

Abstract
Ixodes scapularis and Dermacentor andersoni cell lines were stimulated with heat killed Escherichia coli and Micrococcus luteus to investigate whether infection by Rickettsia peacockii, an endosymbiont of D. andersoni, modifies humoral immune responses. Radial diffusion assays, western blotting, flow cytometry, and quantitative reverse-transcription PCR were used to determine if expression of bacteriolytic peptides, including lysozyme and defensin, was upregulated by bacterial stimulation or infection with R. peacockii. The I. scapularis line IDE12 upregulated expression of lysozyme and defensin following stimulation. The D. andersoni cell line DAE15 also expressed defensin and lysozyme, but only lysozyme was upregulated by bacterial stimulation. R. peacockii infection alone, or in cells stimulated with bacteria, did not modify defensin or lysozyme expression in either cell line. These results suggest tick endosymbionts may avoid recognition by the tick immune system, and infection may not affect humoral immune responses to bacteria not normally associated with ticks.
Keywords: endosymbiont, immune response, defensin, lysozyme, tick cell culture, rickettsia, real-time PCR, immune evasion
Introduction
Ticks are important vectors of bacteria that cause disease in humans and animals. Factors underlying tick success as vectors are poorly understood, but the tick immune system may play a role in determining vector capacity (1). Bacterial challenge causes immunocompetent tissues to produce several soluble antimicrobial factors, including defensin and lysozyme. Defensins are small cationic antimicrobial peptides with activity against gram positive bacteria (2, 3) that have been identified in several hard ticks including Dermacentor variabilis (3) and Ixodes scapularis (4, 5). Lysozymes hydrolyze the 1-4 β linkage between the N-acetyl-muramic acid and N-acetyl-D-glucosamine residues that make up peptidoglycan in bacterial cell walls (6). Immunoresponsive c-type lysozymes have been identified in Ornithodoros moubata (7, 8), D. variabilis and a D. andersoni cell line (9). Lysozyme-like immunoreactivity has been reported in I. ricinus hemocytes (10) and protein extracts from several soft and hard ticks (11). Lysozymes may synergize antimicrobial peptides, including defensins, by degrading the peptidoglycan cell wall and permitting better access to the bacterial cell membrane (3, 12, 13).
Ticks are persistently infected with nonpathogenic rickettsia-like bacteria (14, 15). Immune interactions between ticks and endosymbionts are difficult to assess in vivo, and it is not known whether endosymbionts elicit immune responses similar to those induced by Escherichia coli or Micrococcus luteus. Rickettsia peacockii is a rickettsial endosymbiont of D. andersoni, and is found within the interstitial cells of the ovaries, and developing oocytes (16). It is not known why R. peacockii appears to be restricted to the female reproductive tissues, but host immune responses may limit rickettsial distribution. R. peacockii can infect and grow in cell lines from a variety of arthropods (17) and tick cell culture may help determine whether the tick immune system recognizes and responds to endosymbionts in the same way it responds to bacteria that are not normally present within the tick hemocoel.
Previous research demonstrated the cell lines IDE12 and DAE15 from Ixodes scapularis and D. andersoni, respectively, phagocytosed and killed Borrelia burgdorferi, and that prior infection by R. peacockii slightly diminished phagocytic activity (18). In the studies reported here, we investigated whether these tick cell lines produced soluble antimicrobial factors, and examined expression of these factors following bacterial stimulation. Additionally, we examined whether R. peacockii alone induces a humoral response or modifies the humoral response to heat inactivated E. coli and M. luteus. Our results demonstrate E. coli and M. luteus stimulated IDE12 and DAE15 cells upregulated expression of a lysozyme-like protein that was not upregulated by R. peacockii infection. IDE12 cells upregulated defensin expression following stimulation with heat inactivated E. coli and M. luteus, but R. peacockii infection alone did not induce upregulation or modify expression following stimulation. DAE15 cells expressed defensin, but expression was not upregulated by stimulation or infection. These results indicate cultured tick cell lines respond to stimulation by E. coli and M. luteus, but not R. peacockii, with upregulated humoral immune responses. Thus, our results suggest rickettsial endosymbionts avoid activating the humoral immune responses of their tick host.
Materials and Methods
Cell lines and culture conditions
The I. scapularis lines IDE12 (19) and ISE6 (20), and the D. andersoni line DAE15 (17) were grown in 25 cm2 CellStar tissue culture flasks (Greiner Bio-One Inc, Longwood, FL) containing 5 mL L15B (21) supplemented with 5% fetal bovine serum, 5% tryptose phosphate broth (Becton Dickinson, Franklin Lakes, NJ), and 0.1% cholesterol concentrate (MP Biomedicals, Irvine, CA). New cultures were established by 1:5 dilutions (1 part cell-containing medium to 4 parts fresh medium) of confluent cultures into fresh flasks. Cultures were maintained at 34°C and fed fresh medium weekly. IDE cell lines have been shown to be chronically infected with the St. Croix River virus (Reoviridae: Orbivirus) (22). However, this does not appear to impair their phagocytic ability, since the ISE6 line from the same tick species, but which does not carry that virus (H. Attoui, pers. comm.), shows much less phagocytic activity than IDE lines (18).
Cultures were infected with host-cell free preparations of R. peacockii (Rp) made by filtering Rp-containing medium from heavily infected ISE6 cultures through a 5.0 μm syringe filter (Millex-SV) (Millipore, Bedford, MA). Isolated rickettsiae were added to uninfected cultures one week after subculture with an estimated multiplicity of infection of 100 rickettsiae per cell. The infection status of cultures was monitored by comparing the proportion of infected cells to uninfected cells in Giemsa-stained Cytospin preparations (Shandon, Pittsburg, PA).
E. coli (D21) and M. luteus were grown in LB medium (MP Biomedicals, Irvine, CA) overnight in a shaking water bath at 37°C. Stock cultures of E. coli and M. luteus were collected by centrifugation, the pellets were washed with Hanks’ balanced saline solution (HBBS) (Invitrogen, Carlsbad, CA) and resuspended in 10 mL of PBS. Bacteria were inactivated by autoclaving cultures for 20 minutes at 121°C. Autoclaved bacteria were washed once more as described above and resuspended in HBSS (pH 6.8), counted using a Petroff-Hauser counting chamber, and the density adjusted to 1×109/mL. Bacterial stocks were stored at 4°C.
Bacterial challenge and harvest of cells
Cultures were seeded into Falcon® 12.5 cm2 flasks (Becton-Dickinson) at a density of approximately 5×105 cells per mL and maintained at 32°C in 2.5 mL of medium. One week later, half of the cultures were infected with R. peacockii as described above. Uninfected and infected cultures were grown for one week and the infection status assessed. A 2 × 2 factorial design with three replicates per treatment was used to determine the effects of bacterial stimulation and R. peacockii on expression of antimicrobial peptides. Cultures were divided into four groups: unstimulated and uninfected cultures (control), unstimulated and infected cultures (Rp infected), uninfected and bacteria stimulated cultures (stimulated), and R. peacockii infected and bacteria stimulated cultures (Rp infected and stimulated). Cultures were fed with 2.0 mL of fresh medium the day prior to stimulation. Cells were counted by comparing the number of cells per five randomly chosen microscopic fields per flask to ensure each flask contained similar numbers of cells. Cultures were inoculated with 10 E. coli and 10 M. luteus per cell for a total of 20 bacteria per cell and incubated 48 hours at 32°C.
Antimicrobial factors present in spent culture medium and cell lysate were measured by radial diffusion assays, flow cytometry, and quantitative real-time PCR (qRT-PCR). Spent medium was removed 48 hours post stimulation and transferred to microfuge tubes. Medium samples were centrifuged at 21,000 × g for 20 minutes at 4°C, and filtered through 0.22 μm syringe filter prior to concentration using YM-3 Centricon centrifugal filters (Millipore, Billerica, MA). Samples were centrifuged at 4°C for 110 minutes at 4890 × g in a refrigerated centrifuge (Hettich, Tuttlingen, Germany), concentrating them from 1.2 mL to approximately 200 μL. Concentrated medium samples were stored at -20°C. Remaining cell layers were washed with PBS and resuspended in 3 mL of PBS with a blunt-tip, 14 g cannula attached to a 5 mL syringe. Aliquots containing 0.25 mL of cells, equivalent to 3.5×105 cells, were removed from each culture. The 0.25 mL aliquots were pooled within replicates for a total volume of 0.75 mL per treatment, centrifuged at 300 × g for 8 minutes and the supernatant was removed. Cells were lysed by vortexing in 1 mL of sterile water, and centrifuged at 21,000 × g for 20 minutes at 4°C to pellet insoluble cell fragments. Clarified lysate was stored at -20°C until concentrated as above, and the concentrated lysate was stored at -20°C. A 1 mL aliquot of cells in PBS was removed from each flask, centrifuged at 300 × g for 8 minutes, and the PBS was removed. The cells were resuspended in 50 μL of RNAlater (Ambion, Austin, TX) and frozen at -70°C. The remaining volume of cells in PBS (approximately 1.75 mL) was kept on ice until fixed, permeablized and stained for assay by flow cytometry.
Assays for antimicrobial activity in cells and culture medium
Radial diffusion assays
An overnight culture of M. luteus was measured at 600 nm and diluted to an optical density of 0.01 in 0.8% LB-agarose equilibrated at 45°C. Plates were prepared by pouring 15 mL of bacteria-containing LB-agarose into 150 mm Petri dishes. Wells were punched in the solidified agarose with the sterilized tip of a 14 g cannula attached to a 5 mL syringe, and 4 μL of concentrated 48 hr medium or cell lysate was added to each well. Plates were incubated overnight at 37°C and examined the following day. Wells surrounded by circular zones free of bacterial colonies were considered to be positive for antimicrobial factors. Images of the plates were taken using a Cohu CCD camera (Cohu Inc., San Diego, CA) and the diameters of the bacteria-free zones measured with Photoshop (Adobe Systems Inc., San Jose, CA). The diameter of bacteria-free zones was determined by comparing the diameter of the M. luteus-free zone to the diameter of the cannula-punched well (2 mm). Data from media samples are expressed as the mean diameter of zones from three replicate cultures ± standard deviation.
Nonreducing gel electrophoresis with M. luteus overlay
Nonreducing gel electrophoresis using a M. luteus overlay was adapted from protocols described by Cytrynska et al (23). Briefly, lysate from 1×105 uninfected cells stimulated with bacteria, was prepared as described above and concentrated in a Savant Speed Vac (Thermo Electron Corporation, Waltham, MA) to 10 μL volume. Samples were mixed with 3 μL of 5X nonreducing loading buffer (50% glycerol, 31% 1M Tris-HCL [pH 6.8], 14% deionized water, and 5% of 1% bromophenol blue) and run on a 4-20% tris-tricine precast polyacrylamide gel (ISC BioExpress, Kayesville, UT) that had been run for 15 minutes at 150 V prior to addition of samples. Prestained molecular weight markers were omitted because they had deleterious effects on M. luteus growth. Electrophoresis running buffer included (w/v) 0.61% tris, 0.9% tricine, and 0.05% SDS. Lysates were loaded onto the gel and electrophoresed until the dye front had reached the bottom of the gel. The gel was washed for 30 minutes in 150 mL of 0.1% Triton-X-100 to remove the sodium dodecyl sulphate from the gel, proteins were refolded with one 30 minute incubation in 50 mM Tris-HCl (pH 7.0) at room temperature, and incubated in LB medium for 30 minutes. Gels were placed in a 150 mm Petri dish and overlayed with a thin layer of M. luteus-containing LB agarose (previously described) and incubated overnight at 37°C. Areas containing antimicrobial activity in the gel were indicated by zones without M. luteus growth.
Western blotting
Aliquots containing 1×106 cells were resuspended in 15 μL of 2X protein extraction buffer (50% deionized water, 20% of 10% SDS solution, 12% of 0.5M Tris HCl [pH 6.8], 10% glycerol, 4% B-mercaptoethanol, and 4% of a 1% bromophenol solution), lysed by vortexing, and solubilized by heating in boiling water for 5 minutes. Insoluble material was removed by centrifugation at 21,000 × g for 15 minutes at 4°C. Sample volumes equivalent to 1×105 cells were loaded into 4-20% tris-tricine precast gels (ISC BioExpress) and run at room temperature until the dye front reached the bottom of the gel. Proteins were transferred to Immobilon-P membranes (Millipore) and blocked with 3% powdered milk-PBS overnight at 4°C. A rabbit anti-lysozyme antibody (Lab Vision, Fremont, CA) was chosen for western blotting because it cross reacts with lysozyme from a broad range of vertebrate taxa, as well as chicken egg lysozyme (Fisher Scientific, Waltham, MA), and the lysozyme standard in the SeeBlue Plus2 prestained molecular weight marker (Invitrogen, Carlsbad, CA) (not shown). Membranes were reacted with antibodies diluted 1:1000 in 3% bovine serum albumin PBS (BSA-PBS) followed by several washes with PBS. Membranes were incubated with horseradish peroxidase-conjugated goat anti-rabbit antibody (Kirkegaard & Perry Laboratories, Gaithersburg, MD) diluted 1:1000 in BSA-PBS and developed using the 3C Peroxidase Detection Kit (Kirkegaard & Perry Laboratories). Images of membranes were acquired on a flat bed scanner (UMax, Dallas, TX) at 300 dpi.
Flow cytometric detection of intracellular lysozyme
Cells were harvested as described above, resuspended in PBS, and transferred to microfuge tubes for fixation using the Fix and Perm Kit (Invitrogen) with methanol modification. Briefly, 50 μL of fixative was added to each tube, followed by 300 μL of ice cold absolute methanol, and the cells were incubated on ice for 15 minutes. The fixed cells were pelleted, the fixative was removed, and the cells were washed once with 1.5 mL of PBS. Cells were treated with 50 μL of permeabilization reagent containing primary antibody against lysozyme (diluted 1:50 in permeabilization buffer). Cells were incubated with antibody for 30 minutes at 37°C, washed once with 1.5 mL PBS, and stained for 30 minutes at 37°C with a 1:50 dilution of goat anti-rabbit FITC-conjugated secondary antibody (Pierce, Rockford, IL) in permeablization buffer. The cells were washed once with 1.5 mL of PBS and the pellets were resuspended in 1 mL of PBS for analysis. Samples were run on a FACSCalibur flow cytometer (Becton Dickinson). Forward scatter and side scatter were used to gate on the main cell cloud and exclude debris and aggregated cells. The fluorescence of 10,000 events in the Fl1 channel was recorded and data were analyzed in CellQuest Pro (Becton Dickinson). Cytospin preparations were made of the remaining cells. Cell spots were sealed under glass cover slips using VectaShield Mounting Medium with 4’,6-diamidino-2-phenylindole (DAPI) (Vector Labs, Burligame, CA) to limit photobleaching and to counterstain the cell nuclei. Cells were examined on an Eclipse 400 fluorescence microscope (Nikon, Melville, NY) to visualize staining patterns. Images were taken using a DXM-1200 digital camera (Nikon) at 100× using separate filters for FITC and DAPI.
RNA isolation and DNase treatment
The cells stored at -70°C in RNAlater were diluted 1:5 with PBS, centrifuged for 10 min at 400 × g, and the supernatant was removed. Total RNA was isolated using the RNeasy RNA Isolation Kit (Qiagen, Valencia, CA) following kit protocols. RNA was eluted from the column with 35 μL of RNase-free water and quantified by UV spectrophotometry with a BioPhotometer (Eppendorf AG, Hamburg, Germany). DNase treatment was done using the DNAfree DNase treatment kit (Ambion) following kit protocols. DNase-treated RNA was diluted to 15 ng/μL in DNAse and RNase-free water and stored at -70°C.
Sequencing of the defensin from the DAE15 cell line
Primers for reverse-transcription PCR (RT-PCR) were designed with the PrimerQuest application on the Integrated DNA Technology website (http://scitools.idtdna.com/Primerquest/) (Table 2). RT-PCR was done with DvDef fw and DvDef Rev primers developed from the sequence of varisin (Genebank accession AY181027), a defensin from D. variabilis to amplify a 543 bp sequence of the D. andersoni defensin. Amplification was done using DNase-treated RNA as template according to Access RT-PCR kit protocol (Promega, Madison, WI). Reactions were made to 25 μL using 2.5 U AMV reverse transcriptase, 2.5 U Tfl polymerase, 5X buffer, 0.2 mM dNTP mix, 1.5 mM MgSO4, 0.25 μM each primer and 30 ng total RNA template. Reactions were run on a Robocycler PCR Cycler (Stratagene, La Jolla, CA) with a heated top using the following program: 45 min at 48°C (reverse transcription), 5 min at 94°C (denaturation), 35 cycles of 94°C (30 s), 50°C (45 s), 72°C (1 min) and a final extension at 72°C for 5 minutes. Products were run on a 1.5% agarose gel stained with ethidium bromide, and a band of the predicted size excised and purified using a MinElute Gel Extraction Kit (Qiagen). Eluted products were cloned using the TOPO-TA Cloning Kit for Sequencing PCR Products (Invitrogen) and sequenced in the forward and reverse directions on an ABI 3700 automated sequencer (BioMedical Genomics Center, University of Minnesota). The 5’ and 3’ ends of the defensin cDNA were determined using the GeneRacer Kit (Invitrogen) according to kit protocols. The sequences of GeneRacer kit supplied primers are available at www.invitrogen.com/content/sfs/manuals/3prime_race_man.pdf. The 5’ end was determined by nested PCR reactions on GeneRacer (GR) ligated cDNA using the GR forward (fw) primer and var157 reverse (rev) primer followed by a second reaction using the nested GR fw primer and DaDefInt Rev primer to amplify a 190 bp fragment. The 3’ sequence was determined by performing nested PCR on GR 3’ oligonucleotide-ligated cDNA using the DaDefInt fw primer in conjuction with the GR 3’ rev primer followed by a nested reaction using var157 fw and nested GR rev primers. The 5’ and 3’ ends were cloned using the supplied Zero Blunt TOPO Cloning kit and sequenced at least six times. The 543 bp product produced by the DvDef fw and DvDef rev pimer set was cloned and sequenced three times in the forward and reverse directions. Sequences were aligned and the consensus sequence, and deduced amino acid sequences generated using the BioEdit software package (24). Protein identity and similarity to other tick defensins was determined by BLAST (25), and aligned with BioEdit.
Table 2.
Primers used for sequencing and qRT-PCR.
| Primer name | Primer (5’ - 3’)a | Product
size (bp) |
|---|---|---|
| Var157 fw | TCT GGC ATC ATC AAG CAG AC | 157 |
| Var157 rev | CTG CAA GTA TTC CGG GGT TA | |
| DvDef fw | TGA GAC GAC AAA GAA GGT CGG CCA | 543 |
| DvDef rev | AAC ATG GAG TCG CAG TAG CGT TGT | |
| DaDefInt fw | CTT GTT TGC GGT CTT GTA TC | --- |
| DaDefInt Rev | TCG ACG AAC ACG GAG GTG AG | --- |
| DaDef rev2 | GGC AGA TGC AAA GTC CAC GCA TTT | --- |
| DaDef_85RL | CGC ACC AAG GCA GAT GCA AAG TC[JOE] G | 55 |
| DaDef_85RL/74FU | TTT GGG ACT GAG TGA AAC TTG C | |
| IscapAct_25FL | CGA ACG ATG ACC CAG ATC ATG TTC[FAM] G | 87 |
| IscapAct_25FL/67RU | GGA GGC GTA CAG GGA GAG CAC | |
| IsDef_101FL | CGA TCA TCA TTG CTG TTA CCT TGA TC[JOE] G | 70 |
| IsDef_101FL/124RU | CTC TTG TGC GCT GGA AGT CAT A |
The fluorescent dye on the fluorophore-labeled LUX primer, either carboxyfluorescein (FAM) or 6-carboxy-4’, 5’-dichloro-2’, 7’-dimethoxyfluorescein (JOE), is listed inside brackets.
Quantitative reverse transcription PCR
LUX primers (Invitrogen) for quantitative RT-PCR (qRT-PCR) were generated using the D-LUX designer software (https://orf.invitrogen.com/lux/) from the cDNA sequences of the D. andersoni defensin (this report), I. scapularis defensin (AY660970) and I. scapularis β-actin (AF426178) as template sequences. Primers were tested using standard RT-PCR to find primer sets that worked in multiplexed applications to simultaneously detect defensin and β-actin cDNA (internal control). Products were resolved on 3% NuSieve GTG agarose gels (BMA, Rockland, MD) stained with SYBR green (Cambrex Bioscience, Rockland, ME). Primer sets producing extra amplicons, or that yielded an amplicon intensity in multiplexed reactions which was different from individual primer-pair reactions, were not used. The primers selected for the I. scapularis defensin and β-actin were located within the protein-encoding open reading frame (ORF) of the corresponding gene. The fluorophore-labeled primer for the D. andersoni defensin was located within the 5’ end of the protein-encoding ORF and the unlabeled primer was located in the non-coding cDNA sequence preceding the protein-coding ORF.
A master mix was prepared using the One-Step Supermix kit with ROX (Invitrogen) as described by the kit protocol and included 5 μM each primer (unlabeled and fluorophore-labeled defensin and β-actin primers), 2× buffer, polymerase cocktail and DNase-treated RNA template for a final reaction volume of 20 μL. Reactions for DAE15 defensin used 30 ng of total RNA template whereas IDE12 reactions used 60 ng of total RNA template. Duplicate reactions were prepared using Taq polymerase (Promega) alone as a no reverse-transcriptase control to ensure template RNA was not contaminated with DNA and only the target was being measured. Plates were run on an ABI 9700HT real time PCR cycler (Applied Biosystems, Inc., Foster City, CA) using the following program: a reverse transcription step of 15 min at 50°C, 2 min at 94°C, and 50 cycles of 94°C (30s denaturation), 60°C (30s hybridization and extension). Melting curve analysis followed amplification cycles to ensure a single product was amplified for each primer set and to confirm the absence of primer dimers. Data were analyzed using the Sequence Detection System 2.1 software package (Applied Biosystems) and expressed as the fold change in relative transcript abundance from the untreated control using the method described by Livak and Schmittgen (26).
Statistical analysis
Statistical analysis was performed with SigmaStat (SysStat Software, Inc., Point Richmond, CA). Comparisons of two means were done using the Student t-test. Comparisons of multiple treatments against uninfected, unstimulated controls were done using one way analysis of variance (one way ANOVA). Data with p < 0.05 were considered statistically significant.
Results
Infection of IDE12 and DAE15 cells with R. peacockii
Preliminary experiments were performed to determine culture parameters suitable for detecting and measuring humoral immune responses due to R. peacockii infection or bacterial stimulation. Expression of lysozyme and defensin by confluent or overgrown DAE15 cell layers was less uniform than that observed for less dense cultures (not shown). In addition, IDE12 cells were found to lift off the culture flask when grown past confluency. Consequently, we challenged cultures that contained approximately 1×106 cells per mL and were 80-90% confluent.
One week post R. peacockii inoculation, at least 60% of IDE12 cells and 80% of DAE15 cells were infected when stimulated with the E. coli-M. luteus bacterial suspension. IDE12 cells contained fewer rickettisae per cell than DAE15 cells. Cells infected with R. peacockii did not show of loss of adhesion or visible cytopathic effects. Cell-free R. peacockii were present in the medium of both lines at the time of inoculation with the E. coli-M. luteus bacterial suspension.
Partial characterization of intracellular and secreted antibacterial proteins
We established four treatments to examine expression of bacteriolytic factors: control cultures of uninfected, unstimulated cells, R. peacockii infected cultures to test for the rickettsial effect on constitutive expression, R. peacockii infected cultures stimulated with heat killed E. coli and M. luteus to determine whether intracellular bacteria modulated the response to extracellular bacteria, and uninfected, bacteria stimulated cultures to examine the response of the cell lines to bacteria in the absence of rickettsiae. Nonreducing gel electrophoresis and gel overlays containing live M. luteus enabled us to detect intracellular antimicrobial peptides expressed by IDE12 and DAE15 cells. Lysates from IDE12 and DAE15 cells produced large bacteria-free zones (Fig. 1A). The large bacteria-free area was taken as evidence for the presence of lysozyme-like peptides (see below). The DAE15 lysate contained an additional faintly visible band with higher electrophoretic mobility than the main zone of inhibition (Fig. 1A, arrow). A 4.2 kDa defensin has been identified in D. variabilis (3, 27) and this second zone may be caused by a defensin-like protein in DAE15 cells. No similar zone was detected in the IDE12 lysate.
Figure 1. Lysate from IDE12 and DAE15 cells contains antimicrobial activity and lysozyme-like immunoreactivity.

A. Lysate from IDE12 and DAE15 cells were electrophoresed under nonreducing conditions on a polyacrylamide gel with M. luteus overlay. Proteins with antibacterial activity produced bacteria-free zones in both lines that can be seen as clear zones in the bacterial lawn. The image contrast has been inverted to enhance the visibility of the small area of bacteriolytic activity located in the DAE15 lane (arrow). B. Western blotting of IDE12 and DAE15 lysate using polyclonal anti-lysozyme antibodies shows an immunoreactive protein at 14-16 kDa. Images are from independent, representative experiments. Lane 1. Coomassie Blue stained SeeBlue Plus2 molecular weight marker in polyacrylamide gel. Lane 2. Chicken egg lysozyme. Lane 3. IDE12 cell lysate. Lane 4. DAE15 cell lysate.
A 14-16 kDa factor with immunologic identity to lysozyme was identified in both cell lines by Western blotting (Fig. 1B). This factor comigrated with the lysozyme molecular weight marker (not shown) and with recombinant hen egg lysozyme (Fig. 1B). The electrophoretic mobility of the lysozyme in Western blots was similar to the patterns seen in nonreducing gel electrophoresis, suggesting the large zones may have been caused by a bacteriolytic lysozyme-like protein.
Antimicrobial activity in uninfected and R. peacockii infected tick cells
Radial diffusion assays were used to examine expression of soluble antimicrobial factors following bacterial stimulation or R. peacockii infection and whether these factors were secreted or retained. No zones of inhibition were produced by concentrated culture medium controls or deionized water (not shown). Zones of inhibition were produced by concentrated 48 hour medium and lysate from both IDE12 and DAE15 cells (Table 1). Differences were observed, however, in the diameters of bacteria-free zones and whether the majority of antimicrobial factors were present in the culture medium or cell lysate. Small zones of equivalent diameters were produced by uninfected and R. peacockii infected IDE12 cultures, and significantly larger zones were produced by uninfected and R. peacockii infected cultures stimulated with heat-killed bacteria (one-way ANOVA, F=17.04, p < 0.001) (Fig. 2). A detectable bacteria-free zone was produced only by lysate from stimulated IDE12 cells (Table 1). Concentrated culture medium and cell lysate from DAE15 cells also produced bacteria-free zones. Culture medium produced small zones with diameters that showed significant decreases in diameter when uninfected and R. peacockii infected cells were stimulated with bacteria (t-test, t = 4.480, dof=4, P = 0.011 and t = 4.832, dof = 4, P = 0.008, respectively). Statistically significant differences in zone diameter were not observed between uninfected and R. peacockii infected cells in the absence of heat killed E. coli and M. luteus (Fig. 2). Lysate from DAE15 cells produced zones of equivalent diameters that did not vary significantly by treatment.
Table 1.
Zone diameters and ratio of concentrated lysate and media from IDE12 and DAE15 cultures.
| IDE12
|
DAE15
|
|||||
|---|---|---|---|---|---|---|
| Sample | lysatea,b | mediuma,c | ratio | lysatea,b | mediuma,c | ratio |
| Control | 3.15 | 6.57 ± 0.95 | 0.48 | 8.49 | 6.38 ± 0.29 | 1.33 |
| Rp infected | 3.15 | 7.77 ± 1.35 | 0.41 | 7.65 | 4.49 ± 2.00 | 1.70 |
| Rp infected, stimulated | 3.09 | 11.18 ± 1.24 | 0.28 | 8.02 | 5.44 ± 0.18 | 1.47 |
| Stimulated | 6.83 | 12.37 ± 1.02 | 0.55 | 7.37 | 4.97 ± 0.47 | 1.48 |
Zone diameters are given in millimeters.
Lysate zones were produced by lysate from three replicates that had been pooled.
Media-produced zones diameters include the standard deviation of three replicates.
Figure 2. Antibacterial activity of secreted antimicrobial factors from IDE12 and DAE15 cultures.
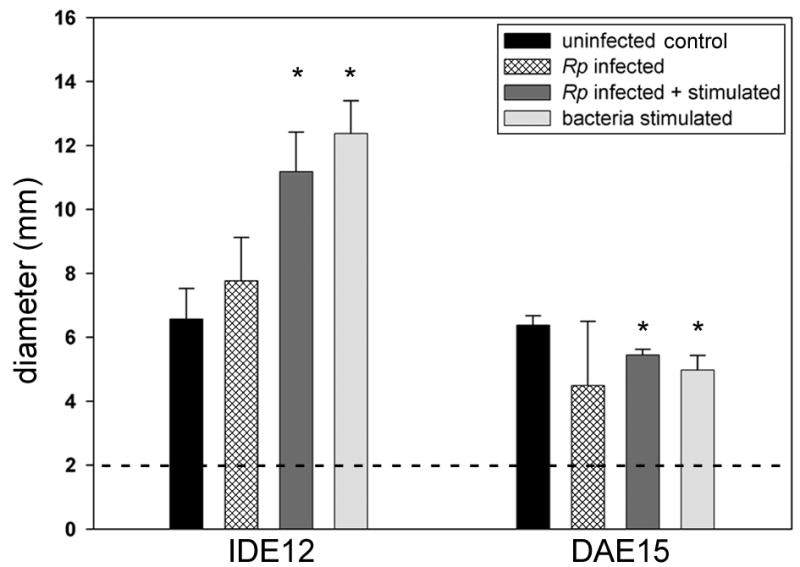
Samples of concentrated medium from each line were loaded into the same dish to minimize inter-plate variation. The diameter of the cannula (2 mm) is indicated by the dotted line. The diameter of the inhibited M. luteus growth was determined from three replicate cultures and expressed as the mean diameter ± standard deviation. Treatments showing statistically significant differences from uninfected controls are indicated with asterisks.
Lysate:medium ratios were calculated by comparing the diameters of the zone produced by lysate (including the well diameter) with the diameters of wells produced by medium to determine whether cells secreted or retained the majority of their antimicrobial factors. We found IDE12 cells secreted most of their antimicrobial factors while most of the antimicrobial factors produced by DAE15 cells were retained intracellularly (Table 1). R. peacockii infection did not change where the majority of the antibacterial activity was located.
Relative measurement of intracellular lysozyme by flow cytometry
Changes in intracellular lysozyme content in cells stained with an anti-lysozyme antibody, and then labeled with a FITC-conjugated secondary antibody, were measured by flow cytometry. Increase in cellular fluorescence relative to uninfected and unstimulated control cultures were taken an indicator of upregulated lysozyme expression. The fluorescence profiles showed uninfected and R. peacockii-infected IDE12 cells stimulated with bacteria contained significantly more lysozyme than unstimulated controls (F = 18.220, p <0.001), but there was no significant difference between uninfected or R. peacockii infected cells in either line (Fig. 3). There was significantly more lysozyme in uninfected and R. peacockii infected DAE15 cells after bacterial stimulation than was present in unstimulated R. peacockii infected cells (F = 4.789, p = 0.034). Cells stained for flow cytometry and viewed by microscope had small cytoplasmic granules that stained with anti-lysozyme antibodies, and these granules increased in size and number when the cells were stimulated with heat-killed bacteria (not shown).
Figure 3. Intracellular lysozyme content in IDE12 and DAE15 cells.
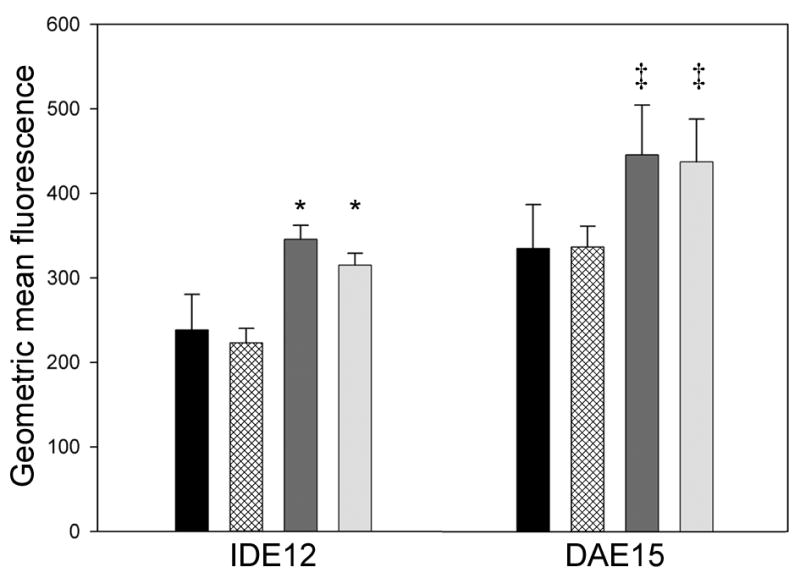
Bars represent the geometric mean fluorescence (arbitrary units) of cells from three replicate cultures ± standard deviation. Treatments are indicated as in the legend of Fig. 2. Treatments showing statistically significant differences from the uninfected IDE12 controls are indicated by asterisks. Statistically significant differences were not observed between uninfected and R. peacockii infected cells in either line in the absence of bacterial stimulation. Double daggers (‡) indicate statistically significant differences between unstimulated, R. peacockii infected DAE15 cells and R. peacockii-infected and uninfected DAE15 cells stimulated with bacteria.
Sequencing of the D. andersoni defensin and qRT-PCR measurement of expression
A 629 bp defensin-like transcript with similarity to varisin, a defensin from D. variabilis (3), was identified in the DAE15 line. A cDNA with 96% identity at the nucleotide level, containing an open reading frame encoding a 74 amino acid preprodefensin with 100% identity to the D. variabilis defensin, was sequenced (Fig. 4).
Figure 4. Alignment of the translated amino acid sequence of the D. andersoni defensin with other tick defensins.

The defensin from D. andersoni was aligned against the sequences of D. variabilis preprodefensin (AAO24323), Haemaphysalis longicornis defensin (BAD93183), Boophilus microplus defensin (AAO48943), I. scapularis preprodefensin (AAV74387), and the Ornithodoros moubata defensin isoforms D (BAC22073), B (BAB41027), C (BAC22074), and A (BAB41028). GenBank accession numbers are indicated in parentheses. Similar residues are indicated with grey shading and identical residues indicated with black shading. The positions of conserved cysteine residues are indicated by asterisks.
We used qRT-PCR to examine expression of defensins in IDE12 and DAE15 cells using fluorophore-labeled primers based on the defensin sequence from the DAE15 line, and an I. scapularis defensin (5). Both lines constitutively expressed defensins. When related to the quantity of total RNA used for qRT-PCR, twice the amount of IDE12 RNA was used to attain Ct values similar to those for DAE15 defensin, indicating DAE15 cells contained more defensin transcript than IDE12 cells. We did not observe significant differences in defensin transcription between unstimulated and R. peacockii-infected IDE12 cultures (t = 1.815, df = 4, P = 0.144) (Fig. 5). Bacterial stimulation significantly upregulated defensin transcription in R. peacockii-infected (t = 4.863, df = 4, P = 0.008) and uninfected IDE12 cultures (t = 8.193, df = 4, P < 0.001) by 8- and 12-fold, respectively, when compared to unstimulated IDE12 cultures (Fig. 5). Transcription of defensin in stimulated DAE15 cells was not uniform between replicate cultures, and was not significantly different between control, R. peacockii infected, and stimulated R. peacockii-infected cultures. Downregulation of defensin by stimulated DAE15 cells was statistically significant (t-test, t = 4.456, dof = 4, P = 0.011), but transcript abundance in stimulated R. peacockii infected cells was unchanged from controls, suggesting defensin downregulation is not caused by R. peacockii but is a response to E. coli and M. luteus that is specific to this cell line.
Figure 5. Defensin transcript abundance in IDE12 cells increases following bacterial stimulation.
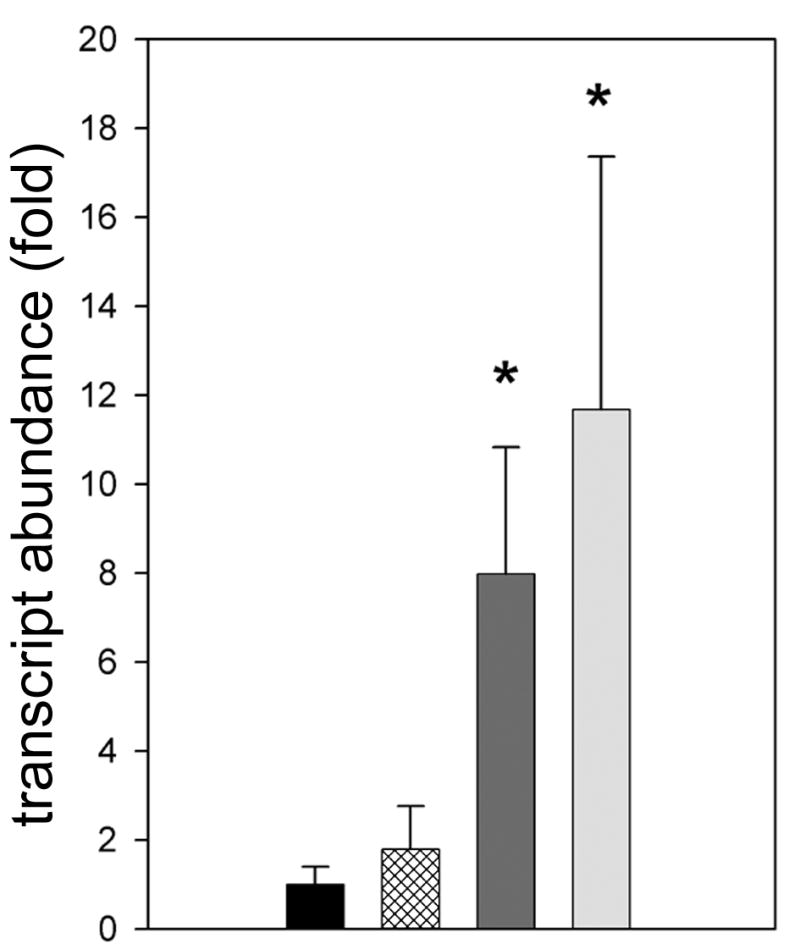
Bars represent the fold change in defensin transcript relative to defensin transcript abundance in uninfected, unstimulated controls ± the standard deviation. Treatments are indicated as in the legend of Fig. 2. Statistically significant differences from uninfected control cultures are indicated with asterisks.
Discussion
The effects of rickettsia-like endosymbionts on tick immune responses to bacteria are unknown. We used tick cell culture to examine whether two tick cell lines that can phagocytose B. burgdorferi (18) also produce soluble antibacterial factors. Additionally, we investigated whether infection by R. peacockii, an endosymbiont of D. andersoni, elicited an immune response, or modified responses to heat inactivated E. coli and M. luteus. We found the IDE12 and DAE15 cell lines constitutively expressed antibacterial factors including a protein with immunologic identity to lysozyme as well as defensin-like transcripts. IDE12 cells secreted most of their antibacterial factors whereas DAE15 cells retained most of their antibacterial factors. Both lines upregulated expression of lysozyme when stimulated, but expression was unaffected by rickettsial infection. Transcripts of defensin-like genes were found in both cell lines and bacterial stimulation upregulated expression of defensin by uninfected and R. peacockii infected IDE12 cultures. In comparison, defensin expression by bacteria-stimulated DAE15 cultures was downregulated slightly, while transcript abundance in unstimulated cultures infected with R. peacockii, and R. peacockii infected cultures stimulated with E. coli and M. luteus was unchanged from uninfected controls. While we focused on two factors, lysozyme and defensin, that are known to have immune functions in ticks, we expect other less characterized antimicrobial factors may be present and may have been be important in the immune response. Moreover, the tissues represented by cells in tick cell lines are unknown and tick cell lines may not generate the global immune response to rickettsial infection in the same way as in immunocompetent tick tissues. Given this caveat, we view these cell lines as models of generalized humoral responses, without specific focus on immunocompetent tick cells such as hemocytes or midgut cells.
There are few reports examining the effects of endosymbiotic bacteria on host immune responses despite their prevalence in arthropods. Bourtzis et al (28) found that Wolbachia, an endosymbiotic bacterium that infects a wide range of arthropods, did not appear to induce or suppress defensin expression in Drosophila simulans or Aedes albopictus. It is not known how Wolbachia avoids eliciting immune responses, but living inside cells may be an important adaptation that hides Wolbachia from arthropod immune systems (29). In nature, R. peacockii inhabits the ovaries of female D. andersoni and is rarely found in hemolymph (16). The ovaries of D. variablis, a tick closely related to D. andersoni, contain little lysozyme transcript relative to the abundant lysozyme transcript found in D. variablis hemocytes (9), suggesting R. peacockii inhabits lysozyme-poor tissues as part of a strategy of avoiding contact with more immunoresponsive tick tissues. Furthermore, the presence of a disrupted rickA gene (30), involved with rickettsial motility, would restrict the rickettsiae to the ovarian tissues and sequestered away from the immune responses found in the gut and hemocytes.
Although R. peacockii is an obligate intracellular bacterium, rickettsiae are exocytosed by cells (17), and cell-free rickettsiae were present in the medium of infected IDE12 and DAE15 cell cultures. These cell-free rickettsiae did not induce upregulation of lysozyme in IDE12 or DAE15 cells, or defensin in IDE12 cells. Furthermore, the lack of responses by the I. scapularis line IDE12 against an endosymbiont from a phylogenetically distant tick, suggests coevolution between the tick host and endosymbiont is not responsible for this lack of activity against endosymbionts. This suggests an uncharacterized aspect of R. peacockii’s biology enables it to go unnoticed by immunoresponsive tick cells, thus not stimulating an immune response.
Bacterial pathogens can limit immune responses in host cells by interfering with intracellular signaling cascades that induce transcription and synthesis of antimicrobial peptides (31). It did not appear that R. peacockii constrained lysozyme or defensin expression by infected tick cells, as infection did not influence expression by infected but otherwise unstimulated cells or decrease the responsiveness of IDE12 or DAE15 cells to bacterial stimulation. Similarly, Spiroplasma infecting D. melanogaster did not upregulate expression of antimicrobial peptides, or prevent upregulation of antimicrobial peptides after stimulation by inactivated E. coli and spores of the entomopathogenic fungus Beauveria bassiana (29). The tick-endosymbiont relationship may be synonymous to the Drosophila response to Spiroplasma. Exocytosed R. peacockii were present in the culture medium without inducing upregulation of defensin or lysozyme from host cells, but not preventing host cells from responding to the stimuli presented by E. coli and M. luteus. R. peacockii is less susceptible to killing by antimicrobial peptides than are some free-living bacteria (13), and may rely on this intrinsic property to protect themselves against host cell immune responses rather than interfering with host cell responses.
Rickettsial mechanisms of immune evasion and microbial recognition systems used by ticks are uncharacterized. Eukaryotic cells recognize extracellular and intracellular bacteria by detecting highly conserved surface antigens, such as peptidoglycan, with pattern recognition receptors (PRRs) (32, 33). The lack of responses against R. peacockii may indicate rickettsial surfaces lack molecular determinants present on the surfaces of E. coli and M. luteus, as was likely to be the case with the Spiroplasma infecting Drosophila (29). Rickettsial surface antigens are poorly characterized, although rickettsial peptidoglycan resembles the peptidoglycan from other gram-negative bacteria (34), and would be predicted to stimulate immune responses. Alternatively, rickettsial avoidance of the arthropod immune system may rely on a coating of host proteins to prevent recognition by host PRRs. A similar behavior may occur in Wolbachia, where these bacteria closely associate with proteins from the host cell (35). Taken together, it appears that molecular mimicry may hide R. peacockii from detection by PRRs on the tick cells, leaving the host cells capable of mounting normal immune responses when stimulated by other bacterial challenges.
Tick-endosymbiont interactions are frequently overlooked aspects of tick biology. A better understanding of these interactions may clarify how bacteria pathogenic to vertebrates evade the tick immune system. The results presented here used tick cell culture to characterize an aspect of the tick response to rickettsial endosymbiosis.
Acknowledgments
Funding provided in part by grants from the National Institutes of Allergy and Infectious Diseases (5R01 AI049424 to U.G.M.) and the Minnesota Agricultural Experiment Station. This work was part of J. Mattila’s doctoral dissertation.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Johns R, Ohnishi J, Broadwater A, Sonenshine DE, De Silva AM, Hynes WL. Contrasts in tick innate immune responses to Borrelia burgdorferi challenge: immunotolerance in Ixodes scapularis versus immunocompetence in Dermacentor variabilis (Acari: Ixodidae) J Med Entomol. 2001;38:99–107. doi: 10.1603/0022-2585-38.1.99. [DOI] [PubMed] [Google Scholar]
- 2.Nakajima Y, van der Goes van Naters-Yasui A, Taylor D, Yamakawa M. Two isoforms of a member of the arthropod defensin family from the soft tick, Ornithodoros moubata (Acari: Argasidae) Insect Biochem Mol Biol. 2001;31:747–51. doi: 10.1016/s0965-1748(01)00066-2. [DOI] [PubMed] [Google Scholar]
- 3.Johns R, Sonenshine DE, Hynes WL. Identification of a defensin from the hemolymph of the American dog tick, Dermacentor variabilis. Insect Biochem Mol Biol. 2001;31:857–65. doi: 10.1016/s0965-1748(01)00031-5. [DOI] [PubMed] [Google Scholar]
- 4.Valenzuela JG, Francischetti IM, Pham VM, Garfield MK, Mather TN, Ribeiro JM. Exploring the sialome of the tick Ixodes scapularis. J Exp Biol. 2002;205:2843–64. doi: 10.1242/jeb.205.18.2843. [DOI] [PubMed] [Google Scholar]
- 5.Hynes WL, Ceraul SM, Todd SM, Seguin KC, Sonenshine DE. A defensin-like gene expressed in the black-legged tick, Ixodes scapularis. Med Vet Entomol. 2005;19:339–44. doi: 10.1111/j.1365-2915.2005.00579.x. [DOI] [PubMed] [Google Scholar]
- 6.Bachali S, Jager M, Hassanin A, Schoentgen F, Jolles P, Fiala-Medioni A, Deutsch JS. Phylogenetic analysis of invertebrate lysozymes and the evolution of lysozyme function. J Mol Evol. 2002;54:652–64. doi: 10.1007/s00239-001-0061-6. [DOI] [PubMed] [Google Scholar]
- 7.Kopacek P, Vogt R, Jindrak L, Weise C, Safarik I. Purification and characterization of the lysozyme from the gut of the soft tick Ornithodoros moubata. Insect Biochem Mol Biol. 1999;29:989–97. doi: 10.1016/s0965-1748(99)00075-2. [DOI] [PubMed] [Google Scholar]
- 8.Grunclova L, Fouquier H, Hypsa V, Kopacek P. Lysozyme from the gut of the soft tick Ornithodoros moubata: the sequence, phylogeny and post-feeding regulation. Dev Comp Immunol. 2003;27:651–60. doi: 10.1016/s0145-305x(03)00052-1. [DOI] [PubMed] [Google Scholar]
- 9.Simser JA, Macaluso KR, Mulenga A, Azad AF. Immune-responsive lysozymes from hemocytes of the American dog tick, Dermacentor variabilis and an embryonic cell line of the Rocky Mountain wood tick, D. andersoni. Insect Biochem Mol Biol. 2004;34:1235–46. doi: 10.1016/j.ibmb.2004.07.003. [DOI] [PubMed] [Google Scholar]
- 10.Kuhn KH, Haug T. Ultrastructural, cytochemical, and immunocytochemical characterization of haemocytes of the hard tick Ixodes ricinus (Acari; Chelicerata) Tissue Research. 1994:493–504. [Google Scholar]
- 11.Podboronov VM. In: Modern Acarology. Dusbabek F, Bukva V, editors. Vol. 2. Prague and SPB Academic Publishing bv; The Hague: 1991. pp. 375–380. [Google Scholar]
- 12.Hancock RE, Scott MG. The role of antimicrobial peptides in animal defenses. Proc Natl Acad Sci U S A. 2000;97:8856–61. doi: 10.1073/pnas.97.16.8856. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Baldridge GD, Kurtti TJ, Munderloh UG. Susceptibility of Rickettsia monacensis and Rickettsia peacockii to Cecropin A, Ceratotoxin A, and lysozyme. Curr Microbiol. 2005;51:233–8. doi: 10.1007/s00284-005-4532-7. [DOI] [PubMed] [Google Scholar]
- 14.Perlman SJ, Hunter MS, Zchori-Fein E. The emerging diversity of Rickettsia. Proceedings of the Royal Society B: Biological Sciences. 2006 doi: 10.1098/rspb.2006.3541. http://dx.doi.org/10.1098/rspb.2006.3541. [DOI] [PMC free article] [PubMed]
- 15.Noda H, Munderloh UG, Kurtti TJ. Endosymbionts of ticks and their relationship to Wolbachia spp. and tick-borne pathogens of humans and animals. Appl Environ Microbiol. 1997;63:3926–32. doi: 10.1128/aem.63.10.3926-3932.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Niebylski ML, Schrumpf ME, Burgdorfer W, Fischer ER, Gage KL, Schwan TG. Rickettsia peacockii sp. nov., a new species infecting wood ticks, Dermacentor andersoni, in western Montana. Int J Syst Bacteriol. 1997;47:446–52. doi: 10.1099/00207713-47-2-446. [DOI] [PubMed] [Google Scholar]
- 17.Kurtti TJ, Simser JA, Baldridge GD, Palmer AT, Munderloh UG. Factors influencing in vitro infectivity and growth of Rickettsia peacockii (Rickettsiales: Rickettsiaceae), an endosymbiont of the Rocky Mountain wood tick, Dermacentor andersoni (Acari, Ixodidae) J Invertebr Pathol. 2005;90:177–86. doi: 10.1016/j.jip.2005.09.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Mattila J, Munderloh UG, Kurtti TJ. Cellular basis of tick immunity to bacteria: Phagocytosis of Borrelia burgdorferi by Ixodes scapularis and Dermacentor andersoni cells infected with an endosymbiont, Rickettsia peacockii. doi: 10.1673/031.007.5801. submitted. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Munderloh UG, Liu Y, Wang M, Chen C, Kurtti TJ. Establishment, maintenance and description of cell lines from the tick Ixodes scapularis. J Parasitol. 1994;80:533–43. [PubMed] [Google Scholar]
- 20.Munderloh UG, Jauron SD, Fingerle V, Leitritz L, Hayes SF, Hautman JM, Nelson CM, Huberty BW, Kurtti TJ, Ahlstrand GG, Greig B, Mellencamp MA, Goodman JL. Invasion and intracellular development of the human granulocytic ehrlichiosis agent in tick cell culture. J Clin Microbiol. 1999;37:2518–24. doi: 10.1128/jcm.37.8.2518-2524.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Munderloh UG, Kurtti TJ. Formulation of medium for tick cell culture. Exp Appl Acarol. 1989;7:219–29. doi: 10.1007/BF01194061. [DOI] [PubMed] [Google Scholar]
- 22.Attoui H, Stirling JM, Munderloh UG, Billoir F, Brookes SM, Burroughs JN, de Micco P, Mertens PP, de Lamballerie X. Complete sequence characterization of the genome of the St Croix River virus, a new orbivirus isolated from cells of Ixodes scapularis. J Gen Virol. 2001;82:795–804. doi: 10.1099/0022-1317-82-4-795. [DOI] [PubMed] [Google Scholar]
- 23.Cytrynska M, Zdybicka-Barabas A, Jablonski P, Jakubowicz T. Detection of antibacterial polypeptide activity in situ after sodium dodecyl sulfate-polyacrylamide gel electrophoresis. Anal Biochem. 2001;299:274–6. doi: 10.1006/abio.2001.5422. [DOI] [PubMed] [Google Scholar]
- 24.Hall T. BioEdit: a user-friendly biological sequence alignment editor and analysis program for Windows 95/98/NT. Nucl Acids Symp Ser. 1999:95–98. [Google Scholar]
- 25.Altschul SF, Madden TL, Schaffer AA, Zhang J, Zhang Z, Miller W, Lipman DJ. Gapped BLAST and PSI-BLAST: a new generation of protein database search programs. Nucleic Acids Res. 1997;25:3389–402. doi: 10.1093/nar/25.17.3389. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Livak KJ, Schmittgen TD. Analysis of relative gene expression data using real-time quantitative PCR and the 2(-Delta Delta C(T)) Method. Methods. 2001;25:402–8. doi: 10.1006/meth.2001.1262. [DOI] [PubMed] [Google Scholar]
- 27.Sonenshine DE, Ceraul SM, Hynes WE, Macaluso KR, Azad AF. Expression of defensin-like peptides in tick hemolymph and midgut in response to challenge with Borrelia burgdorferi, Escherichia coli and Bacillus subtilis. Exp Appl Acarol. 2002;28:127–34. doi: 10.1023/a:1025354326877. [DOI] [PubMed] [Google Scholar]
- 28.Bourtzis K, Pettigrew MM, O’Neill SL. Wolbachia neither induces nor suppresses transcripts encoding antimicrobial peptides. Insect Mol Biol. 2000;9:635–9. doi: 10.1046/j.1365-2583.2000.00224.x. [DOI] [PubMed] [Google Scholar]
- 29.Hurst GD, Anbutsu H, Kutsukake M, Fukatsu T. Hidden from the host: Spiroplasma bacteria infecting Drosophila do not cause an immune response, but are suppressed by ectopic immune activation. Insect Mol Biol. 2003;12:93–7. doi: 10.1046/j.1365-2583.2003.00380.x. [DOI] [PubMed] [Google Scholar]
- 30.Simser JA, Rahman MS, Dreher-Lesnick SM, Azad AF. A novel and naturally occurring transposon, ISRpe1 in the Rickettsia peacockii genome disrupting the rickA gene involved in actin-based motility. Mol Microbiol. 2005;58:71–9. doi: 10.1111/j.1365-2958.2005.04806.x. [DOI] [PubMed] [Google Scholar]
- 31.Sjostedt A. Intracellular survival mechanisms of Francisella tularensis, a stealth pathogen. Microbes Infect. 2006;8:561–7. doi: 10.1016/j.micinf.2005.08.001. [DOI] [PubMed] [Google Scholar]
- 32.Ausubel FM. Are innate immune signaling pathways in plants and animals conserved? Nat Immunol. 2005;6:973–9. doi: 10.1038/ni1253. [DOI] [PubMed] [Google Scholar]
- 33.Akira S, Uematsu S, Takeuchi O. Pathogen recognition and innate immunity. Cell. 2006;124:783–801. doi: 10.1016/j.cell.2006.02.015. [DOI] [PubMed] [Google Scholar]
- 34.Pang H, Winkler HH. Analysis of the peptidoglycan of Rickettsia prowazekii. J Bacteriol. 1994;176:923–6. doi: 10.1128/jb.176.3.923-926.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Braig HR, Zhou W, Dobson SL, O’Neill SL. Cloning and characterization of a gene encoding the major surface protein of the bacterial endosymbiont Wolbachia pipientis. J Bacteriol. 1998;180:2373–8. doi: 10.1128/jb.180.9.2373-2378.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]