permissive window” for aggregating stretches
Whereas the P3 and P4 variants of apoMb1-29 have high values of Sagg and correspondingly high values of kagg compared to the wild-type peptide, the P1 and P2 variants have only marginally lower values of Sagg (Table 1). The 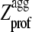 of the P1 variant is lower than 1.0 throughout the sequence (Fig. 1 A), causing its Sagg value to be the lowest possible, that is 0 (Table 1). However, the kagg values of P1 and P2 apoMb1-29 are similar to that determined for the wild-type peptide (Table 2), indicating that the dispersion of the amyloidogenic residues from the center to the edges of the sequence and the subsequent suppression of the central aggregating region does not reduce dramatically aggregation. It therefore appears that maintenance of an aggregating segment within a sequence does not lead to severe consequences in terms of aggregation, provided that the aggregation propensity of such segment is not too marked.
of the P1 variant is lower than 1.0 throughout the sequence (Fig. 1 A), causing its Sagg value to be the lowest possible, that is 0 (Table 1). However, the kagg values of P1 and P2 apoMb1-29 are similar to that determined for the wild-type peptide (Table 2), indicating that the dispersion of the amyloidogenic residues from the center to the edges of the sequence and the subsequent suppression of the central aggregating region does not reduce dramatically aggregation. It therefore appears that maintenance of an aggregating segment within a sequence does not lead to severe consequences in terms of aggregation, provided that the aggregation propensity of such segment is not too marked.
In addition, it was shown that a delicate tradeoff between positive and negative selections exists: the requirement for a proper and stable fold acquired in a reasonable time on the one hand, and the necessity to avoid aggregation on the other hand, two parameters that cannot be optimized simultaneously (50,51). Mutations decreasing the aggregation potential of an aggregating segment within a normally folded protein generally results in a destabilization of the native structure (52). These independent observations suggest that the sequence segments with relatively high aggregation propensity existing in globular proteins are indeed essential for the achievement of a three-dimensional structure, and are easily tolerated because they do not represent a severe problem in aggregation.
Different strategies have been developed by natural evolution to counteract the aggregation of a protein, both by using dedicated cellular machineries, such as chaperones and quality control mechanisms (53,54), and at the sequence level, such as gatekeeper residues, negative design of the β-sheets, etc. (49). This arsenal may be redundant, which could allow the cell to deal with sequences containing short stretches of aggregation-prone residues, necessary for a proper folding.
Overall, our data suggest the existence of a “permissive window” for stretches of aggregation-prone residues: highly aggregating stretches are prevented, but moderately aggregating ones are tolerated. This permissibility might be of fundamental importance in the evolutionary search of modified or novel sequences, as it would guarantee a certain degree of freedom in the search of novel biologically relevant sequences with no significant consequences on protein aggregation.
SUPPLEMENTARY MATERIAL
To view all of the supplemental files associated with this article, visit www.biophysj.org.
Supplementary Material
Acknowledgments
This work was supported by grants from the EMBO Young Investigator Program of the European Molecular Biological Organisation and the Italian Ministero dell'Istruzione, Università e'Ricerca (FIRB Projects No. RBIN04PWNC and RBNE03PX83 and PRIN Project No. 2005027330).
Abbreviations used: apoMb, horse heart apomyoglobin; CD, circular dichroism; FTIR, Fourier transform infrared; SD, standard deviation; SE, standard error; ThT, thioflavin T; UV, ultraviolet; WT, wild-type.
Editor: Heinrich Roder.
References
- 1.Dobson, C. M. 2003. Protein folding and misfolding. Nature. 426:884–890. [DOI] [PubMed] [Google Scholar]
- 2.Chiti, F., and C. M. Dobson. 2006. Protein misfolding, functional amyloid, and human disease. Annu. Rev. Biochem. 75:333–366. [DOI] [PubMed] [Google Scholar]
- 3.Fowler, D. M., A. V. Koulov, W. E. Balch, and J. W. Kelly. 2007. Functional amyloid—from bacteria to humans. Trends Biochem. Sci. 32:217–224. [DOI] [PubMed] [Google Scholar]
- 4.Berson, J. F., A. C. Theos, D. C. Harper, D. Tenza, G. Raposo, and M. S. Marks. 2003. Proprotein convertase cleavage liberates a fibrillogenic fragment of a resident glycoprotein to initiate melanosome biogenesis. J. Cell Biol. 161:521–533. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Fowler, D. M., A. V. Koulov, C. Alory-Jost, M. S. Marks, W. E. Balch, and J. W. Kelly. 2006. Functional amyloid formation within mammalian tissue. PLoS Biol. 4:e6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Chien, P., J. S. Weissman, and A. H. DePace. 2004. Emerging principles of conformation-based prion inheritance. Annu. Rev. Biochem. 73:617–656. [DOI] [PubMed] [Google Scholar]
- 7.Bieler, S., L. Estrada, R. Lagos, M. Baeza, J. Castilla, and C. Soto. 2005. Amyloid formation modulates the biological activity of a bacterial protein. J. Biol. Chem. 280:26880–26885. [DOI] [PubMed] [Google Scholar]
- 8.Selkoe, D. J. 2003. Folding proteins in fatal ways. Nature. 426:900–904. [DOI] [PubMed] [Google Scholar]
- 9.Hamada, D., I. Yanagihara, and K. Tsumoto. 2004. Engineering amyloidogenicity towards the development of nanofibrillar materials. Trends Biotechnol. 22:93–97. [DOI] [PubMed] [Google Scholar]
- 10.Rajagopal, K., and J. P. Schneider. 2004. Self-assembling peptides and proteins for nanotechnological applications. Curr. Opin. Struct. Biol. 14:480–486. [DOI] [PubMed] [Google Scholar]
- 11.Chiti, F., M. Stefani, N. Taddei, G. Ramponi, and C. M. Dobson. 2003. Rationalization of the effects of mutations on peptide and protein aggregation rates. Nature. 424:805–808. [DOI] [PubMed] [Google Scholar]
- 12.Fernandez-Escamilla, A.-M., F. Rousseau, J. Schymkowitz, and L. Serrano. 2004. Prediction of sequence-dependent and mutational effects on the aggregation of peptides and proteins. Nat. Biotechnol. 22:1302–1306. [DOI] [PubMed] [Google Scholar]
- 13.Tartaglia, G. G., A. Cavalli, R. Pellarin, and A. Caflisch. 2004. The role of aromaticity, exposed surface, and dipole moment in determining protein aggregation rates. Protein Sci. 13:1939–1941. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Sanchez de Groot, N., F. X. Aviles, J. Vendrell, and S. Ventura. 2006. Mutagenesis of the central hydrophobic cluster in Abeta42 Alzheimer's peptide. Side-chain properties correlate with aggregation propensities. FEBS J. 273:658–668. [DOI] [PubMed] [Google Scholar]
- 15.Conchillo-Solé, O., N. Sanchez de Groot, F. X. Avilès, J. Vendrell, X. Daura, and S. Ventura. 2007. AGGRESCAN: a server for the prediction and evaluation of “hot spots” of aggregation in polypeptides. BMC Bioinformatics. 8:65. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.DuBay, K. F., A. P. Pawar, F. Chiti, J. Zurdo, C. M. Dobson, and M. Vendruscolo. 2004. Prediction of the absolute aggregation rates of amyloidogenic polypeptide chains. J. Mol. Biol. 341:1317–1326. [DOI] [PubMed] [Google Scholar]
- 17.Tartaglia, G. G., A. Cavalli, R. Pellarin, and A. Caflisch. 2005. Prediction of aggregation rate and aggregation-prone segments in polypeptide sequences. Protein Sci. 14:2723–2734. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Pawar, A. P., K. F. Dubay, J. Zurdo, F. Chiti, M. Vendruscolo, and C. M. Dobson. 2005. Prediction of “aggregation-prone” and “aggregation-susceptible” regions in proteins associated with neurodegenerative diseases. J. Mol. Biol. 350:379–392. [DOI] [PubMed] [Google Scholar]
- 19.Sanchez de Groot, N., I. Pallares, F. X. Aviles, J. Vendrell, and S. Ventura. 2005. Prediction of “hot spots” of aggregation in disease-linked polypeptides. BMC Struct. Biol. 5:18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Thompson, M. J., S. A. Sievers, J. Karanicolas, M. I. Ivanova, D. Baker, and D. Eisenberg. 2006. The 3D profile method for identifying fibril-forming segments of proteins. Proc. Natl. Acad. Sci. USA. 103:4074–4078. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Galzitskaya, O. V., S. O. Garbuzynskiy, and M. Y. Lobanov. 2007. Prediction of amyloidogenic and disordered regions in protein chains. PLoS Computational Biology. 2:e177. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Trovato, A., F. Chiti, A. Maritan, and F. Seno. 2007. Insight into the structure of amyloid fibrils from the analysis of globular proteins. PLoS Computational Biology. 2:e170. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Ventura, S., J. Zurdo, S. Narayanan, M. Parreno, R. Mangues, B. Reif, F. Chiti, E. Giannoni, C. M. Dobson, F. X. Aviles, and L. Serrano. 2004. Short amino acid stretches can mediate amyloid formation in globular proteins: the Src homology 3 (SH3) case. Proc. Natl. Acad. Sci. USA. 101:7258–7263. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Esteras-Chopo, A., L. Serrano, and M. Lopez de la Paz. 2005. The amyloid stretch hypothesis: recruiting proteins toward the dark side. Proc. Natl. Acad. Sci. USA. 102:16672–16677. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Bemporad, F., G. Calloni, S. Campioni, G. Plakoutsi, N. Taddei, and F. Chiti. 2006. Sequence and structural determinants of amyloid fibril formation. Acc. Chem. Res. 39:620–627. [DOI] [PubMed] [Google Scholar]
- 26.Fandrich, M., M. A. Fletcher, and C. M. Dobson. 2001. Amyloid fibrils from muscle myoglobin. Nature. 410:165–166. [DOI] [PubMed] [Google Scholar]
- 27.Fandrich, M., G. Zandomeneghi, M. R. Krebs, M. Kittler, K. Buder, A. Rossner, S. H. Heinemann, C. M. Dobson, and S. Diekmann. 2006. Apomyoglobin reveals a random-nucleation mechanism in amyloid protofibril formation. Acta Histochem. 108:215–219. [DOI] [PubMed] [Google Scholar]
- 28.Jamin, M. 2005. The folding process of apomyoglobin. Protein Pept. Lett. 12:229–234. [DOI] [PubMed] [Google Scholar]
- 29.Nishimura, C., M. A. Lietzow, H. J. Dyson, and P. E. Wright. 2005. Sequence determinants of a protein folding pathway. J. Mol. Biol. 351:383–392. [DOI] [PubMed] [Google Scholar]
- 30.Picotti, P., G. De Franceschi, E. Frare, B. Spolaore, M. Zambonin, F. Chiti, P. Polverino de Laureto, and A. Fontana. 2007. Amyloid fibril formation and disaggregation of fragment 1–29 of apomyoglobin: insights into the effect of pH on protein fibrillogenesis. J. Mol. Biol. 367:1237–1245. [DOI] [PubMed] [Google Scholar]
- 31.Nielsen, L., R. Khurana, A. Coats, S. Frokjaer, J. Brange, S. Vyas, V. N. Uversky, and A. L. Fink. 2001. Effect of environmental factors on the kinetics of insulin fibril formation: elucidation of the molecular mechanism. Biochemistry. 40:6036–6046. [DOI] [PubMed] [Google Scholar]
- 32.Ptitsyn, O. B., and K. L. Ting. 1999. Non-functional conserved residues in globins and their possible role as a folding nucleus. J. Mol. Biol. 291:671–682. [DOI] [PubMed] [Google Scholar]
- 33.Fandrich, M. 2007. Absolute correlation between lag time and growth rate in the spontaneous formation of several amyloid-like aggregates and fibrils. J. Mol. Biol. 365:1266–1270. [DOI] [PubMed] [Google Scholar]
- 34.Chiti, F., N. Taddei, F. Baroni, C. Capanni, M. Stefani, G. Ramponi, and C. M. Dobson. 2002. Kinetic partitioning of protein folding and aggregation. Nat. Struct. Biol. 9:137–143. [DOI] [PubMed] [Google Scholar]
- 35.Ritter, C., M. L. Maddelein, A. B. Siemer, T. Luhrs, M. Ernst, B. H. Meier, S. J. Saupe, and R. Riek. 2005. Correlation of structural elements and infectivity of the HET-s prion. Nature. 435:844–848. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Petkova, A. T., W. M. Yau, and R. Tycko. 2006. Experimental constraints on quaternary structure in Alzheimer's beta-amyloid fibrils. Biochemistry. 45:498–512. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Heise, H., W. Hoyer, S. Becker, O. C. Andronesi, D. Riedel, and M. Baldus. 2005. Molecular-level secondary structure, polymorphism, and dynamics of full-length alpha-synuclein fibrils studied by solid-state NMR. Proc. Natl. Acad. Sci. USA. 102:15871–15876. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Liu, Y., H. Wei, J. Wang, J. Qu, W. Zhao, and H. Tao. 2007. Effects of randomizing the Sup35NM prion domain sequence on formation of amyloid fibrils in vitro. Biochem. Biophys. Res. Commun. 353:139–146. [DOI] [PubMed] [Google Scholar]
- 39.Istrail, S., R. Schwartz, and J. King. 1999. Lattice simulations of aggregation funnels for protein folding. J. Comput. Biol. 6:143–162. [DOI] [PubMed] [Google Scholar]
- 40.Hall, D., N. Hirota, and C. M. Dobson. 2005. A toy model for predicting the rate of amyloid formation from unfolded protein. J. Mol. Biol. 351:195–205. [DOI] [PubMed] [Google Scholar]
- 41.Wetzel, R. 2006. Kinetics and thermodynamics of amyloid fibril assembly. Acc. Chem. Res. 39:671–679. [DOI] [PubMed] [Google Scholar]
- 42.Yan, Y., and C. Wang. 2006. Aβ42 is more rigid than Aβ40 at the C terminus: implications for Aβ aggregation and toxicity. J. Mol. Biol. 364:853–862. [DOI] [PubMed] [Google Scholar]
- 43.Uversky, V. N. 2002. Natively unfolded proteins: a point where biology waits for physics. Protein Sci. 11:739–756. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Schwartz, R., S. Istrail, and J. King. 2001. Frequencies of amino acid strings in globular protein sequences indicate suppression of blocks of consecutive hydrophobic residues. Protein Sci. 10:1023–1031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Schwartz, R., and J. King. 2006. Frequencies of hydrophobic and hydrophilic runs and alternations in proteins of known structure. Protein Sci. 15:102–112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Patki, A. U., A. C. Hausrath, and M. H. Cordes. 2006. High polar content of long buried blocks of sequence in protein domains suggests selection against amyloidogenic non-polar sequences. J. Mol. Biol. 362:800–809. [DOI] [PubMed] [Google Scholar]
- 47.Broome, B. M., and M. H. Hecht. 2000. Nature disfavors sequences of alternating polar and non-polar amino acids: implications for amyloidogenesis. J. Mol. Biol. 296:961–968. [DOI] [PubMed] [Google Scholar]
- 48.Rousseau, F., L. Serrano, and J. W. Schymkowitz. 2006. How evolutionary pressure against protein aggregation shaped chaperone specificity. J. Mol. Biol. 355:1037–1047. [DOI] [PubMed] [Google Scholar]
- 49.Monsellier, E., and F. Chiti. 2007. Prevention of amyloid-like aggregation as a driving force of protein evolution. EMBO Rep. 8:737–742. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Bastolla, U., A. Moya, E. Viguera, and R. C. Van Ham. 2004. Genomic determinants of protein folding thermodynamics in prokaryotic organisms. J. Mol. Biol. 343:1451–1466. [DOI] [PubMed] [Google Scholar]
- 51.Bastolla, U., and L. Demetrius. 2005. Stability constraints and protein evolution: the role of chain length, composition and disulfide bonds. Protein Eng. Des. Sel. 18:405–415. [DOI] [PubMed] [Google Scholar]
- 52.Sanchez, I. E., J. Tejero, C. Gomez-Moreno, M. Medina, and L. Serrano. 2006. Point mutations in protein globular domains: contributions from function, stability and misfolding. J. Mol. Biol. 363:422–432. [DOI] [PubMed] [Google Scholar]
- 53.Young, J. C., V. R. Agagshe, K. Siegers, and F. U. Hartl. 2004. Pathways of chaperone-mediated protein folding in the cytosol. Nat. Rev. Mol. Cell Biol. 5:781–791. [DOI] [PubMed] [Google Scholar]
- 54.Bukau, B., J. Weissman, and A. Horwich. 2006. Molecular chaperones and proteins quality control. Cell. 125:443–451. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.