

Abstract
Inactivation is an inherent property of most voltage-gated K+ channels. While fast N-type inactivation has been analyzed in biophysical and structural details, the mechanisms underlying slow inactivation are yet poorly understood. Here, we characterized a slow inactivation mechanism in various KCNQ1 pore mutants, including L273F, which hinders entry of external Ba2+ to its deep site in the pore and traps it by slowing its egress. Kinetic studies, molecular modeling, and dynamics simulations suggest that this slow inactivation involves conformational changes that converge to the outer carbonyl ring of the selectivity filter, where the backbone becomes less flexible. This mechanism involves acceleration of inactivation kinetics and enhancement of Ba2+ trapping at elevated external K+ concentrations. Hence, KCNQ1 slow inactivation considerably differs from C-type inactivation where vacation of K+ from the filter was invoked. We suggest that trapping of K+ at s1 due to filter rigidity and hindrance of the dehydration-resolvation transition underlie the slow inactivation of KCNQ1 pore mutants.
INTRODUCTION
Potassium channels catalyze fast transfer of K+ ions across the cell membrane at nearly diffusion-limit rates while selecting K+ over Na+ by >100-fold. Structure determination at atomic resolution of bacterial and more recently mammalian K+ channels has proven to be a landmark achievement in the field of ion channels (1,2). K+ channels comprise four subunits arranged symmetrically around an ion-conducting pore. In voltage-gated K+ channels (Kv), each subunit consists of six transmembrane segments, including an S5–S6 region encompassing the aqueous pore and a peripheral S1–S4 voltage sensor domain (3). The narrowest part of the pore is the selectivity filter which achieves a remarkably fast and selective permeation of K+ ions. The filter's geometry, a tunnel-like structure of 12 Å length and 3 Å diameter, provides an efficient permeation pathway for dehydration, diffusion, and rehydration of K+ ions (4). To this end, the selectivity filter of K+ channels is endowed with four K+ coordination sites. The first three sites s1–s3 are formed by four rings of backbone carbonyl oxygen atoms, while the inner s4 site is formed by both backbone carbonyl oxygens and side-chain hydroxyl oxygens (5,6). The K+ coordination geometry in the selectivity filter mimics the coordination of K+ ions by water molecules. Under physiological K+ concentrations, two K+ ions reside in the selectivity filter at a given time and move in a highly concerted fashion between two isoenergetic configurations (7).
Several lines of evidence suggest that the selectivity filter has also the ability to act as a gate by switching between conducting and nonconducting conformations (8–12). Most recent studies revealed that the selectivity filter of the bacterial KcsA K+ channel can undergo large conformational excursions associated with transitions between closed, open, and inactivated states (13,14).
Inactivation of Kv channels can occur by N-type or/and C-type mechanisms. The fast N-type inactivation involves an intracellular peptide domain at the N-terminus of α- or β-subunits which occludes the channel central cavity and prevents ion permeation (15–17). The slow C-type inactivation was suggested to involve structural rearrangements in the outer pore leading to a loss of K+ coordination sites in the selectivity filter (18–21). The biophysical hallmarks of C-type inactivation are reflected by its inhibition by high external K+ or external tetraethylammonium (15,22,23). These features have been interpreted as a foot-in-the-door mechanism, in which occupancy of an ion binding site by K+ or TEA at the external filter entrance slows or prevents the conformational changes required for C-type inactivation (19). It should be noted that in Shaker channels, a Ba2+ site was identified topologically below the C-type inactivation gate (24).
KCNQ1 channels (Kv7.1) belong to a subfamily of voltage-gated K+ channels, Kv7, and coassemble with KCNE1 β-subunits to generate the IKS potassium current that is critical for normal repolarization of the cardiac action potential (25–28). Mutations in either KCNQ1 or KCNE1 genes produce the long QT syndrome (LQT), a human ventricular arrhythmia (26,27,29). Inactivation of KCNQ1 channels does not exhibit the hallmarks of N- and C-type inactivation. Inactivation of wild-type (WT) KCNQ1 channels is invisible macroscopically but can be revealed by hooked tail currents which reflect recovery from an inactivation process (30–32). Here, we characterized KCNQ1 pore mutants, including the LQT mutation L273F, which exhibit a voltage-dependent slow inactivation. We found that this slow inactivation delays entry of Ba2+ ions to the pore and trap them by slowing their exit from the selectivity filter. External potassium ions accelerate inactivation kinetics and exacerbate barium trapping. Our experimental data together with molecular modeling suggest that the voltage-dependent slow inactivation arises from a reduced flexibility of the carbonyl ring of Tyr315 and a stronger coordination of K+ at filter site s1.
MATERIALS AND METHODS
Channel expression into Xenopus oocytes
Female Xenopus Laevis frogs were purchased from Xenopus 1 (Dexter, MI). The procedures followed for surgery and maintenance of frogs were approved by the animal research ethics committee of Tel Aviv University and in accordance with the Guide for the Care and Use of Laboratory Animals (1996, National Academy of Sciences, Washington, DC). Frogs were anaesthetized with 0.15% tricaine (Sigma, St. Louis, MO). Pieces of the ovary were surgically removed and digested with 1 mg/ml collagenase (type IA, Sigma) in Ca2+-free ND96 for ∼1 h, to remove follicular cells. Stage V and VI oocytes were selected for cRNA or DNA injection and maintained at 18°C in ND96 (96 mM NaCl, 2 mM KCl, 1.8 mM CaCl2, 1 mM MgCl2 and 5 mM HEPES titrated to pH = 7.5 with NaOH), supplemented with 1 mM pyruvate and 50 μg/ml gentamycin. The human KCNQ1 cDNA (in pGEM vector) was linearized by Not1. Capped complementary RNA was transcribed by the T7 RNA polymerase, using the mMessage mMachine transcription kit (Ambion, Austin, TX). The cRNA size and integrity was confirmed by formaldehyde-agarose gel electrophoresis. Homomeric expression of human KCNQ1 was performed by injecting 40 nl per oocyte (5 ng cRNA) using a Nanoject injector (Drummond Scientific, Broomall, PA). Several expression experiments were also carried out by microinjecting a recombinant DNA vector (pcDNA3) encoding the human KCNQ1 cDNA directly into Xenopus oocyte nuclei (1 ng into10 nl).
Electrophysiology
Standard two-electrode voltage-clamp measurements were performed at room temperature (22–24°C) 2–5 days after cRNA or DNA microinjection. Oocytes were placed into a 100 μl recording chamber and superfused with a modified ND96 solution (containing 0.1 mM CaCl2) using a fast perfusion system which operates under controlled N2 pressure allowing constant perfusion velocity of 3.9–4.2 ml/min. The exchange of solutions was performed by computer-controlled pinch valves (ALA-VM8; ALA Scientific Instruments, Westbury, NY). A homemade manifold having virtually no void volume and very narrow connecting tubes prevented backward flow upon valve switch. The bath solution was completely replaced within 1.5 s, allowing a solution exchange time of ∼25 ms around the oocyte. Whole-cell currents were recorded using a GeneClamp 500 amplifier (Axon Instruments, Foster City, CA). Stimulation of the preparation and data acquisition were performed using the pCLAMP 6.02 software (Axon Instruments) and a 586 personal computer interfaced with a Digidata 1200 interface (Axon Instruments). Glass microelectrodes (A-M Systems, Carlsborg, WA) were filled with 3 M KCl and had tip resistances of 0.2–0.5 MΩ. Current signals were digitized at 1 kHz and low-pass-filtered at 0.2 kHz. The holding potential was −80 mV. Leak subtraction was performed off-line, using steps from −120 to −90 mV, assuming that the channels are closed at −80 mV and below. Errors introduced by the series resistance of the oocytes were not corrected and were minimized by keeping expression of the currents below 10 μA. All BaCl2 solutions were prepared in modified ND96 (containing 0.1 mM CaCl2) and were isoosmotically changed for NaCl. Modified ND96 solutions containing high K+ concentrations (50 mM) were also isoosmotically changed for NaCl.
Data analyses
Data analysis was performed using the Clampfit program (pCLAMP 8, Axon Instruments), Microsoft Excel 2002 (Microsoft, Redmond, WA), SigmaPlot 8.0 (SPSS, New York, NY) and Prism 4 (GraphPad Software, San Diego, CA). To analyze the voltage dependence of channel activation, a double exponential fit was applied to the tail currents at −60 mV or −120 mV and the slow exponential component was extrapolated to the beginning of the repolarizing step. Chord conductance (G) was calculated by using the equation
 |
(1) |
where I corresponds to the extrapolated tail current, and Vrev is the reversal potential measured in each experiment, which was −98 ± 2 mV (n = 10). G was estimated at the tail voltage V and then, normalized to a maximal conductance value, Gmax. Activation curves were fitted to a Boltzmann distribution of
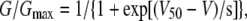 |
(2) |
where V50 is the voltage at which the current is half-activated and s is the slope factor. For a measure of inactivation kinetics, we used a three-pulse protocol where the membrane potential was stepped to a +30 mV conditioning prepulse for increasing durations (200 ms increments) to progressively activate and inactivate the channel; then, a brief (15 ms) hyperpolarizing interpulse to −130 mV was used to allow channel recovery from inactivation before a +30 mV test pulse was applied to reopen and reinactivate KCNQ1 channels. The decaying current of the third test pulse (reinduction of inactivation) could be fitted by a single exponential function. For a quantitative measure of fractional inactivation that takes into account the electrochemical driving force, G was deduced from tail currents at either −60 mV or −120 mV where tail relaxations reveal a recovery from inactivation (hook) faster than deactivation. Currents recorded at the voltage V were first converted to conductance G. The time course of the tail conductance was then fitted to a triple exponential function of the form
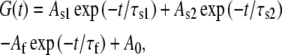 |
(3) |
where a fast time constant τf reflects the recovery from inactivation at the tail potential and two slow time constants τs1 and τs2 describe the deactivation process and A0 is a steady-state current. At the tail potential, the amplitude Af is directly related to the degree of inactivation and the ratio Af/(As1+As2) is a good indicator of the probability of the channels being in the inactivated state.
The voltage-dependence of Ba2+ block was calculated according to the Woodhull's model (33) and the voltage dependence of the dissociation constant is given by
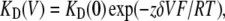 |
(4) |
where KD(0) is the dissociation constant at zero voltage, z is the ion valency, F is the Faraday constant, δ is the fractional potential drop at the binding site (fractional electrical distance), V is the membrane potential (in volts), R is the gas constant, and T is the absolute temperature. All data were expressed as mean ± SE. Statistically significant differences between paired groups were assessed by Student's t-test.
Kinetic model
Initially, we calculated the conductance time course by dividing the current values observed during voltage step from −80 mV to +20 mV by the driving force assuming that the reversal potential for K+ in Xenopus oocytes is −98 mV.
The time course of activation of WT KCNQ1 could be described as a Markov process (31,34,35) by Scheme I, in which k values are the rate constants in s−1 and C1 to I1 are the corresponding state occupancies. Only O1 and O2 are assumed to conduct. Scheme I is sufficient for the description of macroscopic behavior of KCNQ1 channels (31), assuming that the stochastic behavior of a single channel is reflected in the macroscopic behavior of a channel population (36).
SCHEME I.

k-values are the rate constants in s−1 and C1 to I1 are the corresponding state occupancies. Only O1 and O2 are assumed to conduct.
The time course of activation of L273F mutant was characterized by two inactivation processes: 1), fast inactivation, similar to that of the wild-type; and 2), slow inactivation. Among several possible schemes of activation we have chosen Scheme II, which includes both inactivation processes and assumes that the inactivation states can be accessed from different open states (see Results).
SCHEME II.

Characterized by two inactivated states: 1), fast inactivated state I1, similar to that of the wild-type; and 2), slow inactivated state I2, which can be accessed from two different open states O2 and O1, respectively.
We generated a set of differential equations based on Scheme II and fitted it to the observed time-course of the L273F mutant activation (to obtain the transition rate constants and relative occupancies of each state). For simplicity, we assumed that no openings occur when the cell is voltage-clamped to −80 mV (the holding potential) and consequently we fixed initial values of all states to zero, except C1.
According to Scheme II, the open channel probability can be calculated from
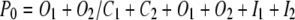 |
(5) |
and the relative state occupancy (φ) is defined as
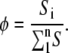 |
(6) |
We fixed the k4 and k−4 values (which are assumed to be voltage-independent) and also the initial C1 to those obtained from the previous procedure, and then fitted the activation from the holding potential (−80 mV) to the −60-mV voltage step.
For the simulation, we used the averaged values of the rate constants and the relative state occupancies obtained from the fit of the activation time course during the +20-mV voltage step and allowed the simulation to run for predefined time intervals. Thereafter, we used the simulated values and changed the rate constants to those corresponding to the −60-mV voltage step.
Time-course fitting and simulation were done with Berkeley Madonna software (Ver. 8.0.2 for Windows; Kagi Shareware, Perth, Western Australia).
Model building
As a first step, the sequence of the S5-pore-S6 segment of the human KCNQ1 channel (Ile257–Gln357; SWISS-PROT entry P51787) was submitted for searching a homologous template in the SWISS-MODEL repository (http://swissmodel.expasy.org/SWISS-MODEL.html). The search for homologous sequences of known three-dimensional structure scored the mammalian Shaker Kv1.2 potassium channel (PDB ID code 2A79) with the highest probability to match as a structural template. The KCNQ1 sequence was therefore aligned with the Ala323–Arg419 sequence of the Shaker Kv1.2 channel using the program T-COFFEE (37) (Supplementary Material Fig. 3), and the alignment was submitted to automated comparative protein modeling via the SWISS-PROT alignment interface (38). The root mean-square difference between the KCNQ1 structural model and the template (Shaker Kv1.2) was 0.11 Å for 96 Cα atoms of the aligned amino acids (0.27 Å for 384 backbone atoms). Note that this superposition does not include amino acids 290–294, which form an extra loop structure owing to an alignment gap in the turret region. Four identical subunit models were organized around the axis of K+ conduction by three-dimensional superposition of 188 backbone atoms of amino acids Thr311–Gln357 onto the homologous segment, Thr373–Arg419, of the Shaker Kv1.2 x-ray crystal structure. Energy minimization of the tetrameric KCNQ1 model was performed in vacuum with GROMOS96. No clashes within the individual subunit or at the subunit interfaces have been observed before or after energy minimization. The models of KCNQ1 mutants were elaborated by introducing the specific mutation in all subunits of the tetrameric KCNQ1 structural model before subjecting the models to energy minimization. In the case of mutant L273F, the side chain of Phe was oriented in the same direction as the side chain of the replaced Leu residue. For Trp substituted at position 273, only one side-chain conformation was energetically favored, as all other side-chain rotamers drastically clashed with the pore helix, even after a step of energy minimization. For the models of the other mutants, see Supplementary Material.
Molecular dynamics simulations
The primary hydration shell method (39) is a method that provides both an efficient representation of solvation effects and a flexible nonspherical restraining potential. To replace the bulk representation of the solvent, a restraining force that balances the instantaneous pressure inside the primary solvent shell is applied. The molecular dynamics (MD) simulations were performed using CHARMM, version 26 (40). We used structurally restrained MD simulations that maintain the structural integrity of the helical TM domain. Specifically, we imposed NOE type restraints on α-helical backbone distances as well as harmonic restraints on the Cα atoms of residues in the outer helices. The region we examined was comprised of the extracellular loops and the selectivity filter. This region was surrounded by a 30 Å sphere of water molecules whose origin was located at its center of mass. Water molecules located within 2.6 Å or further than 15 Å from these residues were deleted and the combined system was subjected to energy minimization. Minimizations were performed using the steepest-descent and the adopted-basis Newton Raphson algorithms. After removing all waters located 5.5 Å from the examined region, the system was further minimized. Three K+ ions were then placed at sites s0, s2, and s4 of the selectivity filter, and the system was minimized again. The structure was then equilibrated and an MD simulation was carried out for 1.5 ns with a time-step of 0.5 fs, and with a restraining force of 0.95 Kcal/mole Å. The analysis was done using structures recorded every 0.5 ps with Origin6.1. Statistical analysis of the third and fourth moments was performed using JPM6.0.0 (statistical discovery, SAS Institute, Cary, NC), which is based on the definition of Fisher's skewness and kurtosis (41).
RESULTS
Two discrete inactivation states coexist in the KCNQ1 pore mutant L273F
Inactivation of WT KCNQ1 is invisible (hidden) in macroscopic inspection but the presence of hooked tail currents suggests a recovery from an inactivation process (30–32,35). However, several KCNQ1 pore mutants also display a voltage-dependent slow inactivation (42–45). Here we examined the inactivation properties of the KCNQ1 long QT mutation L273F residing in the S5 segment (45). In both WT and mutant L273F channels, the tail currents exhibit ascending and descending phases which reflect the fast recovery from a fast inactivation process and channel deactivation, respectively (Fig. 1, A and B, and Fig. 2, A and C). In addition, the L273F mutant generates a voltage-dependent slow inactivation which develops at depolarized potentials above −10 mV (Fig. 2 B), with no change in K+ selectivity (at 2 mM external K+, mutant L273F Erev = −91 ± 4 mV; n = 6, and WT Erev = −93 ± 5 mV; n = 8). This raises the question of whether the fast and slow inactivation of the L273F mutant originate from the same process. To address this issue, careful kinetic analyses of the relaxation and recovery from inactivation were performed.
FIGURE 1.

Two distinct inactivation states coexist in the KCNQ1 mutant L273F. (A) Representative trace from a triple pulse protocol reveals the fast hidden inactivation produced by WT KCNQ1 (dashed inset). (B) Representative trace of slow inactivation of L273F at +30 mV, with hooked tail currents at −60 mV. (C) Single-exponential time constants of fast inactivation for WT and L273F channels (left) measured as in panel A (+30 mV), and of slow inactivation of L273F (right) (+30 mV); note the different scales. (D) Representative trace of a twin pulse protocol measuring recovery from slow inactivation of L273F. (E) Recovery from slow inactivation of L273F as a function of time at −60 mV. (F) Time constants of recovery from fast inactivation of WT and L273F measured from hooked tail currents at −60 mV (left) and from slow inactivation of L273F (right), measured as in panel C. Data shown are mean ± SE (n = 6–8).
FIGURE 2.

Discrimination between fast and slow inactivations by barium. (A and B) Representative I-V traces of WT (A) and L273F (B) in the absence (control) and presence of 10 mM Ba2+. Currents were evoked by depolarizing steps from −70 mV to +30 mV in 10 mV increments (holding potential, −80 mV; tail potentials −60 mV). (C) Normalized tail currents of WT KCNQ1 obtained at −60 mV and −120 mV in the absence and presence of Ba2+ after a +30 mV prepulse (n = 8). (D) Simulation of current traces of WT and mutant L273F, according to gating Schemes I and II, respectively. (E) Simulation of recovery from slow inactivation of mutant L273F according to Scheme II.
The relaxation time of fast inactivation could be revealed by a triple pulse protocol (Fig. 1 A): a first depolarizing pulse to +30 mV opens the channels of which a yet undetected portion inactivates; then, a brief (15 ms) hyperpolarizing interpulse at −130 mV enables recovery from channel inactivation; a third depolarizing pulse at +30 mV reopens and reinactivates the channel population that previously underwent inactivation, leading to an initial extra-current. The fast relaxation measured at the third pulse corresponds to the kinetics of fast inactivation with single-exponential time constants of τ = 18 ± 1 ms and τ = 57 ± 5 ms for WT and mutant L273F, respectively (Fig. 1 A, inset, and C, left; n = 6–8). In contrast, the relaxation time course of slow inactivation in mutant L273F was much slower with τ = 2554 ± 264 ms (Fig. 1, B and C, right; n = 6; p < 0.001).
The distinction between the two types of inactivation was also revealed by the kinetics of recovery from channel inactivation. Recovery from fast inactivation was a relatively fast process with τ = 40 ± 4 ms and τ = 113 ± 9 ms for WT and L273F, respectively (at −60 mV) (Fig. 1, B and F, left; n = 6–8). In contrast, recovery from slow inactivation of mutant L273F was a much slower process with a time constant τ = 3340 ± 210 ms, as measured by a twin pulse protocol at −60 mV (Fig. 1, D–F; n = 6; p < 0.001). These data indicate that two distinct inactivation states (fast and slow) coexist in mutant L273F.
The effect of external Ba2+ ions on pore properties allowed us to further discriminate between the two types of inactivation. We previously showed that steady-state Ba2+ binding prevents the fast inactivation of WT and L273F (this study) by suppressing both the hook of the tail currents (Fig. 2 C) and the fast relaxation measured by the triple pulse protocol (34). In contrast, steady-state Ba2+ binding barely affects the extent of slow inactivation elicited by mutant L273F which displays, at +30 mV, a fractional slow inactivation of 0.65 ± 0.05 and 0.55 ± 0.05 in the absence and presence of 10 mM Ba2+, respectively (Fig. 2 B; n = 6). Notably, we observed similar affinity and fractional electrical distance of Ba2+ block in WT and L273F channels (Kd−40 mV = 0.37 ± 0.08 mM; δ = 0.33 and Kd−40 mV = 0.33 ± 0.06 mM; δ = 0.33, respectively, n = 8) (Fig. 3). The latter results suggest that the binding of Ba2+ deep in the pore (filter site s4) (46) is similar in the inactivating L273F mutant and the WT.
FIGURE 3.

Steady-state Ba2+ block of mutant L273F channels. (A) Current-voltage relations of mutant L273F in the absence (squares), and presence of Ba2+ at 2 mM (up-triangles), 10 mM (down-triangles), and 20 mM (diamonds). (B) Ba2+ inhibition curves of fractional channel unblock plotted as a function of Ba2+ concentrations, measured at −40 mV (squares), −30 mV (up-triangles), and −20 mV (down-triangles). KD values were obtained from the fit of a sigmoidal dose-response function: KD−40mV = 0.33 ± 0.06 mM; KD−30mV = 0.35 ± 0.04 mM; and KD−20mV = 0.47 ± 0.06 mM. (C) Ba2+ inhibition curves expressed as in panel B and measured at −10 mV (open up-triangles), 0 mV (open diamonds), +10 mV (open down-diamonds), and +20 mV (solid diamonds). KD−10mV = 0.63 ± 0.07 mM; KD 0mV = 0.87 ± 0.09 mM; KD+10mV = 1.28 ± 0.12 mM; and KD+20mV = 1.52 ± 0.16 mM. (D) KD values obtained from a single-site fit of the Ba2+ inhibition curves plotted as a function of membrane voltage and yielding δ = 0.33. Data shown are mean ± SE (n = 8).
Unlike the previously described gating behavior of WT KCNQ1, which accounts for only one inactivation state as shown in Scheme I (31,34,35), we elaborated a kinetic model that accounts for two distinct inactivation states in mutant L273F, as shown in Scheme II. The latter assumes two closed states (C1 and C2), two voltage-dependent open states (O1 and O2), a fast voltage-independent inactivated state I1 (fast inactivation) and a slow voltage-dependent inactivated state I2 (slow inactivation) (Supplementary Material Table 1). Simulating the gating behavior of mutant L273F according to Scheme II, generates current kinetics that closely matches the experimental traces with coexistence of slow and fast inactivation (Fig. 2 D). Consistently, Scheme II closely simulates the experimental data measuring the kinetics of recovery from slow inactivation (Figs. 1 D and 2 E).
As native cardiac IKS-channels result from the coassembly of KCNQ1 and KCNE1 subunits, we coexpressed the mutant L273F with KCNE1. Notably, KCNE1 could not completely abolish the marked inactivation of mutant IKS-channels which leads to a significant decrease of the current amplitude (56% inhibition, p < 0.01; Supplementary Material Fig. 1).
Slow inactivation alters kinetics of Ba2+ block and leads to ion trapping
Ba2+ and K+ have similar ionic radii (1.35 Å and 1.33 Å, respectively), a physical attribute extensively used to dissect K+ channel permeation. To probe the pore properties of KCNQ1 mutants displaying voltage-dependent slow inactivation, we used a fast perfusion system (34,47) and monitored the kinetics of Ba2+ block and unblock after an activation step that opens and inactivates the channels. Each cell was subjected to the same kinetic protocol before and after Ba2+ superfusion, which allowed us to superimpose the current traces (Fig. 4, A–C).
FIGURE 4.

Kinetics of Ba2+ block and unblock in WT KCNQ1 and mutants L273F and L273W. (A–C) After depolarization from −80 mV to −20 mV for 24 s, 10 mM Ba2+ was applied to oocytes expressing WT or mutant channels for 12 s and then was washed out for 48 s, while keeping the membrane at −20 mV. (D) Fast (triangles) and slow (squares) time constants of Ba2+ block in WT KCNQ1 (black) and mutant L273F (blue) with their respective slow component amplitude (E) plotted as a function of voltage (As and Af are the amplitudes of the slow and fast components, respectively). (F) Fast (open triangles) and slow (open squares) time constants of Ba2+ unblock in WT KCNQ1 (black) and mutant L273F (blue) with their respective slow component amplitudes (G) plotted as a function of voltage. Data shown are mean ± SE (n = 6–8).
We previously showed that in WT KCNQ1, Ba2+ block and unblock exhibit fast and slow kinetic components (34) (Fig. 4 A). When similar kinetic experiments are performed on the L273F mutant channels, well after they relax into slow inactivation, the rate of Ba2+ block and unblock is considerably slower than that of the WT (Fig. 4 B). The kinetics of the fast component of Ba2+ block is much slower in the mutant compared to WT, with a difference that increases with depolarization (Fig. 4 D, triangles). This difference becomes even more profound with depolarization when considering that the amplitude of the slow kinetic component increases in L273F and decreases in the WT (Fig. 4 E). Strikingly, both fast and slow components of Ba2+ unblock of L273F are far slower than in WT (Fig. 4, F and G). Superimposition of the current traces shows that unblock and recovery of the current upon Ba2+ washout is so slow that it leads to a current deficit at the end of the protocol (Fig. 4 B versus 4 A). This feature suggests that during slow inactivation, the selectivity filter assumes a conformation that traps Ba2+ and delays its egress from the deep pore. Compared to L273F mutant channels, the trapping of Ba2+ ions is not significant in WT KCNQ1 (Fig. 4 B versus 4 A). The slow kinetics of Ba2+ unblock and the degree of current deficit depend on the time L273F channels spend in the slow inactivated state. Indeed, when Ba2+ is applied shortly after activation, kinetics of Ba2+ block and unblock is significantly faster than that obtained when Ba2+ is applied long after L273F channels relax into slow inactivation (Fig. 5).
FIGURE 5.

Dependence of Ba2+ trapping on the time mutant L273F spends in slow inactivation. Representative traces recorded without or with 10 mM Ba2+ that was applied for 12 s after 24 s (A) or 0.9 s (B) of depolarization to −20 mV, followed by washout (48 s). (C) Slow (open bars) and fast (solid bars) time constants of Ba2+ unblock after two different application times with (D) their respective slow (open bars) and fast (solid bars) amplitude components. Data shown are mean ± SE (n = 5; * p < 0.005).
The link between Ba2+ trapping in the pore and the slow inactivation of mutant L273F is highly significant. Indeed, in mutant L273W, where no slow inactivation is observed, the kinetics of Ba2+ block and unblock is similar to that displayed by WT KCNQ1 (Fig. 4 C and Supplementary Material Fig. 2). Further correlation between slow inactivation and Ba2+ trapping is provided by the properties of other KCNQ1 pore mutants such as E295A in the turret, V310G in the pore helix, and D317A in the channel outer vestibule. We found that these pore mutants display a voltage-dependent slow inactivation which correlates with Ba2+ trapping (Fig. 6). Previous studies showed that substitution of the pore helix residue Val310 with smaller amino acids like glycine or alanine produced voltage-dependent slow inactivation, with no change in K+ selectivity (44). We examined the kinetics of Ba2+ block and unblock in the inactivating mutant V310G. While modest slow inactivation and small current deficit are observed at 0 mV (fractional inactivation = 0.07 ± 0.01; relative current deficit = 0.14 ± 0.02; n = 6), substantial slow inactivation accompanied by a large current deficit are detected at +40 mV (fractional inactivation = 0.61 ± 0.02; relative current deficit = 0.32 ± 0.01; n = 6) (Fig. 6, A–C). The kinetics of Ba2+ block and unblock in the E295A mutant is very slow (Fig. 6, D and E). At 0 mV, only one exponential time constant describes the slow Ba2+ block (τ = 4.63 ± 0.48 s, n = 7), while a very slow unblock causes strong Ba2+ trapping (relative current deficit = 0.48 ± 0.04, n = 7). Similarly to the turret (E295A) and pore helix (V310G) mutants, the outer vestibule mutant D317A generates a voltage-dependent slow inactivation which correlates with Ba2+ trapping. Interestingly, the D317A mutant slowly recovers from inactivation during a sustained depolarizing step (Fig. 6 F). Compared to WT, the kinetics of Ba2+ block of D317A is much slower, with the slow kinetic component being predominant. At +20 mV, only one exponential time constant describes the slow Ba2+ unblock of D317A, leading to pronounced Ba2+ trapping (Fig. 6, F–I).
FIGURE 6.

Slow inactivation of KCNQ1 mutants causes Ba2+ trapping. Representative traces of mutant V310G at 0 mV (A) or at +40 mV (B) showing Ba2+ block (10 mM, 12 s) and unblock upon washout for 48 s. (C) Current deficit upon Ba2+ washout (open bars) and fractional inactivation (solid bars) measured in mutant V310G at 0 mV, +20 mV, and +40 mV. Current deficit = (Y-Z)/Y, where Z is the current measured after 48 s Ba2+ washout and Y is the current at the same time point in the absence of Ba2+. (D) Representative traces of mutant E295A at 0 mV showing Ba2+ block (12 s) and unblock upon 48 s washout. (E) Time constant of Ba2+ block in E295A channels at 0 mV (left) and current deficit measured after 48 s washout (right). (F) Representative traces of mutant D317A at +20 mV showing Ba2+ block (12 s) and unblock upon 48 s washout. (G) Slow (open bars) and fast (solid bars) time constants of Ba2+ block (10 s) in WT and D317A channels at +20 mV, with their respective slow component amplitudes (H, I) Slow (open bars) and fast (solid bars) time constants of Ba2+ unblock in WT and mutant D317A. Data shown are mean ± SE (n = 5–7; * p < 0.001).
Mutant L273F displays slower deactivation than WT KCNQ1 while external K+ speeds up kinetics of slow inactivation with stronger Ba2+ trapping
In 2 mM external K+, mutant L273F exhibits significant slower deactivation than WT. At a tail potential of −120 mV, deactivation kinetics could be fitted by a two exponential function with τfast = 54 ± 10 ms, τslow = 308 ± 65 ms (relative slow component amplitude As/(Af + As) = 0.25 ± 0.05) and τfast = 104 ± 9 ms, τslow = 527 ± 85 ms (As/(Af + As) = 0.44 ± 0.03) for WT and L273F, respectively (n = 6–8, p < 0.01; Fig. 7, A–C). In contrast, the noninactivating mutant L273W displays similar deactivation kinetics to those of WT with τfast = 61 ± 11 ms and τslow = 304 ± 45 ms (As/Af + As = 0.26 ± 0.04; n = 6). In 50 mM external K+, mutant L273F closes more slowly than it does in 2 mM K+ and yet shows slower deactivation than WT with τfast = 197 ± 21 ms, τslow = 1593 ± 521 ms (As/(Af + As) = 0.51 ± 0.08) (n = 8, p < 0.01, Fig. 7, D–F).
FIGURE 7.

Mutant L273F exhibits slower deactivation kinetics than WT. (A) Representative traces of WT (upper panel) and L273F channels (lower panel) recorded in 2 mM external K+ with depolarizing steps from −70 mV to +40 mV in 10 mV increments (holding potential −80 mV; tail potentials −120 mV). (B) Fast and slow deactivation time constants obtained from the biexponential fit of the deactivating tail currents in 2 mM external K+ and (C) their corresponding relative slow component amplitude As/(As + Af). (D) Representative traces of WT (upper panel) and L273F channels (lower panel) recorded in 50 mM external K+. (E) Fast and slow deactivation time constants measured in 50 mM external K+ and (F) their corresponding relative slow component amplitude. Data shown are mean ± SE (n = 6–8; * p < 0.01).
Increasing external K+ concentration significantly accelerates the kinetics of slow inactivation of mutant L273F (Fig. 8, A and B). In virtually 0 K+, a single slow relaxation time constant is observed while at 50 mM external K+ both fast and slow time constants are apparent, with the slow kinetic component being even much faster than that seen in the absence of K+ (Fig. 8 C). The fastest inactivation is observed at 50 mM Rb+ ions (not shown). Interestingly, the speedup of the inactivation time course is coupled to an acceleration of the activation kinetics, a link specific for K+ ions. In high external K+ (50 mM K+), the time to peak of the L273F channel activation is faster (at +30 mV, t = 99 ± 8 ms; n = 5, p < 0.01) than in high external Na+ (t = 199 ± 15 ms, at 98 mM Na+, n = 5). The slowest channel activation is obtained with high external Li+ (50 mM) whose time to peak value is t = 240 ± 23 ms (n = 6; p < 0.01). The acceleration of activation and inactivation kinetics of L273F by external K+ is also found in other slowly inactivating KCNQ1 pore mutants (not shown). Notably, the kinetics of Ba2+ block and unblock in mutant L273F are significantly slower in high than in low external K+ (Fig. 8, D–F).
FIGURE 8.

Effect of external K+ on kinetics of activation, inactivation, and Ba2+ trapping in mutant L273F. (A) Representative I-V traces of L273F channels recorded in 50 mM external K+ with depolarizing steps from −70 mV to +30 mV in 10 mV increments (holding potential −80 mV; tail potentials −120 mV). (B) Representative normalized traces of L273F slow inactivation recorded from the same cell at +30 mV in external solutions containing: 50 mM Li+ and 48 mM Na+; 2 mM K+ and 96 mM Na+; and 50 mM K+ and 48 mM Na+. (C) Time constants of slow inactivation displayed by L273F channels under the indicated external solutions. An additional fast time constant (solid bars) could be measured in solutions containing 50 mM K+. (D) Slow (open bars) and fast (solid bars) time constants of Ba2+ block in L273F channels measured at +30 mV in 2 and 50 mM external K+. (E) Slow (open bars) and fast (solid bars) time constants of Ba2+ unblock in L273F channels measured at +30 mV in 2 and 50 mM external K+, with (F) their respective slow (open bars) and fast (solid bars) amplitude components. Data shown are mean ± SE (n = 6–8; * p < 0.05, ** p < 0.01).
Homology modeling and molecular dynamics simulations of WT KCNQ1 and pore mutants
In an attempt to understand how the L273F pore mutation leads to slow inactivation, we first constructed homology models in vacuum for the WT KCNQ1 and its pore mutants using the atomic coordinates of Kv1.2 as a template (48) (Fig. 9 and Supplementary Material Fig. 3). Comparison between the models of the WT KCNQ1 and the L273F mutant suggests that the mutation affects only the selectivity filter site s1 and does not result in any conformational changes at sites s2, s3, and s4. In this model, the phenylalanine is very close to the pore helix where it forms multiple short-distance hydrophobic (<3 Å) and van der Waals (3–4 Å) interactions with the main and side chains of Leu303 (Fig. 9 B and Supplementary Material data). Consequently, the backbone flanking Leu303 moves together with Trp304. In both WT and mutant models, the Nɛ atom of Trp304 forms a hydrogen bond with the carboxyl of Asp317, while its aromatic side chain also forms van der Waals interactions with the aromatic moiety of Tyr315. As such, Trp304, D317, and Y315 move together and this movement introduces torsion in the backbone between positions 317 and 315. Consequently, the carbonyl oxygens of the four equatorial Tyr315 residues in mutant L273F become closer to the axis of ion conduction, leading to constriction of the outer carbonyl ring of the selectivity filter. That is, the center-to-center distance between two facing carbonyl oxygens is 5.09 Å versus 5.78 Å in the L273F mutant versus WT, respectively.
FIGURE 9.

Structural models of WT and pore mutants of KCNQ1 channels. (A) Homology model of the pore domain of WT KCNQ1. For clarity, the frontal subunit was removed and ribbons of the other differently colored three subunits are shown from within the membrane. (B) Two diagonally facing subunits of mutant L273F (green) superposed on the homologous subunits of WT KCNQ1 (Corey Pauling, Kultin (CPK) colors: carbon, nitrogen, and oxygen atoms in light gray, blue, and red, respectively). The backbone of the selectivity filter and the indicated side chains are shown as sticks while part of S5 and the pore helix are shown as a Cα trace (in thinner lines). Other segments were removed for better viewing. Site s1 corresponds to the place where a K+ ion is inferred to be trapped. Magnification of the backbone carbonyls of site s1 is shown in black frame. (C) Superposition of mutant L273W (green) on the WT KCNQ1 (CPK) as in panel B. Dotted black lines correspond to distances of 2.6–2.9 Å (hydrogen bonding between D317 and W304 and hydrophobic interactions between F273 and L303 in panel B). Dotted gray lines correspond to distances of ∼3.7 Å (van der Waals interactions).
In the L273W mutant model, the side chain of Trp273 points downward and neither interacts with Leu303 nor changes the structure of the pore helix and thus does not lead to constriction of the outer carbonyl ring of the selectivity filter (Fig. 9 C). These observations are consistent with the inability of the L273W mutation to produce slow inactivation (Fig. 4 C). Interestingly, in all other in vacuum mutant models (D317A, E295A, and V310G), the structural changes emerging from the loss of the bond network of the replaced amino acids ultimately lead to constriction of the carbonyl ring at position 315 (see Supplementary Material Fig. 4 and Supplementary Data).
It should be noted that the distances measured in vacuo across the selectivity filter reflect a static and limited picture of one given minimized structure. In an attempt to get better insight into the filter's conformational changes that might accompany K+ conductance in WT KCNQ1 and the various pore mutants, we ran molecular dynamics simulations using the aforementioned models in the presence of water and potassium ions (Fig. 10 and Table 1). We found that the distances obtained in the dynamics simulations are within the range of those displayed by the in vacuum models. However, the simulations show that the mean distance between opposite carbonyls at position Y315 in WT and the inactivating mutants are not significantly different (Table 1). Notably, this mean distance does not reflect the extent of the breathing motions of the selectivity filter. In this regard, the maximal distance between facing carbonyl oxygens is more meaningful. Indeed, the simulations show that in all slow inactivating mutants this maximal distance is smaller than that of the WT and the noninactivating mutant L273W (Table 1). The distribution of the distances between the carbonyls of Tyr315 is shifted in the L273F mutant relative to the WT (Fig. 10 D). The corresponding distribution for the L273W mutant does not change significantly compared to the WT (Fig. 10 E). Notably, the range of the fluctuations in the carbonyl distance of Tyr315 in the slow inactivating mutants L273F, E295A, V310G, and D317A decreases compared to the WT and the L273W mutant (Table 1). This suggests that the L273F, E295A, V310G, and D317A mutations also lead to a substantial decrease in the flexibility of this region of the selectivity filter.
FIGURE 10.

Molecular dynamics simulation with a primary hydration shell and three K+ ions. (A–C) Distances between the carbonyls of Y315 in two opposite facing subunits as a function of time for (A) WT KCNQ1, (B) the L273F, and (C) L273W mutants. (D and E) Distribution of the distances between the carbonyls of Y315 in two opposite facing subunits of the WT KCNQ1 and the (D) L273F and (E) L273W mutants.
TABLE 1.
Statistical analysis of the simulation results for the distribution of the distance between the carbonyls of Y315 in two opposite facing subunits for the WT KCNQ1 and for the mutants L273F, L273W, E295A, V310G, and D317A
| Mean distance (Å) | Standard deviation (Å) | Standard error (Å) | Minimal distance (Å) | Maximal distance (Å) | Variance (Å2) | Skewness | Kurtosis | |
|---|---|---|---|---|---|---|---|---|
| KCNQ1 | 5.161 | 0.231 | 0.004 | 4.516 | 6.612 | 0.053 | 0.691 | 1.319 |
| KCNQ1 L273F | 4.998 | 0.223 | 0.004 | 4.278 | 6.042 | 0.049 | 0.268 | 0.525 |
| KCNQ1 L273W | 5.198 | 0.229 | 0.004 | 4.578 | 6.548 | 0.052 | 0.690 | 1.356 |
| KCNQ1 E295A | 5.222 | 0.227 | 0.004 | 4.622 | 6.291 | 0.051 | 0.565 | 0.696 |
| KCNQ1 V310G | 5.126 | 0.215 | 0.004 | 4.373 | 6.194 | 0.046 | 0.329 | 0.519 |
| KCNQ1 D317A | 5.061 | 0.200 | 0.004 | 4.111 | 6.094 | 0.039 | 0.070 | 0.723 |
Quantitative indication concerning the effect of the slow inactivating mutants on the flexibility of the selectivity filter is obtained through the analysis of the third and fourth moments about the mean of the distribution of the distances between the carbonyls of Tyr315 (Table 1). While the first moment, the mean, and the second moment, the variance, characterize the location and the variability of a distribution, respectively, the third and fourth moments characterize the shape of a distribution. The third moment, the skewness, provides an indication about the departure of a distribution from symmetry. Compared with a normal distribution that has a skewness of 0, all distributions were only slightly positively skewed. The extent to which both the WT and the L273W mutant were skewed was, however, higher than that of the slow inactivating mutants (L273F, E295A, V310G, and D317A). For both the WT and the noninactivating mutant L273W, the skewness was 0.69, while for the L273F mutant, it was 0.27. This difference is significant as the standard error of skewness is estimated as 0.05 according to  where n = 3000 is the number of structures included in the analysis (49). This is a quantitative indication that the distribution of the distances between the carbonyls of Tyr315 in the WT and the L273W mutant has an asymmetric tail extending further to larger distances than that of the slow inactivating L273F mutant. The fourth moment, the kurtosis, is a measure of the peakedness of the distribution and of the heaviness of its tails (50). Since a normal distribution has kurtosis of three, the excess kurtosis relative to normal distribution is used and obtained by subtracting three from the value of the fourth moment around the mean. The higher the kurtosis of a distribution, the more distinct its peak close to the mean, the more rapid its decline, and the heavier its tails. As the variance for the distributions is roughly the same, we have proceeded to compare the excess kurtosis of these distributions. At constant variance, a higher kurtosis is an indication of a larger number of cases that deviate further away from the mean. Comparison of the excess kurtosis shows that while for the WT and the noninactivating mutant L273W, the value is approximately the same, and equal to 1.32 and 1.36, respectively, the value for the slow inactivating mutants (L273F, E295A, V310G, and D317A) is smaller (Table 1). For example, excess kurtosis equals to 0.53 for mutant L273F. In view of the large number of structures (n = 3000) that are included in the analysis, the standard error of kurtosis can be estimated as 0.09 according to
where n = 3000 is the number of structures included in the analysis (49). This is a quantitative indication that the distribution of the distances between the carbonyls of Tyr315 in the WT and the L273W mutant has an asymmetric tail extending further to larger distances than that of the slow inactivating L273F mutant. The fourth moment, the kurtosis, is a measure of the peakedness of the distribution and of the heaviness of its tails (50). Since a normal distribution has kurtosis of three, the excess kurtosis relative to normal distribution is used and obtained by subtracting three from the value of the fourth moment around the mean. The higher the kurtosis of a distribution, the more distinct its peak close to the mean, the more rapid its decline, and the heavier its tails. As the variance for the distributions is roughly the same, we have proceeded to compare the excess kurtosis of these distributions. At constant variance, a higher kurtosis is an indication of a larger number of cases that deviate further away from the mean. Comparison of the excess kurtosis shows that while for the WT and the noninactivating mutant L273W, the value is approximately the same, and equal to 1.32 and 1.36, respectively, the value for the slow inactivating mutants (L273F, E295A, V310G, and D317A) is smaller (Table 1). For example, excess kurtosis equals to 0.53 for mutant L273F. In view of the large number of structures (n = 3000) that are included in the analysis, the standard error of kurtosis can be estimated as 0.09 according to  (49), indicating that this difference is significant. As a numerical measure, the excess kurtosis further confirms that the extent to which the distance between the carbonyls of Tyr315 can increase further away from the mean is larger for the WT and the noninactivating mutant L273W than for the slow inactivating mutants L273F, E295A, V310G, and D317A.
(49), indicating that this difference is significant. As a numerical measure, the excess kurtosis further confirms that the extent to which the distance between the carbonyls of Tyr315 can increase further away from the mean is larger for the WT and the noninactivating mutant L273W than for the slow inactivating mutants L273F, E295A, V310G, and D317A.
In all, the dynamics simulations indicate that compared to the WT and the noninactivating mutant L273W, the slow inactivating mutants exhibit both a reduced maximal distance between the facing carbonyls of Y315 and smaller values of excess kurtosis. This implies that in the slow inactivating pore mutants, the upper filter has a reduced capacity to fluctuate, namely, an increased rigidity.
DISCUSSION
Inactivation of WT KCNQ1 is invisible from macroscopic inspection but a careful analysis indicates that it is an intrinsically fast and voltage-independent process (30–32). Interestingly, KCNQ1 pore mutants generate an additional voltage-dependent slow inactivation (42–44). Here we show that these two types of inactivation are discrete processes, displaying distinct kinetics and pore properties. The pore mutant L273F exhibits a fast inactivation similar to the WT, with fast relaxation and recovery time courses. In addition, this channel mutant displays a voltage-dependent slow inactivation, with very slow relaxation and recovery kinetics. Further distinctions between these two inactivation types is provided by their differential impact on kinetics of Ba2+ block and unblock, which revealed that Ba2+ trapping deep in the pore arises primarily from the slow inactivation.
Inactivation of KCNQ1 channels does not share the characteristic features of N- and C-type inactivation. The lack of an N-terminal ball structure within KCNQ1 and the persistence of both types of inactivation in an N-terminally truncated short isoform of mutant L273F (not shown) indicate that KCNQ1 inactivation differs from N-type inactivation. Moreover, the simulations based on the kinetic model that we elaborated here, clearly show that the gating behavior of mutant L273F gives rise to an additional inactivation state (I2), which reflects the slow inactivation. The latter is accelerated by elevated concentrations of external K+, unlike C-type inactivation of Shaker K+ channels which is inhibited by increased external K+ concentration (15,23).
Ba2+ has a large enthalpy of hydration (ΔH = −328 kcal/mol) (51) and, like K+, must be dehydrated before reaching its deep binding site in the selectivity filter and must be resolvated upon its egress from the pore to the external solution. Our experimental results suggest that during slow inactivation the upper part of the selectivity filter obstructs the path of Ba2+ to and from its deep site. This interpretation is supported by the findings that during slow inactivation, Ba2+ block and unblock are markedly delayed despite the steady-state affinity and the electric distance for Ba2+ block are similar in WT and mutant L273F. It would be interesting to investigate the relationship between inactivation and Ba2+ trapping at the single-channel level. However, this is hampered by the very small unitary conductance of KCNQ1 (52–54).
Several lines of evidence point to a tight link between slowdown of Ba2+ entry, Ba2+ trapping, and slow inactivation:
Slowdown of Ba2+ entry to and exit from the pore parallels with the time L273F channels spend in the slow inactivated state (Fig. 5).
Mutation of the same position into tryptophan (L273W) does not generate slow inactivation, does not delay Ba2+ entry, and does not lead to Ba2+ trapping (Fig. 4 C).
The properties of KCNQ1 pore mutants, including L273F in S5, E295A in the turret, D317A in the outer pore vestibule, and V310G in the pore helix closely associate slow inactivation with slowdown of Ba2+ entry and Ba2+ trapping (Fig. 6).
A tight connection exists between the voltage dependence of slow inactivation and the degree of current deficit obtained after depolarization (Fig. 6, A–C).
Elevation of external K+ accelerates slow inactivation kinetics and increases Ba2+ trapping.
Trapping of Ba2+ ions is minimal in WT KCNQ1 channels, raising the question of what might be the physical determinants of fast inactivation. The inactivation of WT KCNQ1 was suggested to relate to fast flickering of the open channel, which may reflect instability of a fast voltage-independent pore gate (35,55). The fast inactivation of WT KCNQ1 does not lead to a large current decline, unlike the conductance fall-down produced by the slow inactivation of the L273F pore mutant described here. The fast voltage-independent flicker possibly reflects small-scale fluctuating motions of the selectivity filter that are not coupled to the sensor movement.
The L273F mutant is a typical example of a group of human inherited mutations which produce a voltage-dependent slow inactivation and are responsible for the long QT syndrome. Indeed, we recently showed that LQT mutations located in the proximal C terminus of KCNQ1 (R366W, A371T, S373P, and W392R) impair calmodulin binding and alter channel gating by generating a voltage-dependent slow inactivation (56). A similar slow inactivation gating behavior is recurrently found in mutated residues of many regions of KCNQ1. Notably, mutations in the voltage sensor S4 (L233W and Q244W) produce a voltage-dependent slow inactivation (Supplementary Material Fig. 5), which suggests that the movement of the voltage sensor promotes the slow inactivation. In Shaker-related Kv channels, it appears that slow C-type inactivation is also coupled to voltage sensor movement (57–62).
No conformational changes are observed at filter sites s2, s3, and s4 in the model of L273F, in line with our results showing that the electrical distance and affinity of Ba2+ to its innermost filter site s4 are similar in mutant L273F and the WT. In contrast, the in vacuum model predicts conformational changes that converge to the upper part of the selectivity filter. We suggest that a decreased flexibility at this part of the filter, as revealed by the dynamics simulations, leads to tighter coordination of a K+ ion in s1. This, in turn, causes hindrance of the dehydration-resolvation transition, impairing thereby high throughput conduction. Our interpretations are in line with the acceleration of slow inactivation and the slower kinetics of Ba2+ block and unblock observed upon elevation of external K+.
The KCNQ1 slow inactivation is largely different from the foot-in-the-door mechanism described for C-type inactivation where the pore is presumed to adopt a nonconductive conformation when permeant ions vacate the selectivity filter (19–21). Interestingly, other channel types also exhibit inactivation properties that diverge from the properties originally described for C-type in Shaker. In rapidly inactivating Kv4 channels, elevation of extracellular K+ concentration promotes inactivation and inhibits recovery of the channels from inactivation (63–65). Similarly, Kv2.1 inactivation is accelerated at elevated concentrations of extracellular K+ or TEA+ (66). In contrast, for C-type inactivation, increase in extracellular K+ is considered to be slow depletion of K+ ions from the selectivity filter and thus to prevent inactivation. However, a coherent picture of C-type inactivation has not emerged yet. The x-ray crystal structure of KcsA determined at low K+ concentration was suggested to reflect a nonconducting filter conformation (4,67). Recent molecular dynamics simulations of KcsA indicate that under low K+ occupancy, the selectivity filter changes its conformation to a nonconducting state of broken fourfold symmetry with widening of the filter, loss of K+ coordination at s2, and stronger K+ coordination at s3 (68). Along this view, it is assumed that, in KcsA, the hydrogen bonding between Tyr78 of the GYG motif or Asp80 of the outer mouth and the aromatic cuff residues (Trp67 and Trp68) is very important for stabilization of the conducting conformation of the selectivity filter (19,68). On the other hand, combined structural and functional studies have recently challenged this view by showing that in KcsA the hydrogen bond between Asp80 and Trp67 and the carboxy-carboxylate interaction between Asp80 and Glu71 destabilize the conducting conformation of the KcsA selectivity filter (14). Disrupting these interactions stabilizes a noninactivating, high-open probability channel state. According to this view, the low K+ crystal structure of KcsA could not represent the C-type inactivated state of the channel (14). Our results show that disruption of the hydrogen bonding between Trp304 and Asp317 in KCNQ1 (mutant D317A) destabilizes a conducting filter conformation and produces slow inactivation associated with Ba2+ trapping (Fig. 6, F–I).
Molecular modeling and dynamics simulations indicate that even minimal changes can already be effective by reducing the flexibility of the upper part of the selectivity filter. The ability of the WT and the noninactivating mutant L273W to extend to wider distances may facilitate the entry to and exit of Ba2+ ions from the pore, thereby preventing Ba2+ trapping. The fluctuations rather than the average distance between the carbonyls of Tyr315 appear to account for the difference between WT and the slow inactivating pore mutants. In other words, a decrease in breathing motions of the filter leads to an increase in the time K+ ions reside in the filter. Our experimental data are consistent with this notion of altered pore occupancy. We showed that the inactivating mutant L273F exhibits slower deactivation than the WT and that high external K+ further slows down channel closure (Fig. 7). This slower deactivation reflects longer residency time of K+ in the pore of mutant L273F (via O1), which ultimately feeds back to the slow inactivation state I2. This feature may also explain why increase in extracellular K+ accelerates inactivation. However, it is also possible that in the outer channel pore, there is a K+-selective binding site that modulates inactivation gating, thereby affecting the conformation of the selectivity filter. In all, decreased flexibility at filter site s1 plausibly underlies the mechanism of slow inactivation in KCNQ1 pore mutants.
SUPPLEMENTARY MATERIAL
To view all of the supplemental files associated with this article, visit www.biophysj.org.
Supplementary Material
Acknowledgments
This work is supported by the Israel Science Foundation (grant No. ISF 672/05), the Israel Ministry of Science “Tashtiot” program, and the Keren Wolfson funds to B.A.
Gilad Gibor, Daniel Yakubovich, and Avia Rosenhouse-Dantsker contributed equally to this work.
Editor: Toshinori Hoshi.
References
- 1.Doyle, D. A., J. Morais Cabral, R. A. Pfuetzner, A. Kuo, J. M. Gulbis, S. L. Cohen, B. T. Chait, and R. MacKinnon. 1998. The structure of the potassium channel: molecular basis of K+ conduction and selectivity. Science. 280:69–77. [DOI] [PubMed] [Google Scholar]
- 2.MacKinnon, R. 2004. Potassium channels and the atomic basis of selective ion conduction (Nobel Lecture). Angew. Chem. Int. Ed. Engl. 43:4265–4277. [DOI] [PubMed] [Google Scholar]
- 3.Swartz, K. J. 2004. Towards a structural view of gating in potassium channels. Nat. Rev. Neurosci. 5:905–916. [DOI] [PubMed] [Google Scholar]
- 4.Zhou, Y., J. H. Morais-Cabral, A. Kaufman, and R. MacKinnon. 2001. Chemistry of ion coordination and hydration revealed by a K+ channel-Fab complex at 2.0 Å resolution. Nature. 414:43–48. [DOI] [PubMed] [Google Scholar]
- 5.Morais-Cabral, J. H., Y. Zhou, and R. MacKinnon. 2001. Energetic optimization of ion conduction rate by the K+ selectivity filter. Nature. 414:37–42. [DOI] [PubMed] [Google Scholar]
- 6.Zhou, Y., and R. MacKinnon. 2004. Ion binding affinity in the cavity of the KcsA potassium channel. Biochemistry. 43:4978–4982. [DOI] [PubMed] [Google Scholar]
- 7.Zhou, Y., and R. MacKinnon. 2003. The occupancy of ions in the K+ selectivity filter: charge balance and coupling of ion binding to a protein conformational change underlie high conduction rates. J. Mol. Biol. 333:965–975. [DOI] [PubMed] [Google Scholar]
- 8.Alagem, N., S. Yesylevskyy, and E. Reuveny. 2003. The pore helix is involved in stabilizing the open state of inwardly rectifying K+ channels. Biophys. J. 85:300–312. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Chapman, M. L., H. M. VanDongen, and A. M. VanDongen. 1997. Activation-dependent subconductance levels in the drk1 K channel suggest a subunit basis for ion permeation and gating. Biophys. J. 72:708–719. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Kiss, L., J. LoTurco, and S. J. Korn. 1999. Contribution of the selectivity filter to inactivation in potassium channels. Biophys. J. 76:253–263. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Lu, T., A. Y. Ting, J. Mainland, L. Y. Jan, P. G. Schultz, and J. Yang. 2001. Probing ion permeation and gating in a K+ channel with backbone mutations in the selectivity filter. Nat. Neurosci. 4:239–246. [DOI] [PubMed] [Google Scholar]
- 12.Proks, P., J. F. Antcliff, and F. M. Ashcroft. 2003. The ligand-sensitive gate of a potassium channel lies close to the selectivity filter. EMBO Rep. 4:70–75. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Cordero-Morales, J. F., L. G. Cuello, and E. Perozo. 2006. Voltage-dependent gating at the KcsA selectivity filter. Nat. Struct. Mol. Biol. 13:319–322. [DOI] [PubMed] [Google Scholar]
- 14.Cordero-Morales, J. F., L. G. Cuello, Y. Zhao, V. Jogini, D. M. Cortes, B. Roux, and E. Perozo. 2006. Molecular determinants of gating at the potassium-channel selectivity filter. Nat. Struct. Mol. Biol. 13:311–318. [DOI] [PubMed] [Google Scholar]
- 15.Hoshi, T., W. N. Zagotta, and R. W. Aldrich. 1990. Biophysical and molecular mechanisms of Shaker potassium channel inactivation. Science. 250:533–538. [DOI] [PubMed] [Google Scholar]
- 16.Zagotta, W. N., T. Hoshi, and R. W. Aldrich. 1990. Restoration of inactivation in mutants of Shaker potassium channels by a peptide derived from ShB. Science. 250:568–571. [DOI] [PubMed] [Google Scholar]
- 17.Zhou, M., J. H. Morais-Cabral, S. Mann, and R. MacKinnon. 2001. Potassium channel receptor site for the inactivation gate and quaternary amine inhibitors. Nature. 411:657–661. [DOI] [PubMed] [Google Scholar]
- 18.Consiglio, J. F., P. Andalib, and S. J. Korn. 2003. Influence of pore residues on permeation properties in the Kv2.1 potassium channel. Evidence for a selective functional interaction of K+ with the outer vestibule. J. Gen. Physiol. 121:111–124. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Kurata, H. T., and D. Fedida. 2006. A structural interpretation of voltage-gated potassium channel inactivation. Prog. Biophys. Mol. Biol. 92:185–208. [DOI] [PubMed] [Google Scholar]
- 20.Starkus, J. G., L. Kuschel, M. D. Rayner, and S. H. Heinemann. 1997. Ion conduction through C-type inactivated Shaker channels. J. Gen. Physiol. 110:539–550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Yellen, G. 1998. The moving parts of voltage-gated ion channels. Q. Rev. Biophys. 31:239–295. [DOI] [PubMed] [Google Scholar]
- 22.Choi, K. L., R. W. Aldrich, and G. Yellen. 1991. Tetraethylammonium blockade distinguishes two inactivation mechanisms in voltage-gated K+ channels. Proc. Natl. Acad. Sci. USA. 88:5092–5095. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Lopez-Barneo, J., T. Hoshi, S. H. Heinemann, and R. W. Aldrich. 1993. Effects of external cations and mutations in the pore region on C-type inactivation of Shaker potassium channels. Receptors Channels. 1:61–71. [PubMed] [Google Scholar]
- 24.Harris, R. E., H. P. Larsson, and E. Y. Isacoff. 1998. A permeant ion binding site located between two gates of the Shaker K+ channel. Biophys. J. 74:1808–1820. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Barhanin, J., F. Lesage, E. Guillemare, M. Fink, M. Lazdunski, and G. Romey. 1996. KvLQT1 and IsK (minK) proteins associate to form the IKs cardiac potassium current. Nature. 384:78–80. [DOI] [PubMed] [Google Scholar]
- 26.Jentsch, T. J. 2000. Neuronal KCNQ potassium channels: physiology and role in diseases. Nat. Neurosci. 1:21–30. [DOI] [PubMed] [Google Scholar]
- 27.Nerbonne, J. M., and R. S. Kass. 2005. Molecular physiology of cardiac repolarization. Physiol. Rev. 85:1205–1253. [DOI] [PubMed] [Google Scholar]
- 28.Sanguinetti, M. C., M. E. Curran, A. Zou, J. Shen, P. S. Spector, D. L. Atkinson, and M. T. Keating. 1996. Coassembly of KvLQT1 and minK (IsK) proteins to form cardiac IKs potassium channel. Nature. 384:80–83. [DOI] [PubMed] [Google Scholar]
- 29.Roden, D. M. 2006. Long QT syndrome: reduced repolarization reserve and the genetic link. J. Intern. Med. 259:59–69. [DOI] [PubMed] [Google Scholar]
- 30.Abitbol, I., A. Peretz, C. Lerche, A. E. Busch, and B. Attali. 1999. Stilbenes and fenamates rescue the loss of IKS channel function induced by an LQT5 mutation and other IsK mutants. EMBO J. 18:4137–4148. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Pusch, M., R. Magrassi, B. Wollnik, and F. Conti. 1998. Activation and inactivation of homomeric KvLQT1 potassium channels. Biophys. J. 75:785–792. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Tristani-Firouzi, M., and M. C. Sanguinetti. 1998. Voltage-dependent inactivation of the human K+ channel KvLQT1 is eliminated by association with minimal K+ channel (minK) subunits. J. Physiol. (Lond.). 510:37–45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Woodhull, A. M. 1973. Ionic blockage of sodium channels in nerve. J. Gen. Physiol. 61:687–708. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Gibor, G., D. Yakubovich, A. Peretz, and B. Attali. 2004. External barium affects the gating of KCNQ1 potassium channels and produces a pore block via two discrete sites. J. Gen. Physiol. 124:83–102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Seebohm, G., C. M. Sanguinetti, and M. Pusch. 2003. Tight coupling of rubidium conductance and inactivation in human KCNQ1 potassium channels. J. Physiol. 552:369–378. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Blunck, R., U. Kirst, T. Riessner, and U. Hansen. 1998. How powerful is the dwell-time analysis of multichannel records? J. Membr. Biol. 165:19–35. [DOI] [PubMed] [Google Scholar]
- 37.Notredame, C., D. G. Higgins, and J. Heringa. 2000. T-COFFEE: a novel method for fast and accurate multiple sequence alignment. J. Mol. Biol. 302:205–217. [DOI] [PubMed] [Google Scholar]
- 38.Schwede, T., J. Kopp, N. Guex, and M. C. Peitsch. 2003. SWISS-MODEL: an automated protein homology-modeling server. Nucleic Acids Res. 31:3381–3385. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Beglov, D., and B. Roux. 1995. Dominant solvation effects from primary shell of hydration: approximation for molecular dynamics simulations. Biopolymers. 35:171–178. [Google Scholar]
- 40.Brooks, B. R., R. Bruccoleri, B. Olafson, D. States, S. Swaminathan, and M. Karplus. 1983. CHARMM: a program for macromolecular energy, minimization, and dynamics calculations. J. Comput. Chem. 4:187–217. [Google Scholar]
- 41.Fisher, R. A. 1970. Statistical Methods for Research Workers, 14th Ed. Oliver and Boyd, Edinburgh, Scotland.
- 42.Peretz, A., H. Schottelndreier, L. Ben Aharon-Shamgar, and B. Attali. 2002. Modulation of homomeric and heteromeric KCNQ1 channels by external acidification. J. Physiol. 545:751–766. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Seebohm, G., C. R. Scherer, A. E. Busch, and C. Lerche. 2001. Identification of specific pore residues mediating KCNQ1 inactivation. A novel mechanism for long QT syndrome. J. Biol. Chem. 276:13600–13605. [DOI] [PubMed] [Google Scholar]
- 44.Seebohm, G., P. Westenskow, F. Lang, and M. C. Sanguinetti. 2005. Mutation of colocalized residues of the pore helix and transmembrane segments S5 and S6 disrupt deactivation and modify inactivation of KCNQ1 K+ channels. J. Physiol. 563:359–368. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Shalaby, F. Y., P. C. Levesque, W. P. Yang, W. A. Little, M. L. Conder, T. Jenkins-West, and M. A. Blanar. 1997. Dominant-negative KvLQT1 mutations underlie the LQT1 form of long QT syndrome. Circulation. 96:1733–1736. [DOI] [PubMed] [Google Scholar]
- 46.Jiang, Y., and R. MacKinnon. 2000. The barium site in a potassium channel by x-ray crystallography. J. Gen. Physiol. 115:269–272. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Paas, Y., G. Gibor, R. Grailhe, N. Savatier-Duclert, V. Dufresne, M. Sunesen, L. P. de Carvalho, J. P. Changeux, and B. Attali. 2005. Pore conformations and gating mechanism of a Cys-loop receptor. Proc. Natl. Acad. Sci. USA. 102:15877–15882. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Long, S. B., E. B. Campbell, and R. Mackinnon. 2005. Crystal structure of a mammalian voltage-dependent Shaker family K+ channel. Science. 309:897–903. [DOI] [PubMed] [Google Scholar]
- 49.Tabachnick, B. G., and L. S. Fidell. 1996. Using Multivariate Statistics, 2nd Ed. HarperCollins, New York.
- 50.Balanda, K. P., and H. L. MacGillivray. 1988. Kurtosis: a critical review. Am. Stat. 42:111–119. [Google Scholar]
- 51.Hille, B. 2001. Ionic Channels of Excitable Membranes, 3rd Ed. Sinauer, Sunderland, MA.
- 52.Pusch, M. 1998. Increase of the single-channel conductance of KvLQT1 potassium channels induced by the association with mink. Pflugers Archiv. Eur. J. Physiol.. 437:172–174. [DOI] [PubMed] [Google Scholar]
- 53.Sesti, F., and S. A. N. Goldstein. 1998. Single-channel characteristics of wild-type Iks channels and channels formed with two mink mutants that cause long QT syndrome. J. Gen. Physiol. 112:651–663. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Yang, Y., and F. J. Sigworth. 1998. Single-channel properties of IKs potassium channels. J. Gen. Physiol. 112:665–678. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Pusch, M., L. Bertorello, and F. Conti. 2000. Gating and flickery block differentially affected by rubidium in homomeric KCNQ1 and heteromeric KCNQ1/KCNE1 potassium channels. Biophys. J. 78:211–226. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Shamgar, L., L. Ma, N. Schmitt, Y. Haitin, A. Peretz, R. Wiener, J. Hirsch, O. Pongs, and B. Attali. 2006. Calmodulin is essential for cardiac IKs channel gating and assembly: impaired function in long-QT mutations. Circ. Res. 98:1055–1063. [DOI] [PubMed] [Google Scholar]
- 57.Klemic, K. G., G. E. Kirsch, and S. W. Jones. 2001. U-type inactivation of Kv3.1 and Shaker potassium channels. Biophys. J. 81:814–826. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Kurata, H. T., G. S. Soon, and D. Fedida. 2001. Altered state dependence of C-type inactivation in the long and short forms of human Kv1.5. J. Gen. Physiol. 118:315–332. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Larsson, H. P., and F. Elinder. 2000. A conserved glutamate is important for slow inactivation in K+ channels. Neuron. 27:573–583. [DOI] [PubMed] [Google Scholar]
- 60.Loots, E., and E. Y. Isacoff. 2000. Molecular coupling of S4 to a K+ channel's slow inactivation gate. J. Gen. Physiol. 116:623–636. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Olcese, R., D. Sigg, R. Latorre, F. Bezanilla, and E. Stefani. 2001. A conducting state with properties of a slow inactivated state in a Shaker K+ channel mutant. J. Gen. Physiol. 117:149–163. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Ortega-Saenz, P., R. Pardal, A. Castellano, and J. Lopez-Barneo. 2000. Collapse of conductance is prevented by a glutamate residue conserved in voltage-dependent K+ channels. J. Gen. Physiol. 116:181–190. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Bahring, R., L. M. Boland, A. Varghese, M. Gebauer, and O. Pongs. 2001. Kinetic analysis of open- and closed-state inactivation transitions in human Kv4.2 A-type potassium channels. J. Physiol. 535:65–81. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Jerng, H. H., and M. Covarrubias. 1997. K+ channel inactivation mediated by the concerted action of the cytoplasmic N- and C-terminal domains. Biophys. J. 72:163–174. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Jerng, H. H., M. Shahidullah, and M. Covarrubias. 1999. Inactivation gating of Kv4 potassium channels: molecular interactions involving the inner vestibule of the pore. J. Gen. Physiol. 113:641–660. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Immke, D., M. Wood, L. Kiss, and S. J. Korn. 1999. Potassium-dependent changes in the conformation of the Kv2.1 potassium channel pore. J. Gen. Physiol. 113:819–836. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Lenaeus, M. J., M. Vamvouka, P. J. Focia, and A. Gross. 2005. Structural basis of TEA blockade in a model potassium channel. Nat. Struct. Mol. Biol. 12:454–459. [DOI] [PubMed] [Google Scholar]
- 68.Berneche, S., and B. Roux. 2005. A gate in the selectivity filter of potassium channels. Structure. 13:591–600. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.