

Abstract
Thioredoxin reductases (Txnrd)1 maintain intracellular redox homeostasis in most organisms. Metazoans Txnrds also participate in signal transduction. Mouse embryos homozygous for a targeted null mutation of the txnrd1 gene, encoding the cytosolic thioredoxin reductase, were viable at embryonic day 8.5 (E8.5) but not at E9.5. Histology revealed that txnrd1−/− cells were capable of proliferation and differentiation; however, mutant embryos were smaller than wild-type littermates and failed to gastrulate. In situ marker gene analyses indicated primitive streak mesoderm did not form. Microarray analyses on E7.5 txnrd−/− and txnrd+/+ littermates showed similar mRNA levels for peroxiredoxins, glutathione reductases, mitochondrial Txnrd2, and most markers of cell proliferation. Conversely, mRNAs encoding sulfiredoxin, IGF-binding protein 1, carbonyl reductase 3, glutamate cysteine ligase, glutathione S-transferases, and metallothioneins were more abundant in mutants. Many gene expression responses mirrored those in thioredoxin reductase 1-null yeast; however mice exhibited a novel response within the peroxiredoxin catalytic cycle. Thus, whereas yeast induce peroxiredoxin mRNAs in response to thioredoxin reductase disruption, mice induced sulfiredoxin mRNA. In summary, Txnrd1 was required for correct patterning of the early embryo and progression to later development. Conserved responses to Txnrd1 disruption likely allowed proliferation and limited differentiation of the mutant embryo cells.
Keywords: Thioredoxin reductase, gastrulation, gene expression profiling, reductase compensation, peroxiredoxin cycle, sulfiredoxin
Introduction
Thioredoxin reductase (Txnrd) enzymes are flavin-containing NADPH-dependent oxidoreductases that restore oxidized thioredoxin (Txn) to the reduced dithiol state [1–3]. Txn was first discovered as the provider of electrons to ribonucleotide reductase [4] and has subsequently been implicated in numerous enzymatic and regulatory processes [1, 5–7]. In mice, disruption of both copies of either the txn1 gene, encoding the ubiquitous cytosolic Txn1, or the txn2 gene, encoding the mitochondrial Txn2, results in early embryonic lethality [8, 9]. However, yeast and bacteria lacking Txn are viable as long as they retain an intact glutathione pathway [10–12].
Txnrd enzymes have been found in representatives of all phyla [1, 13, 14]. In eukaryotes, the txnrd1 and txnrd2 genes encode cytosolic and mitochondrial Txnrd enzymes, respectively. In mammals, a third gene, txnrd3, encodes a testis-specific isoform [15].
Metazoan Txnrd enzymes generally contain the unusual amino acid selenocysteine (Sec) [16], which, together with the cysteine in the conserved C-terminal sequence Gly-Cys-Sec-Gly, participates in redox activity [17–19]. Exceptions, for example in Drosophila, where two Cys residues, rather than a Cys and a Sec, are found at the C-terminal redox center [18], demonstrate that the Sec residue is not essential. Indeed, recombinant human Txnrd1 in which the Sec is replaced with Cys, retains some reductase activity [20]. Conversely, recombinant truncated human Txnrd that simply ends at the residue preceding Sec has none [20].
In addition to its general roles, Txnrd1-dependent reduction reactions are important in specific cells or situations for limiting signal transduction from several cell surface receptors, including the T cell receptor, the insulin receptor, and the epidermal growth factor receptor [21–27]. Txn1 is also required for DNA-binding activity of the transcription factors NFκB, AP1, steroid hormone receptors, and p53 [1, 28–30]. In yeast exposed to oxidants, dithiol-disulfide exchange reactions between Txn, glutathione peroxidase, and the AP-1 family transcription factor YAP1, results in oxidative activation of YAP1 and induction of downstream genes [31].
To study the functions of mammalian Txnrd1 in vivo, we generated a line of mice bearing a conditional-null txnrd1 allele, converted this to a null allele, and measured the effects on embryonic development of homozygous null embryos. Results showed the mutation was not acutely cytotoxic, as evidenced by the formation of embryos composed of several thousand cells. However, embryonic patterning and gastrulation were disrupted. Microarrays revealed gene expression responses that were similar to those previously observed in txnrd1-null yeast [32], indicating that related compensatory pathways are induced in these two evolutionarily distant organisms. Our results suggest a role for Txnrd1 in allowing correct developmental patterning and indicate that general cell physiological processes are not precluded by disruption of Txnrd1.
Materials and Methods
Mouse lines2,3
Isolation of genomic clone, construction of targeting vector, and production of the mouse lines for this study are described in Supplemental section. The targeting strategy and allele structures are shown in Fig. 1a2. All renewable resources developed in this study, including phage, plasmids, and mouse lines, unless restricted by other parties, are freely available for unrestricted non-profit research use.
Fig. 1.
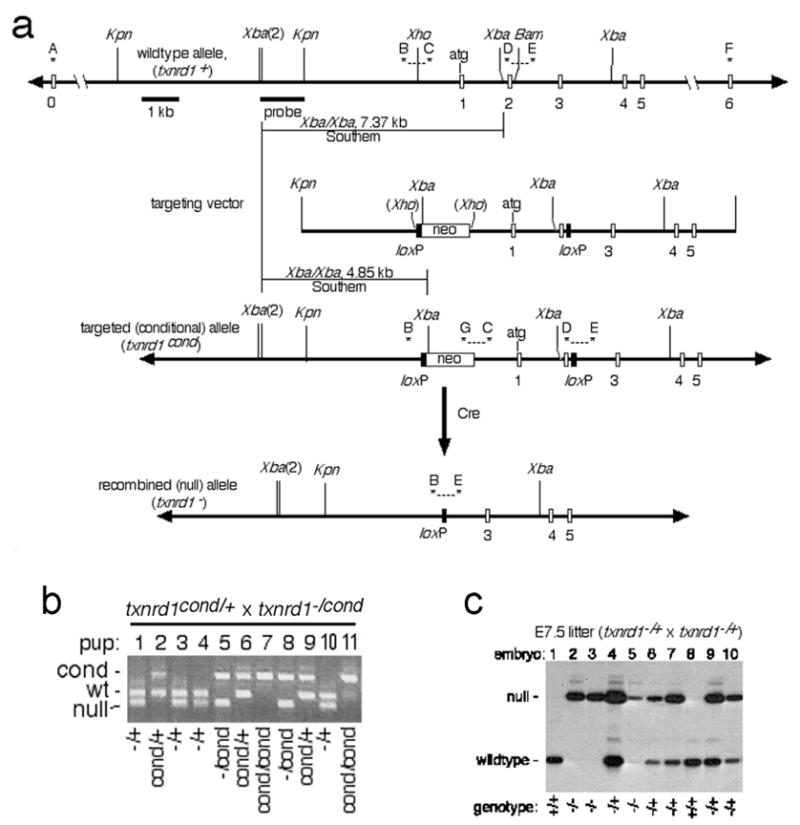
Design and confirmation of mice bearing txnrd1cond and txnrd1− alleles. a, schematic of alleles, targeting vector, and general mutagenesis strategy. Top, map of 5′ region of txnrd1 gene. Exons, represented as open boxes, are labeled below. Uppercase letters and asterisks indicate PCR primers. Probe and expected sizes of Southern blot bands are indicated. Below is the txnrd1-containing region of the targeting vector. Next is the correctly targeted (txnrd1cond) allele, with diagnostic PCR products and Southern blot bands indicated. Below this is the Cre-recombined null (txnrd1−) allele. b, confirmation of allele was determined by genetic analysis of mouse lines developed from ES cells. PCR-based genotyping reactions (primers B, C, E, G combined) were performed on genomic DNA isolated from tail snips at weaning from offspring of the indicated mating. This primer set gives the following allele-specific product sizes: txnrd1+, 338 bp; txnrd1cond, 542 bp; txnrd1−, 258 bp. Four pups had no txnrd1+ allele (pups 7 & 11 were txnrd1cond/cond; pups 5 & 8 were txnrd1−/cond), confirming that the downstream loxP was targeted into the txnrd1 allele. c, genotypic analysis of E7.5 embryos for microarray and RT-PCR analyses. Total nucleic acid was isolated from individual embryos and 10% of the total was used for radioactive PCR with primers B, D, and E. This primer set gives the following allele-specific product sizes: txnrd1+, 204 bp; txnrd1−, 258 bp.
Polysome analyses
Livers were harvested from txnrd1−/+ adult male mice, homogenized, and subjected to sucrose gradient velocity sedimentation, fractionation, and RNA purification, as described previously [33]. An equal proportion (5%) of each fraction was used as template for an oligo(dT)-primed reverse transcriptase reaction and 1% of each cDNA was used as template in PCR reactions as described previously [34] using the primers described in the figure legend and in Table S1.
Embryo harvests and histology
Matings were set up between txnrd1−/+ parents, plugs were checked each morning, and plug-date was defined as E0.5. For DAPI-staining, individual embryos were dissected into phosphate-buffered saline (PBS) containing 0.1% Tween-20 (PBST), fixed in buffered formalin, and stained with 0.01 mg/ml DAPI in PBST prior to evaluation by fluorescent microscopy. After taking photo-micrographs, each embryo was digested with 100μg/ml proteinase K (Sigma) in 1X TES (1% SDS, 10 mM Tris, pH 7.5, 5 mM EDTA) at 52°C overnight. DNA was precipitated with ethanol, resuspended in TE, and evaluated by radioactive PCR amplification using primers Txnrd1-B, -C, and -E (Table S1, Figs 1a, 1c), resolution on a sequencing gel, and autoradiography. For histology, whole pregnant uteri were harvested and fixed in buffered formalin. Uteri were embedded in paraffin and serial-sectioned longitudinally through the entire uterus. All sections were stained with hematoxylin and eosin and evaluated to ensure that differences between embryos were due to developmental progression and not to the plane-of-section.
In situ hybridizations
Detailed protocols are in Supplemental section; probes and methods were from the following sources: brachyury (t) [35]; Cripto [36]; Lim1 [37]; Fgf8 [38]; Snail1 [39].
Single-embryo cDNA production, RT-PCR, real-time PCR, and oligonucleotide array analyses
Pregnant uteri were harvested into ice-cold PBS; embryos were dissected and placed into individual 0.5 ml tubes containing 10 μl 1X TES (10 mM Tris, pH 7.5, 5 mM EDTA, 1% SDS) with 100 μg/ml proteinase K. Tubes were sealed and incubated 2–20 hours at 52°C. Samples received 90 μl of 0.1X TES, 5 μl of 5M NaCl, were extracted with an equal volume of phenol/choloroform/isoamyl alcohol (25:24:1) followed by pure chloroform, and were precipitated with 300 μl ethanol at −20°C overnight. Nucleic acid pellets were resuspended in 10 μl water. A portion (1–3 μl) was used for radioactive PCR-based genotype analyses, as above (Fig. 1c), and the remainder was used for cDNA production. Detailed methods for single embryo genotyping, cDNA production, and array analyses are in the Supplemental section.
Results
Design and production of txnrd1-mutant mice
The mouse txnrd1 gene encompasses 16 major exons, including a nontranslated first exon (“exon 0”) and 15 protein-coding exons, [40–42]. Txnrd1 catalytic activity requires a pair of cysteines near the N terminus of the protein (Cys59 and Cys64) and a C-terminal cysteine and selenocysteine, encoded in exon 15 [16]. Alternatively initiated or spliced Txnrd1 mRNAs have been described [41–43], in particular from testis [40], a tissue that exhibits particularly promiscuous patterns of transcription [44]. Some of these mRNAs could encode versions of Txnrd1 having additional N-terminal extensions; however all isoforms retain exons 1 and 2 [40]. The allele was designed to allow excision of these first two protein-coding exons (Fig. 1a), which encode conserved amino acids 1–30 that contact flavin-adenine dinucleotide (FAD), are an integral part of the Rossman fold common to NADPH-binding proteins [45, 46], and are found in all Txnrd1 mRNA isoforms [40]. Furthermore, when exons 1 and 2 are excised, the first in-frame AUG in the resultant mRNA (Met70) is in a poor context for translation initiation and any polypeptide initiated here would lack both N-terminal active site cysteines (Cys59 and Cys64) (see below).
The txnrd1− allele is functionally null
Animals homozygous for the conditional allele (txnrd1cond/cond) survived and were phenotypically indistinguishable from wild-type littermates at all ages (Fig. 1b and not shown). The txnrd1− allele lacks exons 1 and 2, but retains the promoter region and 5′ UTR-encoding exon 0 at a position roughly 15 kb upstream of the deletion mutation [41]. We predicted that pre-mRNA from the txnrd1− allele would be spliced to join exons 0 and 3, creating an RNA lacking the native start codon and sequences encoding N-terminal FAD-binding and oxidoreductase active site amino acids (see above). RT-PCR analyses using total RNA harvested from adult heterozygous (txnrd1−/+) kidney revealed only a single PCR product, which matched the size predicted for an mRNA in which exon 0 was spliced to exon 3 (not shown). Cloning and sequencing confirmed that this region of the cDNA contained an exon 0–3 splice structure (Fig. 2a). This cDNA contained two out-of-frame ATGs (reading frames of 13 and 8 codons) upstream of the first in-frame ATG (Met70) and no in-frame ATGs in a favorable context for translation initiation [47]. Insertion of the mutant exon 0-3-4-5 cDNA region upstream and in-frame with green fluorescent protein in a cytomegalovirus promoter-driven expression vector inhibited accumulation of green fluorescent protein in transiently transfected cells (not shown). Polysome analyses on heterozygous liver cytoplasm preparations revealed that, while Txnrd1+ mRNA was almost entirely polysomal, Txnrd1− RNA lacking exons 1–2 was not detected in the actively translated polysome fractions, but instead, was in the slowly sedimenting messenger ribonucleoprotein fractions (Fig. 2b). Thus, although RNA lacking exons 1–2 was produced from the mutated allele, this RNA likely was not translated. If residual translation occurred, the resulting predicted polypeptide would be incapable of catalyzing redox reactions. Thus, the txnrd1− allele is functionally null.
Fig. 2.
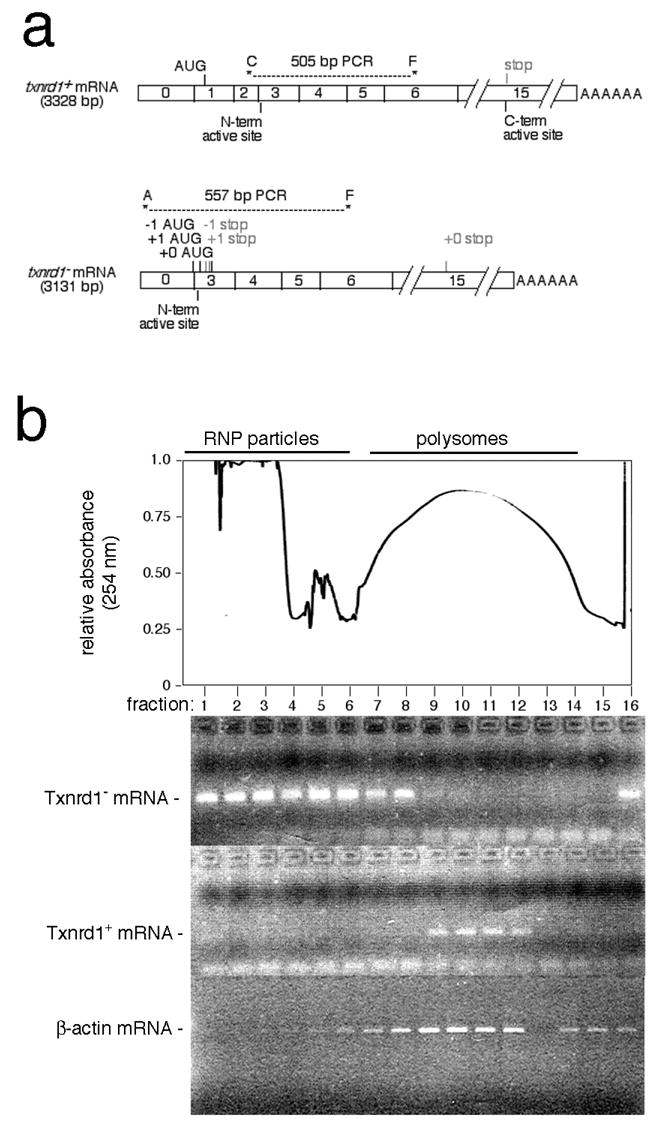
The recombined txnrd1− allele issues a non-translated mRNA. a, schematic. Predicted mRNAs from the wild-type and null alleles are shown. Diagonostic RT-PCR primer pairs and the sizes of expected products are indicated. RT-PCR-based cloning of Txnrd1 cDNAs from heterozygous mouse kidney RNA samples indicated that exon 0 spliced precisely to exon 3 (data not shown). The first two ATGs initiate short out-of-Txnrd1-frame ORFs that would issue predicted oligopeptides of 13 (-1 frame) and 8 (+1 frame) amino acids, respectively (first ATG generated by the exon 0/3 splice junction). The third ATG is in the Txnrd1 reading frame, at Met70 in exon 3. Due to context (AAGCTGATGC), this ATG is predicted to not function as an initiator. b, translational efficiency of Txnrd1+ and Txnrd1− mRNAs in heterozygous mouse liver. At top is shown an absorbance scan of a representative liver polysome gradient with the positions of ribonucleoprotein particles (RNP) and polysomes indicated above and the positions of gradient fractions indicated below. Below are RT-PCR analyses of the positions of Txnrd1 mRNAs in a heterozygous mouse liver polysome gradient using the PCR primer sets indicated in panel a.
Development of txnrd1−/− embryos
At E7.5 and 8.5, the frequencies of txnrd1−/− embryos from matings between txnrd1−/+ parents matched that expected for a randomly segregating allele (Table 1). No txnrd1−/− embryos were recovered at later times.
Table 1.
Genotype-specific survival of embryos from txnrd1−/+ x txnrd1−/+ matings1.
| stage | txnrd1+/+ | txnrd1−/+ | txnrd1−/− | n | litters |
|---|---|---|---|---|---|
| E7.5 | 16 | 27 | 18 | 61 | 5 |
| E8.5 | 30 | 43 | 22 | 95 | 9 |
| E9.5 | 8 | 13 | 0* | 21 | 3 |
| E10.5 | 10 | 14 | 0** | 24 | 3 |
Data is only included from txnrd1−/+ x txnrd1−/+ matings. A larger analysis including matings in which one or both parents were txnrd1−/cond also exhibited near-Mendelian survival of txnrd1−/− embryos until E8.5 with 100% loss of txnrd1−/− embryos by E9.5 (not shown). Where indicated, Chi squared analysis indicates genotype distribution differs significantly from that expected for a randomly segregating allele,
P<0.05;
P<0.01.
Embryos harvested from heterozygous matings at E7.5 were stained with DAPI, photographed using fluorescence microscopy, and then subjected to PCR-based genotypic analysis. Results showed that mutant embryos, although smaller than their wild-type and heterozygous littermates, contained several thousand cells, and that the internuclear-distance (an indicator of cell size) in txnrd1−/− embryos was similar to that in wild-type littermates (not shown). Histological examination of whole pregnant uteri harvested at E7.5 and E8.5 from txnrd1−/+ x txnrd1−/+ matings revealed that mutant embryos had a strikingly defective morphology (Fig. 3). At E7.5, all embryos showed evidence of extensive cell-type differentiation. Mutants showed normal trophectoderm derivatives, including ectoplacental cone (EC), trophoblast giant cells (TGCs), and extraembryonic ectoderm. Primitive endoderm also formed; however the morphology of visceral endoderm (VE) differed from that seen in wild-type embryos. Normally, VE overlaying embryonic tissue (VE(em)) is thin and squamous; VE overlaying extraembyronic tissues (VE(ex)) is composed of cuboidal cells with apical vacuoles and a microvillous brush border (Fig. 3a) [48]. In txnrd1−/− embryos, VE morphology was biased toward the morphology typical of the extraembryonic region (Fig. 3b, c, d). The embryonic ectoderm (epiblast) did not appear to differentiate and no morphologically distinct primitive streak was visible (Figs 3 b, c, d). However, the embryos continued to grow without further differentiation. By E8.5, mutant embryos were 3- to 8-fold larger than they were at E7.5, and the core of the embryo, which appeared to be undifferentiated ectoderm, expanded into a highly disorganized structure (Figs 3f, g, h). TGCs were abundant and the VE with extraembryonic morphology was further expanded, vacuolated, and folded upon itself into a thick layer (Fig. 3f, g, h).
Fig. 3.
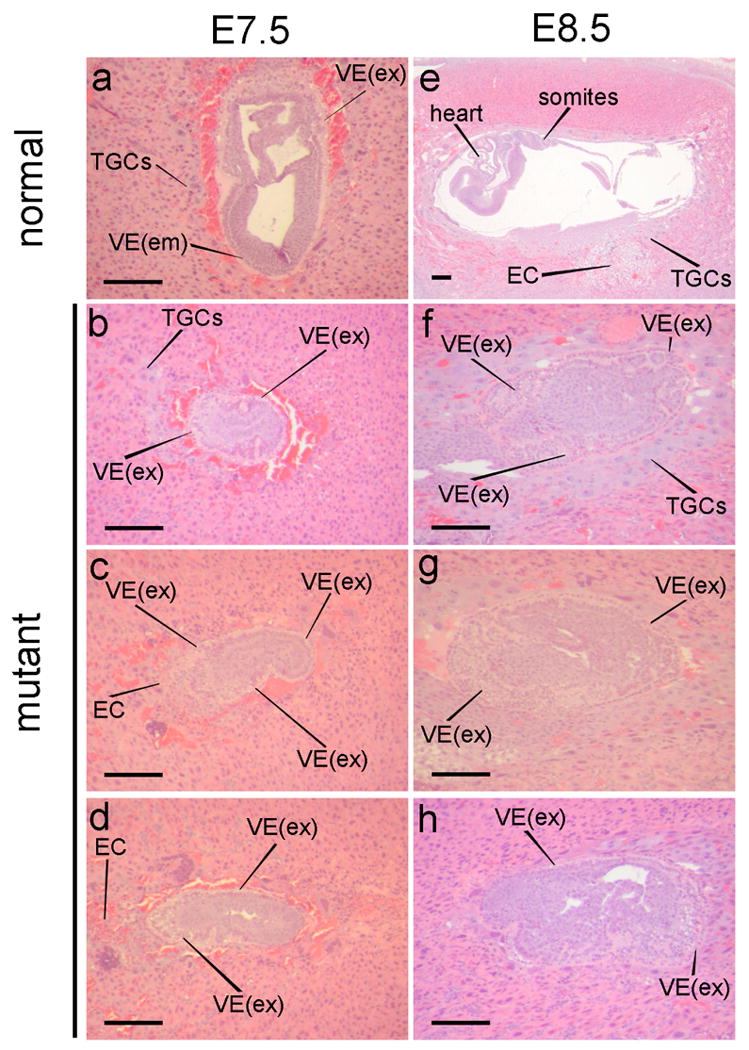
Development of txnrd1−/− embryos. Whole pregnant uteri were harvested from matings between txnrd1−/+ parents and were prepared for histology. Longitudinal serial paraffin-embeded sections were prepared through the entire uterus and were stained with hematoxylin and eosin. Representative E7.5 (a-d) and E8.5 (e-h) embryo-central sections are shown. Abbreviations: EC, ectoderm; PE, parietal endoderm; PS, primitive streak; TGC, trophoblast giant cells; VE, visceral endoderm; VE(em), non-vacuolated squamous visceral endoderm overlying embryonic tissue; VE(ex), vacuolated cuboidal visceral endoderm overlying extraembryonic tissue.
To more accurately determine which embryonic tissues and structures were forming, we harvested E7.5 embryos and looked at marker gene expression patterns by in situ hybridization (Fig. 4). Brachyury (t) and Lim1, which are normally expressed in mesodermal regions of the primitive streak [35, 37], showed little or no expression in mutants. Snail1, which is normally expressed in the ectoplacental cone and pre-streak mesoderm [39] was only expressed in the ectoplacental cone in mutants (Fig. 4). Cripto, which is normally expressed by presumptive mesodermal cells in the epiblast of pre-streak embryos [36], showed a reduced expression domain, consistent with under-representation of embryonic tissues. Fgf8, which is normally expressed in the cells of the primitive streak and to a lesser extent in anterior endoderm, was observed in two reduced domains in the mutants, one corresponding to proximal embryonic ectoderm and one corresponding to the distal VE (Fig. 4). A similar pattern of Fgf8 expression is found in embryos having defective axis formation, including nodal-, lim1-, Otx2-, and Cripto-mutants [49, 50]. These results suggested that development was arrested in a pre-streak stage, mesoderm did not form, embryonic tissues were reduced, and patterning of the embryonic tissues was either arrested or incorrect, resulting in a failure to gastrulate.
Fig. 4.
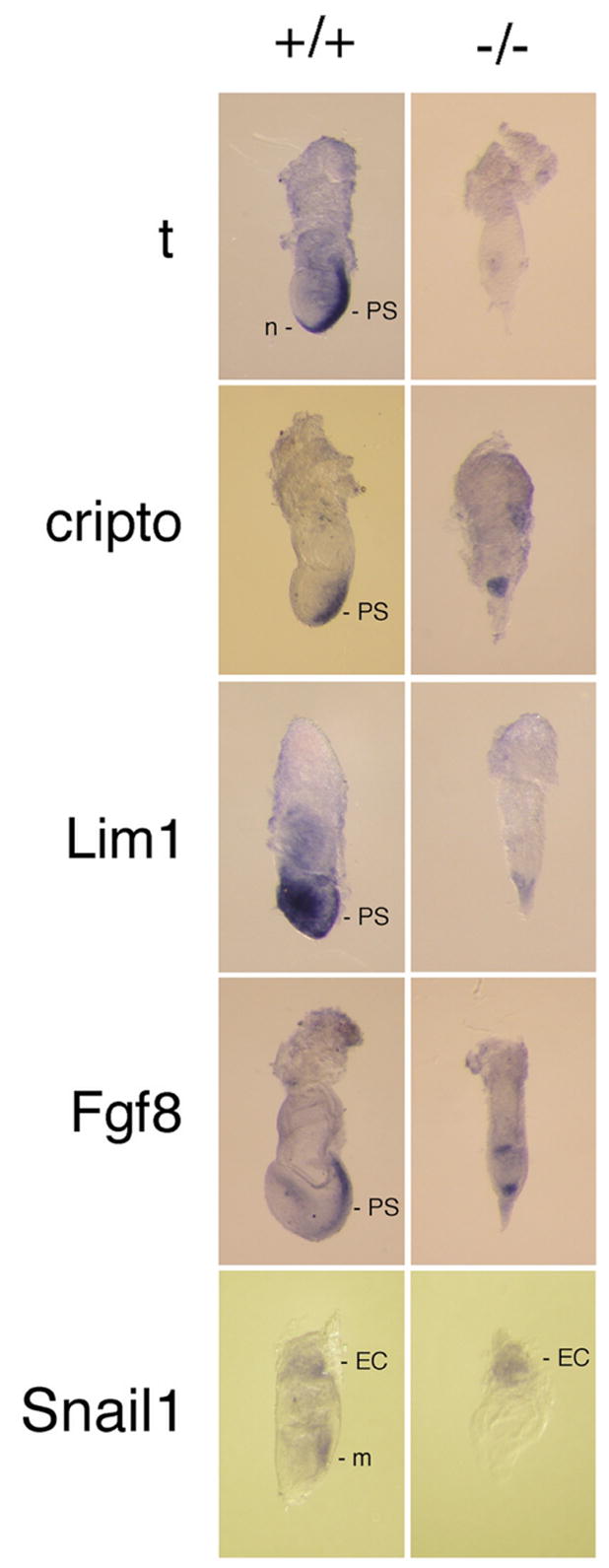
Embryonic gene expression patterning. Embryos were harvested at E7.5 from matings between txnrd1−/+ parents and stained by whole-mount in situ hybridization for the indicated marker genes. After staining and photographing, embryos were digested with proteinase K, nucleic acids were isolated, and genotypes were determined by radioactive PCR (see Fig. 1c). Abbreviations as Fig. 3 except: m, mesoderm; n, node; t, brachyury.
Single-embryo transcriptome profiles
Histological and marker analyses showed that the txnrd1 gene was not required for cell survival or proliferation but was required for embryonic patterning and perhaps to allow differentiation of some tissue types. To better describe the processes that were affected by the mutation, we developed a protocol to compare global mRNA levels (transcriptomes) in individual homozygous mutant and wild-type E7.5 littermates by microarray and RT-PCR (see Supplemental section).
Transcriptome data for three pair of embryos (six arrays) was analyzed statistically using GeneSpring software (Agilent). Signals for annotated probe sets that differed by ≥ 3-fold between mutant and wild-type embryos and whose reproducibility within the three embryo pairs yielded reliable data (P ≤ 0.05) were considered “differentially abundant”. By these criteria, 27 of the 45,101 probe-sets, representing 21 different mRNAs, had a stronger signal in the mutants (Table 2), and 47 probe sets, representing 35 mRNAs, had a weaker signal in the mutants (Table 3).
Table 2.
mRNAs more abundant in txnrd1−/− than in txnrd1+/+ embryos at E7.5.
| mRNA1 | difference2 | −/− signal3 | P-value4 | description | GenBank |
|---|---|---|---|---|---|
| Igfbp1 | 11.7 | 567 | 0.04 | IGF-binding protein 1 | NM_008341 |
| Npn3 (Srxn1) | 10.9 | 474 | 0.00 | Neoplastic progression 3 (sulfiredoxin 1) | BM210600 |
| Gstμ3 | 8.7 | 338 | 0.01 | glutathione S-transferase μ3 | J03953 |
| Mash2 | 8.5 | 3061 | 0.03 | Mash-2, achaete-scute homologue-like 2 | AK010738 |
| Mash2 | 7.2 | 2267 | 0.04 | Mash-2, achaete-scute homologue-like 2 | AK010738 |
| Gstμ1 | 6.9 | 752 | 0.00 | glutathione S-transferase μ1 | NM_010358 |
| Gstμ1 | 6.5 | 1511 | 0.00 | glutathione S-transferase μ1 | NM_010358 |
| Nxf7 | 6.4 | 240 | 0.01 | nuclear RNA export factor 7 | AJ305317 |
| Gstμ1 | 5.4 | 292 | 0.01 | glutathione S-transferase μ1 | J03952 |
| Mt1 | 4.9 | 4410 | 0.01 | metallothionine 1 | NM_013602 |
| Cbr3 | 4.9 | 315 | 0.01 | carbonyl reductase 3 | AK003232 |
| Gclm | 4.4 | 1396 | 0.01 | glutamate-cysteine ligase | NM_008129 |
| Mt2 | 4.3 | 7407 | 0.02 | metallothionine 2 | AA796766 |
| Ltb4dh | 4.3 | 2330 | 0.00 | leukotriene B4 12-dehydrogenase | BC014865 |
| Mgst2 | 3.6 | 233 | 0.01 | microsomal glutathione S-transferase 2 | AV066880 |
| S100A11 | 3.6 | 1144 | 0.02 | S100 calcium-binding protein A11 | BC021916 |
| Ugt1a2 | 3.5 | 926 | 0.02 | UDP glycosyltransferase 1 A6 | D87867 |
| Ugt1a2 | 3.5 | 663 | 0.01 | UDP glycosyltransferase 1 A6 | BC019434 |
| Blvrb | 3.5 | 360 | 0.00 | biliverdin reductase B | BC027279 |
| S100A1 | 3.4 | 1144 | 0.02 | S100 calcium-binding protein A1 | AI266795 |
| Grina | 3.4 | 445 | 0.01 | glutamate receptor-associated 1 | AW212189 |
| Gaa | 3.4 | 760 | 0.01 | glucosidase, alpha, acid | BB357227 |
| Mod1 | 3.3 | 569 | 0.00 | malic enzyme, supernatent | BC011081 |
| Gaa | 3.2 | 533 | 0.00 | glucosidase, alpha, acid | NM_008064 |
| Mod1 | 3.2 | 366 | 0.03 | malic enzyme, supernatent | AK006387 |
| Htatip2 | 3.1 | 636 | 0.05 | HIV-1 tat interactive protein 2 | AF061972 |
| Cln6 | 3.1 | 391 | 0.00 | ceroid-lipofuscinosis, neuronal 6 | BB514333 |
mRNAs having multiple entries were represented by more than one oligonucleotide probe set on the arrays and thus provided independent data readouts. In some cases, probe sets were designed from different GenBank files; in other cases they were designed to a common GenBank file, as indicated in right column.
Only mRNAs having an average abundance difference of three-fold or greater between txnrd1−/− and txnrd1+/+ embryos were included.
Average signal from all three replicates given; only mRNAs in which the higher signal was ≥ 200 units were included.
All data based on statistical analysis of individual arrays from three wild-type and mutant littermate pairs using GeneSpring; only mRNAs showing sufficient signal reproducibility between the three paired array sets to yield a P-value ≤ 0.05 were included.
Table 3.
mRNAs less abundant in txnrd1−/− than in txnrd1+/+ embryos at E7.5.
| mRNA1 | difference | +/+ signal | P-value | description | GenBank |
|---|---|---|---|---|---|
| Nkx1-2 | −21.3 | 639 | 0.04 | Nk1 transcription factor-related 2 | NM_009123 |
| Agtlr1 | −20.1 | 823 | 0.03 | angiotensin receptor-like 1 | BB533323 |
| Pcdh8 | −15.4 | 293 | 0.02 | protocadherin 8 | BB076893 |
| Sox11 | −14.7 | 1580 | 0.01 | SRY-box containing gene 11 | BB656631 |
| Pmp22 | −14.1 | 743 | 0.02 | peripheral myelin protein 22 | NM_008885 |
| Thsd2 | −12.5 | 496 | 0.03 | thrombospondin, type 1, domain 2 | BG072958 |
| Ms4a4d | −12.5 | 322 | 0.04 | membrane-spanning 4, domain A4D | NM_025658 |
| Ebaf | −11.5 | 257 | 0.04 | endometrial bleeding associated factor | AV214969 |
| Ccnd2 | −11.4 | 944 | 0.03 | cyclin D2 | NM_009829 |
| Ccnd2 | −10.9 | 1217 | 0.01 | cyclin D2 | BQ175880 |
| Pcdh18 | −9.5 | 303 | 0.02 | protocadherin 18 | BM218630 |
| Ccnd2 | −8.2 | 383 | 0.02 | cyclin D2 | AK007904 |
| Myl7 | −7.8 | 246 | 0.00 | myosin, light polypeptide 7 | NM_022879 |
| Ccnd2 | −7.7 | 285 | 0.01 | cyclin D2 | NM_009829 |
| Pbx1 | −7.6 | 770 | 0.02 | pre B-cell leukemia factor 1 | BG070361 |
| Fgf13 | −7.5 | 367 | 0.04 | fibroblast growth factor 13 | AF020737 |
| Sox11 | −7.2 | 329 | 0.04 | SRY-box containing gene 11 | BM508495 |
| Dll1 | −6.3 | 340 | 0.01 | delta-like 1 | NM_007865 |
| Gli3 | −6.1 | 478 | 0.04 | GLI-Kruppel family member 3 | BB311687 |
| Frzb | −6.1 | 388 | 0.01 | frizzled-related protein | U91905 |
| Lef1 | −6.0 | 409 | 0.04 | lymphoid enhancer binding factor 1 | AV156352 |
| Dpysl2 | −5.6 | 1834 | 0.03 | dihydropyrimidinase-like 2 | BQ174209 |
| Rhobtb3 | −5.5 | 1090 | 0.02 | Rho-related BTB domain 3 | BM942043 |
| Rhoe | −5.4 | 875 | 0.03 | ras homologue gene family, member E | BC009002 |
| Mpdz | −4.5 | 678 | 0.01 | multiple PDZ domain protein | AK019164 |
| Ror2 | −4.5 | 230 | 0.02 | receptor YK-like orphan receptor 2 | AV324603 |
| Ndn | −4.5 | 255 | 0.00 | necdin | NM_010882 |
| Tnfrsf19 | −4.4 | 383 | 0.03 | tumor necrosis factor receptor 19 | NM_013869 |
| Tgif | −4.1 | 605 | 0.01 | TG-interacting factor | NM-009372 |
| Zfp334 | −4.0 | 413 | 0.04 | zinc finger protein 334 | AU022425 |
| Ndn | −4.0 | 955 | 0.03 | necdin | AW743020 |
| Ccnd1 | −4.0 | 1376 | 0.04 | cyclin D1 | NM_007631 |
| Ndn | −4.0 | 866 | 0.04 | necdin | AV124445 |
| Ptbp2 | −3.9 | 1174 | 0.03 | polypyrimidine binding protein 2 | BB076855 |
| Ndn | −3.8 | 732 | 0.04 | necdin | AW743020 |
| Wnt5a | −3.8 | 404 | 0.05 | wingless-related 5a | BC018425 |
| Marks | −3.8 | 525 | 0.01 | myristoylated ala-rich PKC substrate | AW546141 |
| Prkcm | −3.6 | 489 | 0.04 | protein kinase C μ | AV297026 |
| Hmga2 | −3.6 | 1093 | 0.03 | high mobility group AT-hook 2 | X58380 |
| Prkcm | −3.6 | 489 | 0.04 | protein kinase C μ | NM_008858 |
| Cdh2 | −3.6 | 2808 | 0.05 | cadherin 2 | BC022107 |
| Ets1 | −3.5 | 284 | 0.01 | E26 avian leukemia oncogene 1 | BB151715 |
| Tcfl5 | −3.5 | 2041 | 0.04 | procollagen type IX, alpha 3 | AV044715 |
| Hmgb3 | −3.4 | 4111 | 0.02 | high mobility group box 3 | NM_008253 |
| Marks | −3.3 | 328 | 0.04 | myristoylated ala-rich PKC substrate | AW546141 |
| Wnt5a | −3.2 | 522 | 0.04 | wingless-related 5a | BB067079 |
| Marks | −3.0 | 1371 | 0.02 | myristoylated ala-rich PKC substrate | AW546141 |
Notably absent from the list of differentially abundant mRNAs were those encoding most cell proliferation markers (see below). Also absent was the gene encoding the mitochondrial Txnrd2 protein. mRNAs encoding only five other reductases showed evidence of mRNA overabundance in the mutants. These were sulfiredoxin 1 (Srxn1), carbonyl reductase 3 (Cbr3), leukotriene B4 12-dehydrogenase (Ltb4dh), malic enzyme 1 (Mod1), and biliverdin reductase b (Blvrb) (Table 2). Many mRNAs that were underrepresented in the mutants encoded proteins not expressed until during or after gastrulation (Table 3), consistent with the mutant embryos failing to gastrulate.
To further test whether mRNAs encoding components participating in proliferative or oxidoreductase pathways were affected by the mutation, we used GeneSpring software to evaluate relative levels of all mRNAs whose signal reproducibility (variance) in the arrays provided reliable data (P ≤ 0.05) for genes involved in these processes (Tables S2 and S3). Of the 88 annotated probe sets meeting these criteria in the cell proliferation set, only those representing cyclins D1 and D2 showed differential mRNA abundance (≥ 3-fold differential) between mutant and wild-type embryos. Probe sets having reproducible yet similar signals between mutant and wild-type embryos in the proliferation set included 24 sets for DNA polymerases or primases, 7 sets for histones, and 3 sets for deoxyribonucleotide synthesis (ribonucleotide reductase, dihydrofolate reductase, and thymidylate synthase)(Table S2). The 64 annotated probe sets giving reliable data in the oxidoreductase set showed no underrepresented mRNAs and only 5 mRNAs (listed above) that were ≥ 3-fold overrepresented in the mutants. Probe sets having reliable yet similar signals between mutant and wild-type embryos in the oxidoreductase set included peroxiredoxins (Prdx) 1, 2, and 4 (Table S3).
Representative mRNAs from the microarray data were chosen for RT-PCR or real-time RT-PCR confirmation analyses using littermate-pairs of single wild-type and mutant E7.5 embryos (Figs 5, S2). All primer pairs were designed to amplify 3′ cDNA sequences spanning an exon/exon junction. Real-time PCR analyses gave data that qualitatively matched the array data and thermal dissociation curves that matched predictions based on the length and G/C-content of the amplified region (Figs 5b, S2). Of particular importance, single-embryo RT-PCR analyses confirmed that, whereas Prdx1, 2, and 4 mRNA levels did not differ substantially between mutant and wild type embryos, Srxn1 mRNA levels were dramatically higher in the mutants (Fig. 5c).
Fig. 5.
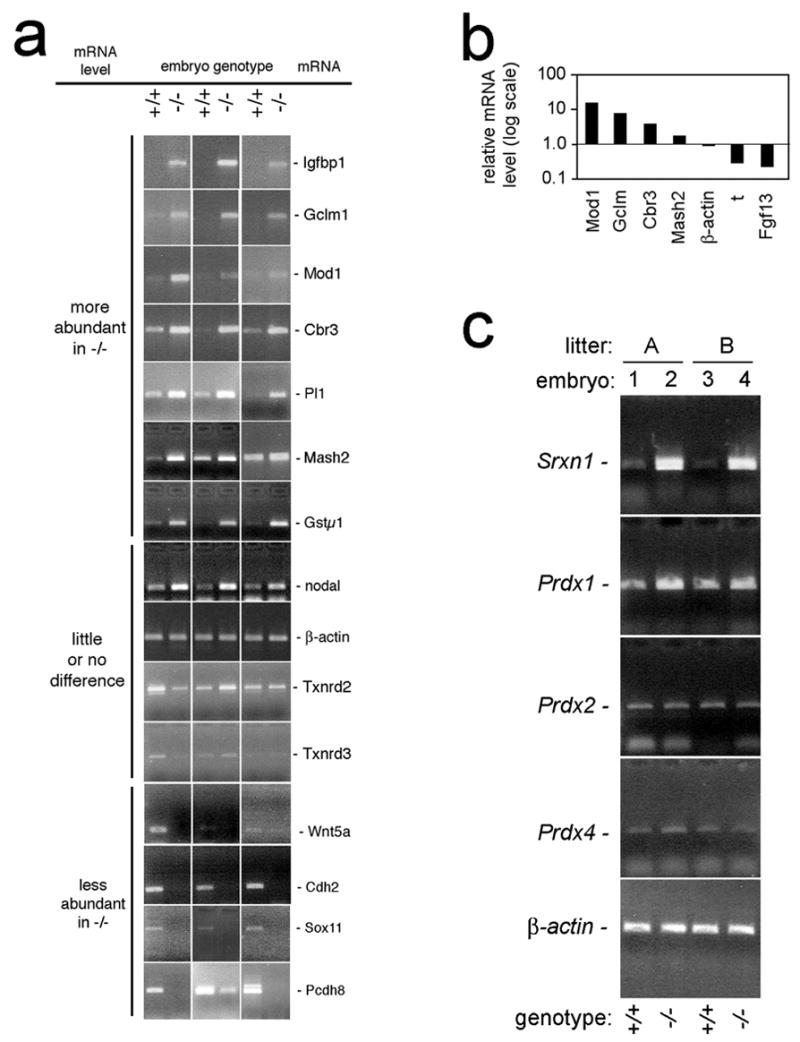
Analyses of gene expression in littermate pairs of single E7.5 txnrd1+/+ and txnrd1−/− embryos. All assays were performed on single E7.5 embryos genotyped as in Fig. 1c. a, RT-PCR confirmation of relative mRNA abundances in paired sets of txnrd1−/− and txnrd1+/+ embryos, as indicated. b, real-time PCR validation of relative mRNA levels for selected genes of interest. Each data point represents the ratio of txnrd1−/− to txnrd1+/+ mRNA in a paired littermate set of single E7.5 embryos. c, single-embryo RT-PCR confirmation of expression differences of Srxn and Prdx mRNAs from E7.5 littermate pairs of two litters, as indicated. Abbreviations: Cbr3, carbonyl reductase 3; Cdh2, cadherin homologue 2; Fgf13, fibroblast growth factor family member 13; Gstμ1, glutathione S-transferase μ1; Gclm, glutamate-cysteine ligase; Igfbp1, insulin-like growth factor-binding protein 1; Mash2, achaete-scute homologue-like 2; Mod1, malic enzyme; Pcdh8, protocadherin 8; Pl1, placental lactogen 1; Prdx, peroxiredoxin; Sox11, SRY box-containing gene 11; Srxn1, sulfiredoxin 1; t, brachyury.
In summary, our data show that disruption of the txnrd1 gene in mice blocked mesoderm formation and patterning of the embryo, but had little effect on zygote-derived extraembryonic cell and tissue types. The mRNA abundance profiles in the mutant embryos were consistent with a failure to form advanced embryonic tissue types and inconsistent with the mutation disrupting proliferation. Many mRNA abundance differences matched those reported in txnrd1-null yeast [32], suggesting that compensatory pathways are generally shared between these evolutionarily distant species. However, notably absent from the list of up-regulated mRNAs in mutant mice were the Prdxs, which have been shown to be strongly up-regulated in thioredoxin reductase-deficient yeast [32]. The implications of these findings are discussed below.
Discussion
Survival, proliferation, and differentiation of cells in txnrd1−/− embryos
Survival of txnrd1−/− embryos to E8.5 indicated that cells lacking Txnrd1 were capable of synthesizing DNA, RNA, lipid, and protein, and performing all basic metabolic functions required for cell survival, growth, and proliferation within the context of early mammalian development. In addition, txnrd1−/− embryos expressed early differentiation markers, including mRNA for Cripto, Fgf8, Snail1, Mash2, and Pl1, some of which were shown by in situ hybridization to be regionally restricted within the embryo (Figs 4, 5). These results indicate that some cell differentiation occurred in the mutants.
Systems affected by Txnrd1 disruption
Yeast lacking a functional Txn system survive due to compensation by the glutathione pathway [12, 51]. In the current study, transcriptome profiling indicated twenty-one annotated mRNAs were substantially overrepresented in txnrd1−/− mouse embryos (Table 1). Interestingly, several of these mRNAs are also upregulated in yeast lacking Txnrd1 [32]. These included mRNAs for glutathione-S-transferases (GSTs) metallothioneins (MTs), and carbonyl reductase 3 (Cbr3) (Table 1). However, unlike in yeast [32], levels of mRNAs encoding glutathione reductases and peroxiredoxins (Prdxs) were not higher in txnrd1−/− as compared to wild-type embryos (Fig. 5c; Tables 2 and S3).
1) GSTs, MTs, and Cbr3
GSTs are detoxifying enzymes that conjugate glutathione to potentially toxic substrates [52]; they are not reductases. Similarly, although MTs contain iron-thiolate centers [53], they cannot sequester ferric or ferrous iron [54], the metals with the highest potential for generating reactive oxygen species in cells [55]. Thus, although both mouse embryos and yeast overaccumulate GST and MT mRNAs in response to disruption of txnrd1 (Table 1) [32], this probably does not directly help cells combat oxidative stress or compensate for a deficit in reducing activity. We suspect these are non-adaptive responses. For example, disruption of the Txn pathway in yeast [51], and perhaps also in mammals, shifts the thiol maintenance burden onto the glutathione pathway. This, in turn, might lead to reduced flux through glutathione-dependent enzymes, including GST, and reduced GST activity might stimulate feedback induction of GST mRNA accumulation [56]. Similarly, the iron-thiolate centers in MTs confer redox sensitivity onto heavy metal-binding by the protein [57–59]. As a result, txnrd1 disruption might antagonize heavy metal chelation by MTs, resulting in elevated levels of free intracellular zinc, a potent inducer of mt gene expression [60, 61], and causing the observed increase in MT mRNA levels. This would suggest that redox-dependent liberation of zinc by MTs might affect transcription of other genes in response to txnrd1 disruption. Interestingly, in Drosophila, GST mRNAs have been shown to be induced in response to zinc [62]. Thus, release of zinc from MTs might also contribute to the strong GST mRNA response observed following txnrd1 disruption in yeast [32] and mouse embryos (this study). Further investigations will be needed to test whether MTs participate in a redox sensor-and-response mechanism and to determine the basis of the GST mRNA response to a Txnrd1 deficiency.
Little is known about the functions of Cbr. Yeast lacking Cbr are viable [63]. Biochemical studies show the enzyme reduces lipid carbonyl groups, which arise from lipid peroxidation during oxidative stress, to alcohols [64], and this may protect neurons from oxidative damage [65]. Upregulation of Cbr3 mRNA in txnrd1-null yeast [32] and mice (Figs 5a, b; Tables 2, S3) might be a reaction to intracellular lipid peroxidation, and thus indicative of intracellular oxidative stress at membrane surfaces. Further studies will be required to determine the pathways by which Cbr3 mRNA levels respond to the loss of Txnrd1.
2) Prdx system
It is of interest that txnrd1−/− mouse embryos did not exhibit increased levels of mRNAs encoding Prdxs (Fig. 5c; Table S3). In yeast, the Tsa1 gene, encoding the major cytosolic form of 2-Cys Prdx, is the most highly induced gene in txn1-null yeast; the Tsa2 gene, encoding a second cytosolic 2-Cys Prdx, is also highly induced [32]. None of the six mammalian 2-Cys Prdxs were substantially induced in txnrd1−/− embryos (Fig. 5c; Table S3).
Prdxs are ubiquitous Txn-dependent peroxidases [66–68]. The 2-Cys family of Prdxs initiate catalysis when a cysteine at the active site reacts with H2O2 to form a sulfenic acid intermediate. The sulfenic acid reacts with a resolving cysteine to form a disulfide, which is subsequently reduced by Txn to return the enzyme to its initial state [66] (Fig. 6). An appendage unique to eukaryotic 2-Cys Prdxs slows the rate of formation of the disulfide [69]. As a result, even at moderate H2O2 concentrations, the sulfenic acid intermediate can react with a second H2O2 molecule to form a sulfinic acid before it can react with the resolving cytseine to form a disulfide (Fig. 6). The sulfinic form is inactive and cannot be reduced by Txn [69]. H2O2 has been implicated as an important signaling molecule in eukaryotic cells [70](see below). It has been hypothesized that eukaryotes evolved 2-Cys Prdxs to serve as “floodgates” that prevent low levels of H2O2 from triggering signaling but allow H2O2 to oxidize downstream substrates once a threshold is exceeded [68]. Srxns, a novel class of slow ATP-dependent enzymes, convert the sulfinic acid form of Prdx back to the active form [67–71] (Fig. 6).
Fig. 6.
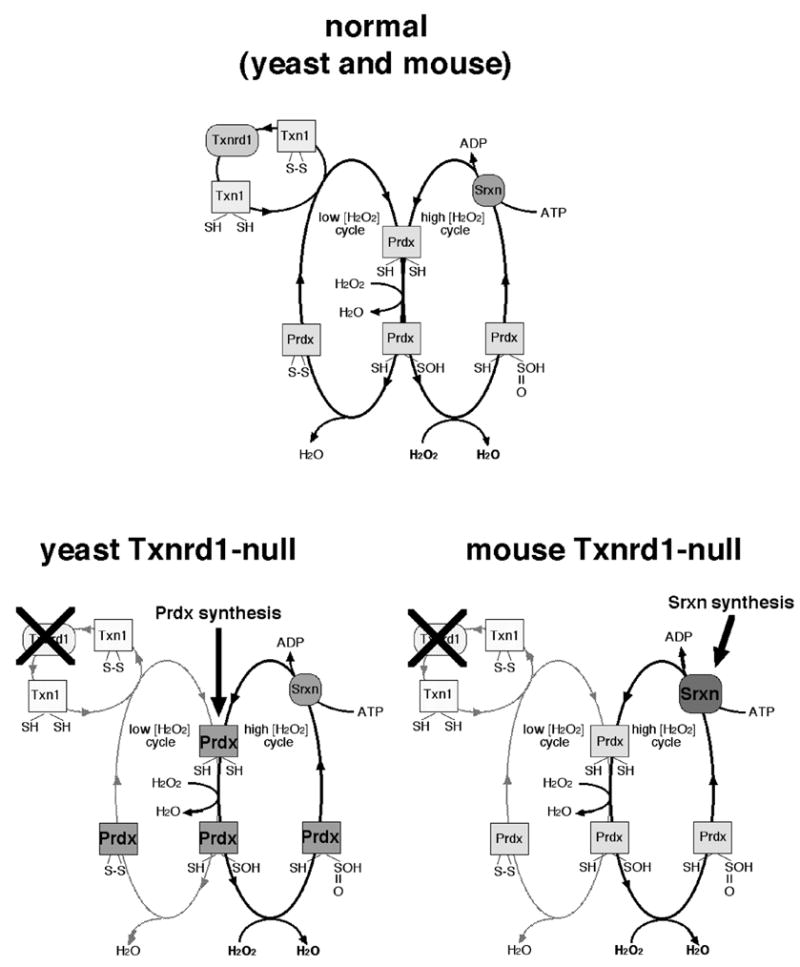
Eukaryotic Prdx catalytic cycles and inferred compensation for Txnrd1 disruption. At top is shown the normal Prdx catalytic cycles [68, 71]. The cycle at left is Txnrd1-dependent and functions to hydrolyze H2O2 at low concentrations. The cycle at right is Txnrd1-independent, and is only active at high H2O2 concentrations, where the Prdx sulfenic acid is further oxidized to a sulfinic acid, which cannot be reduced by Txn [74]. The mechanisms of Srxn catalysis are still under investigation; most models agree it is an ATP-dependent reaction [71, 72]. Below is shown the compensatory shifts expected in yeast [32] (left) and mice (right) in response to Txn system disruption. In both species, disruption of Txnrd1 is expected to disrupt the low [H2O2] Prdx cycle. Yeast strongly upregulate Prdx mRNA levels [32], suggesting compensation involves increasing rates of Prdx protein synthesis. By contrast, mice did not alter Prdx mRNA levels, but strongly upregulated Srxn mRNA (this study). Either response might bolster the high [H2O2] Prdx cycle.
Although the txnrd1−/− embryos did not exhibit increased levels of mRNAs encoding Prdxs, mRNA from the npn3 gene, encoding the mouse homologue of yeast srxnI [72–74], was the second most highly overrepresented mRNA in txnrd1−/− embryos (11-fold; Tables 2, S3; Fig. 5c). This suggests that, like upregulation of mRNAs encoding GSTs, MTs, and Cbr3, augmentation of the Prdx system is a conserved response to an impaired Trx system. However, whereas yeast respond by upregulating Prdx mRNA levels, mice responded by upregulating Srxn, and thereby might more effectively restore the sulfinic form of Prdx to the active form. Interestingly, in both species, this might provide an unexpected ATP-dependent means of H2O2 detoxification through the Txnrd1-independent Srxn-dependent high [H2O2] loop of the Prdx catalytic cycle (Fig. 6). Further studies will be required to test this.
Effects on signaling
Many developmental fate-determining and patterning signals in embryos involve growth factor binding to surface receptors whose activities involve tyrosine phosphorylation. These factors include BMPs, FGFs, IGFs, and others. An intact Txn system may be necessary to attenuate signaling by surface receptors. In response to ligand binding, NADPH-oxidase enzymes associated with the cytosolic aspect of growth factor receptors generate H2O2 [22–24, 26, 75–83]. A conserved catalytic cysteine at the active site of protein tyrosine phosphatases is oxidized by H2O2 to a sulfenic acid, which further reacts with a backbone nitrogen to form a sulfenylamide [68]. Protein tyrosine phosphatase inactivation by H2O2 may facilitate tyrosine kinase signaling by stabilizing phosphotyrosines [26, 70, 82]. Txn1 reduces the sulfenylamide of oxidized protein tyrosine phosphatases and restores enzyme activity [68]. Txn1 is also required for H2O2 decomposition by Prdx at low H2O2 concentrations (Fig. 6; see above). Thus, an intact Txn system may be required to limit H2O2 levels and prevent excessive tyrosine kinase signaling.
Excessive signaling from surface receptors could have diverse effects. For example, if two signaling pathways compete for fate determination in a particular cell, hyper-activation of a tyrosine phosphate-dependent pathway might shift the equilibrium toward having all cells respond only to this signal, thus excluding induction of other cell fates. Alternatively, hyper-activation of a phosphotyrosine-dependent pathway, especially in the absence of effective H2O2 clearance (see above) might kill the target cells, effectively excluding differentiation along these fates. Interestingly, the most dramatically overrepresented mRNA in mutant embryos was that encoding the insulin-like growth factor-binding protein, IGFBP1 (Table 1). In a recent study, Goretta et al. [84] showed that IGFBP3 is up-regulated in cytokine-induced HepG2 human hepatocarcinoma cell cultures that were treated with siRNA targeted to Txnrd1. Increased IGFBP mRNA levels in Txnrd1-deficient cells may be a feedback response to repress excessive signaling from the phosphotyrosine-dependent IGF receptor.
Phenotypic differences between txnrd1−/− mouse lines
While the study described here was in progress, another mouse line bearing a disrupted txnrd1 gene was described that exhibits a substantially different phenotype [85]. Homozygous embryos in the Jakupoglu et al. [85] study did not have a gastrulation defect. Instead, these mice arrested at a later stage, just before turning, with a neural tube and head folds, somites, and a functional heart [85]. At this stage, all cell types except cardiac muscle underwent cataclysmic death.
Although both the allele described in the present study and that of Jakupoglu et al. [85] are predicted to be functionally null in the recombined state (each eliminates a different set of essential active site amino acids, for example), the phenotype we report here is far more severe. The allele developed in the current study eliminates exons 1 and 2, including all functional ATGs and the two N-terminal active site Cys59 and Cys64 residues (Fig. 1); the other eliminates exon 15, including the C-terminal Cys and Sec amino acids [85]. Jakupoglu et al’s [85] allele would be predicted to produce a functional translatable mRNA encoding a catalytically inactive protein. However they were able to detect only trace amounts of mRNA and no protein accumulation[85], suggesting that this C-terminal truncation destabilizes the entire mRNA. All other reported parameters, including mouse and ES cell strains and care conditions, are similar between the two studies.
Jakupoglu et al. [85] suggested that Txnrd2 compensated partially for absence of Txnrd1 in most cell types, allowing normal embryonic development to the neural fold/somite stage, and completely in cardiac cells, allowing full heart development and function. Cell cycle arrest of non-cardiac cells was implicated in embryonic failure at the neural fold/somite stage. Unlike our study, global gene expression was not measured. By contrast, our transcriptome analysis showed that Txnrd2 mRNA was not more abundant in mutant embryos (Table 2, Fig. 5a), and, like in Txnrd1-null yeast [32] and Txnrd1-knock-down HepG2 cells [84], most proliferation markers, including mRNAs encoding histones, DNA polymerases, and others, were similar in mutant and wild-type embryos (Table S2). Further analyses on both alleles will likely be required to resolve the complex physiological roles of Txnrd1 in mouse embryos and why such different phenotypes might be manifested.
Supplementary Material
Acknowledgments
Supported by a Basil O’Connor New Investigator Award (#5-FY00-520) and a research grant (#6-FY03-61) from the March of Dimes Foundation, an appointment from the Montana Agricultural Experiment Station, a seed grant from the Montana Biomedical Research Opportunities program (5 P20 RR16455-02), a research grant (0090884) and CAREER Award (0446536) from the National Science Foundation, and NIH/NIAID grant (R01 AI55739-01) to EES; by an NIH/NCI grant (R01 CA82633) and an NIH/NIEHS Center Pilot Project grant (P30 ES00210) to GFM; and by the Howard Hughes Foundation to MRC; AMS was supported in part by an undergraduate scholarship from Montana State University Scholars Program; SVI was supported by an undergraduate scholarship from the Montana Network for Biomedical Research; OL was supported in part by an NSF-IGERT graduate training grant. The authors thank C. Lenz, S. Barnett, K. Lustig, J. Kundert, A. Lammers, C. Kioussi, C. Stoner, G. Callis, and K. McInnerney for technical assistance; R. Behringer, W. Shawlot, V. Gladyshev, and A. Boulet for providing plasmids; and Y.-s. Piao for commenting on the manuscript.
Footnotes
Abbreviations: DAPI, 4′,6-diamidino-2-phenyindol; EC, ectoplacental cone; EDTA, ethylenediaminetetraacetic acid; EGF, epidermal growth factor; Em, embryonic; Ex, extraembryonic; FAD, flavin-adenine dinucleotide; FGF, fibroblast growth factor; GST, glutathione S-transferase; NADPH, reduced nicotinamide adenine dinucleotide phosphate; PCR, polymerase chain reaction; PE, parietal endoderm; Prdx, peroxiredoxin; PS, primitive streak; RT-PCR, reverse transcriptase-templated polymerase chain reaction; SDS, sodium dodecylsulfate; SEC, selenocysteine; Srxn, sulfiredoxin; TGC, trophoblast giant cells; Txn, thioredoxin; Txnrd, thioredoxin reductase; VE, visceral endoderm.
For tables and figures, the prefix “S” refers to exhibits in the Supplemental section; absence of a prefix refers to exhibits in the manuscript.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Arner ES, Holmgren A. Physiological functions of thioredoxin and thioredoxin reductase. Eur J Biochem. 2000;267:6102–109. doi: 10.1046/j.1432-1327.2000.01701.x. [DOI] [PubMed] [Google Scholar]
- 2.Holmgren A. Thioredoxin. Annu Rev Biochem. 1985;54:237–71. doi: 10.1146/annurev.bi.54.070185.001321. [DOI] [PubMed] [Google Scholar]
- 3.Moore EC, Reichard P, Thelander L. Enzymatic Synthesis Of Deoxyribonucleotides. V. Purification And Properties Of Thioredoxin Reductase From Escherichia Coli B. J Biol Chem. 1964;239:3445–452. [PubMed] [Google Scholar]
- 4.Laurent TC, Moore EC, Reichard P. Enzymatic Synthesis Of Deoxyribonucleotides. Iv. Isolation And Characterization Of Thioredoxin, The Hydrogen Donor From Escherichia Coli B. J Biol Chem. 1964;239:3436–444. [PubMed] [Google Scholar]
- 5.Nakamura H. Thioredoxin and its related molecules: update 2005. Antioxid Redox Signal. 2005;7:823–28. doi: 10.1089/ars.2005.7.823. [DOI] [PubMed] [Google Scholar]
- 6.Nordberg J, Arner ES. Reactive oxygen species, antioxidants, and the mammalian thioredoxin system. Free Radic Biol Med. 2001;31:1287–312. doi: 10.1016/s0891-5849(01)00724-9. [DOI] [PubMed] [Google Scholar]
- 7.Powis G, Montfort WR. Properties and biological activities of thioredoxins. Annu Rev Biophys Biomol Struct. 2001;30:421–55. doi: 10.1146/annurev.biophys.30.1.421. [DOI] [PubMed] [Google Scholar]
- 8.Matsui M, Oshima M, Oshima H, Takaku K, Maruyama T, Yodoi J, Taketo MM. Early embryonic lethality caused by targeted disruption of the mouse thioredoxin gene. Dev Biol. 1996;178:179–85. doi: 10.1006/dbio.1996.0208. [DOI] [PubMed] [Google Scholar]
- 9.Nonn L, Williams RR, Erickson RP, Powis G. The absence of mitochondrial thioredoxin 2 causes massive apoptosis, exencephaly, and early embryonic lethality in homozygous mice. Mol Cell Biol. 2003;23:916–922. doi: 10.1128/MCB.23.3.916-922.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Aslund F, Ehn B, Miranda-Vizuete A, Pueyo C, Holmgren A. Two additional glutaredoxins exist in Escherichia coli: glutaredoxin 3 is a hydrogen donor for ribonucleotide reductase in a thioredoxin/glutaredoxin 1 double mutant. Proc Natl Acad Sci U S A. 1994;91:9813–9817. doi: 10.1073/pnas.91.21.9813. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Muller EG. Thioredoxin deficiency in yeast prolongs S phase and shortens the G1 interval of the cell cycle. J Biol Chem. 1991;266:9194–9202. [PubMed] [Google Scholar]
- 12.Muller EG. A glutathione reductase mutant of yeast accumulates high levels of oxidized glutathione and requires thioredoxin for growth. Mol Biol Cell. 1996;7:1805–1813. doi: 10.1091/mbc.7.11.1805. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Gleason FK, Holmgren A. Thioredoxin and related proteins in procaryotes. FEMS Microbiol Rev. 1988;4:271–297. doi: 10.1111/j.1574-6968.1988.tb02747.x. [DOI] [PubMed] [Google Scholar]
- 14.Yamawaki H, Berk BC. Thioredoxin: a multifunctional antioxidant enzyme in kidney, heart and vessels. Curr Opin Nephrol Hypertens. 2005;14:149–153. doi: 10.1097/00041552-200503000-00010. [DOI] [PubMed] [Google Scholar]
- 15.Gladyshev VN, Hatfield DL. Selenocysteine-containing proteins in mammals. J Biomed Sci. 1999;6:151–160. doi: 10.1007/BF02255899. [DOI] [PubMed] [Google Scholar]
- 16.Gladyshev VN, Jeang KT, Stadtman TC. Selenocysteine, identified as the penultimate C-terminal residue in human T-cell thioredoxin reductase, corresponds to TGA in the human placental gene. Proc Natl Acad Sci U S A. 1996;93:6146–6151. doi: 10.1073/pnas.93.12.6146. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Bjornstedt M, Kumar S, Bjorkhem L, Spyrou G, Holmgren A. Selenium and the thioredoxin and glutaredoxin systems. Biomed Environ Sci. 1997;10:271–279. [PubMed] [Google Scholar]
- 18.Gromer S, Johansson L, Bauer H, Arscott LD, Rauch S, Ballou DP, Williams CH, Jr, Schirmer RH, Arner ES. Active sites of thioredoxin reductases: why selenoproteins? Proc Natl Acad Sci U S A. 2003;100:12618–12623. doi: 10.1073/pnas.2134510100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Mustacich D, Powis G. Thioredoxin reductase. Biochem J. 2000;346(Pt 1):1–8. [PMC free article] [PubMed] [Google Scholar]
- 20.Holmgren A. Antioxidant function of thioredoxin and glutaredoxin systems. Antioxid Redox Signal. 2000;2:811–820. doi: 10.1089/ars.2000.2.4-811. [DOI] [PubMed] [Google Scholar]
- 21.Hanna IR, Taniyama Y, Szocs K, Rocic P, Griendling KK. NAD(P)H oxidase-derived reactive oxygen species as mediators of angiotensin II signaling. Antioxid Redox Signal. 2002;4:899–914. doi: 10.1089/152308602762197443. [DOI] [PubMed] [Google Scholar]
- 22.Jackson SH, Devadas S, Kwon J, Pinto LA, Williams MS. T cells express a phagocyte-type NADPH oxidase that is activated after T cell receptor stimulation. Nat Immunol. 2004;5:818–827. doi: 10.1038/ni1096. [DOI] [PubMed] [Google Scholar]
- 23.Kwon J, Devadas S, Williams MS. T cell receptor-stimulated generation of hydrogen peroxide inhibits MEK-ERK activation and lck serine phosphorylation. Free Radic Biol Med. 2003;35:406–417. doi: 10.1016/s0891-5849(03)00318-6. [DOI] [PubMed] [Google Scholar]
- 24.Mahadev K, Motoshima H, Wu X, Ruddy JM, Arnold RS, Cheng G, Lambeth JD, Goldstein BJ. The NAD(P)H oxidase homolog Nox4 modulates insulin-stimulated generation of H2O2 and plays an integral role in insulin signal transduction. Mol Cell Biol. 2004;24:1844–1854. doi: 10.1128/MCB.24.5.1844-1854.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Peus D, Beyerle A, Vasa M, Pott M, Meves A, Pittelkow MR. Antipsoriatic drug anthralin induces EGF receptor phosphorylation in keratinocytes: requirement for H(2)O(2) generation. Exp Dermatol. 2004;13:78–85. doi: 10.1111/j.0906-6705.2004.00119.x. [DOI] [PubMed] [Google Scholar]
- 26.Rhee SG, Chang TS, Bae YS, Lee SR, Kang SW. Cellular regulation by hydrogen peroxide. J Am Soc Nephrol. 2003;14:S211–215. doi: 10.1097/01.asn.0000077404.45564.7e. [DOI] [PubMed] [Google Scholar]
- 27.Ushio-Fukai M, Tang Y, Fukai T, Dikalov SI, Ma Y, Fujimoto M, Quinn MT, Pagano PJ, Johnson C, Alexander RW. Novel role of gp91(phox)-containing NAD(P)H oxidase in vascular endothelial growth factor-induced signaling and angiogenesis. Circ Res. 2002;91:1160–1167. doi: 10.1161/01.res.0000046227.65158.f8. [DOI] [PubMed] [Google Scholar]
- 28.Arrigo AP. Gene expression and the thiol redox state. Free Radic Biol Med. 1999;27:936–944. doi: 10.1016/s0891-5849(99)00175-6. [DOI] [PubMed] [Google Scholar]
- 29.Hainaut P, Mann K. Zinc binding and redox control of p53 structure and function. Antioxid Redox Signal. 2001;3:611–623. doi: 10.1089/15230860152542961. [DOI] [PubMed] [Google Scholar]
- 30.Toone WM, Morgan BA, Jones N. Redox control of AP-1-like factors in yeast and beyond. Oncogene. 2001;20:2336–2346. doi: 10.1038/sj.onc.1204384. [DOI] [PubMed] [Google Scholar]
- 31.Delaunay A, Pflieger D, Barrault MB, Vinh J, Toledano MB. A thiol peroxidase is an H2O2 receptor and redox-transducer in gene activation. Cell. 2002;111:471–481. doi: 10.1016/s0092-8674(02)01048-6. [DOI] [PubMed] [Google Scholar]
- 32.Carmel-Harel O, Stearman R, Gasch AP, Botstein D, Brown PO, Storz G. Role of thioredoxin reductase in the Yap1p-dependent response to oxidative stress in Saccharomyces cerevisiae. Mol Microbiol. 2001;39:595–605. doi: 10.1046/j.1365-2958.2001.02255.x. [DOI] [PubMed] [Google Scholar]
- 33.Schmidt EE, Hanson ES, Capecchi MR. Sequence-independent assembly of spermatid mRNAs into messenger ribonucleoprotein particles. Mol Cell Biol. 1999;19:3904–3915. doi: 10.1128/mcb.19.5.3904. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Schmidt EE, Bondareva AA, Radke JR, Capecchi MR. Fundamental cellular processes do not require vertebrate-specific sequences within the TATA-binding protein. J Biol Chem. 2003;278:6168–6174. doi: 10.1074/jbc.M211205200. [DOI] [PubMed] [Google Scholar]
- 35.Herrmann BG, Labeit S, Poustka A, King TR, Lehrach H. Cloning of the T gene required in mesoderm formation in the mouse. Nature. 1990;343:617–622. doi: 10.1038/343617a0. [DOI] [PubMed] [Google Scholar]
- 36.Dono R, Scalera L, Pacifico F, Acampora D, Persico MG, Simeone A. The murine cripto gene: expression during mesoderm induction and early heart morphogenesis. Development. 1993;118:1157–1168. doi: 10.1242/dev.118.4.1157. [DOI] [PubMed] [Google Scholar]
- 37.Barnes JD, Crosby JL, Jones CM, Wright CV, Hogan BL. Embryonic expression of Lim-1, the mouse homolog of Xenopus Xlim-1, suggests a role in lateral mesoderm differentiation and neurogenesis. Dev Biol. 1994;161:168–178. doi: 10.1006/dbio.1994.1018. [DOI] [PubMed] [Google Scholar]
- 38.MacArthur CA, Shankar DB, Shackleford GM. Fgf-8, activated by proviral insertion, cooperates with the Wnt-1 transgene in murine mammary tumorigenesis. J Virol. 1995;69:2501–2507. doi: 10.1128/jvi.69.4.2501-2507.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Smith DE, Franco del Amo F, Gridley T. Isolation of Sna, a mouse gene homologous to the Drosophila genes snail and escargot: its expression pattern suggests multiple roles during postimplantation development. Development. 1992;116:1033–1039. doi: 10.1242/dev.116.4.1033. [DOI] [PubMed] [Google Scholar]
- 40.Rundlof AK, Janard M, Miranda-Vizuete A, Arner ES. Evidence for intriguingly complex transcription of human thioredoxin reductase 1. Free Radic Biol Med. 2004;36:641–656. doi: 10.1016/j.freeradbiomed.2003.12.004. [DOI] [PubMed] [Google Scholar]
- 41.Su D, Gladyshev VN. Alternative splicing involving the thioredoxin reductase module in mammals: a glutaredoxin-containing thioredoxin reductase 1. Biochemistry. 2004;43:12177–12188. doi: 10.1021/bi048478t. [DOI] [PubMed] [Google Scholar]
- 42.Sun QA, Kirnarsky L, Sherman S, Gladyshev VN. Selenoprotein oxidoreductase with specificity for thioredoxin and glutathione systems. Proc Natl Acad Sci U S A. 2001;98:3673–3678. doi: 10.1073/pnas.051454398. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Vlamis-Gardikas A, Holmgren A. Thioredoxin and glutaredoxin isoforms. Methods Enzymol. 2002;347:286–296. doi: 10.1016/s0076-6879(02)47028-0. [DOI] [PubMed] [Google Scholar]
- 44.Schmidt EE. Transcriptional promiscuity in testes. Curr Biol. 1996;6:768–769. doi: 10.1016/s0960-9822(02)00589-4. [DOI] [PubMed] [Google Scholar]
- 45.Biterova EI, Turanov AA, Gladyshev VN, Barycki JJ. Crystal structures of oxidized and reduced mitochondrial thioredoxin reductase provide molecular details of the reaction mechanism. Proc Natl Acad Sci U S A. 2005;102:15018–15023. doi: 10.1073/pnas.0504218102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Sandalova T, Zhong L, Lindqvist Y, Holmgren A, Schneider G. Three-dimensional structure of a mammalian thioredoxin reductase: implications for mechanism and evolution of a selenocysteine-dependent enzyme. Proc Natl Acad Sci U S A. 2001;98:9533–9538. doi: 10.1073/pnas.171178698. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Kozak M. Point mutations define a sequence flanking the AUG initiator codon that modulates translation by eukaryotic ribosomes. Cell. 1986;44:283–292. doi: 10.1016/0092-8674(86)90762-2. [DOI] [PubMed] [Google Scholar]
- 48.Batten BE, Haar JL. Fine structural differentiation of germ layers in the mouse at the time of mesoderm formation. Anat Rec. 1979;194:125–141. doi: 10.1002/ar.1091940109. [DOI] [PubMed] [Google Scholar]
- 49.Tsang TE, Kinder SJ, Tam PP. Experimental analysis of the emergence of left-right asymmetry of the body axis in early postimplantation mouse embryos. Cell Mol Biol (Noisy-le-grand) 1999;45:493–503. [PubMed] [Google Scholar]
- 50.Vincent SD, Dunn NR, Hayashi S, Norris DP, Robertson EJ. Cell fate decisions within the mouse organizer are governed by graded Nodal signals. Genes Dev. 2003;17:1646–1662. doi: 10.1101/gad.1100503. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Merwin JR, Mustacich DJ, Muller EG, Pearson GD, Merrill GF. Reporter gene transactivation by human p53 is inhibited in thioredoxin reductase null yeast by a mechanism associated with thioredoxin oxidation and independent of changes in the redox state of glutathione. Carcinogenesis. 2002;23:1609–1615. doi: 10.1093/carcin/23.10.1609. [DOI] [PubMed] [Google Scholar]
- 52.Pool-Zobel B, Veeriah S, Bohmer FD. Modulation of xenobiotic metabolising enzymes by anticarcinogens -- focus on glutathione S-transferases and their role as targets of dietary chemoprevention in colorectal carcinogenesis. Mutat Res. 2005;591:74–92. doi: 10.1016/j.mrfmmm.2005.04.020. [DOI] [PubMed] [Google Scholar]
- 53.Werth MT, Johnson MK. Magnetic circular dichroism and electron paramagnetic resonance studies of iron(II)-metallothionein. Biochemistry. 1989;28:3982–3988. doi: 10.1021/bi00435a053. [DOI] [PubMed] [Google Scholar]
- 54.Kojima N, Young CR, Bates GW. Failure of metallothionein to bind iron or act as an iron mobilizing agent. Biochim Biophys Acta. 1982;716:273–275. doi: 10.1016/0304-4165(82)90278-1. [DOI] [PubMed] [Google Scholar]
- 55.Puntarulo S. Iron, oxidative stress and human health. Mol Aspects Med. 2005;26:299–312. doi: 10.1016/j.mam.2005.07.001. [DOI] [PubMed] [Google Scholar]
- 56.Hayes JD, Pulford DJ. The glutathione S-transferase supergene family: regulation of GST and the contribution of the isoenzymes to cancer chemoprotection and drug resistance. Crit Rev Biochem Mol Biol. 1995;30:445–600. doi: 10.3109/10409239509083491. [DOI] [PubMed] [Google Scholar]
- 57.Maret W. Cellular zinc and redox states converge in the metallothionein/thionein pair. J Nutr. 2003;133:1460S–1462S. doi: 10.1093/jn/133.5.1460S. [DOI] [PubMed] [Google Scholar]
- 58.Maret W, Vallee BL. Thiolate ligands in metallothionein confer redox activity on zinc clusters. Proc Natl Acad Sci U S A. 1998;95:3478–3482. doi: 10.1073/pnas.95.7.3478. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Roschitzki B, Vasak M. Redox labile site in a Zn4 cluster of Cu4,Zn4-metallothionein-3. Biochemistry. 2003;42:9822–9828. doi: 10.1021/bi034816z. [DOI] [PubMed] [Google Scholar]
- 60.Haq F, Mahoney M, Koropatnick J. Signaling events for metallothionein induction. Mutat Res. 2003;533:211–226. doi: 10.1016/j.mrfmmm.2003.07.014. [DOI] [PubMed] [Google Scholar]
- 61.Samson SL, Gedamu L. Molecular analyses of metallothionein gene regulation. Prog Nucleic Acid Res Mol Biol. 1998;59:257–288. doi: 10.1016/s0079-6603(08)61034-x. [DOI] [PubMed] [Google Scholar]
- 62.Yepiskoposyan H, Egli D, Fergestad T, Selvaraj A, Treiber C, Multhaup G, Georgiev O, Schaffner W. Transcriptome response to heavy metal stress in Drosophila reveals a new zinc transporter that confers resistance to zinc. Nucleic Acids Res. 2006;34:4866–4877. doi: 10.1093/nar/gkl606. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Magdolen V, Oechsner U, Trommler P, Bandlow W. Transcriptional control by galactose of a yeast gene encoding a protein homologous to mammalian aldo/keto reductases. Gene. 1990;90:105–114. doi: 10.1016/0378-1119(90)90445-w. [DOI] [PubMed] [Google Scholar]
- 64.Doorn JA, Maser E, Blum A, Claffey DJ, Petersen DR. Human carbonyl reductase catalyzes reduction of 4-oxonon-2-enal. Biochemistry. 2004;43:13106–13114. doi: 10.1021/bi049136q. [DOI] [PubMed] [Google Scholar]
- 65.Maser E. Neuroprotective role for carbonyl reductase? Biochem Biophys Res Commun. 2006;340:1019–1022. doi: 10.1016/j.bbrc.2005.12.113. [DOI] [PubMed] [Google Scholar]
- 66.Dietz KJ, Horling F, Konig J, Baier M. The function of the chloroplast 2-cysteine peroxiredoxin in peroxide detoxification and its regulation. J Exp Bot. 2002;53:1321–1329. [PubMed] [Google Scholar]
- 67.Rhee SG, Chae HZ, Kim K. Peroxiredoxins: a historical overview and speculative preview of novel mechanisms and emerging concepts in cell signaling. Free Radic Biol Med. 2005;38:1543–1552. doi: 10.1016/j.freeradbiomed.2005.02.026. [DOI] [PubMed] [Google Scholar]
- 68.Wood ZA, Poole LB, Karplus PA. Peroxiredoxin evolution and the regulation of hydrogen peroxide signaling. Science. 2003;300:650–653. doi: 10.1126/science.1080405. [DOI] [PubMed] [Google Scholar]
- 69.Wood ZA, Schroder E, Robin Harris J, Poole LB. Structure, mechanism and regulation of peroxiredoxins. Trends Biochem Sci. 2003;28:32–40. doi: 10.1016/s0968-0004(02)00003-8. [DOI] [PubMed] [Google Scholar]
- 70.Rhee SG. Cell signaling. H2O2, a necessary evil for cell signaling. Science. 2006;312:1882–1883. doi: 10.1126/science.1130481. [DOI] [PubMed] [Google Scholar]
- 71.Jacob C, Holme AL, Fry FH. The sulfinic acid switch in proteins. Org Biomol Chem. 2004;2:1953–1956. doi: 10.1039/b406180b. [DOI] [PubMed] [Google Scholar]
- 72.Chang TS, Jeong W, Woo HA, Lee SM, Park S, Rhee SG. Characterization of mammalian sulfiredoxin and its reactivation of hyperoxidized peroxiredoxin through reduction of cysteine sulfinic acid in the active site to cysteine. J Biol Chem. 2004;279:50994–51001. doi: 10.1074/jbc.M409482200. [DOI] [PubMed] [Google Scholar]
- 73.Jeong W, Park SJ, Chang TS, Lee DY, Rhee SG. Molecular mechanism of the reduction of cysteine sulfinic acid of peroxiredoxin to cysteine by mammalian sulfiredoxin. J Biol Chem. 2006;281:14400–14407. doi: 10.1074/jbc.M511082200. [DOI] [PubMed] [Google Scholar]
- 74.Woo HA, Jeong W, Chang TS, Park KJ, Park SJ, Yang JS, Rhee SG. Reduction of cysteine sulfinic acid by sulfiredoxin is specific to 2-cys peroxiredoxins. J Biol Chem. 2005;280:3125–3128. doi: 10.1074/jbc.C400496200. [DOI] [PubMed] [Google Scholar]
- 75.Ashida M, Bito T, Budiyanto A, Ichihashi M, Ueda M. Involvement of EGF receptor activation in the induction of cyclooxygenase-2 in HaCaT keratinocytes after UVB. Exp Dermatol. 2003;12:445–452. doi: 10.1034/j.1600-0625.2003.00101.x. [DOI] [PubMed] [Google Scholar]
- 76.Callsen D, Sandau KB, Brune B. Nitric oxide and superoxide inhibit platelet-derived growth factor receptor phosphotyrosine phosphatases. Free Radic Biol Med. 1999;26:1544–1553. doi: 10.1016/s0891-5849(99)00015-5. [DOI] [PubMed] [Google Scholar]
- 77.Catarzi S, Biagioni C, Giannoni E, Favilli F, Marcucci T, Iantomasi T, Vincenzini MT. Redox regulation of platelet-derived-growth-factor-receptor: role of NADPH-oxidase and c-Src tyrosine kinase. Biochim Biophys Acta. 2005;1745:166–175. doi: 10.1016/j.bbamcr.2005.03.004. [DOI] [PubMed] [Google Scholar]
- 78.Nishinaka T, Yabe-Nishimura C. EGF receptor-ERK pathway is the major signaling pathway that mediates upregulation of aldose reductase expression under oxidative stress. Free Radic Biol Med. 2001;31:205–216. doi: 10.1016/s0891-5849(01)00571-8. [DOI] [PubMed] [Google Scholar]
- 79.Ohba M, Shibanuma M, Kuroki T, Nose K. Production of hydrogen peroxide by transforming growth factor-beta 1 and its involvement in induction of egr-1 in mouse osteoblastic cells. J Cell Biol. 1994;126:1079–1088. doi: 10.1083/jcb.126.4.1079. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Peus D, Meves A, Vasa RA, Beyerle A, O’Brien T, Pittelkow MR. H2O2 is required for UVB-induced EGF receptor and downstream signaling pathway activation. Free Radic Biol Med. 1999;27:1197–1202. doi: 10.1016/s0891-5849(99)00198-7. [DOI] [PubMed] [Google Scholar]
- 81.Peus D, Vasa RA, Meves A, Pott M, Beyerle A, Squillace K, Pittelkow MR. H2O2 is an important mediator of UVB-induced EGF-receptor phosphorylation in cultured keratinocytes. J Invest Dermatol. 1998;110:966–971. doi: 10.1046/j.1523-1747.1998.00210.x. [DOI] [PubMed] [Google Scholar]
- 82.Rhee SG, Kang SW, Jeong W, Chang TS, Yang KS, Woo HA. Intracellular messenger function of hydrogen peroxide and its regulation by peroxiredoxins. Curr Opin Cell Biol. 2005;17:183–189. doi: 10.1016/j.ceb.2005.02.004. [DOI] [PubMed] [Google Scholar]
- 83.Thannickal VJ, Aldweib KD, Fanburg BL. Tyrosine phosphorylation regulates H2O2 production in lung fibroblasts stimulated by transforming growth factor beta1. J Biol Chem. 1998;273:23611–23615. doi: 10.1074/jbc.273.36.23611. [DOI] [PubMed] [Google Scholar]
- 84.Gorreta F, Runfola TP, VanMeter AJ, Barzaghi D, Chandhoke V, Del Giacco L. Identification of thioredoxin reductase 1-regulated genes using small interference RNA and cDNA microarray. Cancer Biol Ther. 2005;4:1079–1088. doi: 10.4161/cbt.4.10.1987. [DOI] [PubMed] [Google Scholar]
- 85.Jakupoglu C, Przemeck GK, Schneider M, Moreno SG, Mayr N, Hatzopoulos AK, de Angelis MH, Wurst W, Bornkamm GW, Brielmeier M, Conrad M. Cytoplasmic thioredoxin reductase is essential for embryogenesis but dispensable for cardiac development. Mol Cell Biol. 2005;25:1980–1988. doi: 10.1128/MCB.25.5.1980-1988.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.