

Abstract
Mitochondria are a major source of reactive oxygen species (ROS) and oxidative stress, key contributors to aging and neurodegenerative disorders. We report that gonadal hormones influence brain mitochondrial ROS production in both females and males. Initial experiments showed that estrogen decreases mitochondrial superoxide production in a receptor-mediated manner, as measured by MitoSOX fluorescence in differentiated PC-12 cells. We then assessed in vivo effects of gonadal hormones on brain mitochondrial oxidative stress in female and male rats. Brain mitochondria were isolated to measure a functional indicator of ROS, i.e., activity of the ROS-sensitive mitochondrial enzyme, aconitase. Gonadectomy of both males and females caused a decrease in aconitase activity, suggesting endogenous gonadal hormones influence mitochondrial ROS production in the brain. In vivo treatment of gonadectomized animals with testosterone or dihydrotestosterone (DHT) had no effect, but estrogen replacement significantly increased aconitase activity in brain mitochondria from both female and male rats. This indicates estrogen decreases brain mitochondrial ROS production in vivo. Sex hormone treatments did not affect protein levels of brain mitochondrial uncoupling proteins (UCP-2, 4, and 5). However, estrogen did increase the activity, but not the levels, of manganese superoxide dismutase (MnSOD), the mitochondrial enzyme that catalyzes superoxide radical breakdown, in brain mitochondria from both female and male rats. Thus, in contrast to the lack of effect of androgens on mitochondrial ROS, estrogen suppression of mitochondrial oxidative stress may influence neurological disease incidence and progression in both females and males.
Keywords: estrogen, testosterone, mitochondria, neuroprotection, oxidation, endocrine
1. Introduction
Increasing evidence points to a central role of oxidative stress and mitochondrial dysfunction in neurodegenerative disease pathophysiology (Andersen, 2004; Beal, 2005; Calabrese et al., 2001; Wallace, 2005). Mitochondria are not only responsible for ATP production, but also are a major source of ROS (Turrens, 2003) and a key player in apoptosis (Szewczyk and Wojtczak, 2002). The redox centers of the mitochondrial electron transport chain constitute the major source of cellular superoxide production as a by-product of oxidative phosphorylation (Beyer, 1992; Cadenas and Davies, 2000; Chance et al., 1979; Turrens, 2003; Wallace, 2005). Chronic exposure to mitochondrial ROS leads to inactivation of key mitochondrial enzymes and accumulation of mitochondrial DNA mutations (Wallace, 2005). It has been proposed that aging and age-related disorders are the result of a decline in mitochondrial function due to cumulative oxidative damage (Beckman and Ames, 1998; Harman, 2006; Vina et al., 2003; Wallace, 2005).
Many studies have focused on protective effects of estrogen (Green and Simpkins, 2000; Henderson, 2006; Wise, 2002; Yang et al., 2005) with less attention on the impact of androgens (Liu et al., 2003). However, little is known regarding effects of sex hormones on brain mitochondrial ROS production. Emerging evidence indicates mitochondria are a novel target of estrogen (Duckles et al., 2006; Nilsen and Brinton, 2004; Singh et al., 2006). We recently demonstrated in cerebral blood vessels of female rats that estrogen decreases mitochondrial H2O2 production (Stirone et al., 2005). Estrogen also increased expression of several mitochondrial electron transport chain proteins, including cytochrome c and complex IV subunits, and increased complex IV and citrate synthase enzyme activities.
In the present study, we tested the hypothesis that gonadal hormones affect superoxide production by brain mitochondria. First we examined effects of estrogen using direct measurements of superoxide with the MitoSOX Red mitochondrial-specific dye in nerve growth factor-differentiated PC-12 cells, commonly used to model neurons in culture (Greene and Tischler, 1976). Subsequently, we used in vivo models of chronic hormone loss or replacement to determine the impact of estrogen and androgens on brain mitochondria of male and female rats. Activity of aconitase, a tricarboxylic acid cycle enzyme, was measured as a functional indicator of mitochondrial ROS production under physiological conditions. The iron-sulfur core of aconitase is reversibly oxidized by superoxide leading to progressive inactivation of aconitase as mitochondrial ROS increases (Gardner and Fridovich, 1991; Gardner and Fridovich, 1992; Hausladen and Fridovich, 1994; Kennedy et al., 1983). Furthermore, oxidized aconitase can be reactivated by reducing agents in vitro, allowing confirmation of the mechanism of aconitase inactivation as due to alterations in enzyme activity per se (Patel et al., 1996; Williams et al., 1998).
Our results indicate endogenous gonadal hormones influence ROS production in brain mitochondria. In particular, suppression of ROS by estrogen gives new insight into the role of estrogen in female, as well as male, brains. Long-term mitochondrial protection is a novel neuroprotective mechanism that needs to be considered in the controversy surrounding estrogen impact and therapy in Alzheimer’s disease and other aging disorders of the brain (Henderson, 2006).
2. Results
Estrogen Decreases Superoxide Production in Differentiated PC-12 Cells
Figure 1A–C verifies the mitochondrial targeting of MitoSOX in NGF-differentiated PC-12 cells. The dye shows exclusive co-localization with subunit I of complex IV, a protein encoded by mitochondrial DNA and restricted to mitochondria. As shown in Figure 1D, mitochondrial superoxide production was stimulated by addition of complex I substrates, pyruvate and malate. Further addition of MnTBAP, a superoxide dismutase mimetic used to scavenge superoxide, decreased MitoSOX fluorescence, validating this measurement for superoxide. The substrate-induced rise in fluorescence was slower for 17β-estradiol-treated cells compared to vehicle-treated cells, indicating a lower rate of superoxide production. As summarized in Figure 1E, 10 nM 17β-estradiol significantly reduced the rate of mitochondrial superoxide production in differentiated PC-12 cells, and this effect was blocked by the estrogen receptor antagonist, ICI-182,780. Since estrogen decreased superoxide production in differentiated PC-12 cells, we then determined the in vivo effect of physiological levels of sex hormones on brain mitochondrial ROS production.
Figure 1. Effects of estrogen on superoxide production in differentiated PC-12 cells.
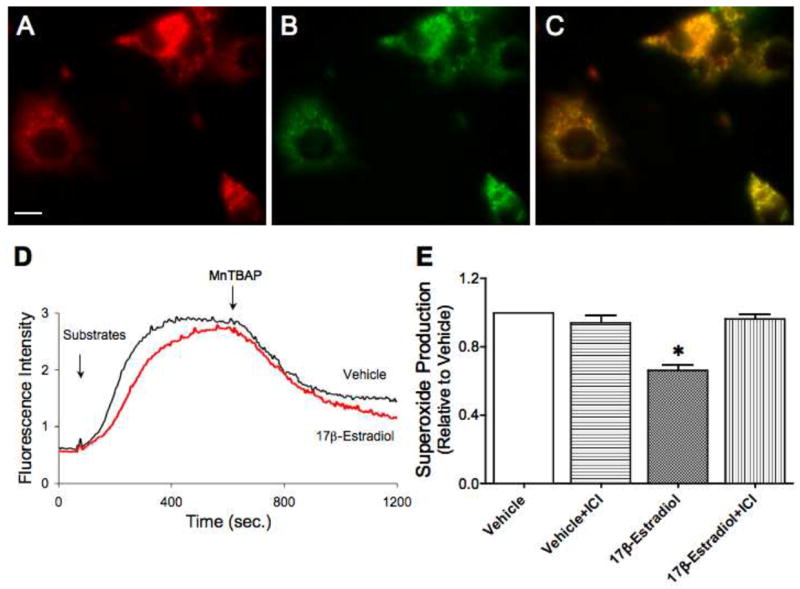
PC-12 cells were differentiated with nerve growth factor for 10 days prior to treatment with either 10 nM 17β-estradiol, 1 μM ICI-182,780 alone or in the presence of 17β-estradiol, or vehicle. Cells were exposed to these treatments for 24 h prior to measurement of superoxide production in live cells using the MitoSOX Red reagent. A. MitoSOX Red mitochondrial superoxide indicator staining shown in red. Scale bar, 10 μm. B. Green fluorescence staining for subunit I of complex IV, a mitochondrial encoded-protein. C. Immunofluorescence co-localization (yellow) of MitoSOX Red and subunit I of complex IV. D. Representative tracing of MitoSOX dye fluorescence intensity reflecting mitochondrial superoxide levels. Tracings for differentiated PC-12 cells pre-treated with 17β-estradiol or vehicle are shown. Complex I substrates, pyruvate and malate (both at 1M), were added to initiate the reaction. After a plateau was reached, MnTBAP, a superoxide dismutase mimetic, was added. E. Mean values of mitochondrial superoxide production, corrected for sample protein concentration and expressed relative to vehicle control, are shown. Values are means ± SEM. *Significantly different from other groups; P ≤ 0.05; n=4.
In Vivo Animal Hormone Treatment
After 3–4 weeks of chronic in vivo hormone treatment, we measured serum hormone levels. As shown in Tables 1 and 2, gonadectomy dramatically lowered circulating levels of 17β-estradiol in female and male rats and also suppressed levels of testosterone and DHT in males. Chronic hormone treatments achieved physiological serum levels of 17β-estradiol, testosterone, and DHT in gonadectomized animals, as we have shown previously (Geary et al., 2000; McNeill et al., 1999; Razmara et al., 2005). Estrogen treatment of OVX females significantly decreased body weight and increased uterine weight compared to OVX rats. Chronic treatment of ORX males with androgens, either testosterone or DHT, significantly increased, while estrogen treatment significantly decreased, body weights compared to ORX animals. These results confirm hormone treatments were biologically active in both female and male rats.
Table 1.
Effect of in vivo hormone treatment on serum levels of 17β-estradiol, body weights, and uterine weights in female rats
| Animal Group | 17β-Estradiol (pg/ml) | Body Weight (g) | Uterine Weight (mg) |
|---|---|---|---|
| Intact Female | 49±8* | 177±2* | 78±5* |
| OVX | 9±1 | 188±3 | 44±6 |
| OVX+E | 65±9* | 178±2* | 111±8* |
Values represent means ± SEM; OVX, ovariectomized; E, 17β-estradiol;
Significantly different than OVX (P ≤ 0.05), n=8–13
Table 2.
Effect of in vivo hormone treatment on serum levels of testosterone, DHT, 17β-estradiol, and body weights in male rats
| Animal Group | Testosterone (ng/ml) | DHT (pg/ml) | 17β-Estradiol (pg/ml) | Body Weight (g) |
|---|---|---|---|---|
| Intact Male | 2.2±0.2* | 476±45* | 27±4* | 287±4 |
| ORX | ND | 49±5 | 8±1 | 292±3 |
| ORX+T | 3.3±0.4* | 214±15* | 6±1 | 317±2* |
| ORX+DHT | ND | 288±24* | 7±1 | 311±4* |
| ORX+E | ND | 49±6 | 32±3* | 268±2* |
Values represent means ± SEM; ORX, orchiectomized; T, testosterone, DHT, dihydrotestosterone; E, 17β-estradiol; ND, not detectable;
Significantly different than ORX (P ≤ 0.05), n=12–20
Estrogen Suppresses Brain Mitochondrial ROS Production in Female and Male Rats
Mitochondrial-derived ROS inactivate aconitase enzymatic activity, but do not alter fumarase activity. Therefore, the activity ratio of mitochondrial aconitase to fumarase (A/F) is a functional indicator of mitochondrial ROS production; higher A/F ratios indicate lower mitochondrial ROS production (Patel et al., 1996). In females, the A/F ratio for brain mitochondria was significantly decreased by ovariectomy, but increased by estrogen treatment (Figure 2A). There were no significant differences in fumarase enzyme activities in the female animal groups (P > 0.05; n=4).
Figure 2. Effects of ovariectomy and estrogen treatment on mitochondrial aconitase activity of female rat brains.
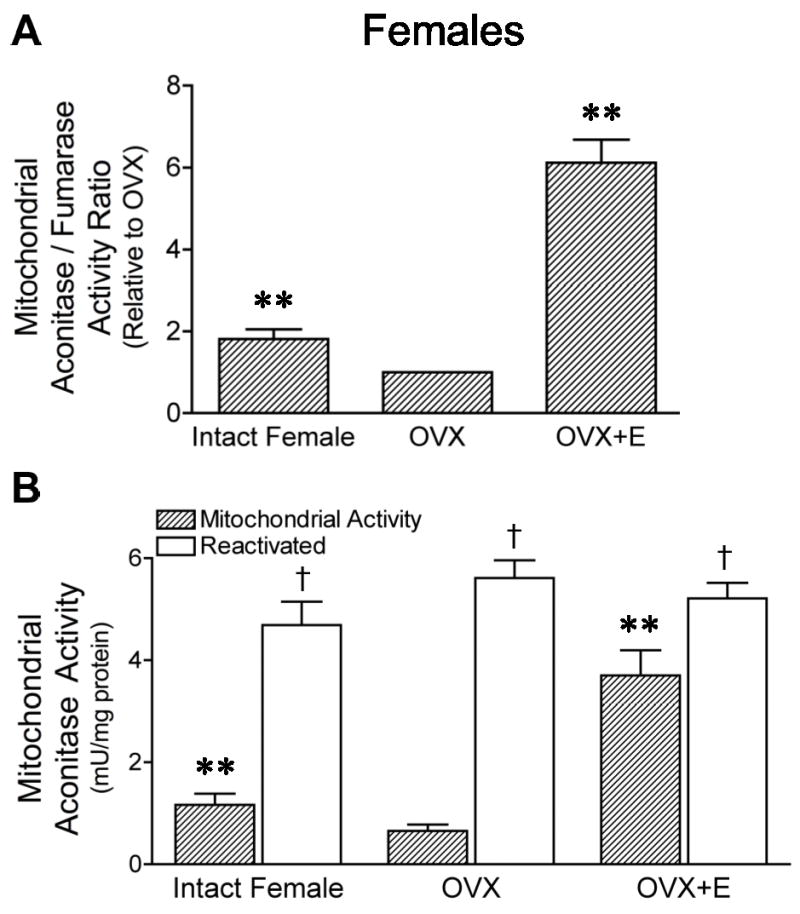
Mitochondria were isolated from intact, ovariectomized (OVX), and estrogen-treated OVX (OVX+E) female rat brains. A. As a functional indicator of mitochondrial ROS production, the ratio of activities of aconitase, a ROS-sensitive enzyme, to fumarase, a ROS-insensitive enzyme, was measured. The activity ratio relative to the OVX group is shown. Values are means ± SEM. **Significantly different from two other groups (P ≤ 0.05; n=4). B. Aconitase activity measured in isolated brain mitochondria before and after total enzyme reactivation with reducing reagents. Values are means ± SEM. **Significantly different from mitochondrial activity of two other groups; P ≤ 0.05; n=4. †Significantly different than activity before reactivation within each group; P ≤ 0.05; n=4.
Figure 2B shows the aconitase activity measured in isolated brain mitochondria compared with the total activity of the enzyme measured after reactivation with reducing agents in vitro. The latter was similar in all female rat groups, indicating estrogen did not affect enzyme protein expression. Compared to total activity, however, aconitase activity in freshly isolated mitochondria was decreased by 76±3% in intact female, 89±2% in OVX, and 29±8% in OVX+E groups, indicating that enzyme inactivation by ROS was less in brain mitochondria from female rats exposed to estrogen.
In males, the mitochondrial A/F ratio was significantly decreased by orchiectomy, compared to intact males (Figure 3A). The A/F ratio was unchanged by testosterone or DHT compared to ORX, but significantly increased by estrogen treatment. There were no significant differences in fumarase enzyme activities in the male animal groups (P > 0.05; n=4). Reducing reagents reactivated aconitase activity to similar levels in all male rat groups (Figure 3B). The degree of mitochondrial aconitase inactivation was 46±6% for intact male, 86±3% for ORX, 80±6% for ORX+DHT, and 36±8% for ORX+E. Aconitase inactivation was significantly lower in estrogen-treated males, reflecting a significant suppression by estrogen of mitochondrial ROS production.
Figure 3. Effect of orchiectomy and treatment with androgens or estrogen on aconitase activity in mitochondria of male rat brains.
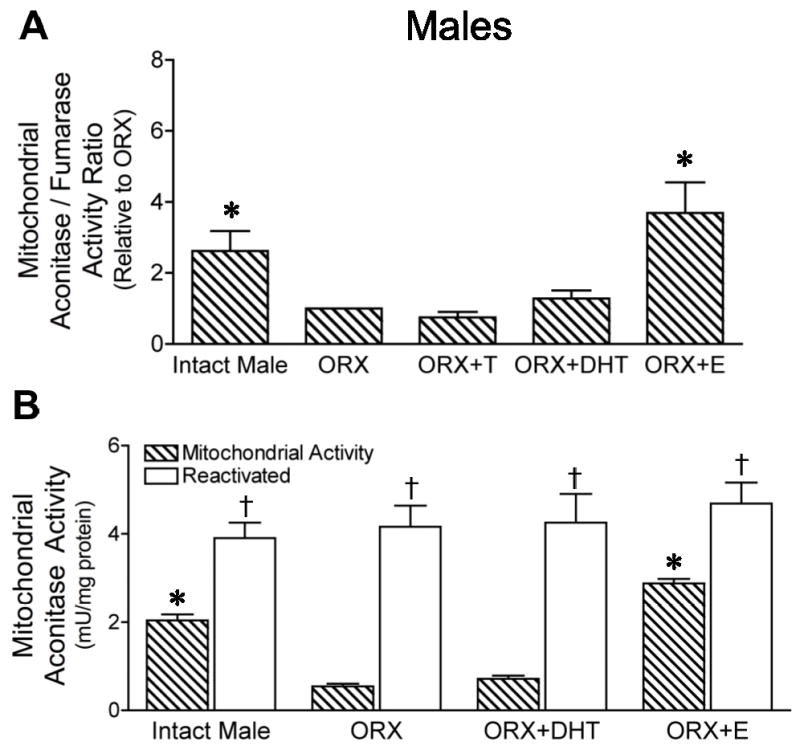
Mitochondria were isolated from intact and orchiectomized (ORX) male rat brains as well as from ORX males treated with either testosterone (ORX+T), dihydrotestosterone (ORX+DHT) or estrogen (ORX+E). A. The ratio of activities of aconitase, a ROS-sensitive enzyme, to fumarase, unaffected by ROS, was measured as a functional indicator of mitochondrial ROS production. The activity ratio relative to the ORX group is shown. Values are means ± SEM. *Significantly different from ORX, ORX+T, and ORX+DHT; P ≤ 0.05; n=4. B. Aconitase activity measured in isolated brain mitochondria before and after total enzyme reactivation with reducing reagents. Values are means ± SEM. *Significantly different from mitochondrial activity of ORX and ORX+DHT; P ≤ 0.05; n=4. †Significantly different from activity before reactivation within each group; P ≤ 0.05; n=4.
Effect of Sex Hormones on Brain Mitochondrial Uncoupling Proteins
Mitochondrial uncoupling proteins (UCPs) significantly modulate mitochondrial ROS production (Cannon et al., 2006; Casteilla et al., 2001; Rousset et al., 2004). We tested the hypothesis that gonadal hormones affect one or more of three isoforms important in the brain: UCP-2, 4 and 5 (Chan et al., 2006; Ho et al., 2006; Mattiasson et al., 2003). Figure 4 shows representative Western blots from brain mitochondrial fractions with bands migrating at 32 kDa for UCP-2, 4, and 5 and 31 kDa for the loading control, porin, in both female (Figure 4A) and male rats (Figure 4B). Corresponding porin bands are only shown for UCP-2 blots since similar bands were obtained from UCP-4 and 5 blots. All three UCP isoforms were detected in brain mitochondria (Figure 4), however, there were no significant differences in protein levels of UCP-2, 4, or 5 among the animal treatment groups (P > 0.05; n=4). Thus, neither gonadectomy nor sex hormone treatments affected brain mitochondrial protein levels of UCPs in either female or male rats.
Figure 4. Levels of uncoupling (UCP) proteins in mitochondria of female and male rat brains.
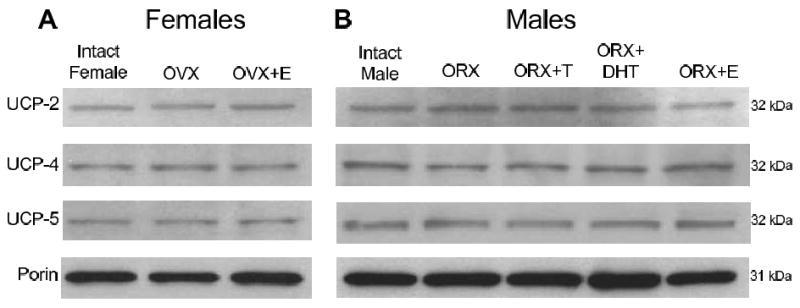
Bands migrating at 32 kDa were detected using antibodies directed against UCP-2, UCP-4, and UCP-5. Porin bands (31 kDa) are shown for the UCP-2 blot, and were run for each blot as a mitochondrial specific protein loading control. A. Representative Western blots show UCP-2, 4 and 5 protein in mitochondria isolated from intact, ovariectomized (OVX), and estrogen-treated OVX (OVX+E) female rat brains. B. Representative Western blots show UCP-2, 4 and 5 protein in mitochondria isolated from intact, orchiectomized (ORX), testosterone-treated ORX (ORX+T), DHT-treated ORX (ORX+DHT), and estrogen-treated ORX (ORX+E) male rat brains.
Effect of Sex Hormones on Brain Mitochondrial MnSOD Protein and Enzyme Activity
We examined effects of gonadectomy and treatment with androgens and estrogen on protein levels of MnSOD, the mitochondrial form of SOD, in female and male rat brain mitochondria (Figure 5). Levels of MnSOD protein were not significantly different among the female treatment groups, intact, OVX, and OVX+E (Figure 5B). In male brain mitochondria, MnSOD protein levels were unaffected by DHT treatment, however testosterone caused a small increase and estrogen caused a small decrease in MnSOD protein levels (Figure 5D).
Figure 5. Levels of manganese superoxide dismutase (MnSOD) protein in mitochondria of female and male rat brains.
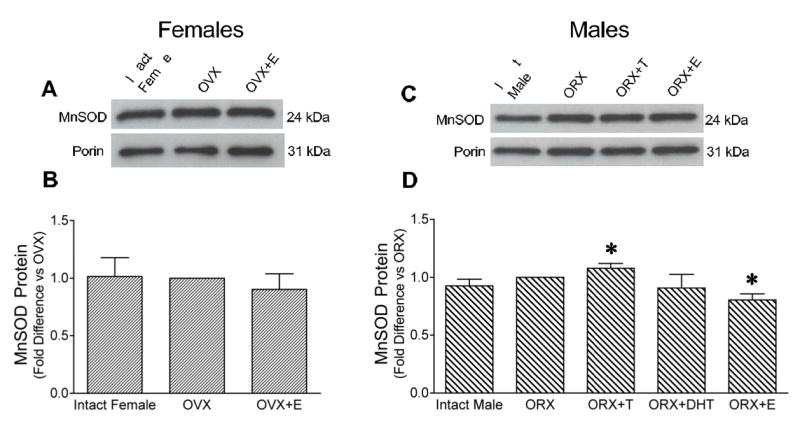
Bands migrating at 24 kDa were detected using an antibody directed against MnSOD, and porin bands (31 kDa) are shown as a mitochondrial specific protein loading control. Densities of MnSOD bands were measured, corrected for porin, and expressed relative to the gonadectomized group run on the same gel. A. Representative Western blot shows MnSOD protein in mitochondria isolated from intact, ovariectomized (OVX), and estrogen-treated OVX (OVX+E) female rat brains. B. Mean MnSOD protein levels in mitochondria isolated from intact, ovariectomized, and OVX+E female rat brains. Values are means ± SEM; P > 0.05; n=4. C. Representative Western blot shows MnSOD protein in mitochondria isolated from intact, orchiectomized (ORX), testosterone-treated ORX (ORX+T), and estrogen-treated ORX (ORX+E) male rat brains. D. Mean MnSOD protein levels, corrected for porin, in male rats, expressed relative to the ORX group. Values are means ± SEM. *Significantly different from all other groups; P ≤ 0.05; n=8 for intact, ORX, and ORX+E; n=4 for ORX+T and ORX+DHT.
The enzyme activity of MnSOD also was measured in brain mitochondria after chronic hormone treatment of female and male rats. Figure 6A shows a significant increase in MnSOD activity in mitochondria from brains of female rats treated with estrogen. Although testosterone and DHT did not alter MnSOD activity, estrogen treatment also caused a significant increase in MnSOD activity in male brain mitochondria (Figure 6B).
Figure 6. Activity of manganese superoxide dismutase (MnSOD) in female and male rat brain mitochondria.
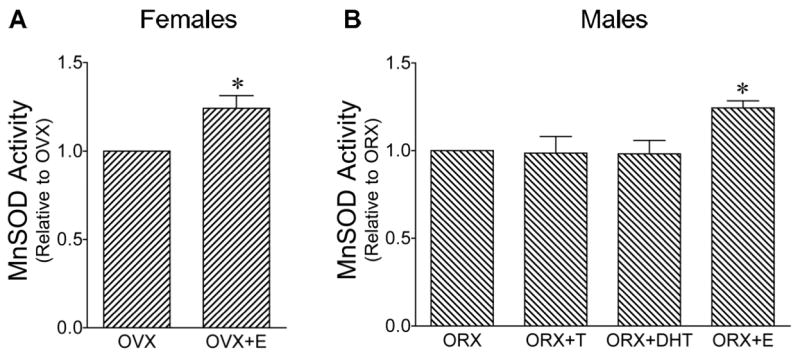
Values are means ± SEM. A. Activity of MnSOD in mitochondria isolated from ovariectomized (OVX) and estrogen-treated OVX (OVX+E) female rat brains expressed relative to the OVX group. *Significantly different from OVX; P ≤ 0.05; n=4. B. MnSOD enzyme activity in mitochondria isolated from orchiectomized (ORX), testosterone-treated ORX (ORX+T), DHT-treated ORX (ORX+DHT), and estrogen-treated ORX (ORX+E) male rat brains expressed relative to the ORX group. *Significantly different from other male groups; P ≤ 0.05; n=4.
3. Discussion
Estrogen was found to profoundly suppress mitochondrial oxidative stress in brains of both female and male rats. Similar effects of estrogen on neuronal-like PC-12 cells confirmed that estrogen inhibits mitochondrial superoxide production in a receptor-mediated manner. Estrogen also increased the activity of the mitochondrial antioxidant enzyme, MnSOD, in both female and male brains, suggesting one mechanism by which this hormone lowers mitochondrial ROS. An important strength of our work is that these striking effects of chronic estrogen treatment were observed using an integrative, whole animal model with exposure to physiological hormone levels, thus emphasizing the relevance of our findings to normal brain function. Interestingly, the androgens, testosterone and DHT, did not affect mitochondrial ROS production. However, castration of male rats significantly increased oxidative stress in brain mitochondria. This effect was reversed by estrogen, but not androgen replacement, suggesting that endogenous estrogen normally suppresses brain mitochondrial ROS production in males as well as in females.
As a major by-product of oxidative phosphorylation, superoxide is generated as electrons leak from the electron transport chain to react with molecular oxygen in the mitochondrial matrix (Cadenas and Davies, 2000; Chance et al., 1979; Wallace, 2005). We directly measured mitochondrial superoxide production in differentiated PC-12 cells, a neuronal model (Greene and Tischler, 1976), using MitoSOX Red. A major advantage of this live-cell permeable dye is its high selectivity for the mitochondrial matrix, where it is oxidized by superoxide and emits a red fluorescence (Robinson et al., 2006). We confirmed the organelle selectivity of MitoSOX Red in PC-12 cells by showing co-localization with subunit I of complex IV, a mitochondrial encoded-protein. The PC-12 cell experiments demonstrate that physiological concentrations of 17β-estradiol decrease superoxide production; and, since ICI-182,780 inhibited this effect, estrogen receptors mediate the suppression of mitochondrial superoxide production. Our recent pharmacological and siRNA data from human brain microvascular endothelial cells also show a role for estrogen receptors in decreasing mitochondrial superoxide production (A. Razmara, L. Sunday, C. Stirone, X. B. Wang, D. N. Krause, S. P. Duckles, and V. Procaccio, unpublished observations). These findings are consistent with receptor-activation by physiological estrogen concentrations, but are in contrast to direct antioxidant actions of the 17β-estradiol molecule that have been reported with high concentrations of this steroid (Behl et al., 1997; Keller et al., 1997; Prokai et al., 2003).
We then used an in vivo model of hormone treatment to investigate effects of physiological levels of gonadal hormones on brain mitochondrial ROS production. Since it was not feasible to use the MitoSOX Red reagent in vivo, we measured a well-accepted functional indicator of mitochondrial ROS, namely the activity of the Krebs cycle enzyme, aconitase (Kennedy et al., 1983; Patel et al., 1996; Tong et al., 2007). Aconitase activity is reversibly decreased when the iron-sulfur core of this enzyme is oxidized by ROS. Since the activity of fumarase is unaffected by ROS, the ratio of activities of mitochondrial aconitase and fumarase (A/F) provides a robust indicator of mitochondrial oxidative stress (Gardner and Fridovich, 1991; Gardner and Fridovich, 1992; Hausladen and Fridovich, 1994; Kennedy et al., 1983; Patel et al., 1996). Our A/F measurements suggest that, in both female and male brains, estrogen, either exogenous or endogenous, decreases mitochondrial ROS production, while androgens have no effect.
After reactivation of aconitase with reducing agents, enzyme activity was not different among the various treatment groups. Thus, it is unlikely that aconitase protein levels were affected by gonadectomy or hormone treatment. Interestingly, there also appears to be higher aconitase activity, and thus lower superoxide production, in both intact female and male rat brains compared to respective gonadectomized animals. These findings reveal that endogenous gonadal hormones can impact the production of ROS production in brain mitochondria. In intact males, the levels of both aconitase activity and serum 17β-estradiol correlated with what was found in estrogen-treated, orchiectomized males. Since androgens had no direct effect on aconitase activity, these findings suggest that endogenous estrogen may normally act to suppress mitochondrial oxidative stress in the male, as well as female, brain. It should be noted that, although the data from the female rats clearly show higher aconitase activity in animals with elevated serum 17β-estradiol, there is not a strict correlation between the intact and estrogen-replaced groups. This likely reflects the fact that the estrous cycle of the intact females was not monitored, and animals were studied randomly throughout the different stages of fluctuating levels of endogenous estrogen.
Testosterone is metabolized to estrogen by aromatase (Bulun et al., 2003) or to DHT through 5α-reductase (Fujimoto et al., 1994), and we have recently shown that both of these metabolic enzymes are present in the male cerebral vasculature (Gonzales et al., 2007). Estrogen clearly impacts male reproductive physiology (Hess et al., 1997; Jones and Simpson, 2000), and there is also strong evidence for non-reproductive effects of estrogen in males. For example, in both sexes local production of estrogen in bone is important in the maintenance of bone mineralization and prevention of osteoporosis (Simpson et al., 2000). During the critical period of brain development, sexual differentiation of the male brain is dependent on endogenous production of estrogen from testosterone by the action of aromatase (Reisert and Pilgrim, 1991). Similarly, local estrogen production in several sites in the brain influences sexual behavior and has been suggested to have a role in cognitive function and Alzheimer’s disease prevention (Simpson et al., 2000). Our study suggests that male brain mitochondria may also be under the influence of estrogen.
Mitochondria play a critical role in neuronal cell survival and maintenance of the high metabolic demands of the brain (Dykens, 1994; Singh et al., 2006). However, high production of ROS as a by-product of oxidative phosphorylation is an inevitable consequence of this high metabolic rate. Since mitochondria have limited DNA protection mechanisms, these cellular power plants are thus vulnerable to oxidative stress (Chance et al., 1979; Clayton, 1984; Wallace, 2005). In fact, excess ROS production (Aliev et al., 2002) leading to mitochondrial dysfunction (Castellani et al., 2002) is among the mechanisms thought to underlie the pathogenesis of neurodegenerative diseases. Recently mitochondria have been suggested as a target of the neuroprotective effects of estrogen (Nilsen and Brinton, 2004; Singh et al., 2006). Protective effects of estrogen also have been observed under different cellular insults through mechanisms involving mitochondrial calcium homeostasis, antiapoptotic pathways, and enhanced mitochondrial viability (Nilsen et al., 2006; Nilsen and Diaz Brinton, 2003). In the present study, we have extended the scope of estrogen action by showing that chronic estrogen treatment is neuroprotective under normal physiologic conditions in males and females through ongoing reduction of mitochondrial ROS production. Suppression of chronic oxidative stress would be expected to protect against mitochondrial dysfunction and delay cellular aging. Several other studies have also shown beneficial effects of estrogen on brain mitochondrial function. For example, in female rats, brain mitochondrial peroxide production was lower than in mitochondria from male rats (Borras et al., 2003). Physiologic concentrations of estrogen can also protect against oxidative damage through modulation of antioxidant enzyme activity, including superoxide dismutase, catalase, and glutathione peroxidase (Azevedo et al., 2001; Murakoshi et al., 1999).
We explored two possible mechanisms by which estrogen treatment might suppress mitochondrial ROS; one involved a decrease in production of superoxide through uncoupling proteins and the other entailed inactivation of superoxide through enzymatic action of MnSOD. Mitochondrial uncoupling proteins are localized to the inner mitochondrial membrane where they translocate protons from the intermembrane space to the mitochondrial matrix and thus reduce the driving force for ATP synthesis, dissipate energy in the form of heat, and decrease production of superoxide (Cannon et al., 2006; Casteilla et al., 2001; Erlanson-Albertsson, 2003; Rousset et al., 2004). Uncoupling proteins show neuroprotective effects, underscoring the importance of reducing mitochondrial ROS for preventing brain damage (Mattiasson et al., 2003) Chan et al., 2006). Three isoforms thought to be important in the brain, UCP-2, 4 and 5 (Chan et al., 2006; Erlanson-Albertsson, 2003; Ho et al., 2006), were detected in brain mitochondrial samples from both female and male rats; however neither gonadectomy nor sex hormone treatments affected protein levels of UCP-2, 4, and 5.
The mitochondrial-specific form of SOD, MnSOD, is pivotal in the removal of superoxide by-product formation. Transgenic heterozygous MnSOD knockout mice demonstrate increased oxidative damage and age-related changes in mitochondrial function (Kokoszka et al., 2001). Overexpression of MnSOD results in neuroprotection by reducing cellular apoptosis and decreasing brain ischemic damage (Keller et al., 1998). Previously, we found that, in cerebrovascular mitochondria from female rats treated with estrogen, protein levels of MnSOD were increased (Stirone et al., 2005). In contrast, in the current study, a similar in vivo estrogen treatment had no effect on protein levels of MnSOD in female brain mitochondria. In male rat brain mitochondria, estrogen and testosterone had small effects on MnSOD levels, but these do not explain the effects we observed on ROS-inactivation of aconitase.
MnSOD protein levels are, however, only one index of superoxide metabolism; evidence suggests MnSOD activity may be regulated as well (Azevedo et al., 2001; Pedram et al., 2006; Strehlow et al., 2003). Interestingly, estrogen enhanced MnSOD activity in both female and male brains, while testosterone and DHT had no effect. This alteration in MnSOD activity by estrogen is in agreement with previous studies in vascular tissue demonstrating increased MnSOD activity after in vitro or in vivo estrogen treatment (Pedram et al., 2006; Strehlow et al., 2003). Thus, in our study, the ability of estrogen to increase MnSOD activity represents one mechanism by which estrogen may reduce brain mitochondrial oxidative stress.
We previously demonstrated novel protective effects of chronic in vivo estrogen treatment on cerebrovascular mitochondria: increases in key mitochondrial proteins important for oxidative phosphorylation with simultaneous decreases in ROS production (Stirone et al., 2005). This enhancement by estrogen of cerebral vascular mitochondrial efficiency appears to be mediated through differential modulation of the peroxisome proliferator-activated receptor-gamma co-activator 1 (PGC-1) family of proteins that are crucial regulators of mitochondrial protein expression (C. Stirone, D. N. Krause, S. P. Duckles, and V. Procaccio, unpublished observations). Regulation of members of the PGC-1 co-activator family of proteins may also be an important mechanism by which estrogen modulates mitochondrial oxidative stress (St-Pierre et al., 2006). Further investigation will be necessary to unravel the complex mechanisms by which estrogen may reduce oxidative stress in brain mitochondria.
In summary, we have demonstrated that physiologic levels of estrogen have profound suppressive effects on brain mitochondrial ROS production, an effect that is not limited to females. In contrast to the lack of effect of androgens, suppression of mitochondrial ROS production by estrogen has the potential to alter the pathophysiology of neurological disease incidence and progression and contribute to neuroprotection in both females and males. It will be important to further explore the molecular mechanisms by which gonadal hormones exert their effects in the metabolically active central nervous system, including defining in more detail the particular receptor involved as well as defining the role of the mitochondrial transcriptional machinery. Understanding the mechanisms underlying neuroprotective actions of estrogen may enable the development of new therapeutic modalities for the prevention of age-related and neurodegenerative diseases.
4. Experimental Procedure
Treatment of PC-12 Cells
Rat pheochromocytoma cells (PC-12) (kindly provided by Dr. Wainer, Emory University) were differentiated in culture by exposure to 100 ng/ml nerve growth factor (NGF; Roche; Indianapolis, IN) for 10 days in Dulbecco’s modified Eagle’s medium (DMEM) supplemented with 1% horse serum and 0.5% fetal bovine serum (FBS) at 37°C in a humidified incubator containing 95% air and 5% CO2. When treated with NGF, the PC-12 cells exhibited slowed proliferation and growth of long neurite processes, which are properties associated with neuronal differentiation of these cells in culture (Greene and Tischler, 1976). Differentiated cells were then treated for 24 h with 10 nM 17β-estradiol (encapsulated in 2-hydroxy-propyl-β-cyclodextrin; Sigma; St. Louis, MO) or an equivalent concentration of 2-hydroxy-propyl-β-cyclodextrin (vehicle control) in hormone-free DMEM (without phenol red or serum) supplemented with 5% FBS (previously stripped of steroid hormones by treatment with dextran-coated charcoal Cocalico Biologicals; Reamstown, PA). In some cases, the estrogen-receptor antagonist, ICI-182,780 (1 μM; Tocris; Ellisville, MI), was added 1 h prior and during the 24-hour treatment with 17β-estradiol or vehicle.
Immunocytochemistry of PC-12 Cells
Differentiated PC-12 cells, grown on poly-D-lysine-coated glass coverslips, were incubated with 2.5 μM MitoSOX Red (Molecular Probes, Eugene, OR) for 45 min at 37°C, and then fixed with 3.7% paraformaldehyde for 15 min at 37°C, permeabilized with 0.2% Triton X-100 (Sigma) for 10 min, and blocked for 1 h in 1% BSA-0.05% Triton X-100. Cells were next incubated overnight at 4°C with antibody against subunit I of complex IV (1:200 dilution; Molecular Probes). The cells were washed with PBS and finally incubated with 1 μg/ml Alexa Fluor 488 goat anti-rabbit IgG (1 μg/ml, Molecular Probes) for 2 h. Images were obtained using a Carl Zeiss Axiovert 200M fluorescent microscope equipped with appropriate filters. Appropriate controls, such as secondary antibody alone, indicated a lack of non-specific staining.
Mitochondrial Superoxide Production
After 24 h hormone treatment, differentiated PC-12 cells were incubated with 2.5 μM MitoSOX Red reagent (Molecular Probes) for 10 min, washed, and then fluorescence was measured for 30 min at 510/580 nm (excitation/emission) using a Perkin Elmer Luminescence Spectrophotometer LS50B. After baseline stabilization, superoxide production was induced by adding mitochondrial complex I substrates (sodium pyruvate and malic acid, 1 M). The superoxide dismutase mimetic, Mn(III) tetrakis (4-benzoic acid) porphyrin (MnTBAP, 1 mM), was used to scavenge superoxide produced in the reaction. Superoxide production was calculated as fluorescence/min/mg protein over the linear range and expressed relative to vehicle control.
In Vivo Animal Hormone Treatments
All experiments on live animals were carried out in accordance with the Institutional Animal Care and Use Committee at the University of California, Irvine. Gonadectomy and hormone treatment protocols were performed on three-month-old female and male Fischer 344 rats (Charles River-SASCO Laboratories; Wilmington, MA) as described previously (Geary et al., 2000; Gonzales et al., 2005; McNeill et al., 1999; Razmara et al., 2005). Female rats were divided into three groups: intact, ovariectomized (OVX), and OVX treated with 17β-estradiol (OVX+E). Five groups of male rats were used: intact, orchiectomized (ORX), ORX treated with testosterone (ORX+T), ORX treated with dihydrotestosterone (ORX+DHT), and ORX treated with 17β-estradiol (ORX+E). At the time of gonadectomy, animals were implanted subcutaneously at the base of the neck with control or hormone slow-release pellets, i.e., Silastic tubing containing either 17β-estradiol 3-benzoate (5 mm in length; Sigma) or testosterone propionate (10 mm in length; Sigma) or commercial pellets containing 25 mg 5α-androstan-17β-ol-3-one (Innovative Research; Sarasota, FL). Surgeries were conducted under anesthesia (46 mg/kg i.p. ketamine and 4.6 mg/kg i.p. xylazine), and all animals received an injection of penicillin (30,000 U, penicillin G benzathine/penicillin G procain). Upon recovery from anesthesia, the animals were returned to a vivarium, where they were housed in a temperature-controlled room in individual cages with ab libitum food and water on a 12:12-hr light-dark cycle. The stage of the estrous cycle was not monitored in intact female rats.
All animals were euthanized 3–4 weeks after surgery and hormone pellet implantation. Prior to decapitation, animals were weighed, anesthesized with CO2, and direct cardiac puncture was used to collect blood samples for radioimmunoassay of 17β-estradiol (Diagnostic Products; Los Angeles, CA) or testosterone (MP Biomedicals; Costa Mesa, CA) or ELISA for DHT (Alpha Diagnostic; San Antonio, TX). Ovaries were removed and dried for weighing. Brains were removed for subsequent mitochondria isolation.
Mitochondria Isolation
Brain mitochondria were freshly isolated using a mitochondrial isolation kit (MITO-ISO1, Sigma), with additional centrifugations to obtain an enriched, purified mitochondrial fraction (Stirone et al., 2005). Cytochrome c and porin proteins, measured by Western blot, were used as markers to confirm the identity of the mitochondrial fraction. Histone H1 protein, a nuclear marker, was used to verify the absence of nuclear contamination.
Aconitase/Fumarase Enzyme Activities and Aconitase Reactivation
The ratio of the specific activities of mitochondrial aconitase (inactivated by ROS) and fumarase (unaffected) was calculated as an indicator of ROS production in brain mitochondria. Enzyme activities of aconitase and fumarase were measured using spectrophotometric rate determination (Gardner and Fridovich, 1992; Gardner et al., 1994). Isolated rat brain mitochondria were freeze-thawed for three cycles, and then centrifuged at 16,000g for 5 minutes. For measurement of aconitase activity, protein-normalized aliquots of the mitochondrial fraction were incubated at 25°C in a reaction buffer containing 154 mM Tris, 5 mM sodium citrate, 0.6 mM MgCl2, 0.2 mM NADP+. Absorbance at 340 nm was measured over time as citrate was converted to α-ketoglutarate using the concomitant reduction of NADP+ to NADPH by 2 units/ml isocitrate dehydrogenase (Krebs and Holzach, 1952). Subsequently, aconitase was fully reactivated by incubation of mitochondrial samples for 5 min with reducing reagents, 2 mM dithiothreitol and 0.2 mM ferrous ammonium sulfate; then the enzyme activity assay was repeated (Gardner and Fridovich, 1992; Patel et al., 1996; Williams et al., 1998). One milliunit of aconitase activity was defined as the amount catalyzing the formation of 1 nmol of isocitrate per min (Gardner et al., 1994). For each animal, the percent inactivation was calculated as the difference between mitochondrial aconitase activity before and after reactivation.
Activity of fumarase was measured by following conversion of malate to fumarate. The mitochondrial fraction was incubated in 0.05 M potassium phosphate buffer, pH 7.4, and 0.05 M sodium L-malate. Specific activity of fumarase was determined by monitoring absorbance at 240 nm as fumarate end-product accumulates (Racker, 1950).
Western Blot Analysis
The mitochondrial pellet was glass homogenized at 4°C in lysis buffer (50 mM β-glycerophosphate, 2 mM MgCl2, 1 mM EGTA, 1 mM DL-dithiothreitol, and 1 mM phenylmethylsulfonyl fluoride, with 100 μM NaVO3, 0.5% Triton X-100, 20 μM pepstatin, 20 μM leupeptin, and 0.1 U/ml aprotinin; all from Sigma). Protein concentrations were determined using the bicinchoninic acid protein assay kit (Pierce, Rockford, IL).
Equal amounts of mitochondrial protein (10 μg/lane), in sodium dodecyl sulfate (SDS) sample buffer were loaded onto 16% Tris-glycine gels (Invitrogen, Carlsbad, CA) and separated by SDS-PAGE. Samples from each hormone treatment group (either male or female) were run together on a single gel for comparison. Broad-range molecular weight markers (Bio-Rad, Hercules, CA) were loaded for protein band identification. Proteins were then transferred to nitrocellulose membranes (Amersham Pharmacia, Piscataway, NJ) and treated with primary antibodies: goat polyclonal anti-UCP-2 (1:200; Santa Cruz Biotech; Santa Cruz, CA), rabbit polyclonal anti-UCP-4 (1:1,000; Alpha Diagnostics; San Antonio, TX), rabbit polyclonal anti-UCP-5 (1:1,000; Alpha Diagnostics), rabbit polyclonal anti-MnSOD (1:1,000; Sigma), or mouse monoclonal anti-porin (1:10,000; Cal-Biochem; San Diego, CA). Membranes were then incubated with the appropriate secondary antibody: goat anti-rabbit IgG-horseradish peroxidase (1:5,000; Santa Cruz), donkey anti-goat IgG-horseradish peroxidase (1:5,000; Santa Cruz), or goat anti-mouse IgG-horseradish peroxidase (1:5,000; Santa Cruz). Protein bands, detected by electrochemiluminescence reagents and exposure to Hyperfilm (Amersham Pharmacia), were analyzed using the UN-SCAN-IT program (Silk Scientific; Orem, UT). Band densities were corrected relative to that of porin, the mitochondrial loading control. The corrected densities were then normalized for each film to that of either the OVX group for females or ORX group for males.
Manganese Superoxide Dismutase Activity Assay
A superoxide dismutase (SOD) assay kit (Cayman Chemical; Ann Arbor, MI) was used to determine MnSOD activity in isolated rat brain mitochondria samples. Potassium cyanide (1 mM) was used to inhibit other forms of cellular SOD (MacMillan-Crow et al., 1996; Marklund, 1980). MnSOD activity per amount of mitochondrial protein was normalized to values of either the OVX or ORX groups.
Statistical Analysis
Data are expressed as means ± SEM. Statistical analysis was performed with GraphPad Prism 4.0 software (San Diego, CA). Because of the variability in values from one assay to the next, every assay included a sample from each of the animal groups; therefore, differences among the groups were assessed by one-way ANOVA with repeated measures. After ANOVA, Newman-Keuls post-hoc analysis was used for pairwise comparisons. Student’s t-test was also used where appropriate. For all comparisons, statistical significance was set at P ≤ 0.05.
Acknowledgments
We thank Jonnie Stevens for animal surgeries and Antonio Davila for technical expertise. We are also grateful to Jennifer Buenzle Dwyer, Michelle Islas, Xiao Bo Wang, Haleh Fazeli-Tehrani, and Michael Yoon for technical assistance. This project was supported by the National Heart, Lung and Blood Institute grant R01 HL-50775.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Aliev G, et al. The role of oxidative stress in the pathophysiology of cerebrovascular lesions in Alzheimer’s disease. Brain Pathol. 2002;12:21–35. doi: 10.1111/j.1750-3639.2002.tb00419.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Andersen JK. Oxidative stress in neurodegeneration: cause or consequence? Nat Med. 2004;10(Suppl):S18–25. doi: 10.1038/nrn1434. [DOI] [PubMed] [Google Scholar]
- Azevedo RB, et al. Regulation of antioxidant enzyme activities in male and female rat macrophages by sex steroids. Braz J Med Biol Res. 2001;34:683–7. doi: 10.1590/s0100-879x2001000500018. [DOI] [PubMed] [Google Scholar]
- Beal MF. Mitochondria take center stage in aging and neurodegeneration. Ann Neurol. 2005;58:495–505. doi: 10.1002/ana.20624. [DOI] [PubMed] [Google Scholar]
- Beckman KB, Ames BN. The free radical theory of aging matures. Physiol Rev. 1998;78:547–81. doi: 10.1152/physrev.1998.78.2.547. [DOI] [PubMed] [Google Scholar]
- Behl C, et al. Neuroprotection against oxidative stress by estrogens: structure-activity relationship. Mol Pharmacol. 1997;51:535–41. [PubMed] [Google Scholar]
- Beyer RE. An analysis of the role of coenzyme Q in free radical generation and as an antioxidant. Biochem Cell Biol. 1992;70:390–403. doi: 10.1139/o92-061. [DOI] [PubMed] [Google Scholar]
- Borras C, et al. Mitochondria from females exhibit higher antioxidant gene expression and lower oxidative damage than males. Free Radic Biol Med. 2003;34:546–52. doi: 10.1016/s0891-5849(02)01356-4. [DOI] [PubMed] [Google Scholar]
- Bulun SE, et al. The human CYP19 (aromatase P450) gene: update on physiologic roles and genomic organization of promoters. J Steroid Biochem Mol Biol. 2003;86:219–24. doi: 10.1016/s0960-0760(03)00359-5. [DOI] [PubMed] [Google Scholar]
- Cadenas E, Davies KJ. Mitochondrial free radical generation, oxidative stress, and aging. Free Radic Biol Med. 2000;29:222–30. doi: 10.1016/s0891-5849(00)00317-8. [DOI] [PubMed] [Google Scholar]
- Calabrese V, et al. Mitochondrial involvement in brain function and dysfunction: relevance to aging, neurodegenerative disorders and longevity. Neurochem Res. 2001;26:739–64. doi: 10.1023/a:1010955807739. [DOI] [PubMed] [Google Scholar]
- Cannon B, et al. Uncoupling proteins: a role in protection against reactive oxygen species--or not? Biochim Biophys Acta. 2006;1757:449–58. doi: 10.1016/j.bbabio.2006.05.016. [DOI] [PubMed] [Google Scholar]
- Casteilla L, et al. Mitochondrial ROS metabolism: modulation by uncoupling proteins. IUBMB Life. 2001;52:181–8. doi: 10.1080/15216540152845984. [DOI] [PubMed] [Google Scholar]
- Castellani R, et al. Role of mitochondrial dysfunction in Alzheimer’s disease. J Neurosci Res. 2002;70:357–60. doi: 10.1002/jnr.10389. [DOI] [PubMed] [Google Scholar]
- Chan SL, et al. Mitochondrial uncoupling protein-4 regulates calcium homeostasis and sensitivity to store depletion-induced apoptosis in neural cells. J Biol Chem. 2006;281:37391–403. doi: 10.1074/jbc.M605552200. [DOI] [PubMed] [Google Scholar]
- Chance B, et al. Hydroperoxide metabolism in mammalian organs. Physiol Rev. 1979;59:527–605. doi: 10.1152/physrev.1979.59.3.527. [DOI] [PubMed] [Google Scholar]
- Clayton DA. Transcription of the mammalian mitochondrial genome. Annu Rev Biochem. 1984;53:573–94. doi: 10.1146/annurev.bi.53.070184.003041. [DOI] [PubMed] [Google Scholar]
- Duckles SP, et al. Estrogen and mitochondria: a new paradigm for vascular protection? Mol Interv. 2006;6:26–35. doi: 10.1124/mi.6.1.6. [DOI] [PubMed] [Google Scholar]
- Dykens JA. Isolated cerebral and cerebellar mitochondria produce free radicals when exposed to elevated CA2+ and Na+: implications for neurodegeneration. J Neurochem. 1994;63:584–91. doi: 10.1046/j.1471-4159.1994.63020584.x. [DOI] [PubMed] [Google Scholar]
- Erlanson-Albertsson C. The role of uncoupling proteins in the regulation of metabolism. Acta Physiol Scand. 2003;178:405–12. doi: 10.1046/j.1365-201X.2003.01159.x. [DOI] [PubMed] [Google Scholar]
- Fujimoto R, et al. Androgen receptors, 5 alpha-reductase activity and androgen-dependent proliferation of vascular smooth muscle cells. J Steroid Biochem Mol Biol. 1994;50:169–74. doi: 10.1016/0960-0760(94)90025-6. [DOI] [PubMed] [Google Scholar]
- Gardner PR, Fridovich I. Superoxide sensitivity of the Escherichia coli aconitase. J Biol Chem. 1991;266:19328–33. [PubMed] [Google Scholar]
- Gardner PR, Fridovich I. Inactivation-reactivation of aconitase in Escherichia coli. A sensitive measure of superoxide radical. J Biol Chem. 1992;267:8757–63. [PubMed] [Google Scholar]
- Gardner PR, et al. Aconitase is a sensitive and critical target of oxygen poisoning in cultured mammalian cells and in rat lungs. Proc Natl Acad Sci U S A. 1994;91:12248–52. doi: 10.1073/pnas.91.25.12248. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Geary GG, et al. Gonadal hormones affect diameter of male rat cerebral arteries through endothelium-dependent mechanisms. Am J Physiol Heart Circ Physiol. 2000;279:H610–8. doi: 10.1152/ajpheart.2000.279.2.H610. [DOI] [PubMed] [Google Scholar]
- Gonzales RJ, et al. Androgenic/estrogenic balance in the male rat cerebral circulation: metabolic enzymes and sex steroid receptors. J Cereb Blood Flow Metab. 2007 doi: 10.1038/sj.jcbfm.9600483. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gonzales RJ, et al. Testosterone treatment increases thromboxane function in rat cerebral arteries. Am J Physiol Heart Circ Physiol. 2005;289:H578–85. doi: 10.1152/ajpheart.00958.2004. [DOI] [PubMed] [Google Scholar]
- Green PS, Simpkins JW. Neuroprotective effects of estrogens: potential mechanisms of action. Int J Dev Neurosci. 2000;18:347–58. doi: 10.1016/s0736-5748(00)00017-4. [DOI] [PubMed] [Google Scholar]
- Greene LA, Tischler AS. Establishment of a noradrenergic clonal line of rat adrenal pheochromocytoma cells which respond to nerve growth factor. Proc Natl Acad Sci U S A. 1976;73:2424–8. doi: 10.1073/pnas.73.7.2424. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harman D. Free radical theory of aging: an update: increasing the functional life span. Ann N Y Acad Sci. 2006;1067:10–21. doi: 10.1196/annals.1354.003. [DOI] [PubMed] [Google Scholar]
- Hausladen A, Fridovich I. Superoxide and peroxynitrite inactivate aconitases, but nitric oxide does not. J Biol Chem. 1994;269:29405–8. [PubMed] [Google Scholar]
- Henderson VW. Estrogen-containing hormone therapy and Alzheimer’s disease risk: understanding discrepant inferences from observational and experimental research. Neuroscience. 2006;138:1031–9. doi: 10.1016/j.neuroscience.2005.06.017. [DOI] [PubMed] [Google Scholar]
- Hess RA, et al. A role for oestrogens in the male reproductive system. Nature. 1997;390:509–12. doi: 10.1038/37352. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ho PW, et al. Knockdown of uncoupling protein-5 in neuronal SH-SY5Y cells: Effects on MPP+-induced mitochondrial membrane depolarization, ATP deficiency, and oxidative cytotoxicity. J Neurosci Res. 2006;84:1358–66. doi: 10.1002/jnr.21034. [DOI] [PubMed] [Google Scholar]
- Jones ME, Simpson ER. Oestrogens in male reproduction. Baillieres Best Pract Res Clin Endocrinol Metab. 2000;14:505–16. doi: 10.1053/beem.2000.0094. [DOI] [PubMed] [Google Scholar]
- Keller JN, et al. 17Beta-estradiol attenuates oxidative impairment of synaptic Na+/K+-ATPase activity, glucose transport, and glutamate transport induced by amyloid beta-peptide and iron. J Neurosci Res. 1997;50:522–30. doi: 10.1002/(SICI)1097-4547(19971115)50:4<522::AID-JNR3>3.0.CO;2-G. [DOI] [PubMed] [Google Scholar]
- Keller JN, et al. Mitochondrial manganese superoxide dismutase prevents neural apoptosis and reduces ischemic brain injury: suppression of peroxynitrite production, lipid peroxidation, and mitochondrial dysfunction. J Neurosci. 1998;18:687–97. doi: 10.1523/JNEUROSCI.18-02-00687.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kennedy MC, et al. The role of iron in the activation-inactivation of aconitase. J Biol Chem. 1983;258:11098–105. [PubMed] [Google Scholar]
- Kokoszka JE, et al. Increased mitochondrial oxidative stress in the Sod2 (+/−) mouse results in the age-related decline of mitochondrial function culminating in increased apoptosis. Proc Natl Acad Sci U S A. 2001;98:2278–83. doi: 10.1073/pnas.051627098. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krebs HA, Holzach O. The conversion of citrate into cis-aconitate and isocitrate in the presence of aconitase. Biochem J. 1952;52:527–8. doi: 10.1042/bj0520527. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu PY, et al. Androgens and cardiovascular disease. Endocr Rev. 2003;24:313–40. doi: 10.1210/er.2003-0005. [DOI] [PubMed] [Google Scholar]
- MacMillan-Crow LA, et al. Nitration and inactivation of manganese superoxide dismutase in chronic rejection of human renal allografts. Proc Natl Acad Sci U S A. 1996;93:11853–8. doi: 10.1073/pnas.93.21.11853. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marklund S. Distribution of CuZn superoxide dismutase and Mn superoxide dismutase in human tissues and extracellular fluids. Acta Physiol Scand Suppl. 1980;492:19–23. [PubMed] [Google Scholar]
- Mattiasson G, et al. Uncoupling protein-2 prevents neuronal death and diminishes brain dysfunction after stroke and brain trauma. Nat Med. 2003;9:1062–8. doi: 10.1038/nm903. [DOI] [PubMed] [Google Scholar]
- McNeill AM, et al. Chronic estrogen treatment increases levels of endothelial nitric oxide synthase protein in rat cerebral microvessels. Stroke. 1999;30:2186–90. doi: 10.1161/01.str.30.10.2186. [DOI] [PubMed] [Google Scholar]
- Murakoshi M, et al. Regulation of prostatic glutathione-peroxidase (GSH-PO) in rats treated with a combination of testosterone and 17 beta-estradiol. J Toxicol Sci. 1999;24:415–20. doi: 10.2131/jts.24.5_415. [DOI] [PubMed] [Google Scholar]
- Nilsen J, Brinton RD. Mitochondria as therapeutic targets of estrogen action in the central nervous system. Curr Drug Targets CNS Neurol Disord. 2004;3:297–313. doi: 10.2174/1568007043337193. [DOI] [PubMed] [Google Scholar]
- Nilsen J, et al. Estrogen protects neuronal cells from amyloid beta-induced apoptosis via regulation of mitochondrial proteins and function. BMC Neurosci. 2006;7:74. doi: 10.1186/1471-2202-7-74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nilsen J, Diaz Brinton R. Mechanism of estrogen-mediated neuroprotection: regulation of mitochondrial calcium and Bcl-2 expression. Proc Natl Acad Sci U S A. 2003;100:2842–7. doi: 10.1073/pnas.0438041100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Patel M, et al. Requirement for superoxide in excitotoxic cell death. Neuron. 1996;16:345–55. doi: 10.1016/s0896-6273(00)80052-5. [DOI] [PubMed] [Google Scholar]
- Pedram A, et al. Functional estrogen receptors in the mitochondria of breast cancer cells. Mol Biol Cell. 2006;17:2125–37. doi: 10.1091/mbc.E05-11-1013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Prokai L, et al. Quinol-based cyclic antioxidant mechanism in estrogen neuroprotection. Proc Natl Acad Sci U S A. 2003;100:11741–6. doi: 10.1073/pnas.2032621100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Racker E. Spectrophotometric measurements of the enzymatic formation of fumaric and cis-aconitic acids. Biochim Biophys Acta. 1950;4:211–4. doi: 10.1016/0006-3002(50)90026-6. [DOI] [PubMed] [Google Scholar]
- Razmara A, et al. Testosterone augments endotoxin-mediated cerebrovascular inflammation in male rats. Am J Physiol Heart Circ Physiol. 2005;289:H1843–50. doi: 10.1152/ajpheart.00465.2005. [DOI] [PubMed] [Google Scholar]
- Reisert I, Pilgrim C. Sexual differentiation of monoaminergic neurons--genetic or epigenetic? Trends Neurosci. 1991;14:468–73. doi: 10.1016/0166-2236(91)90047-x. [DOI] [PubMed] [Google Scholar]
- Robinson KM, et al. Selective fluorescent imaging of superoxide in vivo using ethidium-based probes. Proc Natl Acad Sci U S A. 2006;103:15038–43. doi: 10.1073/pnas.0601945103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rousset S, et al. The biology of mitochondrial uncoupling proteins. Diabetes. 2004;53(Suppl 1):S130–5. doi: 10.2337/diabetes.53.2007.s130. [DOI] [PubMed] [Google Scholar]
- Simpson E, et al. The role of local estrogen biosynthesis in males and females. Trends Endocrinol Metab. 2000;11:184–8. doi: 10.1016/s1043-2760(00)00254-x. [DOI] [PubMed] [Google Scholar]
- Singh M, et al. Novel mechanisms for estrogen-induced neuroprotection. Exp Biol Med (Maywood) 2006;231:514–21. doi: 10.1177/153537020623100505. [DOI] [PubMed] [Google Scholar]
- St-Pierre J, et al. Suppression of reactive oxygen species and neurodegeneration by the PGC-1 transcriptional coactivators. Cell. 2006;127:397–408. doi: 10.1016/j.cell.2006.09.024. [DOI] [PubMed] [Google Scholar]
- Stirone C, et al. Estrogen increases mitochondrial efficiency and reduces oxidative stress in cerebral blood vessels. Mol Pharmacol. 2005;68:959–65. doi: 10.1124/mol.105.014662. [DOI] [PubMed] [Google Scholar]
- Strehlow K, et al. Modulation of antioxidant enzyme expression and function by estrogen. Circ Res. 2003;93:170–7. doi: 10.1161/01.RES.0000082334.17947.11. [DOI] [PubMed] [Google Scholar]
- Szewczyk A, Wojtczak L. Mitochondria as a pharmacological target. Pharmacol Rev. 2002;54:101–27. doi: 10.1124/pr.54.1.101. [DOI] [PubMed] [Google Scholar]
- Tong JJ, et al. Life extension through neurofibromin mitochondrial regulation and antioxidant therapy for neurofibromatosis-1 in Drosophila melanogaster. Nat Genet. 2007;39:476–485. doi: 10.1038/ng2004. [DOI] [PubMed] [Google Scholar]
- Turrens JF. Mitochondrial formation of reactive oxygen species. J Physiol. 2003;552:335–44. doi: 10.1113/jphysiol.2003.049478. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vina J, et al. Mitochondrial theory of aging: importance to explain why females live longer than males. Antioxid Redox Signal. 2003;5:549–56. doi: 10.1089/152308603770310194. [DOI] [PubMed] [Google Scholar]
- Wallace DC. A mitochondrial paradigm of metabolic and degenerative diseases, aging, and cancer: a dawn for evolutionary medicine. Annu Rev Genet. 2005;39:359–407. doi: 10.1146/annurev.genet.39.110304.095751. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Williams MD, et al. Increased oxidative damage is correlated to altered mitochondrial function in heterozygous manganese superoxide dismutase knockout mice. J Biol Chem. 1998;273:28510–5. doi: 10.1074/jbc.273.43.28510. [DOI] [PubMed] [Google Scholar]
- Wise PM. Estrogens and neuroprotection. Trends Endocrinol Metab. 2002;13:229–30. doi: 10.1016/s1043-2760(02)00611-2. [DOI] [PubMed] [Google Scholar]
- Yang SH, et al. Estrogens as protectants of the neurovascular unit against ischemic stroke. Curr Drug Targets CNS Neurol Disord. 2005;4:169–77. doi: 10.2174/1568007053544174. [DOI] [PubMed] [Google Scholar]