

Abstract
Slow inactivated states in voltage-gated ion channels can be modulated by binding molecules both to the outside and to the inside of the pore. Thus, external K+ inhibits C-type inactivation in Shaker K+ channels by a “foot-in-the-door” mechanism. Here, we explore the modulation of a very long-lived inactivated state, ultraslow inactivation (IUS), by ligand binding to the outer vestibule in voltage-gated Na+ channels. Blocking the outer vestibule by a mutant μ-conotoxin GIIIA substantially accelerated recovery from IUS. A similar effect was observed if Cd2+ was bound to a cysteine engineered to the selectivity filter (K1237C). In K1237C channels, exposed to 30 μM Cd2+, the time constant of recovery from IUS was decreased from 145.0 ± 10.2 s to 32.5 ± 3.3 s (P < 0.001). Recovery from IUS was only accelerated if Cd2+ was added to the bath solution during recovery (V = −120 mV) from IUS, but not when the channels were selectively exposed to Cd2+ during the development of IUS (−20 mV). These data could be explained by a kinetic model in which Cd2+ binds with high affinity to a slow inactivated state (IS), which is transiently occupied during recovery from IUS. A total of 50 μM Cd2+ produced an ∼8 mV hyperpolarizing shift of the steady-state inactivation curve of IS, supporting this kinetic model. Binding of lidocaine to the internal vestibule significantly reduced the number of channels entering IUS, suggesting that IUS is associated with a conformational change of the internal vestibule of the channel. We propose a molecular model in which slow inactivation (IS) occurs by a closure of the outer vestibule, whereas IUS arises from a constriction of the internal vestibule produced by a widening of the selectivity filter region. Binding of Cd2+ to C1237 promotes the closure of the selectivity filter region, thereby hastening recovery from IUS. Thus, Cd2+ ions may act like a foot-on-the-door, kicking the IS gate to close.
INTRODUCTION
Voltage-gated Na+ channels are key players in the regulation of excitability in nerve and muscle. One mechanism by which excitability may be modulated is the entry of these channels into slow inactivated states upon depolarization. When slow inactivated channels are repolarized they do not recover until hundreds of milliseconds or even several hundred seconds have elapsed, thereby causing prolonged refractoriness.
Defective slow inactivation in voltage-gated Na+ channels has been associated with a number of disease states of the central nervous system (1–3), skeletal muscle (4–6), and the heart (7,8).
Many compounds alter the function of ion channels by modulation of their gating behavior (9). Modification of gating may arise from high-affinity binding to a specific state of the channel, resulting in stabilization of the high-affinity state with respect to low-affinity states (10,11). One possibility by which such stabilization may be envisioned on a molecular basis was first reported for quaternary ammonium compounds in voltage-gated K+ channels: Clay Armstrong proposed that a compound bound to the inner pore prevented normal closure of the activation gate (“foot-in-the-door”) and that channels could close only after the drug dissociated from its binding site (12). A similar mechanism can also be operative at the outer vestibule of ion channels: In Shaker-type K+ channels (Kv1) external tetraethylammonium and external K+ ions compete with an inactivation process (C-type inactivation) by a foot-in-the-door mechanism, giving rise to the idea that C-type inactivation is produced by a constriction or partial collapse of the outer vestibule of the channel (13–17).
μ-Conotoxin GIIIA (μ-CTX) interacts with voltage-gated Na+ channels by plugging the outer vestibule (18,19). Investigation of possible kinetic effects of this toxin has long been prevented by the slow off-rate of the compound. However, the construction of mutant derivatives of μ-CTX, which act as partial blockers, recently enabled assessment of the gating behavior in the blocked state (20–22). Using this approach we were able to demonstrate that μ-CTX R13Q interacts with a very stable inactivated state, which we refer to as “ultraslow inactivation” (IUS). Thus, when μ-CTX R13Q was bound, both the amplitude and the time constant of ultraslow recovery were significantly reduced (21). Superficially this mode of action was reminiscent of the interaction of K+ and of tetraethylammonium with Shaker K+ channels (15) suggesting that, similar to C-type inactivation in Shaker K+ channels, IUS results from a constriction of the outer vestibule. Other forms of slow inactivation in voltage-gated Na+ channels have also been associated with a closure of the outer vestibule (23–31).
On the other hand, the likelihood of entry into IUS was dramatically enhanced if a key residue in the selectivity filter of the rNaV1.4 channel, K1237, was replaced by a glutamic acid (21). In addition to changing the gating behavior, mutations at this site also severely affected ion selectivity, allowing for the permeation of both K+ and Ca2+ ions as well as enabling the passage of large organic cations like choline (32–37). Hence, the mutation K1237E most likely increased the diameter of the selectivity filter rather than causing a constriction. This prompted us to propose a widening rather than a constriction of the outer vestibule as the molecular mechanism underlying IUS (38): If IUS is indeed produced by a widening of the outer vestibule, then inhibition of IUS by μ-CTX R13Q most likely results from a stabilizing action on the structure of the channel. Hence, the binding of the toxin to the external vestibule may impede the widening of the vestibule by acting as a “splint in the vestibule” (21). However, whereas the mutation which greatly enhanced the likelihood of entry into IUS was located at the selectivity filter of the channel (K1237E), μ-CTX has been shown to mainly interact with residues more external to this site (18,19,39–43), suggesting that IUS encompasses a broad rearrangement of the outer vestibule. In addition, widening of the outer vestibule fails to explain why the channel is nonconducting. A possible explanation is found in experiments showing that lidocaine bound in the inner pore also accelerates recovery from IUS (44). Perhaps the failure of conduction is a tilting of the pore lining such that the inner pore is blocked.
The aim of this study was to explore whether IUS can be modulated by a direct interaction between a molecule and the site 1237. For this purpose we tested the effect of binding Cd2+ ions to a cysteine engineered to this site. We found Cd2+ to substantially accelerate recovery from IUS without affecting entry into this state. We suggest that, during recovery, Cd2+ ions enter the pore in a voltage-dependent manner and bind in a coordinative manner to one or more sites at the level of the selectivity filter. This binding of Cd2+ promotes a constriction of the outer vestibule which, in turn, results in a weakening of the interaction between the selectivity filter and the S6 segment, thereby speeding recovery from IUS. Thus, rather than acting as a foot-in-the-door, Cd2+ ions may “kick the door” and close the gate responsible for IUS (“foot-on-the-door”).
MATERIALS AND METHODS
A detailed description of materials and methods is given in our previous work (22).
Site-directed mutagenesis
Detailed methods for the mutagenesis have been published previously (18,21,45).
Electrophysiological recordings
Two-microelectrode voltage clamp recording in Xenopus oocytes
All experiments except those summarized in Fig. 8 C were performed using the two-electrode voltage clamp technique in Xenopus laevis oocytes. Stage V and VI Xenopus laevis oocytes were isolated from female frogs (NASCO, Ft. Atkinson, WI), washed with Ca2+-free solution (90 mM NaCl, 2.5 mM KCl, 1 mM MgCl2, 1 mM NaHPO4, and 5 mM HEPES titrated to pH 7.6 with 1 N NaOH), treated with 2 mg/ml collagenase (Sigma, St. Louis, MO) for 1.5 h and had their follicular cell layers manually removed. As judged from photometric measurements, ∼50–100 ng of complementary RNA (cRNA) was injected into each oocyte with a Drummond microinjector (Broomall, PA). Either native or mutant α subunit cRNA was mixed with rat brain β1 cRNA at a molar ratio of 1:1. Oocytes were incubated at 17°C for 12 h to 3 days before examination.
FIGURE 8.

(A) Kinetic modeling of the modulation of IUS by Cd2+. Hypothetical scheme of the proposed kinetic states and transitions. Upper row depicts Cd2+-free states: NI = noninactivated, IS = slow inactivated, IUS = ultraslow inactivated. Lower row: Cd2+-bound states. (B) Time course of recovery from slow inactivation. From a holding potential of −120 mV, channels were depolarized to −20 mV for 25 s. Thereafter, the membrane potential was returned to −120 mV for the indicated time periods after which channel availability was tested by a 20-ms pulse to −20 mV. Peak inward currents were normalized to the value after full recovery. The time course of recovery was determined during a control period and during superfusion with 50 μM Cd2+. Data points were fitted with double exponential functions (Eq. 5). The parameters were drug free: A1 = 0.62 ± 0.04, τ1 = 573.2 ± 32.3 ms, A2 = 0.30 ± 0.02, τ2 = 6493.4 ± 56.3 ms; Cd2+: A1 = 0.61 ± 0.07, τ1 = 663 ± 64.3 ms, A2 = 0.29 ± 0.03 (P = n.s.), τ2 = 6828.5 ± 90.4 ms, n = 3; P = n.s. (C) Time course of relief from block by 50 μM Cd2+ in K1237C channels. K1237C channels were heterologously expressed in tsA201 cells and examined by the whole-cell patch-clamp technique. Cells were held at −120 mV, and inward currents were elicited by 10 ms depolarizations to −20 mV at 20-s intervals. Data points were fitted with a monoexponential function (Eq. 6). See text for fitting parameters. (D) Model A: Modulation of IUS by low-affinity binding of Cd2+ to the IUS state. Data points are time courses of recovery during control and during exposure to 20 μM Cd2+ (i.e., a concentration close to the KD), taken from Fig. 2 C. Lines are model calculations assuming rates of unbinding of Cd2+ from the IUS state to be reduced with respect to the unbinding rates from the IS and NI state by factors 0, 2, 5, 10, 100, and 1000 (lines in order from right to left). The lines representing model calculations assuming unbinding rates 100-fold and 1000-fold lower for IUS than for IS and NI are superimposed. (E) Model B: Modulation of IUS by high-affinity binding of Cd2+ to the IS state. The affinity of Cd2+ to the IS state was assumed to be 10 times higher than the affinities for the NI and the IUS states. Data points are the same as in Fig. 2 D. Lines from right to left represent calculations assuming the following concentrations of Cd2+: 0 μM, 10 μM, 20 μM, and 50 μM.
Recordings were made in the two-electrode voltage clamp configuration using a TEC 10CD clamp (npi electronic, Tamm, Germany). For accurate adjustment of the experimental temperature (18°C ± 0.5°C) an oocyte bath cooling system (HE 204, Dagan, Minneapolis, MN) was used. Oocytes were placed in recording chambers in which the bath flow rate was ∼100 ml/h, and the bath level was adjusted so that the total bath volume was <500 μl. Electrodes were filled with 3 M KCl and had resistances of <0.5 MΩ. Using pCLAMP6 software (Axon Instruments, Foster City, CA), data were acquired at 71.4 kHz after low-pass filtration at 2 kHz (−3 dB). Recordings were made in a bathing solution that consisted of (in mM): 90 NaCl, 2.5 KCl, 1 BaCl2, 1 MgCl2, and 5 mM HEPES titrated to pH 7.2 with 1 N NaOH. BaCl2 was used as a replacement for CaCl2 to minimize Ca2+-activated Cl− currents. In some experiments [Na+]o was reduced by equimolar replacement with N-methyl-D-glucamine-chloride. Lidocaine was obtained from Sigma. The derivative of μ-CTX, R13Q, in which arginine 13 is replaced by a glutamine, was made as previously described (46). To examine the modification of K1237C channels by Ag+, AgNO3 was added to the bathing solution at a concentration of 500 nM. Due to high concentration of Cl− ions in the bath solution, the concentration of free Ag+ ions is limited by the low solubility of AgCl. To determine the final concentration of Ag+ we first considered the temperature dependence of the solution product (Ksp) of AgCl. Reported values of the Ksp are 2.54 × 10−11, 7.05 × 10−11, 1.78 × 10−10, 4.16 × 10−10, 6.17 × 10−10, and 1.32 × 10−9 mol2 l−1 at temperatures of 5°C, 15°C, 25°C, 35°C, 40°C, and 50°C, respectively (47). The temperature dependence of these Ksp values could be well fitted (R2 = 0.99) by a single exponential equation of the form
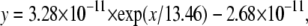 |
(1) |
which yielded a Ksp of 1.18 × 10−10 mol2 l−2 at 20°C. Furthermore, we considered the ionic activities (ai) of the dissolved ions (i), which are given by
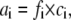 |
(2) |
where fi is the activity coefficient of the ion i. fi can be calculated following the equation of the Debeye-Hückel theory:
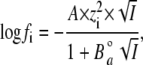 |
(3) |
where  is the ion size parameter and I is the ionic strength (i.e., 103 mM for the bathing solution). For the calculation, the following values were used (47): A = 0.507, B = 0.328,
is the ion size parameter and I is the ionic strength (i.e., 103 mM for the bathing solution). For the calculation, the following values were used (47): A = 0.507, B = 0.328,  (Ag+) = 2.5 Å,
(Ag+) = 2.5 Å,
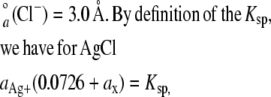 |
(4) |
because ax is small in comparison to 0.0726 M, it can be disregarded. Also, 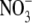 ions were not considered because of their low concentration in comparison to Cl− ions. Rearrangement of Eq. 4 yields an ionic activity of Ag+ of 2.19 nM. This low activity was sufficient for the irreversible modification of the thiolate group of the cysteine side chains.
ions were not considered because of their low concentration in comparison to Cl− ions. Rearrangement of Eq. 4 yields an ionic activity of Ag+ of 2.19 nM. This low activity was sufficient for the irreversible modification of the thiolate group of the cysteine side chains.
Whole-cell patch-clamp recording
To determine the off-rate of Cd2+ from K1237C channels (see Fig. 8 C), experiments were performed using the whole-cell patch-clamp recording technique. TsA201 cells were grown in Dulbecco's modified Eagle's medium supplemented with 10% fetal bovine serum and 20 units/ml each of penicillin and streptomycin (Gibco, Gaithersburg, MD). Cells were maintained at 37°C in a humid atmosphere containing 5% CO2. Before recording, cells were dissociated from their substrate by treatment with a 0.25% trypsin solution (Gibco) for ∼2 min, pelleted, resuspended in bath solution, and allowed to settle to the bottom of the recording chamber. Channel DNA was transiently transfected into tsA201 cells using a calcium phosphate transfection system (Gibco).
Na+ currents were recorded using an Axopatch 200B patch-clamp amplifier (Axon Instruments) as in Zebedin et al. (48). Recording was begun ∼10 min after whole-cell access was attained to minimize time-dependent shifts in gating. Pipettes were formed from aluminosilicate glass (AF150-100-10; Science Products, Hofheim, Germany) with a P-97 horizontal puller (Sutter Instruments, Novato, CA), heat polished on a microforge (MF-830; Narishige, Tokyo, Japan), and had resistances between 1 and 2 MΩ when filled with the recording pipette solution (105 mM CsF, 10 mM NaCl, 10 mM EGTA, 10 mM HEPES, pH = 7.3). Peak current amplitudes equaled −1.2 ± 0.4 nA. Series resistance was minimized (>80%–90%) using the Axoclamp 200B device. As such the uncompensated voltage error across the pipette was calculated to be 3.2 ± 0.3 mV. The bath solution consisted of (in mM) 140 NaCl, 2.5 KCl, 1 CaCl2, 1 MgCl2 10 HEPES. Voltage-clamp protocols and data acquisition were performed with pclamp 6.0 software (Axon Instruments) through a 12-bit A-D/D-A interface (Digidata 1200; Axon Instruments). Data were low-pass filtered at 2 kHz (−3 dB) and digitized at 10–20 kHz.
The external solution containing Cd2+ was applied via a DAD-12 drug application device (Adams & List, Westbury, NY). This superfusion system delivers buffers from 12 reservoirs under pressure (200–400 mm H2O) via a capillary with an inner diameter of ∼100 μm and permits a complete exchange of solutions surrounding the cells under investigation within 100 ms (49).
Data evaluation. The time courses of recovery from IUS of normalized peak inward currents were fit with the double exponential function:
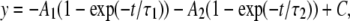 |
(5) |
where τ1 and τ2 are the time constants of distinct components of recovery, A1 and A2 are the respective amplitudes of these components, and C is the final level of recovery.
The time course of entry into IUS was fitted with the monoexponential function:
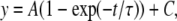 |
(6) |
where τ is the time constants of entry and C is the final level of entry.
To determine the dissociation constant (KD) of Cd2+ from Na+ channels, 10-ms test pulses to −20 mV (Vh = −120 mV) were applied at 20-s intervals during Cd2+-free conditions and during superfusion with various concentrations of Cd2+. Data plots of the fractional block as a function of Cd2+ concentration were then fitted with the Hill equation:
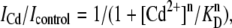 |
(7) |
where ICd and Icontrol are maximum inward currents during exposure to Cd2+ and during control, respectively, KD is the midpoint of the curve, and n is the Hill coefficient.
Steady-state inactivation data were fitted with a Boltzmann function
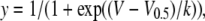 |
(8) |
where V is the voltage of the conditioning prepulse, V0.5 is the voltage at which half-maximum inactivation occurred, and k is the slope factor.
To determine the voltage-dependence of block by Cd2+ ions, the following equation was fitted to the data points in the inset to Fig. 9 B (50):
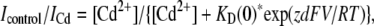 |
(9) |
where KD(0) represents the apparent affinity at 0 mV, z the charge valence of Cd2+, d the fractional electrical distance, and F, R, and T have their usual meanings.
FIGURE 9.

IS is stabilized by Cd2+. (A) Steady-state availability curve for IS. K1237C channels were depolarized for 25 s to different potentials, after which the membrane was hyperpolarized to −120 mV for 500 ms to allow for recovery of fast inactivated channels. Then a 20-ms test pulse to −20 mV was applied to open recovered channels. In each oocyte this protocol was performed under Cd2+-free conditions and after washin of 50 μM Cd2+. The minimum availability (−20 mV) was 0.48 ± 0.1 and was not changed by Cd2+. Data points were normalized for maximum and minimum availability and were fitted with a Boltzmann function (Eq. 8). The parameters were control: V1/2 = −68.9 ± 0.8 mV, k = 7.2 ± 0.9 mV; Cd2+: V1/2 = −76.6 ± 2.3 mV, k = 5.0 ± 2.2 mV, n = 6, P < 0.05). (B) Correction for voltage-dependent block left shifts the midpoint of availability and decreases its slope. Protocol, as in A, in a single oocyte exposed to 50 μM Cd2+. Triangles: uncorrected data points. Squares: data points corrected for voltage-dependent block. Inset: Voltage-dependent block by Cd2+. The data points for the voltages −60 mV, −40 mV, and −20 mV, shown in B were expressed as a fraction of the current before superfusion with Cd2+ and replotted. The line represents the fit of a Woodhull model (Eq. 9) to the data points. Fitting parameters were KD(0) = 100.0 ± 6.9 μM, d = 0.13 ± 0.02, room temperature (20°C). This relationship was used to correct voltage-dependent availability (squares). Data points were normalized for maximum and minimum availability and were fitted with a Boltzmann function (Eq. 8). The parameters were uncorrected (straight line): V1/2 = −73.4 ± 4.14 mV, k = 4.5 ± 3.6 mV; corrected (dotted line): V1/2 = −75.8 ± 1.4 mV, k = 5.6 ± 1.2 mV.
Curve fitting was performed using ORIGIN 7.5 (MicroCal Software, Northampton, MA). Kinetic modeling was carried out using ModelMaker software (Cherwell Scientific, Oxford, UK). Data are expressed as mean ± SE. Statistical comparisons were made using the two-tailed Student's t-tests or one-way ANOVA for comparison of more than two groups. A P < 0.05 was considered significant.
RESULTS
A charge-altering mutation in the selectivity filter of rNaV1.4 induces ultraslow inactivation
Amino acid replacement of the lysine at position 1237 in the selectivity filter of the rNaV1.4 channel by glutamate, serine, and alanine substantially enhances the likelihood of entry into IUS during long depolarizations (>100 s; 21). To further explore the role of the nature of the amino acid and of charge at site 1237 in the modulation of IUS, we engineered a cysteine to site 1237. As shown in Fig. 1, A and B, recovery of peak inward currents from a 300-s conditioning pulse to −20 mV took several minutes. The time course of recovery could be fitted with two exponentials yielding time constants of ∼10 s and ∼100 s (Table 1, “0 μM”). The smaller time constant most likely represents recovery from slow inactivation (IS; 51), whereas the larger time constant is typical for recovery from IUS. The amplitude of the ultraslow component of recovery (A2 in Table 1) suggests that ∼70% of K1237C channels enter into IUS during the 300-s depolarization to −20 mV. These time constants and amplitudes are similar to those previously reported for the other mutations at site 1237, i.e., K1237E, K1237S, and K1237A (21,38). By contrast <20% of wild-type channels, or channels carrying the charge-conservative mutation K1237R, recover from IUS after a 300-s depolarization (21). Therefore, a positive charge at site 1237 appears to be essential for the prevention of entry into IUS.
FIGURE 1.

(A) Ultraslow recovery from inactivation in the mutation K1237C. Original traces of inward currents in an oocyte expressing K1237C channels. From a holding potential of −120 mV, the channels were inactivated by a depolarizing step to −20 mV of 300 s duration. After returning to −120 mV, recovery from inactivation was monitored by repetitive 20-ms steps to −10 mV at 20-s intervals. Peak inward currents were slowly rising approaching a maximum after several minutes. (B) Normalized time course of recovery from IUS. Peak inward currents from nine experiments as shown in A were normalized to the maximum current level after full recovery and plotted as a function of time after the conditioning prepulse. The time course of recovery was fitted with a double exponential function (Eq. 5). (C) Time course of development of ultraslow inactivation. The time course of recovery from IUS after varying prepulse durations was evaluated as in B (n = 4–12). The amplitudes of ultraslow recovery (A2), as determined from fitting Eq. 5 to each time course of recovery, is plotted as a function of the duration of the conditioning prepulse. Data points were fitted with a monoexponential function (Eq. 6).
TABLE 1.
Parameters of recovery from IUS in K1237C
| [Cd2+] | 0 μM (n = 9) | 10 μM (n = 7) | 20 μM (n = 6) | 30 μM (n = 6) | 50 μM (n = 9) |
|---|---|---|---|---|---|
| τ1(s) | 10.24 ± 1.0 | 6.9 ± 0.5 | 6.6 ± 0.7 | 5.2 ± 0.7* | 8.4 ± 1.1 |
| τ2 (s) | 145.0 ± 10.2 | 70.2 ± 4.9† | 55.6 ± 2.9† | 32.5 ± 3.3† | — |
| A1 | 0.27 ± 0.07 | 0.26 ± 0.02 | 0.32 ± 0.03 | 0.76 ± 0.04† | 0.87 ± 0.02† |
| A2 | 0.73 ± 0.07 | 0.74 ± 0.02 | 0.64 ± 0.02 | 0.24 ± 0.04‡ | — |
Development of IUS in K1237C was also extremely slow (τ = 299.0 ± 22.0 s), and IUS approached unity after ∼600 s, indicating that IUS is absorbing in K1237C (Fig. 1 C). Thus, replacement of lysine by cysteine at site 1237 strongly favored entry into IUS.
Recovery from IUS is modified by a mutant μ-CTX
μ-CTX is a potent blocker of the outer vestibule of the skeletal muscle Na+ channel (18), but the mutant μ-CTX R13Q only partially blocks the outer vestibule, allowing for flow of ∼30% residual current in the blocked state (20). This property allows the investigation of kinetic changes when μ-CTX is fully bound to the channel. In an earlier report we presented evidence that binding μ-CTX R13Q to the outer pore substantially accelerated the time course of recovery from IUS after prolonged depolarizations in K1237E (21). Here, we test whether μ-CTX R13Q had a similar effect on IUS in K1237C. In the presence of 27 μM μ-CTX R13Q a significantly smaller fraction of K1237C channels appeared to be in IUS at the end of prolonged depolarizations (Fig. 2 A).
FIGURE 2.

(A) The time course of recovery from IUS is modulated by a mutant μ-CTX. Experiments like those described in Fig. 1, A and B, were carried out before and after addition of 27 μM μ-CTX R13Q to the bath solution. μ-CTX R13Q significantly reduced both the amplitude and the time constant of ultraslow recovery (drug free: A2 = 0.58 ± 0.01, τ2 = 155.9 ± 4.3; μ-CTX R13Q: A2 = 0.51 ± 0.01 (P = 0.03), τ2 = 87.8 ± 2.6 (P = 0.002), n = 4). (B) Cysteine engineered to site 1237 creates a high-affinity binding site for Cd2+. Block by various concentrations of Cd2+ of maximum inward currents elicited by 20-ms depolarizations to −20 mV (Vh = −120 mV) at 20-s intervals was evaluated in wild-type (100 μM: n = 6, 500 μM: n = 5, 1000 μM: n = 4, 2000 μM: n = 3), and K1237C channels (5 μM: n = 5, 10 μM: n = 8, 30 μM: n = 5, 50 μM: n = 9, 70 μM: n = 5, 100 μM: n = 6). Data points were fitted with Eq. 3. The KD for Cd2+ binding was significantly smaller in K1237C than in wild-type. (C) Speeding of recovery from IUS by Cd2+. Time course of peak inward currents during a typical experiment. Data points represent peak inward currents elicited by 20-ms step depolarizations to −20 mV from the indicated holding potential (Vh). During a Cd2+-free control, recovery from a 300-s prepulse to −20 mV was monitored analogously to the protocol described for Fig. 1 A. After recovery from ultraslow inactivation was complete, perfusion with 50 μM Cd2+ was started (arrow, “50 μM Cd2+”). After the reduction of peak Na current by Cd2+ had reached a steady-state level, the inactivation/recovery protocol was repeated. (D) Normalized time courses of recovery from IUS from several experiments at different Cd2+ concentrations. Data points were fitted with Eq. 5. The fitting results are presented in Table 1.
The binding site of μ-CTX involves residues in the P-loops located extracellularly to the selectivity filter (18,19). On the other hand, the mutation that gives rise to IUS is located within the selectivity filter ring (site 1237). The fact that μ-CTX interacts with IUS despite no direct interaction with site 1237 suggests that IUS involves a broad rearrangement of the outer vestibule. Therefore, it appeared of to be of interest whether an interaction at the level of the selectivity filter can also interfere with IUS.
Cysteine at amino acid position 1237 acts as a high-affinity binding site for Cd2+
Cd2+ ions strongly interact with the sulfhydryl groups of cysteine residues (52). Therefore, we sought to investigate whether binding Cd2+ to C1237 results in a modulation of IUS. Such an interaction would tend to restore the positive charge to the selectivity filter that was lost when lysine was removed. First, we tested whether the construct K1237C binds Cd2+ ions with higher affinity than wild-type channels, as would be expected if the engineered cysteine formed a new binding site for Cd2+ ions. Oocytes expressing wild-type and K1237C channels were superfused with different concentrations of Cd2+, and the fractional block of maximal inward Na+ currents at holding potential −120 mV was determined. Short depolarizing steps were used at slow rates to avoid entry of channels into slow or ultraslow inactivated states. Plots of fractional block as a function of Cd2+ concentration were fitted with Hill equations (Eq. 7), yielding KD values of 338.9 ± 10.3 μm for wild-type and 35.0 ± 1.8 μm for K1237C (P < 0.001; Fig. 2 B). The Hill factors were 1.2 ± 0.05 for wild-type and 1.2 ± 0.09 (not significant; n.s.) for K1237C, suggesting a 1:1 binding stochiometry. These dissociation constants are similar to those determined previously in this mutant (34,36). On the other hand, the low KD of block by Cd2+ in the K1237C mutation could have been produced by a steric effect due to replacement of the lysine at site 1237 by a different amino acid. However, this appears unlikely because replacement of lysine 1237 by serine did not give rise to a high-affinity binding site for Cd2+ (see below, Fig. 3 A). Thus, the mutation K1237C created a high-affinity binding site for Cd2+ that resulted in block of current.
FIGURE 3.

(A) Binding of Cd2+ to C1237 mediates the kinetic effect on IUS. Concentration-dependent Cd2+ block in wild-type and in the mutant K1237S. Experimental protocol was analogous to Fig. 2 B. Data points were fitted with Eq. 3. The affinity of Cd2+ to K1237S channels was similar to wild-type (KD = 440.1 ± 33 μm; n = 5; P = n.s. compared with wild-type). Data points and the fitted curve for wild-type are identical to Fig. 2 B. (B) Recovery from IUS in K1237S channels during control and after exposure to 300 μM Cd2+. Experimental protocol was analogous to Fig. 2 C. Data points were fitted with Eq. 5. Cd2+ did not change the time course of recovery from IUS (drug free: A2 = 0.63 ± 0.04, τ2 = 110 ± 15 s, n = 6; Cd2+: A2 = 0.65 ± 0.07, τ2 = 101 ± 17 s, n = 4; P = n.s.).
Cd2+ modulates the time course of recovery from IUS in the construct K1237C
To test whether IUS in K1237C is modulated by binding of Cd2+ ions, we monitored recovery from IUS during a Cd2+-free control and during subsequent superfusion with 50 μM Cd2+(Fig. 2 C). As shown in Fig. 2 D, Cd2+ dramatically altered the time course of recovery from IUS: both amplitude and time constant of recovery from IUS decreased as a function of the concentration of Cd2+ (Fig. 2 D, Table 1). At the highest concentration of 50 μM Cd2+, the time course of recovery after a 300-s depolarizing pulse to −20 mV of K1237 channels was indistinguishable from wild-type (data not shown, but see Hilber et al. 22).
Binding of Cd2+ to cysteine at position 1237 mediates the effect on ultraslow inactivation
The question arises, however, whether the modulation of IUS by Cd2+ ions resulted from interaction with C1237 or with some other superficial binding site. If the modulation of channel kinetics by Cd2+ resulted from interaction with site 1237, then Cd2+ should have had no kinetic effect on mutations at this site that do not increase binding affinity for Cd2+. Therefore, we tested the effect of Cd2+ ions on the mutant K1237S. When depolarized for 300 s, ∼60% K1237S channels entered into IUS as judged from the amplitude of the slower component of recovery (21). The affinity of Cd2+ ions to K1237S channels was similar to wild-type, suggesting that Cd2+ did not bind with high affinity to the selectivity filter of K1237S channels (Fig. 3 A). Superfusion with 300 μM Cd2+ did not affect the time course of recovery from IUS in K1237S (Fig. 3 B). This supports the interpretation that the modulatory action of Cd2+ ions on IUS in K1237C channels indeed occurs by interaction with C1237.
Acceleration of recovery is not caused by use-dependent inhibition by Cd2+
In the experiments shown in Figs. 1, 2, 3, 5, 6, and 7, we assessed the time course of recovery from IUS by repetitive 20-ms test pulses at 20-s intervals after the conditioning prepulse. Superfusion with Cd2+ reduced the time until currents reached steady-state conditions in K1237C channels in a concentration-dependent fashion (Fig. 2, C and D). However, such apparent acceleration of recovery could have been recovery from the cumulative use-dependent block by Cd2+ ions during the test pulses applied to monitor recovery from IUS. The combination of unchanged ultraslow recovery and cumulative block may have artifactually resulted in an acceleration of the recovery process. Therefore we tested whether K1237C channels exposed to 50 μM Cd2+ recovered completely during the interval between test pulses. We determined the time course of recovery from a 1-s depolarization to −20 mV. The duration of this depolarization was substantially longer than the test pulse duration in the protocols that tested recovery from IUS. Thus, even slow binding of Cd2+ to inactivated channels would be detected by this protocol. Fig. 4 shows that recovery from inactivation elicited by a single 1-s pulse was complete after ∼5 s. Thus, the interval of 20 s between test pulses was sufficient to avoid development of cumulative block during the recovery period.
FIGURE 5.

(A) Effect of Ag+ on IUS in K1237C. Time course of peak inward sodium currents during a typical experiment. The experimental protocol was analogous to Fig. 2 C. Because of the irreversible modification of the channels by Ag+, the exposure to Ag+ (∼2 nM) was terminated after the current was reduced to by ∼30%. (B) Normalized time course of recovery from IUS. Peak inward currents from the experiment shown in A were normalized to the respective maximum after full recovery. Superfusion with ∼2 nM Ag+ did not substantially affect the time course of recovery from IUS.
FIGURE 6.

Lowering the concentration of Na+ does not affect the time course of recovery from IUS. Time course of peak inward sodium currents during a typical experiment. The experimental protocol was analogous to Fig. 2 C. At the time indicated by the arrow the bath solution was changed to a solution in which the 90 mM Na+ were replaced by 40 mM Na+ + 50 mM choline. The time course of recovery from IUS was not substantially changed by the solution exchange.
FIGURE 7.

Cd2+ has no effect on the development of IUS but selectively hastens the time course of recovery. To examine which phase of the inactivation process was modulated by Cd2+, the application of Cd2+ was restricted to either the development or the recovery process. (A) Selective application of Cd2+ during development of IUS. Time course of peak inward sodium currents during a typical experiment (voltage clamp protocol as in Fig. 2 C). Superfusion with Cd2+ was started 100 s before the conditioning prepulse to allow for steady-state Cd2+ concentrations to be established at the beginning of the conditioning prepulse. Exposure to Cd2+ was terminated 180 s before the end of the conditioning prepulse to allow for complete washout of Cd2+ before initiation of recovery. (B) Normalized time course of recovery. Four experiments as described in A. Data points were fitted by Eq. 5, yielding the following parameters: IUS (drug free: A2 = 0.58 ± 0.02, τ2 = 163 ± 6, n = 6; Cd2+: A2 = 0.59 ± 0.03, τ2 = 158 ± 4 (n.s.), n = 4). (C) Selective application of Cd2+ during recovery from IUS. Time course of peak inward sodium currents during a typical experiment (voltage clamp protocol as in Fig. 2 C). Superfusion with Cd2+ was started 120 s before the end of the conditioning prepulse to allow the bath concentration of Cd2+ to reach steady state at the time of the start of the recovery period. (D) Normalized time course of recovery. Four experiments as described in C. Data points were fitted to Eq. 5, yielding the following parameters: drug free: A2 = 0.68 ± 0.04, τ2 = 120.0 ± 5.9; Cd2+: A2 = 0.64 ± 0.03 (P < 0.05), τ2 = 64.2 ± 3.8 s, (P < 0.05).
FIGURE 4.

Cd2+ does not affect the time course of recovery from short depolarizations in K1237C. Oocytes were superfused with 50 μM Cd2+. From a holding potential of −120 mV channels were depolarized to −20 mV for 1000 ms. Thereafter, the membrane potential was returned to −120 mV for the indicated time periods, after which channel availability was tested by a 20-ms pulse to −20 mV. Peak inward currents were normalized to the value after full recovery. Data points were fitted with a single exponential function (Eq. 6). The time constant of recovery was 4.7 ± 0.2 s (n = 8).
The kinetic effect of Cd2+ is not due to selective block of a defined population of channels
As shown in Fig. 2, A and B, the time course of recovery of K1237C channels from a 300-s conditioning prepulse can be fitted with two exponentials, representing the recovery of at least two populations of channels (Table 1). If there is no interconversion between these populations (i.e., channels residing in slow and ultraslow inactivation, see above), the acceleration of recovery produced by Cd2+ may be the result of selective block of the fraction of channels determined to enter into IUS. To explore this question we tested the effect of irreversible block of the C1237 site on the time course of recovery from IUS. Ag+ ions react with sulfhydryl groups to form a strong Ag+-S bond (53). If the acceleration of recovery from IUS by Cd2+ resulted from simple elimination of the population of ultraslow inactivating channels, then irreversible block by Ag+ should have reproduced this phenomenon. As shown in Fig. 5, superfusion with ∼2 nM Ag+ did not affect IUS despite blocking ∼50% of channels.
Furthermore, as shown in Fig. 1 C entry into IUS approaches unity after ∼600 s. This indicates that all channels have the potential to enter into IUS, provided the conditioning pulse is long enough. This argues against the presence of distinct channel populations without interconversion. Thus, modulation of IUS results from dynamic interaction of Cd2+ ions with the pore of the K1237C channels. Cd2+ appears to bind and unbind faster than the rates of the inactivation processes, achieving an equilibrium with each inactivation state.
The kinetic effect of Cd2+ is not due to depletion of Na+ ions entering the pore
Lowering the concentration of Na+ ions in the pore has been shown to promote entry into slow (not ultraslow) inactivation (24,54). If we assume a linear kinetic scheme in which IUS is reached via a slow inactivated state (IS), then lowering of the concentration of Na+ ions in the pore may stabilize slow inactivation and prevent further transitions from IS to IUS. If binding Cd2+ ions in the pore results in depletion of Na+ ions from the pore, the reduction in IUS by Cd2+ may be a consequence of this depletion and the resulting stabilization of IS. Therefore, we tested whether depletion of Na+ ions per se altered the likelihood of entry into IUS. Oocytes were first bathed in a solution containing 90 mM Na+ and the time course of recovery from IUS was assessed. Thereafter the concentration of Na+ was changed to 40 mM, which resulted in a reduction of the maximum inward current by ∼50%, which corresponds to the reduction of the Na+ current by ∼30–50 μM Cd2+. As shown in Fig. 6, bathing the oocytes in 40 mM Na+ affected neither the amplitude nor the time constant of recovery from IUS: τ1: 10.4 ± 1.1 s, τ2: 117.5 ± 18.5 s, A1: 0.22 ± 0.06, A2: 0.78 ± 0.07. Therefore, any Na+ depletion in the pore had no effect on entry into IUS.
Cd2+ ions do not prevent entry into IUS by a foot-in-the-door mechanism
In K+ channels C-type inactivation is considered to be prevented by K+ ions in the outer pore by a foot-in-the-door mechanism (15). Analogously, IUS may arise from a collapse of the outer vestibule and Cd2+ ions bound to the selectivity filter may prevent this constriction of the permeation pathway. If Cd2+ ions acted by a foot-in-the-door mechanism, then the time course of development of IUS at depolarized potentials should be prolonged by Cd2+. We tested this prediction by selectively exposing K1237C channels to Cd2+ either only during the conditioning prepulse or only during repolarization after the conditioning prepulse. If Cd2+ prevented IUS by a foot-in-the-door mechanism, then application of Cd2+ during the conditioning prepulse should have resulted in a substantial acceleration of the time course of recovery, whereas application of Cd2+ during recovery should have had no kinetic effect. As shown in Fig. 7, A and B, Cd2+ did not affect the time course of recovery from inactivation when applied during the conditioning prepulse (Vm = −20 mV) but dramatically accelerated recovery when applied during repolarization to −120 mV (Fig. 7, C and D). Therefore, the action of Cd2+ was exclusively during recovery from IUS, not during its development strongly arguing against a foot-in-the-door mechanism.
A kinetic model of the modulation of IUS by Cd2+
The experiment in Fig. 7 suggests that Cd2+ accelerates the time course of recovery from IUS at hyperpolarized potentials. During the 300-s conditioning prepulse K1237C channels enter into IUS even in the presence of Cd2+ ions, but after hyperpolarization Cd2+ ions may bind to and stabilize a conformation that is traversed during recovery, thereby speeding the time course of recovery from IUS. Therefore, a Cd2+-bound IUS state must recover at hyperpolarized potentials much faster than the unbound IUS state. To explore this concept we developed a gating model of recovery from inactivation at −120 mV. We considered three states: noninactivated (NI), slow inactivated (IS), and ultraslow inactivated (IUS). Open, closed, and fast inactivated states were omitted because they are unlikely to contribute to time constants of recovery >1 s. We considered a serial gating model in which the IUS state is reached via the IS state (Fig. 8 A).
During recovery from inactivation the backward rates leading into NI predominate those leading to inactivation. Therefore, the forward rates leading into inactivation were set two orders of magnitude smaller than the respective rates leading to the NI state. kIus→Is was defined as the reciprocal value of τ2 during Cd2+-free conditions (0.0068 s−1, Table 1). To determine kIs→NI we evaluated the time course of recovery from inactivation produced by a 25-s conditioning prepulse to −20 mV. As shown in Fig. 8 B this time course could be well fitted by two exponentials. During depolarizations of longer durations than 25 s, recovery will be dominated by the larger time constant (τ2 = 6493.4 ms). Hence, kIs→NI was set to 1/τ2 = 0.154 s−1. Fig. 8, D and E (squares), shows that the modeled time course of recovery under Cd2+-free conditions fits well the experimental data.
To model recovery during block by Cd2+ we introduced a Cd2+-bound state for the noninactivated and both inactivated states (lower row in Fig. 8 A). To model the transitions between Cd2+-free and Cd2+-bound states we had to determine the on- and off-rates of Cd2+ with K1237C channels.
Determination of off-rates of Cd2+ from the noninactivated channel
Fast removal of a blocking agent in the bath solution will lead to dissociation from the binding site. Under this condition dissociation kinetics will only depend on the microscopic off-rate because the microscopic on-rate is zero. However, the two-electrode voltage clamp method in Xenopus laevis oocytes may not allow for bath exchange rates fast enough to faithfully monitor the off-rate of Cd2+ from K1237C channels. Therefore, we expressed K1237C channels in tsA201 cells and examined the washout of Cd2+ in the whole-cell patch-clamp configuration using a fast application system for rapid bath exchange. Cells were bathed in 50 μM Cd2+ until steady-state block was achieved (Fig. 8 C). Thereafter, Cd2+ was rapidly removed from the bath and the time course of unblock was monitored by sequential depolarizations at 0.2 Hz. We did not use higher test pulse frequencies to avoid possible contamination of the data by potential use-dependent unblock from open or fast inactivated states. Fig. 8 C shows that, upon washout of Cd2+, currents through K1237C channels recovered from block with a time constant of 3.2 ± 0.2 s (n = 8), yielding an off-rate of 0.3125 s−1. Since the KD of binding of Cd2+ to noninactivated channels is 35.0 ± 1.8 μM at −120 mV (Fig. 2 B), the association rate constant (kon) of Cd2+ at that voltage was calculated to be 8930 M−1s−1.
Binding of Cd2+ does not alter the time course of recovery from IS
Channels were exposed to 50 μM Cd2+ and the time course of recovery from a 25-s prepulse was determined. As shown in Fig. 8 B, the time course of recovery from IS was not affected by Cd2+. Therefore, in the model kIs-Cd→NI-Cd was set at the same values as the corresponding rates for the Cd2+-free transition (kIs→NI).
Modeling modulation of IUS by low-affinity binding of Cd2+ to the IUS state (Model A)
For the inactivated states the on- and off-rates of Cd2+ cannot be determined directly. Therefore, we explored the effect of assuming different binding affinities to the IUS and IS states as origin of the acceleration of recovery from IUS by Cd2+. Modulation of the rate constants of transition between states may result if the affinity of binding to a specific state differs from another connected state, resulting in stabilization of the high-affinity state with respect to the connected low-affinity state. Specifically, an acceleration of kIus-Cd→Is-Cd could result from either low-affinity binding of Cd2+ to the IUS state or from high-affinity binding of Cd2+ to the IS state. We first considered low-affinity binding to the IUS state. For the IS state we assumed on- and off-rates of Cd2+ to be equal to the corresponding values for the interaction with the NI state. Fig. 8 D shows the effect of increasing the rate of unbinding from the Cd2+-bound IUS state by factors of 0, 2, 5, 10, 100, and 1000. Microscopic reversibility required that kIus-Cd→Is-Cd be accelerated by the same factor by which the off-rate of Cd2+ from the IUS-Cd2+ state was increased (kIus-Cd→Ius). This procedure resulted in a speeding of the time course of recovery as a function of the decrease in affinity for Cd2+ of the IUS state. However, the increase in kIus-Cd→Ius causes channels to recover preferentially via the Cd2+-free states, setting an upper limit for recovery rate (upper row in Fig. 8 A). This effect produces a saturation of the speed of recovery at very high off-rates from the Cd2+-bound IUS state. Fig. 8 D shows that even with very low affinities of the IUS state for Cd2+, the experimental data cannot be faithfully reproduced.
Modeling modulation of IUS by high-affinity binding of Cd2+ to the IS state (Model B)
In Fig. 8 E, we tested the hypothesis that the acceleration of recovery from IUS by Cd2+ resulted from high-affinity binding of Cd2+ to the IS state. Such high-affinity binding during recovery could arise from a voltage-dependent increase in the on-rate to the IS state at negative recovery potentials (−120 mV vs. −20 mV during the conditioning test pulse). We assumed the kon of Cd2+ binding to the IS state (see Model A) to be 10 times faster than the kon of binding Cd2+ to the NI and IUS states. The off-rates of Cd2+ were defined to be equal for all Cd2+-bound states. The values for kon of Cd2+ to the NI and to the IUS state were assumed to be equal. To preserve microscopic reversibility we accelerated kIus-Cd→Is-Cd and kNI-Cd→Is-Cd by the same factor by which kIs→Is-Cd was increased. This model is similar to a previously published model of mexiletine binding to the fast inactivated state of human skeletal muscle sodium channels. In this model the authors assumed high-affinity binding of mexiletine to fast inactivated channels to occur mainly by a 10-fold higher on-rate of binding to the inactivated state which, in turn, gives rise to a faster development of inactivation (55).
Fig. 8 E shows that Model B is able to reasonably reproduce the concentration-dependent acceleration of recovery from IUS. Thus, high-affinity binding of Cd2+ to IS may account for the observed speeding of recovery from IUS.
Test of the prediction that Cd2+ ions bind with higher affinity to the IS state than to the NI state
If Cd2+ binds to the IS state with higher affinity than it does to the IUS state, then this should produce a depolarizing shift of the steady-state inactivation curve of the IUS state. However, as shown in Table 1, Cd2+ gives rise to a dramatic acceleration of the time course of recovery from IUS, such that the time constants of recovery from IUS and from IS approach each other. Thus, the amplitude of IUS cannot be defined unequivocally at higher concentrations of Cd2+, which precludes the analysis of the effect of Cd2+ on the steady-state level of IUS at a given potential.
Another assumption of the model is that Cd2+ has a higher affinity to the IS state relative to the NI state. This can readily be tested by determination of the steady-state inactivation for the IS state (Fig. 9 A). To this end K1237C channels were depolarized for 25 s to different potentials, after which the membrane was hyperpolarized to −120 mV for 500 ms to allow for recovery of fast inactivated channels. Thereafter, a 20-ms test pulse to −20 mV was applied to open recovered channels. In each oocyte this protocol was performed under Cd2+-free conditions and during exposure to 50 μM Cd2+. Unfortunately, when exposed to Cd2+ during this protocol, currents through K1237C channels exhibited a high degree of rundown. To correct the data for rundown during superfusion with Cd2+ the degree of rundown was monitored at regular intervals during the experiment. In most experiments the maximum inward current decreased as a linear function of time, allowing the currents to be corrected for this linear trend in each experiment. Both the data during the Cd2+-free control and the data during application of Cd2+ were fit with Boltzmann curves.
As shown in Fig. 9 A the effects of Cd2+ on the availability of IS were complex: First, Cd2+ produced an ∼8 mV hyperpolarizing shift of the availability curve, supporting the notion that Cd2+ binds with high affinity to the IS state, thereby stabilizing this state. Second, the slope of the availability curve was increased by Cd2+, and, third, during exposure to Cd2+, availability did not saturate at depolarized voltages but increased at voltages positive to −60 mV. Both the increase in the slope of availability and the increase in availability at voltages positive to −60 mV may reflect the voltage-dependent unblock of Cd2+ from the channels at depolarized voltages. We assume that during the conditioning prepulse a fraction of the channels will enter into fast inactivation and will also be blocked by Cd2+ in a voltage-dependent manner. During the 500-ms interpulse at −120 mV these channels will recover from fast inactivation but may still be blocked by Cd2+ and will add to the nonavailable current produced by the channels in the IS state. The steady-state availability curve may thus reflect a combination of voltage-dependent inactivation and voltage-dependent block. Therefore, we examined the effect of correcting availability for the influence of the voltage-dependent block. Fig. 9 B shows an availability curve in a single oocyte in which K1237C currents exhibited only minimal rundown during superfusion with Cd2+.
As in Fig. 9 A the slope of voltage-dependent availability negative to −60 mV was substantially steeper than in Cd2+-free solution (Fig. 9 A), and currents increased positive to −60 mV. To correct the steady-state availability curve for voltage-dependent block we fitted a Woodhull model (Eq. 9) to the measurements positive to −60 mV (inset of Fig. 9 B). Positive to −60 mV occupancy of IS presumably is maximal, thus the change in availability in this potential range only reflects voltage-dependent block. The Woodhull model shown in the inset of Fig. 9 B was then used to calculate the contribution of voltage-dependent block at the potential range negative to −60 mV. Thereafter, the original data were corrected for the contribution of voltage-dependent block and replotted (squares in Fig. 9 B). If corrected by this procedure, the hyperpolarizing shift in availability imposed by 50 μM was increased and the slope factor of the availability curve was decreased. Hence, the complex effects of Cd2+ on voltage-dependent availability of IS may have resulted from a combination of high-affinity binding to IS with voltage-dependent block of fast inactivated channels. Taken together, the data support the notion that Cd2+ may bind with high affinity to the IS state, thereby stabilizing this state.
IUS in K1237C channels involves a conformational change at the inner vestibule of the channel
Local anesthetics modulate Na channel gating by binding to the inner vestibule of the channel. In a recent report we demonstrated that the local anesthetic lidocaine prevented channels in which K1237 was replaced by a glutamic acid from entering into IUS by a foot-in-the-door mechanism (44). A similar mechanism has recently been reported for the interaction of a pore blocker with the Kv1.5 channel (56). This suggested that IUS encompasses a conformational change of the inner vestibule of the channel and that binding of lidocaine may interfere with this conformational change. It appeared to be of interest whether a similar mechanism may account for IUS in K1237C. Fig. 10 shows that superfusion of K1237C channels with 300 μM lidocaine significantly decreased the fraction of channels dwelling in the IUS state at the end of a 300-s depolarizing pulse to −20 mV. Hence, IUS in K1237C may involve a conformational change of the inner vestibule of the channel.
FIGURE 10.

Modulation of IUS by lidocaine. The protocol was analogous to Fig. 2 A. After control oocytes were superfused with 300 μM lidocaine, which resulted in a significant acceleration of recovery from IUS. Data points were fitted with Eq. 5. Fitting parameters were drug free: A1 = 0.29 ± 0.07, τ1 = 9.8 ± 1.3 s, A2 = 0.71 ± 0.07, τ2 = 149.1 ± 6.8 s; lidocaine: A1 = 0.64 ± 0.04 (P < 0.05), τ1 = 10.0 ± 0.7 s, n.s., A2 = 0.35 ± 0.04 (P < 0.05), τ2 = 159.72 ± 16 s, n.s., n = 3).
DISCUSSION
The major finding of this study is that both the binding of a mutant μ-CTX to the outer vestibule and the binding of Cd2+ to a cysteine engineered into the selectivity filter of the voltage-gated Na channel dramatically accelerates recovery from ultraslow inactivation.
Previously, we reported that replacement of K1237 with E or S substantially increased the fraction of channels entering into IUS during long depolarizations (21). The same holds true for the replacement of K1237 by cysteine (Fig. 1). Also, similar to K1237E, binding of the mutant μ-CTX R13Q to K1237C decreases the fraction of channels recovering from IUS (Fig. 2 A). However, an even more dramatic effect is observed when K1237C channels are exposed to Cd2+. As shown in Table 1 and Fig. 2, C and D, Cd2+ reduces both the longer time constant (τ2) and the amplitude of recovery with τ2 (A2) in a concentration-dependent fashion. τ1 and τ2 most likely represent the time courses of recovery from slow (IS) and ultraslow (IUS) inactivation. The fact that Cd2+ reduced the fraction of channels recovering with τ2 after a 300-s depolarization may indicate that the development of IUS is prolonged by Cd2+. However, the dramatic acceleration of τ2 with a higher concentration of Cd2+ suggests that Cd2+ may exert most of its effect on the time course of recovery. Furthermore, given the dual action of Cd2+ on both amplitude and time course, the fitting procedure may be quite equivocal.
To clarify whether the time course of development of IUS is affected by Cd2+, we exposed K1237C channels to Cd2+ either during a depolarizing pulse (during which recovery develops) or during the following hyperpolarization (during which channels recover from IUS). Fig. 7 demonstrates that Cd2+ failed to affect recovery from IUS when the channels were exposed to Cd2+ only during the phase of development of IUS, whereas recovery from IUS was accelerated when channels were exposed to Cd2+ during recovery. The absence of an effect on development of IUS strongly argues against a static foot-in-the-door or a splint-in-the-vestibule mechanism as molecular underpinning of the modulation of IUS by Cd2+. Instead Cd2+ appears to act as a catalyst, decreasing the energy barrier which has to be surmounted to recover from IUS, resulting in an acceleration of the rate constants leading from the IUS to the NI state. A similar mechanism has recently been suggested for the hastening of recovery from inactivation of hERG K+ channels by Na+ ions (57). The authors presented a kinetic model in which both the rate constants of recovery from inactivation and entry into inactivation were accelerated by Na+.
Because the rate constants of recovery dominate those for entry at hyperpolarized potentials, such an acceleration of rate constants, although preserving microscopic reversibility, reproduces a speeding of recovery. Although such a model could formally reproduce our data, it falls short in elucidating the molecular mechanism of the acceleration of IUS recovery by Cd2+. Furthermore, the energy driving the catalytic activity of Cd2+ can only be derived from the energy of binding to the K1237C site, most likely to a state transiently occupied during recovery. Therefore, we considered two kinetic models in which a difference in binding energy to two distinct inactivated states is translated into the acceleration of the rate constants driving recovery from IUS. In Model A IUS is considered a low-affinity state by decreasing the off-rate of Cd2+ from the IUS state, and, to preserve microscopic reversibility, increasing the rate of transition from the Cd2+-bound IUS state to the Cd2+-bound IS state (Fig. 8 D). Although the model can qualitatively reproduce the concentration-acceleration of recovery, the effect is only small and saturates if large differences in affinity of Cd2+ binding between IUS and IS states are assumed. Model B tested the assumption that the IS state has a 10-fold higher affinity for Cd2+ than the IUS and the NI states. Here, a higher affinity was the result of an increase in the association rate constant to the IS state. Again, to preserve microscopic reversibility the rates of recovery from IUS to the IS state were accelerated. As shown in Fig. 8 E this model closely reproduced the concentration-dependent acceleration of recovery from IUS.
For the reasons given above it is not possible to provide experimental evidence for a 10-fold difference in affinity for Cd2+ binding between the IUS and the IS state. However, as shown in Fig. 9, 50 μM Cd2+ produce an ∼9 mV hyperpolarizing shift of the steady-state inactivation curve for the IS state with respect to the NI, consistent with a higher affinity of the IS state for Cd2+. According to Bean et al. (58), in the case of state-dependent binding to the inactivated state (in this case IS) with respect to the noninactivated state, the amount of V1/2 shift produced by this high-affinity binding is given by
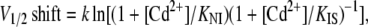 |
(10) |
where k represents the slope of the steady-state inactivation curve and KNI and KIS represent the respective affinities of the NI and the IS state (58,59). If we consider values of 30 μM and 3 μM for KNI and KIS, respectively (i.e., a 10-fold higher affinity of Cd2+ to the IS state as assumed in Model B), and a value of 7.2 mV for k (slope of the steady-state inactivation curve in Fig. 11 A), Eq. 10 yields a V1/2 shift of −13.6 mV, which is larger than the experimentally observed shift by ∼−9 mV (Fig. 9). However, the data in Fig. 9 suggest that block by Cd2+ is voltage dependent. Thus, at −120 mV, i.e., the voltage at which the time course of recovery from IUS was assessed, block of channels in the IS state would be enhanced by the negative voltage. This may explain why Cd2+ did not affect the time course development of IUS at −20 mV, whereas the recovery from IUS at −120 mV was dramatically accelerated (Fig. 7). Hence, upon recovery, Cd2+ ions may preferentially associate with channels in the IUS state, because the IUS state has a higher affinity for Cd2+ than other connected states and because of the negative voltage. This high-affinity binding to the IS state may increase the rate constants leading into the Cd2+-bound IS state (kIus-Cd→Is-Cd, kNI-Cd→IS-Cd), resulting in an acceleration of recovery from IUS. It may be argued that Cd2+ should have accelerated the time course of recovery from IS, which was not the case (Fig. 8 B). However, the time course of recovery from IS is dominated by kIS-Cd→NI-Cd, so that modifications of kNI-Cd→IS-Cd have little effect on the time course of recovery from IS.
FIGURE 11.

Molecular model of the interaction of Cd2+ with IS and IUS. Shown are the S6 segments of two domains. The selectivity filter is indicated by a ring separating the outer vestibule (funnel-like shape) from the inner vestibule (central cavity). The kinetic scheme is analogous to Fig. 8 A. Slow inactivation (IS) is considered to occur by a collapse of the outer vestibule at the level of the selectivity filter. A further rearrangement, consisting of a widening of the external vestibule and a simultaneous constriction of the inner vestibule, gives rise to IUS. The Cd2+-bound states are depicted in the lower row (Cd2+ is indicated by the ball). Cd2+ binds to all states, but the binding is strongest with the IS state, producing a constriction of the selectivity filter ring. Hence, Cd2+ exposure during the IUS state (wide selectivity filter) promotes the formation of IS (narrow selectivity filter) thereby speeding recovery from IUS.
A molecular model of the modulation of IUS by Cd2+
Which molecular events may underlie the modulation of IUS by Cd2+ ions? Recently we proposed a model of IUS in which IUS occurs by a conformational change of the cytoplasmic vestibule of the channel. This model was based on experimental findings showing that the binding of the inactivation particle to the cytoplasmic vestibule reduces the propensity of channels to enter IUS (60). Furthermore, lidocaine, by binding to residues in the cytoplasmic vestibule, opposes IUS by a foot-in-the-door mechanism (44). The modulation of IUS by lidocaine in the K1237C mutant suggests that in this construct IUS occurs by a similar mechanism as in K1237E. The experiment in Fig. 10 shows that lidocaine substantially reduced the fraction of channels recovering from IUS after a 300-s conditioning pulse without effect on the time course of recovery from IUS. This suggests that lidocaine prolonged entry into IUS, consistent with a foot-in-the-door mechanism, as reported for the mutation K1237E (44). Similarly, in Shaker K+ channels C-type inactivation—although initially thought to arise from a closure of the external vestibule—may also involve conformational changes at the internal side of the membrane (61). But how is the collapse of the internal vestibule, which presumably generates the IUS state, linked to the mutation at position 1237 in the selectivity filter?
We suggest that the mutations at site 1237 produce an increase in diameter of the selectivity filter, which is supported by ample experimental evidence demonstrating the fact that mutations of K1237 allow the permeation of cations which are substantially larger than Na (37). How is the widening of the selectivity filter transmitted to the internal vestibule to promote entry into IUS? Recently Cordero-Morales et al. presented electron paramagnetic resonance-spectroscopic evidence for an interaction between the selectivity filter and the adjacent pore helix in the KcsA channel (62). This interaction determines the propensity of the channel to inactivate. A similar interaction may account for the modulation of IUS by molecular events at the selectivity filter and in the internal vestibule in the K1237C mutation. We propose that the widening of the external vestibule associated with the replacement of K1237 with cysteine may result in an interaction of C1237 with the adjacent DIV S6 segment. Such an interaction between the selectivity filter and the adjacent S6 segment is supported by the finding that mutation of I1575 in DIV S6 to E abolishes IUS in K1237E (63). The interaction between C1237 and the adjacent S6 segment may result then in a conformational change of DIV S6 during inactivation.
This conformational change of DIV S6 then gives rise to the very stable IUS state. On the other hand, slow inactivation (IS) has been suggested to result from a collapse of the outer vestibule (28). Structural evidence for the selectivity filter as inactivation gate has recently been found in the KcsA channel (64). In Fig. 11, we propose a model in which the idea that IUS and IS represent drastically different molecular conformations is reconciled with the kinetic effects of Cd2+: To explain the acceleration of recovery from IUS in K1237C, we envision Cd2+ ions entering the pore during recovery from inactivation (lower row in Fig. 11). This entry may be favored by the negative holding potential of −120 mV during recovery from IUS. The development of IUS occurs at −20 mV, a potential at which a substantially smaller number of Cd2+ ions may enter the pore, accounting for the fact that washin of Cd2+ ions during the development of IUS does not prolong entry into IUS.
Group 12 ions are known to bind to cysteines, histidines, and aspartates (52). Cd2+ ions can coordinate at least three cysteines (65,66). In the absence of cysteines and histidines, glutamate and aspartates can serve as sites of action (52). Hence, it is possible that Cd2+ ions form coordinated binding with residues in the selectivity filter region. Candidate residues may be C1237, E755, D400, but also residues in the second ring of charge may be involved (E403, E758, D1241, D1532). This coordinated binding may result in a conformational change of the binding site, giving rise to a constriction of the pore and the formation of IS (IS-Cd2+ in Fig. 11). The reduction in the diameter of the pore at the level of the selectivity filter may then abolish the interaction between C1237 and the adjacent S6 segment, presumably with I1575, thereby accelerating recovery from IUS (Fig. 11). In the gating Model B (Fig. 8 D) the shaping of the outer vestibule by the Cd2+ ions entering the pore is reflected by the higher on-rate from the Cd2+-bound IUS state to the Cd2+-bound IS state. A similar modification of the Cd2+ binding site by binding of Cd2+ to the channel pore has been suggested previously: in inwardly rectifying  channels, the binding of Cd2+ to cysteines engineered into various sites within the pore was slowed down when the open state was stabilized by PIP2.
channels, the binding of Cd2+ to cysteines engineered into various sites within the pore was slowed down when the open state was stabilized by PIP2.
The authors suggested that Cd2+ first enters the open channel, after which the helices collapse onto the ion in the process of coordination (67).
The idea that the outer vestibule of an ion channel adapts its structure to the ion in the permeation pathway is supported by recent crystallographic data obtained in the KcsA channel: The selectivity filter of the KcsA channel assumes a conductive conformation when exposed to high [K+] but becomes constricted in high [K+] (68). In the case of the interaction of Cd2+ ions with K1237C channels, we envision Cd2+ ions entering the pore during recovery from the IUS state, thereby producing an induced fit of the outer vestibule resulting in the formation of a high-affinity state (IS), thereby speeding recovery from IUS. As shown in Fig. 7 D recovery from IUS was accelerated when the channels were exposed to Cd2+ ions during the recovery phase. This suggests that C1237 is accessible for Cd2+ ions when channels dwell in the IUS state, supporting the contention that IUS is not associated with a collapse of the external vestibule.
The modulation of recovery from slow inactivated states by cations may not be confined to voltage-gated Na channels. In voltage-gated K+ channels recovery from slow inactivation is modulated by cation binding to both extracellular sites (14,69–71) and to more intracellular sites (72,73) along the pathway of permeation. One of the modulatory sites is located at the intracellular end of the selectivity filter, i.e., at a site close to the homologous position of K1237 in the rNaV1.4 channel (74). Hence the border between the external vestibule and the central cavity may be of pivotal importance for the regulation of slow inactivated states in voltage-gated ion channels.
As shown in Fig. 2 A, binding the mutant μ-CTX R13Q to K1237C channels resulted in a substantial acceleration of recovery from IUS. This effect was qualitatively similar to the hastening of recovery from IUS by Cd2+ ions. This suggests that binding of this large peptide toxin may produce a structural rearrangement of the outer vestibule, similar to the induced fit by Cd2+. Conformational changes in the toxin binding site in the outer pore of ion channels was previously suggested using computational methods (75,76) and was more recently confirmed by Lange and colleagues (77): Using high-resolution solid-state NMR spectroscopy, these authors demonstrated that high-affinity binding of the scorpion toxin kaliotoxin to a chimeric K+ channel was associated with significant structural rearrangements in both molecules.
Although both μ-CTX and Cd2+ produced a substantial acceleration of recovery from IUS, the effect was more dramatic with Cd2+ than with μ-CTX. This discrepancy may be explained by the fact that μ-CTX does not bind directly to the selectivity filter but has a more external binding site in the second ring of charge. Thus, the residue R13 of the toxin interacts with amino acid residues downstream of the selectivity filter (18,78,79). Interestingly, this region has recently been implicated in the generation of slow inactivation (80). Given the fact that the P-loops are highly flexible structures (81), it is conceivable that conformational changes due to toxin binding to residues may be transmitted downstream to site 1237. Furthermore, the fact that ligands at two different albeit close binding sites exert similar effects on the kinetics of IUS suggests that the conformational change which underlies IUS represents a broad rearrangement of the outer vestibule.
In summary, our data appear to be inconsistent with a collapse of the outer vestibule as the molecular mechanism of IUS. This seems at variance with the common notion that slow inactivation in Na channels as well as C-type inactivation in K+ channels result from a pore collapse. It is unclear, however, whether slow inactivation may have the same molecular underpinnings as ultraslow inactivation. As a matter of fact, in our proposed model Cd2+ ions favor a constriction of the outer vestibule corresponding to the formation of a slow inactivated state, thereby forcing channels to leave the ultraslow inactivated state. In other words, Cd2+ ions may act as a foot-on-the-door, “kicking” the IS inactivation gate, thereby forcing the channels to leave IUS. Hence, slow inactivated states may represent an unexpected diversity of a quite heterogeneous class of molecular conformations.
Acknowledgments
Evelyne Gross, Sandra Bolzer, and Martin Horvath are acknowledged for animal care. We thank Mr. Rene Cervenka, Drs. John Kyle (University of Chicago) and Birgit Latzenhofer (Medical University of Vienna) for advice and support, and Mr. Anton Karel for technical assistance. Dr. R. J. French (University of Calgary, Calgary, Canada) generously provided μ-CTX R13Q.
This work was supported by grants P13961-B05 and P17509-B11 provided by the Austrian Science Fund.
Julia Szendroedi and Walter Sandtner contributed equally to this work.
Editor: Richard W Aldrich.
References
- 1.Lossin, C., T. H. Rhodes, R. R. Desai, C. G. Vanoye, D. Wang, S. Carniciu, O. Devinsky, and A. L. George Jr. 2003. Epilepsy-associated dysfunction in the voltage-gated neuronal sodium channel SCN1A. J. Neurosci. 23:11289–11295. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Black, J. A., S. Dib-Hajj, D. Baker, J. Newcombe, M. L. Cuzner, and S. G. Waxman. 2000. Sensory neuron-specific sodium channel SNS is abnormally expressed in the brains of mice with experimental allergic encephalomyelitis and humans with multiple sclerosis. Proc. Natl. Acad. Sci. USA. 97:11598–11602. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Abdulla, F. A., and P. A. Smith. 2002. Changes in Na+ channel currents of rat dorsal root ganglion neurons following axotomy and axotomy-induced autotomy. J. Neurophysiol. 88:2518–2529. [DOI] [PubMed] [Google Scholar]
- 4.Cannon, S. C. 1996. Ion-channel defects and aberrant excitability in myotonia and periodic paralysis. Trends Neurosci. 19:3–10. [DOI] [PubMed] [Google Scholar]
- 5.Takahashi, M. P., and S. C. Cannon. 1999. Enhanced slow inactivation by V445M: a sodium channel mutation associated with myotonia. Biophys. J. 76:861–868. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Rich, M. M., and M. J. Pinter. 2003. Crucial role of sodium channel fast inactivation in muscle fibre inexcitability in a rat model of critical illness myopathy. J. Physiol. 547:555–566. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Veldkamp, M. W., P. C. Viswanathan, C. Bezzina, A. Baartscheer, A. A. Wilde, and J. R. Balser. 2000. Two distinct congenital arrhythmias evoked by a multidysfunctional Na+ channel. Circ. Res. 86:E91–E97. [DOI] [PubMed] [Google Scholar]
- 8.Wang, D. W., N. Makita, A. Kitabatake, J. R. Balser, and A. L. George Jr. 2000. Enhanced Na+ channel intermediate inactivation in Brugada syndrome. Circ. Res. 87:E37–E43. [DOI] [PubMed] [Google Scholar]
- 9.Hille, B. 1992. Ionic Channels of Excitable Membranes. Sinauer Associates, Sunderland, MA.
- 10.Hille, B. 1977. Local anesthetics: hydrophilic and hydrophobic pathways for the drug-receptor reaction. J. Gen. Physiol. 69:497–515. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Hondeghem, L. M., and B. G. Katzung. 1977. Time- and voltage-dependent interactions of antiarrhythmic drugs with cardiac sodium channels. Biochim. Biophys. Acta. 472:373–398. [DOI] [PubMed] [Google Scholar]
- 12.Armstrong, C. M. 1971. Interaction of tetraethylammonium ion derivatives with the potassium channels of giant axons. J. Gen. Physiol. 58:413–437. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Grissmer, S., and M. Cahalan. 1989. TEA prevents inactivation while blocking open K+ channels in human T lymphocytes. Biophys. J. 55:203–206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Pardo, L. A., S. H. Heinemann, H. Terlau, U. Ludewig, C. Lorra, O. Pongs, and W. Stühmer. 1992. Extracellular K+ specifically modulates a rat brain K+ channel. Proc. Natl. Acad. Sci. USA. 89:2466–2470. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Choi, K. L., R. W. Aldrich, and G. Yellen. 1991. Tetraethylammonium blockade distinguishes two inactivation mechanisms in voltage-activated K+ channels. Proc. Natl. Acad. Sci. USA. 88:5092–5095. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Lopez-Barneo, J., T. Hoshi, S. H. Heinemann, and R. W. Aldrich. 1993. Effects of external cations and mutations in the pore region on C-type inactivation of Shaker potassium channels. Receptors Channels. 1:61–71. [PubMed] [Google Scholar]
- 17.Baukrowitz, T., and G. Yellen. 1995. Modulation of K+ current by frequency and external [K+]: a tale of two inactivation mechanisms. Neuron. 15:951–960. [DOI] [PubMed] [Google Scholar]
- 18.Dudley, S. C. Jr., H. Todt, G. Lipkind, and H. A. Fozzard. 1995. A μ-conotoxin-insensitive Na+ channel mutant: possible localization of a binding site at the outer vestibule. Biophys. J. 69:1657–1665. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Dudley, S. C., N. Chang, J. Hall, G. Lipkind, H. A. Fozzard, and R. J. French. 2000. μ-Conotoxin GIIIA interactions with the voltage-gated Na+ channel predict a clockwise arrangement of the domains. J. Gen. Physiol. 116:679–690. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.French, R. J., E. Prusak-Sochaczewski, G. W. Zamponi, S. Becker, A. S. Kularatna, and R. Horn. 1996. Interactions between a pore-blocking peptide and the voltage sensor of the sodium channel: an electrostatic approach to channel geometry. Neuron. 16:407–413. [DOI] [PubMed] [Google Scholar]
- 21.Todt, H., S. C. J. Dudley, J. W. Kyle, R. J. French, and H. A. Fozzard. 1999. Ultra-slow inactivation in μ1 Na+ channels is produced by a structural rearrangement of the outer vestibule. Biophys. J. 76:1335–1345. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Hilber, K., W. Sandtner, O. Kudlacek, I. W. Glaaser, E. Weisz, J. W. Kyle, R. J. French, H. A. Fozzard, S. C. Dudley, and H. Todt. 2001. The selectivity filter of the voltage-gated sodium channel is involved in channel activation. J. Biol. Chem. 276:27831–27839. [DOI] [PubMed] [Google Scholar]
- 23.Townsend, C., H. A. Hartmann, and R. Horn. 1997. Anomalous effect of permeant ion concentration on peak open probability of cardiac Na+ channels. J. Gen. Physiol. 110:11–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Townsend, C., and R. Horn. 1997. Effect of alkali metal cations on slow inactivation of cardiac Na+ channels. J. Gen. Physiol. 110:23–33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Tomaselli, G. F., N. Chiamvimonvat, H. B. Nuss, J. R. Balser, M. T. Perez-Garcia, R. H. Xu, D. W. Orias, P. H. Backx, and E. Marban. 1995. A mutation in the pore of the sodium channel alters gating. Biophys. J. 68:1814–1827. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Balser, J. R., H. B. Nuss, N. Chiamvimonvat, M. T. Perez-Garcia, E. Marban, and G. F. Tomaselli. 1996. External pore residue mediates slow inactivation in μ1 rat skeletal muscle sodium channels. J. Physiol. 494:431–442. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Kambouris, N. G., L. A. Hastings, S. Stepanovic, E. Marban, G. F. Tomaselli, and J. R. Balser. 1998. Mechanistic link between lidocaine block and inactivation probed by outer pore mutations in the rat μ1 skeletal muscle sodium channel. J. Physiol. 512:693–705. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Ong, B. H., G. F. Tomaselli, and J. R. Balser. 2000. A structural rearrangement in the sodium channel pore linked to slow inactivation and use dependence. J. Gen. Physiol. 116:653–662. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Vilin, Y. Y., E. Fujimoto, and P. C. Ruben. 2001. A single residue differentiates between human cardiac and skeletal muscle Na+ channel slow inactivation. Biophys. J. 80:2221–2230. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Zhang, Z., Y. Xu, P. H. Dong, D. Sharma, and N. Chiamvimonvat. 2003. A negatively charged residue in the outer mouth of rat sodium channel determines the gating kinetics of the channel. Am. J. Physiol. Cell Physiol. 284:C1247–C1254. [DOI] [PubMed] [Google Scholar]
- 31.Xiong, W., R. A. Li, Y. Tian, and G. F. Tomaselli. 2003. Molecular motions of the outer ring of charge of the sodium channel: do they couple to slow inactivation? J. Gen. Physiol. 122:323–332. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Heinemann, S. H., H. Terlau, W. Stühmer, K. Imoto, and S. Numa. 1992. Calcium channel characteristics conferred on the sodium channel by single mutations. Nature. 356:441–443. [DOI] [PubMed] [Google Scholar]
- 33.Favre, I., E. Moczydlowski, and L. Schild. 1996. On the structural basis for ionic selectivity among Na+, K+, and Ca2+ in the voltage-gated sodium channel. Biophys. J. 71:3110–3125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Tsushima, R. G., R. A. Li, and P. H. Backx. 1997. Altered ionic selectivity of the sodium channel revealed by cysteine mutations within the pore. J. Gen. Physiol. 109:463–475. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Chiamvimonvat, N., M. T. Perez Garcia, G. F. Tomaselli, and E. Marban. 1996. Control of ion flux and selectivity by negatively charged residues in the outer mouth of rat sodium channels. J. Physiol. 491:51–59. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Perez-Garcia, M. T., N. Chiamvimonvat, E. Marban, and G. F. Tomaselli. 1996. Structure of the sodium channel pore revealed by serial cysteine mutagenesis. Proc. Natl. Acad. Sci. USA. 93:300–304. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Sun, Y., I. Favre, L. Schild, and E. Moczydlowski. 1997. On the structural basis for size-selective permeation of organic cations through the voltage-gated sodium channel: effect of alanine mutations at the DEKA locus on selectivity, inhibition by Ca2+ and H+, and molecular sieving. J. Gen. Physiol. 110:693–715. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Hilber, K., W. Sandtner, T. Zarrabi, E. Zebedin, O. Kudlacek, H. A. Fozzard, and H. Todt. 2005. Selectivity filter residues contribute unequally to pore stabilization in voltage-gated sodium channels. Biochemistry. 44:13874–13882. [DOI] [PubMed] [Google Scholar]
- 39.Li, R. A., R. G. Tsushima, R. G. Kallen, and P. H. Backx. 1997. Pore residue critical for μ-CTX binding to rat skeletal muscle Na+ channels revealed by cysteine mutagenesis. Biophys. J. 73:1874–1884. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Li, R. A., I. L. Ennis, T. Xue, H. M. Nguyen, G. F. Tomaselli, A. L. Goldin, and E. Marban. 2003. Molecular basis of isoform-specific μ-conotoxin block of cardiac, skeletal muscle, and brain Na+ channels. J. Biol. Chem. 278:8717–8724. [DOI] [PubMed] [Google Scholar]
- 41.Li, R. A., I. I. Ennis, R. J. French, S. C. Dudley, G. F. Tomaselli, and E. Marban. 2001. Clockwise domain arrangement of the sodium channel revealed by μ-conotoxin (GIIIA) docking orientation. J. Biol. Chem. 276:11072–11077. [DOI] [PubMed] [Google Scholar]
- 42.Hui, K., G. Lipkind, H. A. Fozzard, and R. J. French. 2002. Electrostatic and steric contributions to block of the skeletal muscle sodium channel by μ-conotoxin. J. Gen. Physiol. 119:45–54. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Chahine, M., J. Sirois, P. Marcotte, L. Chen, and R. G. Kallen. 1998. Extrapore residues of the S5–S6 loop of domain 2 of the voltage-gated skeletal muscle sodium channel (rSkM1) contribute to the μ-conotoxin GIIIA binding site. Biophys. J. 75:236–246. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Sandtner, W., J. Szendroedi, T. Zarrabi, E. Zebedin, K. Hilber, I. Glaaser, H. A. Fozzard, S. C. Dudley, and H. Todt. 2004. Lidocaine: a foot in the door of the inner vestibule prevents ultra-slow inactivation of a voltage-gated sodium channel. Mol. Pharmacol. 66:648–657. [DOI] [PubMed] [Google Scholar]
- 45.Sunami, A., S. C. Jr. Dudley, and H. A. Fozzard. 1997. Sodium channel selectivity filter regulates antiarrhythmic drug binding. Proc. Natl. Acad. Sci. USA. 94:14126–14131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Becker, S., E. Prusak-Sochaczewski, G. Zamponi, A. G. Beck-Sickinger, R. D. Gordon, and R. J. French. 1992. Action of derivatives of μ-conotoxin GIIIA on sodium channels. Single amino acid substitutions in the toxin separately affect association and dissociation rates. Biochemistry. 31:8229–8238. [DOI] [PubMed] [Google Scholar]
- 47.Dean, J. A. 1992. Lange's Handbook of Chemistry. McGraw-Hill, New York.
- 48.Zebedin, E., W. Sandtner, S. Galler, J. Szendroedi, H. Just, H. Todt, and K. Hilber. 2004. Fiber type conversion alters inactivation of voltage-dependent sodium currents in mouse C2C12 skeletal muscle cells. Am. J. Physiol Cell Physiol. 287:C270–C280. [DOI] [PubMed] [Google Scholar]
- 49.Boehm, S. 1999. ATP stimulates sympathetic transmitter release via presynaptic P2X purinoceptors. J. Neurosci. 19:737–746. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Woodhull, A. M. 1973. Ionic blockage of sodium channels in nerve. J. Gen. Physiol. 61:687–708. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Featherstone, D. E., J. E. Richmond, and P. C. Ruben. 1996. Interaction between fast and slow inactivation in Skm1 sodium channels. Biophys. J. 71:3098–3109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Elinder, F., and P. Arhem. 2003. Metal ion effects on ion channel gating. Q. Rev. Biophys. 36:373–427. [DOI] [PubMed] [Google Scholar]
- 53.Kellenberger, S., T. Scheuer, and W. A. Catterall. 1996. Movement of the Na+ channel inactivation gate during inactivation. J. Biol. Chem. 271:30971–30979. [DOI] [PubMed] [Google Scholar]
- 54.Chen, Z., B. H. Ong, N. G. Kambouris, E. Marban, G. F. Tomaselli, and J. R. Balser. 2000. Lidocaine induces a slow inactivated state in rat skeletal muscle sodium channels. J. Physiol. 524:37–49. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Takahashi, M. P., and S. C. Cannon. 2001. Mexiletine block of disease-associated mutations in S6 segments of the human skeletal muscle Na+ channel. J. Physiol. 537:701–714. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Decher, N., P. Kumar, T. Gonzalez, B. Pirard, and M. C. Sanguinetti. 2006. Binding site of a novel Kv1.5 blocker: a “foot in the door” against atrial fibrillation. Mol. Pharmacol. 70:1204–1211. [DOI] [PubMed] [Google Scholar]
- 57.Mullins, F. M., S. Z. Stepanovic, R. R. Desai, A. L. George Jr., and J. R. Balser. 2002. Extracellular sodium interacts with the HERG channel at an outer pore site. J. Gen. Physiol. 120:517–537. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Bean, B. P., C. J. Cohen, and R. W. Tsien. 1983. Lidocaine block of cardiac sodium channels. J. Gen. Physiol. 81:613–642. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Balser, J. R., H. B. Nuss, D. N. Romashko, E. Marban, and G. F. Tomaselli. 1996. Functional consequences of lidocaine binding to slow-inactivated sodium channels. J. Gen. Physiol. 107:643–658. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Hilber, K., W. Sandtner, O. Kudlacek, B. Schreiner, I. Glaaser, W. Schutz, H. A. Fozzard, S. C. Dudley, and H. Todt. 2002. Interaction between fast and ultra-slow inactivation in the voltage-gated sodium channel. Does the inactivation gate stabilize the channel structure? J. Biol. Chem. 277:37105–37115. [DOI] [PubMed] [Google Scholar]
- 61.Jiang, X., G. C. Bett, X. Li, V. E. Bondarenko, and R. L. Rasmusson. 2003. C-type inactivation involves a significant decrease in the intracellular aqueous pore volume of Kv1.4 K+ channels expressed in Xenopus oocytes. J. Physiol. 549:683–695. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Cordero-Morales, J. F., L. G. Cuello, Y. Zhao, V. Jogini, D. M. Cortes, B. Roux, and E. Perozo. 2006. Molecular determinants of gating at the potassium-channel selectivity filter. Nat. Struct. Mol. Biol. 13:311–318. [DOI] [PubMed] [Google Scholar]
- 63.Zarrabi, T., W. Sandtner, J. Szendroedi, E. Zebedin, H. A. Fozzard, K. Hilber, and H. Todt. 2005. The structure of the pore of the voltage-gated sodium channel is stabilized by an interaction between the selectivity filter and the DIV-S6 segment. Biophys. J. 88:94a. (Abstr.) [Google Scholar]
- 64.Blunck, R., J. F. Cordero-Morales, L. G. Cuello, E. Perozo, and F. Bezanilla. 2006. Detection of the opening of the bundle crossing in KcsA with fluorescence lifetime spectroscopy reveals the existence of two gates for ion conduction. J. Gen. Physiol. 128:569–581. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Liu, Y., M. Holmgren, M. E. Jurman, and G. Yellen. 1997. Gated access to the pore of a voltage-dependent K+ channel. Neuron. 19:175–184. [DOI] [PubMed] [Google Scholar]
- 66.Swartz, K. J. 2004. Towards a structural view of gating in potassium channels. Nat. Rev. Neurosci. 5:905–916. [DOI] [PubMed] [Google Scholar]
- 67.Loussouarn, G., E. N. Makhina, T. Rose, and C. G. Nichols. 2000. Structure and dynamics of the pore of inwardly rectifying KATP channels. J. Biol. Chem. 275:1137–1144. [DOI] [PubMed] [Google Scholar]
- 68.Valiyaveetil, F. I., M. Leonetti, T. W. Muir, and R. MacKinnon. 2006. Ion selectivity in a semisynthetic K+ channel locked in the conductive conformation. Science. 314:1004–1007. [DOI] [PubMed] [Google Scholar]
- 69.Levy, D. I., and C. Deutsch. 1996. Recovery from C-type inactivation is modulated by extracellular potassium. Biophys. J. 70:798–805. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Levy, D. I., and C. Deutsch. 1996. A voltage-dependent role for K+ in recovery from C-type inactivation. Biophys. J. 71:3157–3166. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Rasmusson, R. L., M. J. Morales, S. M. Wang, S. G. Liu, D. L. Campbell, M. V. Brahmajothi, and H. C. Strauss. 1998. Inactivation of voltage-gated cardiac K+ channels. Circ. Res. 82:739–750. [DOI] [PubMed] [Google Scholar]
- 72.Fedida, D., N. D. Maruoka, and S. Lin. 1999. Modulation of slow inactivation in human cardiac Kv1.5 channels by extra- and intracellular permeant cations. J. Physiol. 515:315–329. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Ogielska, E. M., and R. W. Aldrich. 1999. Functional consequences of a decreased potassium affinity in a potassium channel pore. Ion interactions and C-type inactivation. J. Gen. Physiol. 113:347–358. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Ray, E. C., and C. Deutsch. 2006. A trapped intracellular cation modulates K+ channel recovery from slow inactivation. J. Gen. Physiol. 128:203–217. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Eriksson, M. A., and B. Roux. 2002. Modeling the structure of agitoxin in complex with the Shaker K+ channel: a computational approach based on experimental distance restraints extracted from thermodynamic mutant cycles. Biophys. J. 83:2595–2609. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Huang, X., F. Dong, and H. X. Zhou. 2005. Electrostatic recognition and induced fit in the κ-PVIIA toxin binding to Shaker potassium channel. J. Am. Chem. Soc. 127:6836–6849. [DOI] [PubMed] [Google Scholar]
- 77.Lange, A., K. Giller, S. Hornig, M. F. Martin-Eauclaire, O. Pongs, S. Becker, and M. Baldus. 2006. Toxin-induced conformational changes in a potassium channel revealed by solid-state NMR. Nature. 440:959–962. [DOI] [PubMed] [Google Scholar]
- 78.Chang, N. S., R. J. French, G. M. Lipkind, H. A. Fozzard, and S. Dudley. 1998. Predominant interactions between μ-conotoxin Arg-13 and the skeletal muscle Na+ channel localized by mutant cycle analysis. Biochemistry. 37:4407–4419. [DOI] [PubMed] [Google Scholar]
- 79.Li, R. A., I. L. Ennis, P. Velez, G. F. Tomaselli, and E. Marban. 2000. Novel structural determinants of μ-conotoxin (GIIIB) block in rat skeletal muscle (μ1) Na+ channels. J. Biol. Chem. 275:27551–27558. [DOI] [PubMed] [Google Scholar]
- 80.Xiong, W., Y. Z. Farukhi, Y. Tian, D. DiSilvestre, R. A. Li, and G. F. Tomaselli. 2006. A conserved ring of charge in mammalian Na+ channels: a molecular regulator of the outer pore conformation during slow inactivation. J. Physiol. 576:739–754. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Benitah, J. P., R. Ranjan, T. Yamagishi, M. Janecki, G. F. Tomaselli, and E. Marban. 1997. Molecular motions within the pore of voltage-dependent sodium channels. Biophys. J. 73:603–613. [DOI] [PMC free article] [PubMed] [Google Scholar]