

Abstract
HMO1 is a high-mobility group B protein that plays a role in transcription of genes encoding rRNA and ribosomal proteins (RPGs) in Saccharomyces cerevisiae. This study uses genome-wide chromatin immunoprecipitation to study the roles of HMO1, FHL1, and RAP1 in transcription of these genes as well as other RNA polymerase II-transcribed genes in yeast. The results show that HMO1 associates with the 35S rRNA gene in an RNA polymerase I-dependent manner and that RPG promoters (138 in total) can be classified into several distinct groups based on HMO1 abundance at the promoter and the HMO1 dependence of FHL1 and/or RAP1 binding to the promoter. FHL1, a key regulator of RPGs, binds to most of the HMO1-enriched and transcriptionally HMO1-dependent RPG promoters in an HMO1-dependent manner, whereas it binds to HMO1-limited RPG promoters in an HMO1-independent manner, irrespective of whether they are transcribed in an HMO1-dependent manner. Reporter gene assays indicate that these functional properties are determined by the promoter sequence.
The yeast ribosome is composed of four rRNAs and 79 ribosomal proteins (RPs) (58, 73). Yeast rRNA genes occur as a tandem repeat of approximately 150 copies. The 25S, 18S, and 5.8S RNAs are transcribed by RNA polymerase I (Pol I), 5S RNA is transcribed by RNA Pol III, and the 138 RP genes (RPGs) are transcribed by RNA Pol II (58, 73). In a rapidly growing cell, transcription of rRNA and RPGs accounts for approximately 60% of total transcription and 50% of Pol II-mediated transcription, respectively (73), representing a large fraction of the total energy consumption of the cell. Little is known about the regulatory mechanisms involved in coordinating the transcription of rRNA and RPGs under various growth conditions. Recent studies showed that the TOR complex 1 (TORC1) plays a central role in regulating transcription of rRNA and RPGs in response to changes in the abundance of extracellular nutrients (44). Under favorable nutrient conditions, TORC1 is localized to the nucleus and directly binds to the 35S rDNA promoter to activate transcription by Pol I (36). TORC1 also indirectly regulates Pol II-mediated RPG transcription by recruiting IFH1, a coactivator for FHL1 (45, 58, 60, 72). FHL1 was originally identified as a suppressor of a Pol III mutant (24) and was later shown to be important for RPG transcription (27, 34, 45, 58, 60, 72). Under poor nutrient conditions, TORC1 is exported from the nucleus to the cytoplasm, so that the synthesis of 35S rRNA is substantially diminished (36). Concurrently, CRF1, a corepressor for FHL1, displaces IFH1 from RPG promoters to inhibit RPG transcription (45).
HMO1 is a member of the high-mobility group B (HMGB) protein family, which include nonhistone proteins that bind to and have diverse roles in eukaryotic chromatin. HMGB proteins contain one or more distinctive DNA-binding motifs known as “HMG boxes” (11, 69). The HMG box is a conserved protein structural motif, in which three alpha helices are arranged in an L shape (55, 74). As the HMG box domain binds to the minor groove of DNA, one or two hydrophobic residues partially intercalate in between stacked base pairs in the double-stranded DNA (69). The HMG box domain interacts preferentially with distorted DNA, such as four-way junctions, minicircles, and cisplatinated DNA. HMGB proteins are involved in diverse biological processes, such as transcription, recombination, and DNA repair; they also have the ability to facilitate assembly of nucleoprotein complexes (1, 22, 56).
Saccharomyces cerevisiae contains seven HMGB proteins, known as HMO1, HMO2 (also called NHP10), NHP6A, NHP6B, ABF2, ROX1, and IXR1. The first four proteins are nuclear proteins that play roles in chromatin architecture but do not act as sequence-specific transcription factors. NHP6A/B participates in Pol II- and Pol III-mediated transcription (32, 53) and weakly associates with SPT16 and POB3 (SPN [18] or yFACT [9]), which play roles in the initiation and elongation of transcription (7, 46). HMO2 is a component of the INO80 chromatin remodeling complex that mediates Pol II-dependent transcription and the repair of double-strand breaks (62, 63). HMO1, which is less well characterized than NHP6A/B and HMO2, is primarily localized to the nucleolus and is involved in the transcription and/or processing of rRNA (19). Thus, yeast HMO1 may be a functional equivalent of the mammalian upstream binding factor (UBF) (19).
Recently, we used the Sos recruitment system (3) to show that HMO1 binds to the N-terminal domain of TAF1 and to the TATA box binding protein, both of which are subunits of the general transcription factor TFIID (unpublished data). In addition, HMO1 interacts genetically with TFIIA/TFIIB and appears to be required for the transcription of several class II genes (unpublished data). Hall et al. also recently demonstrated that HMO1 associates specifically with many RP and non-RP genes and the rRNA locus (23). These observations indicate that HMO1 is involved in both Pol I- and Pol II-mediated transcription.
In this study, genome-wide chromatin immunoprecipitation (ChIP) was used to analyze the roles of HMO1, FHL1, RAP1, and SFP1 in transcription of rRNA and RPGs (20, 23, 27, 43, 45, 58, 60, 72, 78). The results show that target genes of HMO1, FHL1, and RAP1 overlap significantly and that very few target genes bind to SFP1. In contrast to a previous observation (23), these results indicate that FHL1 binds to some RPG promoters in an HMO1-dependent manner and to others in an HMO1-independent manner. Furthermore, HMO1 binds to RPG promoters in a sequence-specific manner. Thus, we propose that RPGs are regulated by multiple protein factors and multiple mechanisms, rather than by a unified mechanism as previously thought.
MATERIALS AND METHODS
Yeast strains, medium, and cultures.
Standard techniques were used for the growth and transformation of the yeast (2). Yeast strains used in this study are listed in Table S1 in the supplemental material.
The yeast strains Y13.2, H2450, and H2451 used in this study were previously described (31). The yeast strain YKK74 was generated using the protocol of Puig et al. (54). In brief, a DNA fragment encoding the tandem affinity purification (TAP) tag at the carboxy terminus of HMO1 was amplified from pBS1479 (54) using PCR and the primer pair TK4585-TK4586. Oligonucleotides used in this study are listed in Table S2 in the supplemental material. Subsequently, the PCR product was used to transform Y13.2 yeast cells. The recombinants were selected on a synthetic medium lacking tryptophan. Similarly, YTK8475, YTK8416, and YTK8409 were generated by transforming Y13.2 with PCR fragments encoding the TAP tag at the carboxy termini of FHL1, RAP1, and SFP1, which were amplified using the primer pairs TK8209-TK8210, TK4466-TK4467, and TK8341-TK8342, respectively.
YKK291 was generated from Y13.2 by replacing pYN1/TAF1 (31) with pM1169/TAF1 (68) using a plasmid shuffle technique and then transforming the yeast cells with the pM5032/RPA135 plasmid (all plasmids constructed in this study are described below). Using kanMX as the selectable marker, targeted disruption of RPA135 was performed in the YKK291 strain using PCR-based gene deletion (39) with the primer pair TK5860-TK5861. This generated a new yeast strain, YKK69. Again using a plasmid shuffle technique, YKK72 was generated from YKK69 by replacing pM5032/RPA135 with the multicopy helper plasmid pM5057/35S rDNA.
Targeted disruption of HMO1 was performed on the following strains by PCR-based gene deletion using the primer pair TK4022-TK4023. The HMO1-disrupted strain YTK8276 was generated from H2450 using TRP1 as the selectable marker. HMO1-disrupted strains YKK70 and YKK100 were generated from YKK69 and YKK72, respectively, using HIS3 as the selectable marker. Subsequently, YTK8475 and YTK8276 were crossed and dissected to obtain the new strains YTK8434, YTK8436, YTK8439, and YTK8443. Similarly, two other sets of parental strains, YTK8416 and YTK8276 or YTK8409 and YTK8276, were crossed and dissected to obtain YTK8663 and YTK8665 or YTK8876 and YTK8877, respectively.
YTK8866 and YTK8867 were generated by transforming YKK100 with pM5032/RPA135 plus pM5459/HMO1-FLAG or pRS316 (65) plus pM5459/HMO1-FLAG, respectively. The transformants were selected on a synthetic medium lacking uracil and containing aureobasidin A (0.2 μg/ml).
YTK8573 and YTK8574 were generated by transforming YKK74 with BstPI-digested pM5457 (RPS5 promoter-driven mini-CLN2/pAUR101) and pM5458 (RPL10 promoter-driven mini-CLN2/pAUR101), respectively. The recombinants were selected on a rich medium containing aureobasidin A (0.2 μg/ml). YTK8575 and YTK8579 were generated from YTK8443 in the same way as described for YTK8573 and YTK8574, respectively.
YTK8868 and YTK8869 were generated by transforming YTK8575 with pRS315 and pM2782/HMO1-FLAG, respectively. YTK8870 and YTK8871 were generated from YTK8579 in the same way as described for YTK8868 and YTK8869, respectively. Similarly, YTK8872 and YTK8873 were generated by transforming YTK8443 with pM2777/HMO1 and pRS315 (65), respectively.
YTK8534 was generated by transforming H2451 with a PCR fragment encoding HMO1 and a C-terminal PK tag (67), which was amplified from pM4375/pUC19-3×PK-HIS3 (35) using the primer pair TK8731-TK8732.
Construction of the plasmids for the genetic studies. (i) HMO1.
A 1.5-kb fragment encoding the promoter and the entire open reading frame (ORF) of HMO1 and a 0.5-kb fragment encoding the terminator of HMO1 were each amplified from genomic DNA by PCR using the primer pairs TK4028-TK4031 and TK4030-TK4029, respectively. These fragments were mixed and used as templates for a second round of PCR using the primer pair TK4028-TK4029 to amplify the entire HMO1 gene (2.0 kb) containing an XhoI site at the carboxy terminus located just before the stop codon. This fused PCR fragment was digested with PstI-SalI and ligated into the PstI-XhoI-digested pRS315 plasmid to generate pM2777. A 105-bp XhoI-SalI fragment encoding three copies of the FLAG peptide (3×FLAG) was ligated into the XhoI site of pM2777, which generated the plasmid pM2782 (HMO1-3×FLAG/pRS315).
The 2.0- and 2.1-kb SacII-KpnI fragments from pM2777 and pM2782 (KpnI-digestion was partial) were ligated into the SacII-KpnI-digested pM5140 and pM5138, which generated the plasmids pM5320 and pM5459, respectively. pM5140 and pM5138 were constructed by ligating a 2.2-kb MscI-PvuII fragment containing AUR1-C from pAUR101 (TaKaRa) into the Ecl136II-digested plasmids pRS316 and pRS314 (65), respectively.
(ii) RPA135 and the helper plasmid.
A 5.2-kb blunt-ended DNA fragment containing RPA135 was amplified by PCR with the primer pair TK5676-TK5677 and then ligated into the SmaI site of pRS316, which generated the plasmid pM5032 (RPA135/pRS316).
pM5049 was constructed by ligating the 0.73-kb KpnI-SacI fragment containing the expression cassette of pRS424-TEF (50) into a similarly digested pRS425 (12), whose KpnI site in the LEU2 marker had been disrupted by site-specific mutagenesis (33). A 7.5-kb fragment containing the 35S rDNA was amplified from pNOY353 (51) by PCR using the primer pair TK5824-TK5825 and then digested and ligated into the BamHI-XhoI sites of pM5049, generating the plasmid pM5057.
(iii) Mini-CLN2 reporter plasmid.
A 0.7-kb fragment encoding the RPS5 promoter was amplified from genomic DNA by PCR using the primer pair TK8769-TK8770. A 0.7-kb fragment encoding the mini-CLN2 reporter gene was amplified from the plasmid pM1452 (70) by PCR using the primer pair TK8768-TK8771. These two PCR fragments were mixed and used as templates for a second round of PCR using the primer pair TK8770-TK8771 to amplify a fragment encoding the mini-CLN2 reporter gene, which is driven by the RPS5 promoter. This fused PCR fragment was digested with EagI-SalI and ligated into the similarly digested plasmid pRS315, generating the plasmid pM5388. A 0.6-kb fragment encoding the RPL10 promoter was amplified by PCR with the primer pair TK8914-TK8915 and digested with EagI-XhoI, ligated into the similarly digested plasmid pM5388, generating the plasmid pM5390. pM5457 and pM5458 were constructed by ligating the 1.4-kb SacI-KpnI fragments from pM5388 and pM5390, respectively, into the similarly digested plasmid pAUR101.
ChIP analyses.
ChIP analysis was conducted according to the Hahn laboratory protocol (http://www.fhcrc.org/science/labs/hahn/methods/mol_bio_meth/hahnlab_ChIP_method.html), with minor modifications. A detailed protocol is available upon request.
Briefly, the PCR amplification conditions were as follows: 94°C for 1.5 min; 19 to 20 cycles (rDNA) or 26 to 29 cycles (Pol II genes) of 94°C for 15 s, 55°C for 30 s, and 72°C for 30 s; and 72°C for 7 min. The PCR products were separated using a 5% nondenaturing polyacrylamide gel electrophoresis gel and stained with SYBR Green I (Invitrogen). Each band was quantified using an LAS-1000 plus image analyzer (Fuji Film), and the ratio of the immunoprecipitate/input was calculated.
As outlined in Table S2 in the supplemental material, the following PCR primer pairs were used for amplification (a, b, c, and d indicate amplified gene regions): 35S rDNA region 1, TK9115-TK6123; region 2, TK6837-TK5947; region 3, TK6838-TK5826; region 4/P, TK9075-TK9076; region 5, TK5909-TK5612; region 6, TK5911-TK8138; 35S region 7, TK5913-TK8139; region 8, TK5915-TK5661; region 9, TK5917-TK8140; region 10, TK5919-TK8141; region 11, TK5921-TK8142; region 12, TK5923-TK8143; region 13, TK5925-TK8144; region 14/T, TK9077-TK9078; helper plasmid-P, TK2878-TK9076; helper plasmid-T, TK9077-TK2652; helper plasmid-I, TK9084-TK9116; RPS5-a, TK8935-TK4243; RPS5-b, TK8018-TK8019; RPS5-c, TK8020-TK8021; HIS4-a, TK8649-TK2272; HIS4-b, T8187-TK8188; HIS4-c, TK8949-TK8950; PHO84-a, TK7892-TK7893; PHO84-b, TK8951-TK7941; PHO84-c, TK3684-TK8952; TEF2-a, TK7980-TK7981; TEF2-b, TK8953-TK8954; TEF2-c, TK8955-TK8956; ADE3-a, TK7297-TK8959; ADE3-b, TK8960-TK8961; ADE3-c, TK8962-TK7296; ADE2-a, TK8540-TK8541; ADE2-b, TK8963-TK8964; ADE2-c, TK7053-TK7054; ADH1-a, TK7616-TK2766; ADH1-b, TK8022-TK8023; ADH1-c, TK8024-TK5851; ADH1-d, TK8957-TK8958; RPS24B, TK8995-TK8996; RPL27B, TK8997-TK8998; RPL23B, TK8993-TK8994; RPL13B/RPS16A, TK8987-TK8988; RPS24A, TK8425-TK8426; RPS18A, TK8991-TK8992; RPS14B, TK8989-TK8990; RPL8A, TK8970-TK8971; RPL7A, TK8974-TK8975; RPL14B, TK8976-TK8977; RPS10B, TK8979-TK8980; RPS20, TK8981-TK8982; RPS30A, TK8983-TK8984; RPS27A, TK8985-TK8986; RPS31, TK4692-TK4625; RPL10, TK5031-TK5032; RPL3, TK5035-TK5036; RPL22B, TK8978-TK6429; RPL1B, TK8972-TK8973; RPS5-miniCLN2, TK8935-TK8933; RPL10-miniCLN2, TK5413-TK8933; and the subtelomeric region on chromosome V (indicated by an asterisk in Fig. 3 to 5 and 7), TK7894-TK7977.
FIG. 3.

The in vivo binding of HMO1 with several class II genes. (A) Schematic diagrams of the amplified regions (a, b, c, and d) of RPS5, HIS4, PHO84, TEF2, ADE3, ADE2, and ADH1 genes by ChIP assays conducted as described in for panel B. (B) The raw data of the ChIP assays. The strains expressing the TAP-tagged HMO1 (YKK74) or the untagged HMO1 (Y13.2) were grown in YPD (yeast extract, peptone, dextrose) medium to mid-log phase at 30°C. The cross-linked chromatin from the former (even-numbered lanes; +) or the latter (odd-numbered lanes; −) strains were prepared and precipitated using immunoglobulin G-Sepharose. After the reversal of the cross-linking, PCR was performed and analyzed as described in the legend of Fig. 1A to test for the presence of DNA corresponding to the regions that are summarized in panel A. Each PCR contained a second primer pair that amplified a region (188 bp) of chromosome V that is devoid of ORFs, thus providing an internal background control (asterisk). The lower and upper panels show the results of the PCR conducted with the chromatin before (input) or after immunoprecipitation (IP), respectively. (C) The quantification of the raw data shown in panel B. The signals corresponding to each band were quantified by an image analyzer, after staining with SYBR Green I. The ratio of the precipitated signal (IP) to the input signal derived from the lysates containing the TAP-tagged (gray bar) or untagged (open bar) HMO1 was calculated for the genes indicated (upper panel) and a control region of chromosome V (lower panel, asterisk).
FIG. 5.

The effect of Δhmo1 on the binding of FHL1 with various RPG promoters. (A) The in vivo FHL1 binding to HMO1-enriched RPG promoters described above the panel were investigated by ChIP assays. The HMO1 strains expressing the TAP-tagged (YTK8436) or untagged FHL1 (YTK8434) and the Δhmo1 strains expressing the TAP-tagged (YTK8443) or untagged FHL1 (YTK8439) were grown in YPD (yeast extract, peptone, dextrose) medium to mid-log phase at 30°C. The ChIP assays were conducted as described in the legend of Fig. 3. The primer pairs were the same as described in the legend of Fig. 4A. (B) The quantification of the raw data shown in panel A was performed as described in the legend of Fig. 3C. (C) The in vivo FHL1 binding to HMO1-limited RPG promoters described above the panel were investigated by ChIP assays using the same DNA fractions (input and immunoprecipitate [IP]) as described in panel A. (D) The quantification of the raw data shown in panel C was performed as described in the legend of Fig. 3C. WT, wild type.
FIG. 7.

The HMO1 abundance at and the HMO1 dependence of FHL1 binding to the RPS5 and RPL10 promoters and the HMO1 dependence of the transcription of the RPS5 and RPL10 promoters were determined by the promoter sequences. (A) A schematic diagram of the mini-CLN2 reporter genes integrated at aur1 locus. The arrows indicate the positions of the PCR primers for the ChIP assays conducted shown in panels B and D. (B) The in vivo HMO1 binding to the reporter genes was analyzed by ChIP assays. The strains expressing the TAP-tagged HMO1 and containing the RPS5 promoter (YTK8573)- or the RPL10 promoter (YTK8574)-driven mini-CLN2 reporter gene were grown in YPD (yeast extract, peptone, dextrose) medium to mid-log phase at 30°C. ChIP assays were conducted as described in the legends of Fig. 1A and 3B. (C) The quantification of the raw data shown in panel B was performed as described in the legend of Fig. 3C. (D) The in vivo FHL1 binding to the reporter genes was analyzed by ChIP assays. The HMO1 strains expressing the TAP-tagged FHL1 and containing the RPS5 promoter (YTK8869)- or the RPL10 promoter (YTK8871)-driven mini-CLN2 reporter gene or the Δhmo1 strains expressing the TAP-tagged FHL1 and containing the RPS5 promoter (YTK8868)- or RPL10 promoter (YTK8870)-driven mini-CLN2 reporter gene were grown in SC medium to mid-log phase at 25°C. ChIP assays were conducted as described in the legends of Fig. 1A and 3B. (E) The quantification of the raw data shown in panel D was performed as described in the legend of Fig. 3C. (F) The expression of the chromosomal CLN2 (indicated with a filled triangle to the right), RPS5, RPL10, and TEF2 genes and the mini-CLN2 reporter gene (indicated with an open triangle to the right) were measured by Northern blot analysis. The same set of yeast strains that were described in panel D were grown in SC medium to mid-log phase at 25°C. Total RNA was prepared and separated on the gel and hybridized with the gene-specific probes as described in the legend of Fig. 2A. (G) The quantification of the raw data shown in panel F was performed as described in the legend of Fig. 6B. IP, immunoprecipitate; WT, wild type.
Northern blot analyses.
The analysis of the expression of several endogenous genes and the analysis of the poly(A)+ RNA were performed by Northern blotting and slot blot analysis, respectively, as previously described (70).
For detection of HIS4, ADE2, ADE3, RPS5, RPL3, RPS31, RPL10, HSP12, PHO84, PHO12, ADH1, ACT, TEF2, CLN2, 5S rRNA, and SNR6, DNA fragments were amplified by PCR from the yeast genomic DNA, purified, and then 32P-labeled using random priming. The PCR primers used for ACT1, ADH1, RPS5, and HIS4 were previously described (68, 70). Other primer pairs used were the following: for PHO84, TK1043-TK1044; PHO12, TK1045-TK1046; ADE2, TK3787-TK3788; ADE3, TK1175-TK1176; RPL3, TK5035-TK5036; RPS31, TK4464-TK4465; RPL10, TK4438-TK4439; HSP12, TK247-TK248; TEF2, TK6965-TK6966; CLN2, TK1079-TK1080; 5S rRNA, TK6123-TK6124; and SNR6, TK6147-TK5168.
RPS24B, RPL27B, RPL23B, RPS16A, RPL13B, RPS24A, RPS18A, RPS14B, RPL8A, RPL1B, RPL7A, RPL14B, RPS10B, RPS30A, RPS27A, 25S rRNA, 5.8S rRNA, and tRNAArg were also detected using Northern blot analysis. The probes were generated by 5′ end labeling gene-specific oligonucleotides with [32P]ATP using T4 polynucleotide kinase (59). The oligonucleotides used were the following: for RPS24B, TK8996; RPL27B, TK8998; RPL23B, TK8994; RPS16A, TK9045; RPL13B, TK9046; RPS24A, TK9047; RPS18A, TK8992; RPS14B, TK8990; RPL8A, TK8971; RPL1B, TK8973; RPL7A, TK8975; RPL14B, TK8977; RPS10B, TK8980; RPS30A, TK8984; RPS27A, TK8986; 25S rRNA, TK5611; 5.8S rRNA, TK9119; tRNAArg, TK2761.
The preparation of the probe to detect poly(A)+ RNA by slot blot analysis was previously described (70).
Genome-wide ChIP analyses.
The yeast strains expressing the TAP-tagged FHL1 (YTK8872 and YTK8873), TAP-tagged RAP1 (YTK8663 and YTK8665), TAP-tagged SFP1 (YTK8876 and YTK8877), or PK-tagged HMO1 (YTK8534) were used for the genome-wide ChIP analyses. The cell lysates and the ChIP DNA were prepared as described for the ChIP analyses. S. cerevisiae whole-genome tiling arrays (Saccharomyces cerevisiae Tiling 1.0F Array, P/N 520286) were purchased from Affymetrix and used in this study. The amplification of the ChIP DNA, the labeling with biotin-11-ddATP, and hybridization of the ChIP DNA and primary data analyses were performed as previously described (28, 29). The three criteria used to classify positive and negative binding of a specific factor to a specific gene were described previously (38). Briefly, the reliability of signal strength was evaluated based on “detection P value” for each locus (P value of 0.001%). Then, the reliability of the binding ratio was evaluated based on “change P value” (P value of 0.001%). Finally, clusters consisting of at least 500-bp contiguous loci that satisfied the above two criteria were selected.
RESULTS
Pol I-dependent association of HMO1 with the 35S rRNA gene.
Previous studies demonstrated that HMO1 is primarily localized to nucleoli (19) and that it binds to the entire Pol I-transcribed region of the rRNA gene cluster (23). To confirm this observation, ChIP analysis was performed using 14 primer sets covering the entire rRNA locus (Fig. 1A and B). As previously described, HMO1 bound throughout the 35S rRNA gene and was enriched in the promoter and 5′ external transcribed spacer regions of the 35S rRNA gene but was not associated with the 5S rRNA gene. This distribution of HMO1 suggests that the binding of HMO1 might correlate with the activity or binding of Pol I along the 35S rRNA gene. This possibility was tested using yeast strain Δrpa135, which has a deletion in the second largest subunit of Pol I and expresses 35S rRNA from a multicopy helper plasmid under the control of the TEF1 promoter. If HMO1 binds to the 35S rRNA gene in a Pol I-dependent manner, it is predicted that HMO1 binding should be strongly reduced in the Δrpa135 strain.
FIG. 1.

HMO1 associates throughout the 35S rRNA gene in a Pol I-dependent manner. (A) In vivo binding of HMO1 to various regions of the rRNA gene was analyzed by ChIP assays. The yeast strain expressing the TAP-tagged HMO1 (YKK74) was grown in YPD (yeast extract, peptone, dextrose) medium to mid-log phase at 30°C. Cross-linked chromatin from this strain was prepared and precipitated with either immunoglobulin G (IgG)-Sepharose 6 FastFlow (+IgG) or Sepharose 6 FastFlow (−IgG; negative control) beads. After the reversal of the cross-linking, PCR was performed to test for the presence of DNA corresponding to regions 1 to 14 (indicated schematically in panel B). The PCR products were separated by 5% polyacrylamide gel electrophoresis and stained with SYBR Green I. The top, middle, and bottom (input DNA) panels indicate the results of the PCR conducted for the chromatin after precipitation with or without IgG, or before precipitation, respectively. (B) The quantification of the raw data shown in panel A. The signals corresponding to each band were quantified by an image analyzer. The ratio of the IgG-precipitated signal (IP) to the input signal was calculated for the regions 1 to 14. 4/P and 14/T regions are overlapped with the promoter (P) and terminator (T) of the 35S RNA gene, respectively. (C) The effect of Δhmo1 and/or Δrpa135 on the growth of the yeast cells. The Δrpa135 and Δhmo1 Δrpa135 strains carrying the plasmid pM5032 (encoding RPA135) were designated HMO1 RPA135 (YKK69) and Δhmo1 RPA135 (YKK70), respectively. Similarly, the Δrpa135 and Δhmo1 Δrpa135 strains carrying the helper plasmid pM5057 expressing 35S rRNA from the TEF1 promoter (schematically indicated in panel G) were designated HMO1 Δrpa135 (YKK72) and Δhmo1 Δrpa135 (YKK100), respectively. These four strains were spotted onto YPD plates at three dilutions and grown at 30°C for 5 days. (D) The strains described in panel C were grown in YPD medium to mid-log phase at 30°C and total RNA was obtained from 1 × 107 cells, then separated by 1% agarose gel electrophoresis and stained with ethidium bromide. The positions of 25S and 18S rRNA are indicated on the left. The asterisk may correspond to 5S/5.8S rRNA and tRNA. Further analyses on this band were described in Fig. S1 in the supplemental material. (E) In vivo binding of HMO1 to the promoter (4/P) and terminator (14/T) regions of the chromosomal 35S rRNA gene were analyzed by ChIP assays in the HMO1 RPA135 (YTK8866, indicated as WT) and HMO1 Δrpa135 (YTK8867, indicated as Δ) strains expressing the FLAG-tagged HMO1. The strains were grown in synthetic complete (SC) medium to mid-log phase at 25°C. The cross-linked chromatin was prepared and precipitated with (+) or without (−) anti-FLAG monoclonal antibodies. After reversal of the cross-linking, PCR was performed and analyzed as described in panel A to test for the presence of DNA corresponding to the regions of 4/P and 14/T, both of which are modified (thereby were not contained) in the helper plasmid as shown in panels B and G. Left panel (input) shows the results of the PCR conducted on the chromatin before precipitation. (F) The quantification of the raw data shown in panel E. The signals corresponding to each band were quantified by an image analyzer. The ratio of the precipitated signal (IP) to the input signal was calculated for the regions 4/P and 14/T. (G) A schematic diagram of the helper plasmid pM5057. The regions that were amplified by PCR in the ChIP assays (H and I) are indicated as P, T, and I. (H) In vivo binding of HMO1 to the promoter (P), terminator (T) and unrelated intermediate (I) regions of the 35S rRNA gene on the helper plasmid were analyzed by ChIP assays using the same DNA fractions (input and IP) as described in panel E. (I) The quantification of the raw data shown in panel H was performed as described in panel F. WT, wild type.
The growth profile of wild-type, single (Δhmo1 or Δrpa135) or double (Δhmo1 Δrpa135) mutant strains was examined at 30°C on rich medium (Fig. 1C). As previously shown (19, 40), Δhmo1 cells grow more slowly than wild-type cells at 30°C. In addition, Δrpa135 cells grow poorly, at least in part because transcription of rRNA is not sufficiently robust to support normal growth on rich medium (Fig. 1D). However, Δhmo1 Δrpa135 cells grow even more slowly than Δrpa135 cells (the doubling times of these strains were approximately 1.8 h for wild type, 4.4 h for Δhmo1, 4.7 h for Δrpa135, and 6.4 h for Δhmo1 Δrpa135). Consistent with this, the Δhmo1 Δrpa135 strain had the lowest amount of 25S and 18S rRNAs (Fig. 1D). Since the TEF1 promoter is independent of HMO1 (data not shown), this may be due to low plasmid stability or inefficient maturation of rRNA, both of which may be associated with deficiency in Δhmo1 (19, 23, 40).
Primer sets that specifically target the promoter (4/P region) or the terminator (14/T region) regions of the chromosomal 35S rRNA gene (Fig. 1E and F) were used to analyze the dependence of HMO1 binding to these gene regions on Pol I-mediated transcription. The results showed significantly lower levels of HMO1 in these regions in Δrpa135 cells than in wild-type cells. ChIP analysis was also performed using primer sets that target the TEF1 promoter (Fig. 1G, P), the CYC1 terminator (Fig. 1G, T) and an unrelated region near the LEU2 gene (Fig. 1G, I) of the helper plasmid. These experiments showed that significant amounts of HMO1 bound all regions of the helper plasmid (Fig. 1H and I) in wild-type and Δrpa135 cells. These results suggest that HMO1 binds to all regions of the 35S rRNA gene in a Pol I-dependent manner.
HMO1 is required for expression of a subset of class II genes.
The expression of several class II genes was examined by Northern blotting in wild-type, Δhmo1, Δrpa135, and Δhmo1 Δrpa135 cells cultured at 30°C in rich medium (Fig. 2A). Consistent with the notion that HMO1 plays a role in Pol II-mediated transcription (23; also unpublished observations), the expression of some genes was reduced in Δhmo1 cells (Fig. 2A, lanes 1 and 2). Importantly, similar gene-specific effects of Δhmo1 were observed in the Pol I-deficient strain (Fig. 2A, lanes 3 and 4). In particular, HIS4, ADE2, ADE3, RPS5, and HSP12 were expressed at a lower level in Δhmo1 Δrpa135 cells (lane 4) than in Δrpa135 cells (lane 3). In contrast, RPL3, RPS31, RPL10, ADH1, and ACT1 were expressed at similar levels in Δhmo1 Δrpa135 and Δrpa135 cells. Expression of PHO84 and PHO12 was almost undetectable, even in the Δrpa135 strain, suggesting a functional link between Pol I and the transcription of the PHO genes. In addition, expression of 5S rRNA and tRNAArg, which are transcribed by Pol III, was lower in Δrpa135 cells than in wild-type cells, presumably because of the growth defect in the Δrpa135 cells; transcription of these genes was only slightly lower in Δhmo1 Δrpa135 cells than in Δrpa135 cells. Similarly, expression of 25S and 5.8S rRNAs was lower in Δrpa135 cells than in wild-type cells and slightly lower still in Δhmo1 Δrpa135 cells (Fig. 2A; see Fig. S1 in the supplemental material) (71). These results suggest that deletion of HMO1 reduces expression of several class II genes, independent of the status of Pol I transcription. However, it remains possible that deletion of HMO1 leads to a lower steady-state level of rRNA, which in turn leads to slower growth and lower Pol II transcription.
FIG. 2.

The effect of Δhmo1 and/or Δrpa135 on the transcription of the class II genes. (A) The expression of HIS4, ADE2, ADE3, RPS5, RPL3, RPS31, RPL10, HSP12, PHO84, PHO12, ADH1, ACT1, 25S rRNA, 5.8S rRNA, 5S rRNA, SNR6, and tRNAArg were measured by Northern blot analysis in the same strains as described in the legend of Fig. 1C. Total RNA was prepared and separated on the gel as described in the legend of Fig. 1D. Subsequently, the RNA was blotted onto the membrane and hybridized with the gene-specific probes indicated to the left of the image. The raw data (left panel) were quantified and are presented graphically in the right panel. Values for each transcript were normalized to the maximum expression of that transcript. (B) The effect of Δhmo1 and/or Δrpa135 on total poly(A)+ RNA levels. A slot blot of total RNA was hybridized with a radioactive oligo(dT) probe, and the quantified data are summarized in the lower panel, as described in panel A. WT, wild type.
Notably, the amount of poly(A)+ RNA synthesized in the Δrpa135 and Δhmo1 Δrpa135 strains appeared to be similar (Fig. 2B, lanes 3 and 4). This result suggests that HMO1 plays a specialized role in the Pol II-mediated transcription and is not required for the transcription of most class II genes.
HMO1 binds with high affinity to a subset of RPGs in vivo.
The above results predict that HMO1 will bind to a subset of class II genes. This idea was tested by using ChIP analyses to determine the extent of HMO1 binding to the HMO1-dependent class II genes described above (Fig. 2). ChIP analyses were performed at multiple regions of each locus (Fig. 3A) in yeast strains expressing TAP-tagged or untagged HMO1 (Fig. 3B and C). The results showed that HMO1 was more abundant at the promoters of RPS5, HIS4, and TEF2 than at the promoters of PHO84, ADE3, ADE2, and ADH1 (Fig. 3B and C). However, RPS5, HIS4, ADE3, and ADE2 are transcribed in an HMO1-dependent manner, but the other genes are not (Fig. 2; see also Fig. 6). Although these results are consistent with HMO1 binding to the promoters of some class II genes, they also show that HMO1-dependent transcription does not correlate well with HMO1 binding, at least for this group of class II genes. In fact, binding of HMO1 to ADE2, which is transcribed in an HMO1-dependent manner, was very weak.
FIG. 6.

The effect of Δhmo1 on the transcription of various RPGs. (A) The expression of the HMO-enriched RPG promoters and a few of the non-RP (i.e., TEF2, ACT1, and ADH1) gene promoters was measured by Northern blot analysis. The HMO1 (H2450) or Δhmo1 (YTK8276) strains were grown in YPD (yeast extract, peptone, dextrose) medium to mid-log phase at 25°C. Total RNA was prepared and separated on the gel and hybridized with the gene-specific probes as described in the legend of Fig. 2A. (B) The quantification of the raw data shown in panel A was performed as described in the legend of Fig. 2A. The values for each transcript that were obtained from the Δhmo1 strain were normalized to that the value of the transcript obtained from the HMO1 strain. (C) The expression of the HMO-limited RPG promoters was measured by Northern blot analysis as described for panel A. (D) The quantification of the raw data shown in panel C was performed as described for panel B. WT, wild type.
Of the seven promoters tested, HMO1 binds with highest affinity to the RPS5 promoter (Fig. 3). Therefore, similar ChIP analyses were performed for other RPGs (Fig. 4). The results showed that HMO1 bound to some RPGs more strongly than to others (Fig. 4B, left versus right panels) and that HMO1 bound with very low affinity to a considerable number of RPGs. This is in contrast to a previous study suggesting that few RPG promoters have low affinity for HMO1 (23). These observations prompted a closer investigation of factors that regulate transcription of RPGs.
FIG. 4.

The in vivo binding of HMO1 with various RPG promoters. (A) The ChIP assays were conducted as described in the legend of Fig. 3 except that the primer pairs to amplify the RPG promoters indicated above the panel were used. The RPGs tested here were grouped into two types, the HMO1-enriched (left panel) or the HMO1-limited (right panel) RPGs, according to the signal intensity. (B) The quantification of the raw data shown in panel A was performed as described in the legend of Fig. 3C. IP, immunoprecipitate.
HMO1-dependent and -independent recruitment of FHL1 to RPG promoters.
It is well known that FHL1 and its binding partners, IFH1 and CRF, play a central role in the growth-regulated expression of RPGs (45, 58, 60, 72). Recently, it was shown that HMO1 is required for binding of FHL1 to RPG promoters (23). Results presented above suggest that there are two distinct types of RPG promoters, HMO1-enriched and HMO1-limited (Fig. 4). This prompted us to investigate whether FHL1 is recruited to both types of promoters in an HMO1-dependent manner. To test this, ChIP analyses were performed using wild-type and Δhmo1 cells that express TAP-tagged or untagged FHL1 (Fig. 5). As previously shown (58, 60, 72), FHL1 binds specifically to RPG promoters, although with variable affinity (Fig. 5). Surprisingly, ChIP analyses demonstrated HMO1-dependent recruitment of FHL1 only on HMO1-enriched RP promoters (with the exception of RPS18A) (Fig. 5A and B). The extent of the HMO1 dependence differed for different RP promoters; it was strong at the promoters of RPS5, RPS24B, RPL27B, and RPL23B, and it was weak at promoters of RPL13B-RPS16A, and RPS24A (Fig. 5A and B). In contrast, recruitment of FHL1 to HMO1-limited RP promoters was HMO1 independent (Fig. 5C and D). Taken together, these results strongly support the idea that multiple independent factors regulate transcription of RPGs by multiple mechanisms.
HMO1 is required for expression of a subset of RPGs.
A previous study showed that deletion of HMO1 did not reduce transcription of several RPGs, although it did reduce binding of FHL1 to some RPGs (23). However, data presented above indicate that HMO1 influences transcription of RPS5 more than RPL3, RPS31, and RPL10 (Fig. 2). In addition, RPS5 and RPL3/RPS31/RPL10 interacted differently with HMO1 and FHL1; HMO1 was more enriched on the former than on the latter (Fig. 4), and FHL1 was recruited to the former in an HMO1-dependent manner but to the latter in a HMO1-independent manner (Fig. 5). Thus, we sought to determine whether HMO1 is important for expression of other RPGs and, if so, whether HMO1-dependent transcription correlates with HMO1 binding to the promoter and/or HMO1-dependent recruitment of FHL1.
The expression of 19 RPGs (including RPS5) and three control genes was examined by Northern blotting in wild-type and Δhmo1 cells cultured at 30°C in rich medium (Fig. 6). The results showed that the expression of RPS5, RPS24B, RPS14B, RPL8A, RPL1B, and RPS30A decreased to less than 50% of control in Δhmo1 cells, whereas expression of RPL27B, RPL23B, RPL14B, RPS10B, and RPL10 decreased to less than 80% of the control in Δhmo1 cells; RPL13B, RPS24A, RPL7A, RPS27A, RPL3, and control genes (TEF2, ACT1 and ADH1) were expressed at similar levels in wild-type and Δhmo1 cells. Because deletion of HMO1 reduced RPG expression in a selective and gene-specific manner, it seems unlikely that this is an indirect effect (i.e., due to reduced growth rate). Rather, these data support the idea that HMO1 is directly involved in the expression of a subset of RPGs. Intriguingly, HMO1-dependent transcription was observed for many RPG promoters and does not appear to correlate with HMO1 binding or HMO1-dependent recruitment of FHL1 (Fig. 6), as was observed for non-RPG promoters (Fig. 2 and 3).
HMO1-dependent transcription and FHL1 binding are determined by promoter sequence.
As described above, HMO1 was more enriched at the RPS5 promoter than at the RPL10 promoter (Fig. 4), and the HMO1 dependency of transcription and FHL1 binding were observed more strongly for the RPS5 promoter than for the RPL10 promoter (Fig. 2, 5, and 6). One possible explanation for this result is that HMO1 and FHL1 interact with RPG promoters in a sequence-specific manner. This idea was tested using a mini-CLN2 reporter gene (70) which was integrated into the aur1 locus on the yeast chromosome (Fig. 7A) and transcribed under the control of the promoter region from RPS5 (∼ −690 bp) or RPL10 (∼ −644 bp).
ChIP analyses using yeast strains expressing TAP-tagged HMO1 showed that HMO1 was more enriched on the ectopic RPS5 promoter than on the ectopic RPL10 promoter (Fig. 7B and C). ChIP analyses in wild-type and Δhmo1 strains expressing TAP-tagged FHL1 showed that deletion of HMO1 preferentially reduced binding of FHL1 on the ectopic RPS5 promoter (Fig. 7D and E), which was also observed at the endogenous chromosomal RPS5 promoter (Fig. 5). Northern blot analyses showed that deletion of HMO1 greatly reduced expression of the reporter gene from the RPS5 promoter at aur1 (Fig. 7F and G, open arrowhead or mini-CLN2) and of the endogenous RPS5 gene (Fig. 7F and G). In contrast, deletion of HMO1 only slightly reduced expression of the reporter gene from the RPL10 promoter and had a similar weak effect on the endogenous RPL10 gene (Fig. 7F and G). Furthermore, it should be noted that the endogenous CLN2 and TEF2 genes were similarly affected (CLN2) or unaffected (TEF2) by deletion of HMO1 in all cases (Fig. 7F and G). Therefore, we conclude that the promoter sequence itself may determine the gene-specific transcriptional and factor loading properties of the RPG promoters.
Genome-wide identification of genes targeted by HMO1, FHL1, RAP1, and SFP1.
To investigate the role of HMO1 in transcription of other class II genes, genome-wide ChIP analysis was carried out using cells expressing PK-tagged HMO1 and a high-density oligonucleotide tiling-array (38) (see Fig. S2 in the supplemental material). Similar analyses were also conducted for cells expressing TAP-tagged FHL1, RAP1, or SFP1 (see Fig. S3, S4, and S5 in the supplemental material). Partial results for HMO1, FHL1, and RAP1 are shown in Fig. 8A for a 100-kb segment of chromosome X. Nine chromosomal sites were identified that bound to HMO1, FHL1, or RAP1 (Fig. 8B), but no binding sites for SFP1 were observed in this chromosomal region (see Fig. S5 in the supplemental material).
FIG. 8.

The identification of the in vivo target genes of HMO1, FHL1 and RAP1. (A) The genome-wide ChIP analyses were conducted to identify in vivo target genes of HMO1, FHL1 and RAP1. The strains expressing the PK-tagged HMO1 (YTK8534) or the TAP-tagged RAP1 (YTK8663) were grown in YPD (yeast extract, peptone, dextrose) medium to mid-log phase at 30°C, whereas the strain expressing the TAP-tagged FHL1 (YTK8872) was grown in SC medium to mid-log phase at 30°C. The cross-linked chromatin was prepared and precipitated with IgG-Sepharose (RAP1 and FHL1) or anti-PK tag immunoglobulin G and Dynabeads protein G (HMO1), and then analyzed by GeneChip (Saccharomyces cerevisiae Tiling 1.0F Array; Affymetrix). The orange, green, and blue vertical bars represent the significant binding of HMO1, FHL1, and RAP1 to the region between 600,000 and 700,000 of chromosome X (the coordinates in kilobases are shown at the bottom of each panel). The horizontal small squares with a different color in each panel indicate the ORFs. The bottom panel represents the merged image of the top three panels. The broken vertical lines going through the three panels and with the numbers at the top (1 to 9) indicate the positions of the genes targeted by at least one of the three (HMO1, FHL1, or RAP1) factors. Note that the vertical bars shown in light colors in the top three panels represent signals that were less significant (i.e., clusters that satisfy the requirement for P values are not contiguous) and thereby were not counted in this study. In the merged image, the thick-colored vertical bars are also shown light colors to increase their transparency. (B) The summary of the target genes identified in panel A. The numbers in the left-most column correspond to those of binding sites that are depicted by the broken vertical lines in panel A. The genes on the Watson (W) and Crick (C) strands, whose promoters are bound by HMO1, FHL1, or RAP1, are summarized in this table. (C) A Venn diagram of the genes targeted by HMO1, FHL1, and RAP1. The number of target loci corresponding to each segment identified by the genome-wide ChIP analyses is indicated.
The binding of SFP1 to RPG promoters remains controversial; one report indicates that SFP1 binds to the promoters of several actively transcribing RPGs (43), but other reports indicate that binding of SFP1 at RPG promoters is barely detectable (27, 34). Although SFP1 also regulates expression of the ribosome biogenesis (Ribi) regulon, the binding of SFP1 to the promoters of the Ribi genes has not yet been detected by ChIP (17, 27, 34). Our results suggest that SFP1 does not bind significantly to RPG promoters or Ribi genes, but it does bind weakly to a small number (∼20) of other loci (see Fig. S5 in the supplemental material).
The binding profiles of HMO1, FHL1, and RAP1 for the entire genome were compared, and the common target genes of these three factors were identified (see Fig. S6 in the supplemental material). Partial results for the same 100-kb segment of chromosome X are shown in Fig. 8A (bottom panel). As expected, binding sites for these factors were primarily in gene promoter regions and not in gene coding regions. Binding sites occurred in RPG promoters, as well as in a wide range of other genes (Fig. 8B; see also Table S3 in the supplemental material). Interestingly, of the two RPGs in this region of the chromosome X, RPS5 bound all three factors (Fig. 8B, binding site 5) whereas RPL43B bound only RAP1 (Fig. 8B, binding site 1).
Venn diagrams were generated to describe the overlap between binding sites for each factor (Fig. 8C). These three factors had 177 common targets, representing 37% of HMO1 targets (177/483), 43% of FHL1 targets (177/412), and 36% of RAP1 targets (177/489). Notably, HMO1 and FHL1 binding correlates at more target loci (64% of HMO1 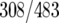 and 75% of FHL1
and 75% of FHL1 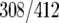 targets) than HMO1 and RAP1 (46% of HMO1
targets) than HMO1 and RAP1 (46% of HMO1 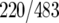 and 45% of RAP1
and 45% of RAP1 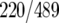 targets), or FHL1 and RAP1 (52% of FHL1
targets), or FHL1 and RAP1 (52% of FHL1 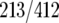 and 44% of RAP1
and 44% of RAP1 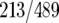 targets) (Fig. 8C). These results indicate that coordinated binding of HMO1 and FHL1, and perhaps other combinations of factors, may play an important role in regulating transcription of RPGs as well as other class II genes.
targets) (Fig. 8C). These results indicate that coordinated binding of HMO1 and FHL1, and perhaps other combinations of factors, may play an important role in regulating transcription of RPGs as well as other class II genes.
Genome-wide survey of the HMO1-dependence of binding of FHL1 and RAP1 to chromosomal targets.
FHL1 binds to some RPG promoters in an HMO1-dependent manner but to others in an HMO1-independent manner (Fig. 5); in addition, FHL1 and/or RAP1 coassociate with HMO1 at several hundred target loci throughout the genome (Fig. 8). The HMO1 dependence of FHL1 and RAP1 promoter binding was investigated using genome-wide ChIP analyses in HMO1 and Δhmo1 cells expressing TAP-tagged FHL1 or TAP-tagged RAP1 (see Fig. S7 and S8 in the supplemental material). Binding data were analyzed, and partial results for the same 100-kb region of chromosome X are shown in Fig. 9. Deletion of HMO1 affected binding of FHL1 and RAP1 at many loci, even those with low levels of HMO1 (Fig. 9A, flag 1, e.g.). Interestingly, HMO1 had a negative effect on recruitment of FHL1 or RAP1 at some target loci (Fig. 9A, flag 1) while it had a positive effect at other loci (Fig. 9A, flags 2, 4, and 5). Overall, the results showed that deletion of HMO1 decreased binding to 75% of the FHL1 sites, whereas it decreased the binding of RAP1 at far fewer sites. However, deletion of HMO1 altered the shapes of the peaks on the binding histogram. For example, of approximately 25% of the peaks became narrower and higher in cells lacking HMO1 (Fig. 9B; affected peaks are indicated by a rectangle composed of blue and red triangles). These changes suggest that deletion of HMO1 alters the apparent distribution of RAP1 along the target gene, probably by changing the compaction state of chromatin but without affecting the total amount of RAP1 bound to the target gene.
FIG. 9.

The effect of Δhmo1 on FHL1 and RAP1 binding to the chromosome. (A) The genome-wide ChIP analyses were conducted as described in the legend of Fig. 8A to examine the effect of Δhmo1 on FHL1 binding to the chromosome. The HMO1 (YTK8872) or Δhmo1 (YTK8873) strains expressing the TAP-tagged FHL1 were grown in SC medium to mid-log phase at 30°C. The orange and blue vertical bars represent the significant binding of FHL1 in YTK8872 (top panel) and YTK8873 (middle panel) to the same chromosomal region as described in the legend of Fig. 8A. The bottom panel represents the merged image of the upper two panels. The broken vertical lines represent that as described in the legend of Fig. 8A. The small red or blue triangle flags at the top of these lines indicate that the FHL1 binding to each target site was increased or decreased by Δhmo1, respectively. (B) The genome-wide ChIP analyses were conducted as described in the legend of Fig. 8A to examine the effect of Δhmo1 on RAP1 binding to the chromosome. The HMO1 (YTK8863) or Δhmo1 (YTK8865) strains expressing the TAP-tagged RAP1 were grown in YPD (yeast extract, peptone, dextrose) medium to mid-log phase at 30°C. The orange and blue vertical bars represent the significant binding of RAP1 in YTK8863 (top panel) and YTK8865 (middle panel), and the bottom panel represents the merged image of the upper two panels, as described for panel A. The broken vertical lines and the attached small flags are as described for panel A. The rectangle composed of two triangles (blue and red) represents the target site where the peak width was narrowed and the peak height was increased by Δhmo1. Notably, such an effect of Δhmo1 on the peak shape was observed much more frequently in panel B than in panel A. WT, wild type.
Similar analyses were conducted for HMO1 and Δhmo1 cells expressing TAP-tagged SFP1 (see Fig. S9 in the supplemental material). The binding of SFP1 to seven sites was similar in Δhmo1 and wild-type cells, but binding of SFP1 to 13 sites was lower in Δhmo1 than in wild-type cells. In addition, SFP1 bound to 29 sites in Δhmo1 cells that were not bound by SFP1 in wild-type cells. These sites include five Ribi genes (MAK16, SRO9, STP4, BMS1, and MRD1) and 13 loci which did not bind HMO1, FHL1, or RAP1 in wild-type cells; the latter group of genes included MAK16, BMS1, and MRD1.
Classification of the RPG promoters based on HMO1 abundance and HMO1 dependence of FHL1 and RAP1 binding.
The studies described above include data on transcriptional regulation of ∼20 RPGs. Here, a genome-wide analysis was undertaken of the abundance of HMO1 and the HMO1 dependence of FHL1 and RAP1 binding at 138 RPG promoters. The genome-wide ChIP data used for this analysis are presented in Fig. S10 in the supplemental material, and the results are summarized in Table 1.
TABLE 1.
Classification of RPGs
| Class | Subclass | No. of RPGs | HMO1 bindinga | HMO1-dependent bindingb
|
Gene name(s) | |
|---|---|---|---|---|---|---|
| FHL1 | RAP1c | |||||
| 1 | A | 24 | ++ | ++ | RPL2A, RPL2B, RPL12A, RPL12B, RPL16B, RPL17B, RPL19B, RPL20B, RPL23B, RPL25, RPL27A, RPL31A, RPL31B, RPL32, RPL34B, RPL40A, RPS6Ad, RPS7A, RPS8A, RPS10A, RPS17A, RPS24B, RPS26A, RPS27Bd | |
| B | 25 | ++ | ++ | + | RPL6Ad,e, RPL13A-RPS16B, RPL14A, RPL16A, RPL17Ae, RPL19Ae, RPL24A-RPL30, RPL24B, RPL27B, RPL33A, RPL33B, RPL34Ae, RPL37Ae, RPL42A, RPS0B, RPS4B, RPS5, RPS8B, RPS11Ae, RPS15/RPP2Ae, RPS18B, RPS19Be | |
| C | 12 | ++ | + | RPL15Bf, RPL21A, RPL36B, RPL42B, RPS1B, RPS4A, RPS6B, RPS7B, RPS11B, RPS21B, RPS23A, RPS24A | ||
| D | 8 | ++ | + | + | RPL13B-RPS16A, RPL20Ae, RPL39-RPS22Ae, RPS0A, RPS14Ae, RPS23B | |
| E | 4 | ++ | RPL35A, RPS17B, RPS18A, RPS25B | |||
| 2 | A | 20 | + | RPL3f, RPL5, RPL8A, RPL9A, RPL9B, RPL11A, RPL28, RPL29, RPL37B, RPL38, RPL41A, RPL41B, RPS1A, RPS2, RPS9A, RPS26B, RPS29A, RPS29B, RPS30B, RPP1B | ||
| B | 4 | + | + | RPL10, RPL18A, RPL18Bg, RPS3 | ||
| 3 | A | 24 | RPL7A, RPL7B, RPL8B, RPL11B, RPL14B, RPL15Af, RPL21B, RPL22B, RPL23A, RPL35B, RPL36A, RPL40B, RPL43A, RPL43Bg, RPS9B, RPS10B, RPS12, RPS14B, RPS19Af, RPS20, RPS25A, RPS27A, RPS30A, RPS31 | |||
| B | 2 | + | RPL1Bg, RPL22A | |||
| C | 1 | + | + | RPP0 | ||
| 4 | A | 2 | N/A | RPL1A, RPS21A | ||
| B | 2 | N/A | + | RPS13, ASC1 | ||
| C | 10 | N/A | N/A | RPL4A, RPL4B, RPL6B, RPL26A, RPL26B, RPS22B, RPS28A, RPS28B, RPP1A, RPP2B | ||
HMO1 binding was marked as ++ and + when signals shown in Fig. S2 in the supplemental material were >log 3 or <log 3, respectively.
HMO1-dependent binding of FHL1 and RAP1 to each RP promoter was evaluated. Note that only positive dependence (i.e., decrease by Δhmo1) was marked as ++ and + in this table. NA, not applicable (no binding; peaks shown in light colors in the figures are included in this group for simplicity).
Because most of the peaks were narrower in the Δhmo1 strain, RAP1 binding was scored as + only when peak height was reduced.
When peak color changed from a dark color to a lighter one, FHL1 binding was judged to be decreased. (See Fig. S7 in the supplemental material.)
When peak color changed from a dark color to a lighter one, RAP1 binding was judged to be decreased. (See Fig. S8 in the supplemental material.)
RAP1 peaks had a light color in both (i.e., wild-type and Δhmo1) strains. (See Fig. S8 in the supplemental material.)
FHL1 peaks had a light color in the wild-type strain. (See Fig. S3 in the supplemental material.)
Based on HMO1 binding, HMO1-enriched RPGs were designated as class 1 and HMO1-limited RPGs were designated class 2. Class 3 RPGs bound little or no HMO1 but did bind other factors, while class 4 RPGs did not bind HMO1, FHL1, or RAP1. Subclasses were assigned depending on the HMO1 dependence of FHL1 and RAP1 binding. However, we should emphasize that HMO1 dependence of FHL1 and RAP1 binding was evaluated in a qualitative manner based on changes in peak height and width in the binding histogram. Furthermore, these changes were often both modest in magnitude and complex (see Fig. S10 in the supplemental material). In addition, this analysis was limited to signals that fulfill three specific criteria (see Materials and Methods) (see Fig. S10 in the supplemental material) and considered only those that were decreased by deletion of HMO1 as HMO1 dependent. Therefore, signals that failed to meet the specified criteria were excluded from the analysis, and signals whose intensity was higher in Δhmo1 cells than in wild-type cells were scored as HMO1 independent.
The most significant findings in this classification were that only the HMO1-enriched RPG promoters showed HMO1 dependence of FHL1 binding, with the exception of RPP0 (class 3C). Conversely, there were only four HMO1-enriched RPG promoters that showed no HMO1 dependence of FHL1 binding (class 1E). As described above (Fig. 5), RPS18A belongs to this minor subclass. We should also emphasize that a considerable number of RPGs, which corresponded to one-third (24 out of 73) of class 1 genes, showed weak or no requirement of HMO1 for FHL1 binding, even though HMO1 was highly enriched at these promoters (classes 1C, D, and E).
HMO1 bound to 97 (70% of total) RPGs, whereas FHL1 and RAP1 bound to 124 (90%) and 128 (93%) RPGs, respectively. Moreover, there were no RPGs that were bound by HMO1 but not by FHL1 or RAP1. These observations suggest that FHL1 and RAP1 play a more general and global role in regulating transcription of RPGs than HMO1.
In this study, genome-wide ChIP analysis was conducted, and results were shown for several representative RPGs (Fig. 10A). Gene-specific ChIP analyses for HMO1 and FHL1 binding were also carried out for these RPGs (Fig. 4 and 5). Importantly, these data are wholly consistent within themselves, and genome-wide data presented here are in agreement with previously reported results (23), suggesting that the methods applied here are reliable and reproducible, even for analyzing subtle changes in binding on a genome-wide basis.
FIG. 10.

The classification of the RPGs based on the HMO1 abundance and the HMO1 dependence of FHL1 and RAP1 binding. (A) The examples of the RPGs that are grouped into classes 1A to E, 2A and B, and 3A and B (see details in the text and Table 1). These were extracted from the data of the genome-wide ChIP analyses (see Fig. S2, S7, and S8 in the supplemental material). The binding of HMO1 in the HMO1 strain and the binding of FHL1 and RAP1 in the HMO1/Δhmo1 strains (middle and bottom panels; these are merged images as shown in Fig. 9A and B, respectively) are shown for each RPG as indicated at the top. Red triangles and asterisks indicate the positions of the peaks and ORFs, respectively. Note that the results obtained in the top and middle panels are consistent with those of ChIP analyses conducted individually and shown in Fig. 4 and 5. (B) A schematic model depicting the two distinct binding modes for FHL1 to the RPG promoters. FHL1 binds to some RPG promoters in an HMO1-dependent manner (e.g., RPS5), whereas FHL1 binds to other RPG promoters in an HMO1-independent manner (e.g., RPL10). WT, wild type.
DISCUSSION
HMO1 associates with the 35S rRNA gene in a Pol I-dependent manner.
It is well established that many factors associate with the hyperphosphorylated form of Pol II, forming a multiprotein transcription complex that translocates along transcriptionally active chromatin and executes multiple tasks that are required for mRNA maturation, e.g., pre-mRNA capping, splicing and 3′ end processing (66). In an analogous manner, the small-subunit processome (21) and Spt4/Spt5 (61) are factors that travel with Pol I and are engaged in both transcription and pre-rRNA processing. This study (Fig. 1) suggests that HMO1 may also travel with Pol I, at least during transcription of the 35S rRNA gene. Furthermore, deletion of HMO1 causes abnormalities in both transcription and pre-rRNA processing (19, 23), and HMO1 associates with some components of the small-subunit processome in vivo (5, 25). Together, these observations suggest that HMO1 may play a role in coregulating transcription of rDNA and pre-rRNA processing.
Alternatively, yeast HMO1 may play a role similar to that proposed for the mammalian counterpart UBF (42, 52). Of note, other components of the Pol I machinery besides UBF are also associated with sequences across the entire rRNA gene in mammalian cells (42). Moreover, UBF apparently recruits the whole Pol I machinery to the heterologous UBF-binding sequences integrated at ectopic sites and can form morphologically indistinguishable nucleolar organizer regions (42). HMO1 may play a similar role and thus contribute to the establishment of an “open” chromatin conformation at active rRNA genes (approximately one-half of ∼150 copies) in growing cells (14). However, binding of UBF is transcription independent (42) whereas binding of HMO1 is Pol I dependent, suggesting a critical difference in how the two factors may function. The fact that the open chromatin conformation can be established on the coding sequences of active rRNA genes by elongating Pol I but not Pol II (14) suggests that HMO1 may be recruited to 35S rDNA after removal of nucleosomes by elongating Pol I; thus, HMO1 may play a role in the maintenance of but not the establishment of open chromatin in the rRNA gene cluster.
How is HMO1 recruited to RPG promoters?
This study shows that 73 (class 1) out of 138 total genome-wide RPG promoters bound substantial amounts of HMO1; 24 RPGs (class 2) were associated with a low level of HMO1 and 41 RPGs (classes 3 and 4) bound no detectable HMO1 (Table 1). This is consistent with the previous results of Hall et al. (23) since eight RPGs that they identified as HMO1 enriched (i.e., RPS19B, RPS23A, RPL30, RPL17B, RPL27A, RPL13A, RPS18B, and RPS21B) belonged to class 1 in our study, whereas eight RPGs that they identified as HMO1 limited (i.e., RPL26A, RPL29, RPL9A, RPL22A, RPL18B, RPS13, RPL26B, and RPS22B) belonged to either class 2, 3, or 4 in our study (Table 1).
This raises the question, what produces such quantitative difference in the binding of HMO1? Reporter gene assays suggest that HMO1 binds to the RPS5 promoter in a sequence-specific manner (class 1B) and that it interacts weakly with the RPL10 promoter (class 2B) (Fig. 7). Importantly, the HMO1 dependence of FHL1 binding and HMO1-dependence of transcription also appeared to be DNA sequence specific (Fig. 7). Further studies are needed to identify specific DNA sequence motifs with which HMO1 interacts directly or indirectly; these studies could potentially involve deletion analyses of the RPS5 promoter or RPS5-RPL10 chimeric promoters.
Previous in vivo and in silico studies suggest that RAP1 might recruit FHL1/IFH1 and HMO1 to the IFHL motif (23, 72, 78). However, mutation of this motif decreased the binding of FHL1/IFH1 and transcription (72) but had little effect on binding of HMO1 (23). Furthermore, this motif is not found at all promoters of HMO1-enriched RPGs (class 1). Thus, it seems unlikely that HMO1 recognizes this motif directly.
Intriguingly, when RAP1 is tethered to DNA by a heterologous DNA binding domain, it fails to recruit FHL1/IFH1 (78). Furthermore, the RAP1 binding site in a glycolytic enzyme gene, which is different from the RAP1 binding sites in RPG promoters (so-called RPG boxes) (41), can recruit RAP1 but not FHL1/IFH1 (78). These observations suggest that a specific RAP1/RPG promoter complex may be required to recruit FHL1/IFH1. Therefore, RAP1 may adopt different conformations when bound to different RPG promoters, such as those of RPS5 and RPL10, and this may influence the DNA binding affinity of HMO1 (4), the extent of HMO1 self-association (15), or the HMO1 dependence of FHL1 binding. In this model, it is expected that different conformations of RAP1 would correlate with different three-dimensional configurations of the RAP1-HMO1-FHL1/IFH1 complex on each RPG promoter.
Paradoxically, it was shown that the ABF1 binding site is required for the expression of RPS28A, but the loss of ABF1 binding from this site does not reduce this expression (76). This suggests that another factor that binds to the ABF1 binding site is required for RPS28A expression. Thus, it remains possible that HMO1 binds to RPG promoters independently of RAP1, even though the RAP1 binding site appears to be required for HMO1 binding (23). Indeed, 263 non-RP target loci of HMO1 do not bind RAP1 (Fig. 8C). In addition, HMO1 binds to CAG repeats in vivo apparently without other factors (30). Additional genetic studies using RAP1 mutants are needed to better understand the role played by RAP1 in regulating binding of HMO1 to RPG promoters and transcription of RPGs.
Multiple pathways for recruitment of FHL1 to RPG promoters.
Genome-wide ChIP analyses demonstrated that HMO1, FHL1, and RAP1 bind to 177 common loci, of which nearly half (∼90) are not RPGs (see Table S3 in the supplemental material). The functions of the non-RP genes are diverse, including characteristic genes, such as G1 cyclin (i.e., CLN1 and CLN3); however, these genes have few GO terms in common (http://db.yeastgenome.org/cgi-bin/GO/goTermFinder), and no common functions could be inferred from GO analysis (8).
Previous studies showed that FHL1 binds with high specificity to RPGs (34, 72). This study shows that FHL1 binds to 124 RPG promoters (Table 1), and that 23 of 36 genes with binding sites for FHL1 and RAP1 but not HMO1 (Fig. 8C) were RPGs (see Table S3 in the supplemental material). However, unlike previous studies, this study provides evidence that FHL1 binds to a significant number of non-RP genes. This discrepancy may be due in part to the sensitivity of the ChIP analyses performed here. In fact, nearly 300 target loci of FHL1 were identified in this study that were not identified in a previous study with a stringent cutoff of threefold enrichment (72). Among the 297 non-RP target loci of FHL1 identified in this study, 130 (44%), 13 (4%), and 90 (30%) loci were common targets for HMO1, RAP1, or both HMO1 and RAP1, respectively. Sixty-four loci (20%) only had binding sites for FHL1. GO analysis showed that 18 of the latter genes participate in carboxylic/organic acid metabolism (P value of <0.001).
One of the most important findings of this study was that FHL1 binds to RPG promoters and possibly other promoters in either an HMO1-dependent or an HMO1-independent manner (Fig. 10B). The previous study showed that FHL1 binds to RPL2B/RPL27B/RPL40A promoters in an HMO1-dependent manner (23). In good agreement with this, the HMO1 dependency of FHL1 binding to these three RPG promoters was scored as the strongest in our study (Table 1, ++). Although FHL1 has been proposed to bind to all RPG promoters in an HMO1-dependent manner (23), we identified a novel subgroup of RPGs that show HMO1-independent FHL1 binding by simply examining all RPG family members for HMO1 dependency (Table 1).
In general, a higher level of HMO1 binding correlated with greater HMO1 dependence of FHL1 binding (Table 1). The molecular mechanisms underlying these observations are not known; however, these results suggest that HMO1 may play different roles on HMO1-enriched and HMO1-limited RPG promoters. As proposed for the 35S rRNA gene, HMO1 may also help maintain an open chromatin conformation after it is established by other factors (75, 78). We propose that HMO1 may stimulate FHL1 binding by this mechanism at HMO1-enriched promoters, while it may play another role at HMO1-limited promoters.
There is substantial overlap in the target loci of HMO1 and FHL1 (Fig. 8C), suggesting that they may cooperate functionally at some promoters. This is consistent with the observation that they copurify as a protein complex with histones H2A and H4 (25) and that their binding to some RPG promoters is reciprocally dependent (unpublished observations) (23). However, the effect of Δhmo1 on transcription did not correlate with the binding of FHL1/IFH1 (Fig. 5 and 6) (23). For instance, Δhmo1 resulted in the loss of FHL1/IFH1 binding at some of the RPG promoters but did not always lead to a decrease in transcription. In addition, when FHL1 was tethered to a promoter, it could recruit IFH1, but it failed to activate transcription (78). These observations suggest that differential assembly of RAP1-HMO1-FHL1/IFH1, and possibly different conformations of these protein complexes, may play specific roles in regulating transcription of specific RPG promoters. If the roles of HMO1 and FHL1/IFH1 varied at different RPG promoters, this would explain differential effects of deletion of HMO1 on recruitment of other protein factors and on transcription.
How does SFP1 regulate transcription?
SFP1 binds to many RPG promoters (43) and plays a significant role in transcription of RPGs (27, 43). In addition, mutating SFP1 (Δsfp1) reduced binding of FHL1/IFH1 to several RPG promoters (27). Since FHL1/IFH1 cannot bind directly to DNA (58), it seems likely that RAP1, HMO1, and/or SFP1 facilitate this process. In contrast to a previous study (43), the genome-wide ChIP analyses presented here indicate that SFP1 does not bind significantly to RPG promoters (see Fig. S5 in the supplemental material). The reasons for this discrepancy are unclear. However, it is possible that SFP1 binding to RPG promoters was below the limit of detection of the ChIP method used here. Alternatively, the different results could be due to use of different yeast strains in the two studies. Indeed, the function of a corepressor of FHL1, CRF1, in the transcription of RPGs is strain specific (78). A previous study showed that carbon starvation induced translocation of SFP1 from the nucleus to the cytoplasm, and FHL1 was relocalized near to the nucleolus but remained bound to the RPG promoters; this result indicates that SFP1 does not play a role in recruiting FHL1 to RPGs under these conditions.
In contrast to HMO1, RAP1, and FHL1, SFP1 binds to ORFs rather than to promoters (see Fig. S5 in the supplemental material). This suggests that SFP1 plays a different role than the other protein factors. GO analysis of the 29 target loci of SFP1 identified in Δhmo1 cells revealed that five genes (CLA4, LAS17, SCD5, VRP1, and YAP1802) had a function related to the cortical actin cytoskeleton (P value of 0.00019). Thus, SFP1 may regulate other genes in this category. Furthermore, it is consistent with the well-established function of SFP1 in cell size homeostasis (26, 27).
What are the molecular functions of HMO1, FHL1, and RAP1 in transcription?
Many factors such as NuA4, RAP1, FHL1/IFH1, SFP1, CRF1, and HMO1 play roles in the transcription of RPGs (23, 27, 43, 45, 47, 57, 58, 60, 64, 72, 78; also the present study). However, a recent in vitro study revealed that RAP1 was sufficient to activate transcription of native or chimeric RPG promoters (20) and that RAP1 interacts directly with several subunits (TAFs) of TFIID. This suggests that RAP1 may be sufficient to recruit TFIID onto the RPG promoter. However, as we discuss above, certain configurations of the RAP1-HMO1-FHL1/IFH1 complex may be important for transcription of RPGs in vivo. Such a specific three-dimensional architecture comprised of several transcription factors is reminiscent of the so-called enhanceosome in mammalian cells (1, 48). In the enhanceosome, the role of each component in the complex is different from its role outside of the complex. The analogy is strengthened by the observation that HMO1 can facilitate formation of an enhanceosome by introducing bends into DNA (49). This in vitro study was conducted using a naked template (20); therefore, it is also possible that other factors are specifically required for transcription on a chromatin template. In addition to the well-established role of NuA4 in modifying histones (16), HMO1 and FHL1 associate with histones H2A and H4 in vivo (25) and may play a role in modulating chromatin structure. This is consistent with the observation that deletion of hmo1 increases chromatin hypersensitivity to digestion by micrococcal nuclease (40).
In this study, genome-wide ChIP analyses identified approximately 190 target loci of RAP1 that were not identified in previous studies (10, 37). The list includes most of the putative direct targets of RAP1 proposed by a different method (76), confirming the sensitivity and reproducibility of the methods used here. Notably, this study shows that RAP1 binds in an HMO1-independent manner to the 35S RNA gene (see Fig. S4 in the supplemental material). Because this result appears to contradict a previous study (23), it was confirmed by ChIP using 14 sets of primer pairs (Fig. 1B); the results showed that only one primer set (35S rDNA region 10) detected strong RAP1 binding (unpublished observations). This region overlaps with the promoter of TAR1, which encodes a mitochondrial protein (13). Further studies are required to confirm whether RAP1 is involved in transcription of this gene.
We originally identified HMO1 as a protein that interacts with TATA box binding protein and the N-terminal domain of TAF1, a large subunit of TFIID (unpublished data). Thus, HMO1 may directly help recruit TFIID to the promoter, as observed for NHP6A and NHP6B (6, 53). On the other hand, RAP1 facilitates GCN4 binding to the HIS4 promoter, which is a common target of HMO1 and RAP1, by overcoming the suppressive effect of the chromatin (77). These observations suggest that HMO1 and RAP1 activate transcription of class II genes via TFIID by multiple mechanisms. Future studies are needed to elucidate the exact roles of these regulators in the transcription of RPGs and non-RP genes.
Supplementary Material
Acknowledgments
We thank H. Iwasaki and other members of our laboratory for advice and comments on this work. We also thank A. G. Hinnebusch, Y. Nakatani, J. Heitman, and M. Nomura for supplying the yeast strains; M. Funk, F. Uhlmann, Y. Tsukihashi, and S. Takahata for the plasmids; and Y. Katou and T. Itoh for their help in genome- wide ChIP analysis. The TAP plasmid was obtained from CellZome (Heidelberg).
This study was supported by grant for the 2006 Strategic Research Project (grant W18013) of Yokohama City University and grants from the Japan Society for the Promotion of Science; the Ministry of Education, Culture, Sports, Science and Technology of Japan; CREST of Japan Science and Technology Corporation; and the Mitsubishi Foundation.
Footnotes
Published ahead of print on 23 July 2007.
Supplemental material for this article may be found at http://mcb.asm.org/.
REFERENCES
- 1.Agresti, A., and M. E. Bianchi. 2003. HMGB proteins and gene expression. Curr. Opin. Genet. Dev. 13:170-178. [DOI] [PubMed] [Google Scholar]
- 2.Amberg, D. C., D. J. Burke, and J. N. Strathern. 2005. Methods in yeast genetics: a Cold Spring Harbor Laboratory course manual. Cold Spring Harbor Laboratory Press, Cold Spring Harbor, NY.
- 3.Aronheim, A., E. Zandi, H. Hennemann, S. J. Elledge, and M. Karin. 1997. Isolation of an AP-1 repressor by a novel method for detecting protein-protein interactions. Mol. Cell. Biol. 17:3094-3102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Bauerle, K. T., E. Kamau, and A. Grove. 2006. Interactions between N- and C-terminal domains of the Saccharomyces cerevisiae high-mobility group protein HMO1 are required for DNA bending. Biochemistry 45:3635-3645. [DOI] [PubMed] [Google Scholar]
- 5.Bernstein, K. A., J. E. Gallagher, B. M. Mitchell, S. Granneman, and S. J. Baserga. 2004. The small-subunit processome is a ribosome assembly intermediate. Eukaryot. Cell 3:1619-1626. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Biswas, D., A. N. Imbalzano, P. Eriksson, Y. Yu, and D. J. Stillman. 2004. Role for Nhp6, Gcn5, and the Swi/Snf complex in stimulating formation of the TATA-binding protein-TFIIA-DNA complex. Mol. Cell. Biol. 24:8312-8321. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Biswas, D., Y. Yu, M. Prall, T. Formosa, and D. J. Stillman. 2005. The yeast FACT complex has a role in transcriptional initiation Mol. Cell. Biol. 25:5812-5822. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Boyle, E. I., S. Weng, J. Gollub, H. Jin, D. Botstein, J. M. Cherry, and G. Sherlock. 2004. GO::TermFinder-open source software for accessing Gene Ontology information and finding significantly enriched Gene Ontology terms associated with a list of genes. Bioinformatics 20:3710-3715. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Brewster, N. K., G. C. Johnston, and R. A. Singer. 2001. A bipartite yeast SSRP1 analog comprised of Pob3 and Nhp6 proteins modulates transcription. Mol. Cell. Biol. 21:3491-3502. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Buck, M. J., and J. D. Lieb. 2006. A chromatin-mediated mechanism for specification of conditional transcription factor targets. Nat. Genet. 38:1446-1451. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Bustin, M. 2001. Revised nomenclature for high mobility group (HMG) chromosomal proteins. Trends Biochem. Sci. 26:152-153. [DOI] [PubMed] [Google Scholar]
- 12.Christianson, T. W., R. S. Sikorski, M. Dante, J. H. Shero, and P. Hieter. 1992. Multifunctional yeast high-copy-number shuttle vectors. Gene 110:119-122. [DOI] [PubMed] [Google Scholar]
- 13.Coelho, P. S., A. C. Bryan, A. Kumar, G. S. Shadel, and M. Snyder. 2002. A novel mitochondrial protein, Tar1p, is encoded on the antisense strand of the nuclear 25S rDNA. Genes Dev. 16:2755-2760. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Dammann, R., R. Lucchini, T. Koller, and J. M. Sogo. 1995. Transcription in the yeast rRNA gene locus: distribution of the active gene copies and chromatin structure of their flanking regulatory sequences. Mol. Cell. Biol. 15:5294-5303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Dolinski, K. J., and J. Heitman. 1999. Hmo1p, a high mobility group 1/2 homolog, genetically and physically interacts with the yeast FKBP12 prolyl isomerase. Genetics 151:935-944. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Doyon, Y., and J. Cote. 2004. The highly conserved and multifunctional NuA4 HAT complex. Curr. Opin. Genet. Dev. 14:147-154. [DOI] [PubMed] [Google Scholar]
- 17.Fingerman, I., V. Nagaraj, D. Norris, and A. K. Vershon. 2003. Sfp1 plays a key role in yeast ribosome biogenesis. Eukaryot. Cell 2:1061-1068. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Formosa, T., P. Eriksson, J. Wittmeyer, J. Ginn, Y. Yu, and D. J. Stillman. 2001. Spt16-Pob3 and the HMG protein Nhp6 combine to form the nucleosome-binding factor SPN. EMBO J. 20:3506-3517. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Gadal, O., S. Labarre, C. Boschiero, and P. Thuriaux. 2002. Hmo1, an HMG-box protein, belongs to the yeast ribosomal DNA transcription system. EMBO J. 21:5498-5507. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Garbett, K. A., M. K. Tripathi, B. Cencki, J. H. Layer, and P. A. Weil. 2007. Yeast TFIID serves as a coactivator for Rap1p by direct protein-protein interaction. Mol. Cell. Biol. 27:297-311. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Granneman, S., and S. J. Baserga. 2005. Crosstalk in gene expression: coupling and co-regulation of rDNA transcription, pre-ribosome assembly and pre-rRNA processing. Curr. Opin. Cell Biol. 17:281-286. [DOI] [PubMed] [Google Scholar]
- 22.Grasser, K. D. 2003. Chromatin-associated HMGA and HMGB proteins: versatile co-regulators of DNA-dependent processes. Plant Mol. Biol. 53:281-295. [DOI] [PubMed] [Google Scholar]
- 23.Hall, D. B., J. T. Wade, and K. Struhl. 2006. An HMG protein, Hmo1, associates with promoters of many ribosomal protein genes and throughout the rRNA gene locus in Saccharomyces cerevisiae. Mol. Cell. Biol. 26:3672-3679. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Hermann-Le Denmat, S., M. Werner, A. Sentenac, and P. Thuriaux. 1994. Suppression of yeast RNA polymerase III mutations by FHL1, a gene coding for a fork head protein involved in rRNA processing. Mol. Cell. Biol. 14:2905-2913. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Ho, Y., A. Gruhler, A. Heilbut, G. D. Bader, L. Moore, S. L. Adams, A. Millar, P. Taylor, K. Bennett, K. Boutilier, L. Yang, C. Wolting, I. Donaldson, S. Schandorff, J. Shewnarane, M. Vo, J. Taggart, M. Goudreault, B. Muskat, C. Alfarano, D. Dewar, Z. Lin, K. Michalickova, A. R. Willems, H. Sassi, P. A. Nielsen, K. J. Rasmussen, J. R. Andersen, L. E. Johansen, L. H. Hansen, H. Jespersen, A. Podtelejnikov, E. Nielsen, J. Crawford, V. Poulsen, B. D. Sorensen, J. Matthiesen, R. C. Hendrickson, F. Gleeson, T. Pawson, M. F. Moran, D. Durocher, M. Mann, C. W. Hogue, D. Figeys, and M. Tyers. 2002. Systematic identification of protein complexes in Saccharomyces cerevisiae by mass spectrometry. Nature 415:180-183. [DOI] [PubMed] [Google Scholar]
- 26.Jorgensen, P., J. L. Nishikawa, B. J. Breitkreutz, and M. Tyers. 2002. Systematic identification of pathways that couple cell growth and division in yeast. Science 297:395-400. [DOI] [PubMed] [Google Scholar]
- 27.Jorgensen, P., I. Rupes, J. R. Sharom, L. Schneper, J. R. Broach, and M. Tyers. 2004. A dynamic transcriptional network communicates growth potential to ribosome synthesis and critical cell size. Genes Dev. 18:2491-2505. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Katou, Y., K. Kaneshiro, H. Aburatani, and K. Shirahige. 2006. Genomic approach for the understanding of dynamic aspect of chromosome behavior. Methods Enzymol. 409:389-410. [DOI] [PubMed] [Google Scholar]
- 29.Katou, Y., Y. Kanoh, M. Bando, H. Noguchi, H. Tanaka, T. Ashikari, K. Sugimoto, and K. Shirahige. 2003. S-phase checkpoint proteins Tof1 and Mrc1 form a stable replication-pausing complex. Nature 424:1078-1083. [DOI] [PubMed] [Google Scholar]
- 30.Kim, H., and D. M. Livingston. 2006. A high mobility group protein binds to long CAG repeat tracts and establishes their chromatin organization in Saccharomyces cerevisiae. J. Biol. Chem. 281:15735-15740. [DOI] [PubMed] [Google Scholar]
- 31.Kokubo, T., M. J. Swanson, J. I. Nishikawa, A. G. Hinnebusch, and Y. Nakatani. 1998. The yeast TAF145 inhibitory domain and TFIIA competitively bind to TATA-binding protein. Mol. Cell. Biol. 18:1003-1012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Kruppa, M., R. D. Moir, D. Kolodrubetz, and I. M. Willis. 2001. Nhp6, an HMG1 protein, functions in SNR6 transcription by RNA polymerase III in S. cerevisiae. Mol. Cell 7:309-318. [DOI] [PubMed] [Google Scholar]
- 33.Kunkel, T. A., J. D. Roberts, and R. A. Zakour. 1987. Rapid and efficient site-specific mutagenesis without phenotypic selection. Methods Enzymol. 154:367-382. [DOI] [PubMed] [Google Scholar]
- 34.Lee, T. I., N. J. Rinaldi, F. Robert, D. T. Odom, Z. Bar-Joseph, G. K. Gerber, N. M. Hannett, C. T. Harbison, C. M. Thompson, I. Simon, J. Zeitlinger, E. G. Jennings, H. L. Murray, D. B. Gordon, B. Ren, J. J. Wyrick, J. B. Tagne, T. L. Volkert, E. Fraenkel, D. K. Gifford, and R. A. Young. 2002. Transcriptional regulatory networks in Saccharomyces cerevisiae. Science 298:799-804. [DOI] [PubMed] [Google Scholar]
- 35.Lengronne, A., J. McIntyre, Y. Katou, Y. Kanoh, K. P. Hopfner, K. Shirahige, and F. Uhlmann. 2006. Establishment of sister chromatid cohesion at the S. cerevisiae replication fork. Mol. Cell 23:787-799. [DOI] [PubMed] [Google Scholar]
- 36.Li, H., C. K. Tsang, M. Watkins, P. G. Bertram, and X. F. Zheng. 2006. Nutrient regulates Tor1 nuclear localization and association with rDNA promoter. Nature 442:1058-1061. [DOI] [PubMed] [Google Scholar]
- 37.Lieb, J. D., X. Liu, D. Botstein, and P. O. Brown. 2001. Promoter-specific binding of Rap1 revealed by genome-wide maps of protein-DNA association. Nat. Genet. 28:327-334. [DOI] [PubMed] [Google Scholar]
- 38.Lindroos, H. B., L. Strom, T. Itoh, Y. Katou, K. Shirahige, and C. Sjogren. 2006. Chromosomal association of the Smc5/6 complex reveals that it functions in differently regulated pathways. Mol. Cell 22:755-767. [DOI] [PubMed] [Google Scholar]
- 39.Longtine, M. S., A. McKenzie III, D. J. Demarini, N. G. Shah, A. Wach, A. Brachat, P. Philippsen, and J. R. Pringle. 1998. Additional modules for versatile and economical PCR-based gene deletion and modification in Saccharomyces cerevisiae. Yeast 14:953-961. [DOI] [PubMed] [Google Scholar]
- 40.Lu, J., R. Kobayashi, and S. J. Brill. 1996. Characterization of a high mobility group 1/2 homolog in yeast. J. Biol. Chem. 271:33678-33685. [DOI] [PubMed] [Google Scholar]
- 41.Mager, W. H., and R. J. Planta. 1991. Coordinate expression of ribosomal protein genes in yeast as a function of cellular growth rate. Mol. Cell Biochem. 104:181-187. [DOI] [PubMed] [Google Scholar]
- 42.Mais, C., J. E. Wright, J. L. Prieto, S. L. Raggett, and B. McStay. 2005. UBF-binding site arrays form pseudo-NORs and sequester the RNA polymerase I transcription machinery. Genes Dev. 19:50-64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Marion, R. M., A. Regev, E. Segal, Y. Barash, D. Koller, N. Friedman, and E. K. O'Shea. 2004. Sfp1 is a stress- and nutrient-sensitive regulator of ribosomal protein gene expression. Proc. Natl. Acad. Sci. USA 101:14315-14322. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Martin, D. E., T. Powers, and M. N. Hall. 2006. Regulation of ribosome biogenesis: where is TOR? Cell Metab. 4:259-260. [DOI] [PubMed] [Google Scholar]
- 45.Martin, D. E., A. Soulard, and M. N. Hall. 2004. TOR regulates ribosomal protein gene expression via PKA and the forkhead transcription factor FHL1. Cell 119:969-979. [DOI] [PubMed] [Google Scholar]
- 46.Mason, P. B., and K. Struhl. 2003. The FACT complex travels with elongating RNA polymerase II and is important for the fidelity of transcriptional initiation in vivo. Mol. Cell. Biol. 23:8323-8333. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Mencia, M., Z. Moqtaderi, J. V. Geisberg, L. Kuras, and K. Struhl. 2002. Activator-specific recruitment of TFIID and regulation of ribosomal protein genes in yeast. Mol. Cell 9:823-833. [DOI] [PubMed] [Google Scholar]
- 48.Merika, M., and D. Thanos. 2001. Enhanceosomes. Curr. Opin. Genet. Dev. 11:205-208. [DOI] [PubMed] [Google Scholar]
- 49.Mitsouras, K., B. Wong, C. Arayata, R. C. Johnson, and M. Carey. 2002. The DNA architectural protein HMGB1 displays two distinct modes of action that promote enhanceosome assembly. Mol. Cell. Biol. 22:4390-4401. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Mumberg, D., R. Muller, and M. Funk. 1995. Yeast vectors for the controlled expression of heterologous proteins in different genetic backgrounds. Gene 156:119-122. [DOI] [PubMed] [Google Scholar]
- 51.Oakes, M., J. P. Aris, J. S. Brockenbrough, H. Wai, L. Vu, and M. Nomura. 1998. Mutational analysis of the structure and localization of the nucleolus in the yeast Saccharomyces cerevisiae. J. Cell Biol. 143:23-34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.O'Sullivan, A. C., G. J. Sullivan, and B. McStay. 2002. UBF binding in vivo is not restricted to regulatory sequences within the vertebrate ribosomal DNA repeat. Mol. Cell. Biol. 22:657-668. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Paull, T. T., M. Carey, and R. C. Johnson. 1996. Yeast HMG proteins NHP6A/B potentiate promoter-specific transcriptional activation in vivo and assembly of preinitiation complexes in vitro. Genes Dev. 10:2769-2781. [DOI] [PubMed] [Google Scholar]
- 54.Puig, O., F. Caspary, G. Rigaut, B. Rutz, E. Bouveret, E. Bragado-Nilsson, M. Wilm, and B. Seraphin. 2001. The tandem affinity purification (TAP) method: a general procedure of protein complex purification. Methods 24:218-229. [DOI] [PubMed] [Google Scholar]
- 55.Read, C. M., P. D. Cary, C. Crane-Robinson, P. C. Driscoll, and D. G. Norman. 1993. Solution structure of a DNA-binding domain from HMG1. Nucleic Acids Res. 21:3427-3436. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Reeves, R., and J. E. Adair. 2005. Role of high mobility group (HMG) chromatin proteins in DNA repair. DNA Repair 4:926-938. [DOI] [PubMed] [Google Scholar]
- 57.Reid, J. L., V. R. Iyer, P. O. Brown, and K. Struhl. 2000. Coordinate regulation of yeast ribosomal protein genes is associated with targeted recruitment of Esa1 histone acetylase. Mol. Cell 6:1297-1307. [DOI] [PubMed] [Google Scholar]
- 58.Rudra, D., Y. Zhao, and J. R. Warner. 2005. Central role of Ifh1p-Fhl1p interaction in the synthesis of yeast ribosomal proteins. EMBO J. 24:533-542. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Sambrook, J., and D. W. Russell. 2001. Molecular cloning: a laboratory manual, 3rd ed. Cold Spring Harbor Laboratory Press, Cold Spring Harbor, NY.
- 60.Schawalder, S. B., M. Kabani, I. Howald, U. Choudhury, M. Werner, and D. Shore. 2004. Growth-regulated recruitment of the essential yeast ribosomal protein gene activator Ifh1. Nature 432:1058-1061. [DOI] [PubMed] [Google Scholar]
- 61.Schneider, D. A., S. L. French, Y. N. Osheim, A. O. Bailey, L. Vu, J. Dodd, J. R. Yates, A. L. Beyer, and M. Nomura. 2006. RNA polymerase II elongation factors Spt4p and Spt5p play roles in transcription elongation by RNA polymerase I and rRNA processing. Proc. Natl. Acad. Sci. USA 103:12707-12712. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Shen, X., G. Mizuguchi, A. Hamiche, and C. Wu. 2000. A chromatin remodelling complex involved in transcription and DNA processing. Nature 406:541-544. [DOI] [PubMed] [Google Scholar]
- 63.Shen, X., R. Ranallo, E. Choi, and C. Wu. 2003. Involvement of actin-related proteins in ATP-dependent chromatin remodeling. Mol. Cell 12:147-155. [DOI] [PubMed] [Google Scholar]
- 64.Shore, D., and K. Nasmyth. 1987. Purification and cloning of a DNA binding protein from yeast that binds to both silencer and activator elements. Cell 51:721-732. [DOI] [PubMed] [Google Scholar]
- 65.Sikorski, R. S., and P. Hieter. 1989. A system of shuttle vectors and yeast host strains designed for efficient manipulation of DNA in Saccharomyces cerevisiae. Genetics 122:19-27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Sims, R. J., III, R. Belotserkovskaya, and D. Reinberg. 2004. Elongation by RNA polymerase II: the short and long of it. Genes Dev. 18:2437-2468. [DOI] [PubMed] [Google Scholar]
- 67.Southern, J. A., D. F. Young, F. Heaney, W. K. Baumgartner, and R. E. Randall. 1991. Identification of an epitope on the P and V proteins of simian virus 5 that distinguishes between two isolates with different biological characteristics. J. Gen. Virol. 72:1551-1557. [DOI] [PubMed] [Google Scholar]
- 68.Takahata, S., H. Ryu, K. Ohtsuki, K. Kasahara, M. Kawaichi, and T. Kokubo. 2003. Identification of a novel TATA element-binding protein binding region at the N terminus of the Saccharomyces cerevisiae TAF1 protein. J. Biol. Chem. 278:45888-45902. [DOI] [PubMed] [Google Scholar]
- 69.Thomas, J. O., and A. A. Travers. 2001. HMG1 and 2, and related “architectural” DNA-binding proteins. Trends Biochem. Sci. 26:167-174. [DOI] [PubMed] [Google Scholar]
- 70.Tsukihashi, Y., T. Miyake, M. Kawaichi, and T. Kokubo. 2000. Impaired core promoter recognition caused by novel yeast TAF145 mutations can be restored by creating a canonical TATA element within the promoter region of the TUB2 gene. Mol. Cell. Biol. 20:2385-2399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Velazquez, E., O. Calvo, E. Cervantes, P. F. Mateos, M. Tamame, and E. Martinez-Molina. 2000. Staircase electrophoresis profiles of stable low-molecular-weight RNA—a new technique for yeast fingerprinting. Int. J. Syst. Evol. Microbiol. 50:917-923. [DOI] [PubMed] [Google Scholar]
- 72.Wade, J. T., D. B. Hall, and K. Struhl. 2004. The transcription factor Ifh1 is a key regulator of yeast ribosomal protein genes. Nature 432:1054-1058. [DOI] [PubMed] [Google Scholar]
- 73.Warner, J. R. 1999. The economics of ribosome biosynthesis in yeast. Trends Biochem. Sci. 24:437-440. [DOI] [PubMed] [Google Scholar]
- 74.Weir, H. M., P. J. Kraulis, C. S. Hill, A. R. Raine, E. D. Laue, and J. O. Thomas. 1993. Structure of the HMG box motif in the B-domain of HMG1. EMBO J. 12:1311-1319. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Yarragudi, A., T. Miyake, R. Li, and R. H. Morse. 2004. Comparison of ABF1 and RAP1 in chromatin opening and transactivator potentiation in the budding yeast Saccharomyces cerevisiae. Mol. Cell. Biol. 24:9152-9164. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Yarragudi, A., L. W. Parfrey, and R. H. Morse. 2007. Genome-wide analysis of transcriptional dependence and probable target sites for Abf1 and Rap1 in Saccharomyces cerevisiae. Nucleic. Acids Res. 35:193-202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Yu, L., and R. H. Morse. 1999. Chromatin opening and transactivator potentiation by RAP1 in Saccharomyces cerevisiae. Mol. Cell. Biol. 19:5279-5288. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Zhao, Y., K. B. McIntosh, D. Rudra, S. Schawalder, D. Shore, and J. R. Warner. 2006. Fine-structure analysis of ribosomal protein gene transcription. Mol. Cell. Biol. 26:4853-4862. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.