

Abstract
Because of the predominant role of skeletal muscle in insulin-stimulated clearance of blood glucose, understanding mechanisms for increasing the ability of muscle to respond to insulin could potentially lead to novel strategies for treatment or prevention of diabetes. Recently, the AMP activated protein kinase (AMPK) has emerged as a promising candidate for potentiation of insulin action. Several antidiabetic drugs have been shown to activate AMPK, cellular stresses such as exercise that increase AMPK activity also increase insulin action, and several downstream targets of AMPK seem to be involved in regulation of insulin action. Although the picture is currently incomplete, it seems possible that AMPK or one of its effectors is a positive regulator of insulin-stimulated glucose transport. In addition to discussion of the latest literature regarding AMPK and insulin action, this review includes a non-technical summary for students, academics from other fields, interested professionals, and the general public.
Introduction
Skeletal muscle is the primary depot for glucose cleared from the blood in the presence of insulin, and resistance of skeletal muscle to stimulation of glucose transport by insulin is a central characteristic of adult onset diabetes (Defronzo, 1988; Petersen et al., 1998; Cline et al., 1999). Understanding mechanisms for increasing insulin sensitivity in skeletal muscle could elucidate potential therapeutic targets in the prevention or treatment of diabetes.
Experts on the effects of insulin and metabolic stress on energy metabolism and glucose transport have published comprehensive reviews on these topics, and these fine reviews will be cited below as background for the central issue of the current review, the synergistic actions of AMPK and insulin.
Insulin stimulates glucose transport in skeletal muscle (figure 1) by increasing cell surface localization of the glucose transporter GLUT4 and possibly by increasing the activity of individual transport proteins [reviewed in (Cushman et al., 1998; Michelle, Poon, & Klip, 2003; Krook, Wallberg-Henriksson, & Zierath, 2004; Thong, Dugani, & Klip, 2005)]. Cellular stresses, including (but not limited to) exercise or muscle contractions, incubation of muscle in hyperosmotic medium, mitochondrial uncoupling, inhibition of cellular respiration, and hypoxic conditions, stimulate glucose transport in muscle in an insulin-independent fashion (Hayashi et al., 2000). This insulin-independent stimulation of glucose transport appears to involve activation of AMPK, although (at least for the case of muscle contractions) there is a growing pool of evidence that AMPK may not be necessary to or may only partially mediate these effects (Mu, Barton, & Birnbaum, 2003; Fujii, Aschenbach, Musi, Hirshman, & Goodyear, 2004; Fujii et al., 2005; Jorgensen et al., 2004). AMPK, introduced to the field of muscle glucose transport in 1998 by William Winder′s and Grahame Hardie′s groups (Merrill, Kurth, Hardie, & Winder, 1997) seems to be a metabolic fuel gauge that acts to combat cellular stress by coordinating suppression of non-essential ATP-dependent pathways and increasing means for ATP production [reviewed in (Fryer & Carling, 2005; Carling, 2004; Winder & Hardie, 1999; Hardie, 2003)].
Figure 1.

Some components of insulin signaling leading to glucose transport. Binding of insulin to its receptor starts a docking and phosphorylation  cascade involving insulin receptor substrate (IRS) and Akt and leads to stimulation of glucose transport. Phosphatidylinositol 3-kinase (PI3K) is a lipid kinase that phosphorylates membrane phosphatidylinositol, allowing recruitment of Akt to the membrane for subsequent phosphorylation and activation of Akt by phosphoinositide dependent kinases (PDKs). See reviews cited below for details.
cascade involving insulin receptor substrate (IRS) and Akt and leads to stimulation of glucose transport. Phosphatidylinositol 3-kinase (PI3K) is a lipid kinase that phosphorylates membrane phosphatidylinositol, allowing recruitment of Akt to the membrane for subsequent phosphorylation and activation of Akt by phosphoinositide dependent kinases (PDKs). See reviews cited below for details.
Synergistic effects of cellular stress and insulin on glucose transport
It has been known for more than 20 years that insulin and cellular stress act synergistically in the stimulation of glucose transport. For example, Defronzo et al found in 1981 that the stimulatory effect of insulin on glucose transport was much greater in exercising muscle than in resting muscle (Defronzo, Ferrannini, Sato, Felig, & Wahren, 1981). Shortly afterward, Richter et al firmly established that exercise acutely increases insulin sensitivity in muscle (Richter, Garetto, Goodman, & Ruderman, 1982). For example, within 30 min after exercise, the stimulatory effect of low physiological insulin concentrations on glucose transport in skeletal muscle roughly doubles compared to effects in non-exercised muscle (Richter et al., 1982). The increased insulin sensitivity after exercise remains up to 18 hours (Cartee et al., 1989) or until glycogen supercompensation occurs, and because of its prolonged presence is likely to be responsible for a large portion of the exercise effect on glucose transport into muscle. Surprisingly, although a few candidate mechanisms for the exercise effect on potentiation of insulin action have come to prominence [reviewed in (Wojtaszewski, Nielsen, & Richter, 2002; Holloszy, 2005; Christ-Roberts & Mandarino, 2004)], the phenomenon seems somewhat understudied in comparison to its potential importance to the understanding of factors that increase insulin action.
Other beneficial effects of exercise that would lead to increases in insulin action occur on a longer time scale than the acute increase in insulin action after exercise. For example, there is a well-known exercise training effect that leads to increased expression of glucose transporters in skeletal muscle (Gulve & Spina, 1995). Additionally, exercise training is associated with decreased body fat content and increased insulin action (Arciero, Vukovich, Holloszy, Racette, & Kohrt, 1999). These long-term mechanisms for potentiation of insulin action (i.e. training effects, as opposed to immediate influences) will not be dealt with further in this review.
Is there a role for AMPK in potentiation of insulin action?
A recently-developed hypothesis is that activation of AMPK increases sensitivity to insulin (figure 3). For example, several agents that normalize blood glucose concentrations and/or improve insulin action, including the adipokines adiponectin (Yamauchi et al., 2002) and leptin (Minokoshi et al., 2002), metformin, (Zhou et al., 2001), phenformin (Lizcano et al., 2004), creatine (Ju, Smith, Oppelt, & Fisher, 2004), rosiglitazone (Fryer, Parbu-Patel, & Carling, 2002), troglitazone (Konrad et al., 2005), and α-lipoic acid (Lee et al., 2005) have been shown to activate or phosphorylate AMPK. It′s tempting to hypothesize that at least a portion of the effects of these agents are mediated through AMPK activation. Intriguingly, the AMPK activator AICAR prevents insulin resistance associated with prolonged hyperglycemia (Kawanaka, Han, Gao, Nolte, & Holloszy, 2001).
Figure 3.

Some AMPK targets. NOS, p38, and AS160 appear to be regulated by both AMPK and insulin signaling, so it seems possible that they could mediate potential synergistic action between insulin signaling and AMPK. AMPK inhibits ACC, that may negatively impact insulin signaling through its production of malonyl CoA. AMPK is upstream of mTOR, that is a negative or positive modulator of insulin action, depending on its binding partner. Legend: negative or inhibitory affect  ; stimulatory affect
; stimulatory affect 
Two research groups independently found that AICAR, an AMPK activator, increases insulin sensitivity in skeletal muscle (Fisher, Gao, Han, Holloszy, & Nolte, 2002; Iglesias et al., 2002). For the case of isolated muscle, the presence of serum during treatment of the tissue with AICAR is a requirement for the subsequent increase in insulin action (Fisher et al., 2002). This requirement for serum is also necessary for induction of insulin sensitivity by hypoxia and muscle contractions (Gao, Gulve, & Holloszy, 1994; Fisher et al., 2002), though the mechanism for the permissive effect of serum remains a mystery. Because hypoxia, exercise, muscle contractions, and AICAR all produce increases in insulin action and all activate AMPK, it seems possible that AMPK could be a mediator of increased insulin action (Fisher et al., 2002). The exercise-related increase in insulin action is not dependent on synthesis of new proteins--it happens in a matter of a few hours and persists in the presence of the protein synthesis inhibitor cycloheximide (Fisher et al., 2002). Thus, if AICAR acts through the same pathways as exercise to increase insulin sensitivity, AICAR effects on insulin action are most likely separate from the known effects of AICAR on increasing expression of the glucose transport and phosphorylation machinery (Ojuka, Nolte, & Holloszy, 2000; Holmes, Kurth-Kraczek, & Winder, 1999).
Unfortunately, AICAR is not necessarily a specific activator of AMPK. AICAR (an adenosine analog) stimulates AMPK after the intracellular phosphorylation of AICAR to ZMP, an AMP analog (Corton, Gillespie, Hawley, & Hardie, 1995). Increased ZMP concentrations in AICAR-treated tissues could affect activities of any AMP-regulated enzymes and would not be limited to activation of AMPK. The recent characterizations of about a dozen AMPK-related kinases (Lizcano et al., 2004), at least some of which are activated by AMP (Suzuki et al., 2003; Lefebvre et al., 2001; Lefebvre & Rosen, 2005), provides fertile ground for future study. A world-leading group of researchers on AMPK and its activation has not been able to find activation of AMPK-related kinases by AICAR, phenformin, or muscle contractions (Sakamoto, Goransson, Hardie, & Alessi, 2004). However, researchers in my lab have found that AMPK-related kinase 5 (ARK5), also known as Nua Kinase 1 (Nuak1) is expressed in muscle and appears to be phosphorylated by Akt after exposure of muscle to insulin (Fisher et al., 2005). ARK5 is so far the only AMPK-related kinase that has been reported to be activated by Akt (Suzuki et al., 2003; Suzuki et al., 2004). Whether or not ARK5 is expressed in muscle and is activated by cellular stress or insulin remains an open question, though at this time opinion should probably lean toward the careful and comprehensive study that demonstrated that in skeletal muscle AMPK itself is the only AMPK family member activated by AICAR, phenformin, and muscle contractions (Sakamoto et al., 2004).
Jill L. Smith and Pankaj B. Patil recently worked out conditions for examining factors that regulate insulin action in C2C12 myotubes (Smith J.L., Patil P.B., & Fisher J.S., 2005). Some groups [e.g. (Tortorella & Pilch, 2002)] have not found C2C12 myotubes to be useful for studying insulin action. However, in our laboratory, C2C12 myotubes contain GLUT4 and are insulin-responsive (in terms of glucose transport) in a GLUT4-dependent manner (Smith J.L. et al., 2005; Smith J.L., Patil, Minteer, Lipsitz, & Fisher J.S., 2005). Under our conditions with C2C12 myotubes, AICAR and exposure to hyperosmotic stress both potentiate stimulation of glucose transport by insulin (Smith J.L. et al., 2005). There needs to be a recovery from the hyperosmotic stress before potentiation of insulin action occurs. Compound C (a somewhat-specific AMPK inhibitor) and iodotubercidin (a general kinase inhibitor that is effective against AMPK) prevented the increase in insulin action caused by hyperosmotic stress. Smith et al (Smith J.L. et al., 2005) were unable to use these AMPK inhibitors to probe for a role of AMPK in AICAR-associated increases in insulin action, because Compound C prevents AICAR uptake and iodotubercidin inhibits adenosine kinase, that is necessary for conversion of AICAR to ZMP.
What lies beyond AMPK?
Unfortunately, this appears to be the extent of information implicating AMPK in the acute regulation (i.e. probably not requiring changes in gene expression) of insulin action. There is even some compelling data demonstrating that activation of AMPK is not sufficient to increase insulin action (Kim, Solis, Arias, & Cartee, 2003; Al Khalili, Krook, Zierath, & Cartee, 2004). These findings suggest that activation of another signaling pathway in addition to activation of AMPK may be necessary to impact insulin action.
If AMPK is really involved in potentiation of insulin action, there seems to be no shortage of potential downstream effectors of AMPK′s effect on insulin sensitivity. A few of the known or probable AMPK targets [this is not necessarily an exhaustive list (Hardie, 2003)] that could potentially alter insulin action after AMPK activation include IRS-1, the p38 mitogen-activated protein kinase (p38), nitric oxide synthase (NOS), acetyl coenzyme A carboxylase (ACC), the mammalian target of rapamycin (mTOR), and the 160 kDa Akt substrate protein (AS160).
IRS-1
AMPK phosphorylates S789 of IRS-1 in vitro, and in myotubes AICAR-stimulated phosphorylation of IRS-1 on S789 is reportedly associated with increased insulin-stimulated PI3K activity (Jakobsen, Hardie, Morrice, & Tornqvist, 2001). However, the role of S789 phosphorylation of IRS-1 in insulin action is unclear. For example, phosphorylation of the same site seems to be associated with insulin resistance in liver (Qiao, Zhande, Jetton, Zhou, & Sun, 2002). Interestingly, in vitro and in adipocytes, the site (or its equivalent) is phosphorylated by salt-inducible kinase 2 (SIK2), an AMPK-related kinase, and there is increased SIK2 expression in white fat of diabetic animals (Horike et al., 2003).
p38
p38 has been implicated in activation of GLUT4 by insulin (Somwar et al., 2001; Somwar et al., 2002), though this point remains controversial (Turban et al., 2005). Anisomycin, an activator of p38, has been reported to induce insulin sensitivity in isolated rat skeletal muscle (Geiger, Wright, Han, & Holloszy, 2004). Inhibitors of p38 have been found to prevent AICAR-stimulated glucose transport, suggesting that p38 lies downstream of AMPK and is important in regulation of glucose transport (Lemieux, Konrad, Klip, & Marette, 2003; Xi, Han, & Zhang, 2001). Consistent with the hypothesis that p38 lies downstream of AMPK, a dominant negative p38 mutant has been reported to prevent AICAR-stimulated glucose transport (Xi et al., 2001). Regardless of whether or not p38 is downstream of AMPK, it appears to be activated by several stimulators of AMPK, including AICAR, mitochondrial uncoupling, and hyperosmotic stress (Xi et al., 2001; Lemieux et al., 2003; Taha, Tsakiridis, McCall, & Klip, 1997). Thus, it is possible that p38 could be a downstream activator of AMPK-induced insulin sensitivity or a co-activator along with AMPK of insulin sensitivity.
NOS
A few lines of evidence suggest that NOS lies downstream of AMPK. For example, AICAR treatment of mouse myotubes increases NOS activity, and L-NAME (a NOS inhibitor) prevents AICAR-stimulated glucose transport for skeletal muscle both in vitro and in vivo (Fryer et al., 2000; Shearer et al., 2004). Sodium nitroprusside (SNP, an NO donor that would lead to activation of guanylate cyclase), and 8-Bromo-cGMP (Br-cGMP, a cell-permeable cGMP analog) mimic the effects of AICAR on stimulation of glucose transport, while LY83583 (a guanylate cyclase inhibitor) blocks the stimulatory effect of AICAR on glucose transport (Fryer et al., 2000). Activating phosphorylation of neuronal NOS (nNOS) occurs in muscle during exercise (Chen et al., 2003), and AICAR reportedly activates nNOS and endothelial NOS (Fryer et al., 2000; Chen et al., 1999). NOS may also play a role in insulin-stimulated glucose transport in muscle (Roy, Perreault, & Marette, 1998) and therefore seems a potential candidate for mediation of insulin sensitivity after activation of AMPK.
ACC
AMPK phosphorylates and inactivates ACC, that catalyzes the conversion of acetyl CoA to malonyl coenzyme A (CoA). It has been suggested that inhibition by malonyl CoA of translocation of long chain fatty acyl (LCFA) groups from the cytosol to the mitochondrial matrix leads to subsequent buildup of LCFA CoAs in the cytosol, activation of protein kinase C, and serine phosphorylation of IRS-1 that is associated with insulin resistance (Ruderman, Saha, Vavvas, & Witters, 1999; Saha et al., 1997; Ruderman & Dean, 1998; Ruderman et al., 1999; Ruderman & Prentki, 2004). For example, glucose infusion causes insulin resistance in muscle that is related to increased malonyl CoA concentrations (Kraegen et al., 2005). Conversely, transgenic animals that lack the form of ACC that is predominant in skeletal muscle have greater insulin sensitivity than wildtype littermates (Abu-Elheiga, Oh, Kordari, & Wakil, 2003). Likewise, long-term treatment of rats with an ACC inhibitor increases insulin action (Harwood, Jr. et al., 2003; Harwood, Jr., 2004). However, it does not appear that any studies have been performed regarding the acute effects (i.e. within a few minutes or hours) of ACC inhibition on insulin action.
mTOR
The mammalian target of rapamycin has been implicated as a negative regulator of insulin action through its (or its downstream effector S6K′s) serine phosphorylation of insulin receptor substrate-1 (IRS-1) (Ozes et al., 2001; Li, DeFea, & Roth, 1999; Um et al., 2004). Recently, two binding partners of mTOR that appear to control substrate specificity have been characterized (Sarbassov, Guertin, Ali, & Sabatini, 2005; Sarbassov et al., 2004; Kim et al., 2002). When mTOR is bound to raptor (rapamycin sensitive partner of mTOR), it is involved in phosphorylation of IRS-1. In contrast, when mTOR is associated with rictor (rapamycin insensitive companion of mTOR), serine 473 of Akt is one of its targets (Sarbassov et al., 2005). Thus, mTOR·rictor is one of the PDKs shown in figures 1 and 2. Phosphorylation of Akt on serine 473 is essential to full activation of Akt (a key component of the insulin-signaling pathway) (Sarbassov et al., 2005), and mTOR·rictor has been shown to be an insulin-stimulated Akt serine 473 kinase (Hresko & Mueckler, 2005). Thus, mTOR·rictor is a positive regulator of insulin action (through activation of Akt), while mTOR·raptor negatively affects insulin signaling (through serine phosphorylation of IRS-1). AMPK has been demonstrated to be an upstream negative regulator of mTOR [reviewed in (Kahn, Alquier, Carling, & Hardie, 2005)], so it seems likely that AMPK activation would decrease the serine phosphorylation of IRS-1 that impedes insulin action. Whether or not AMPK would differentially regulate mTOR·rictor and mTOR·raptor is an open question.
Figure 2.
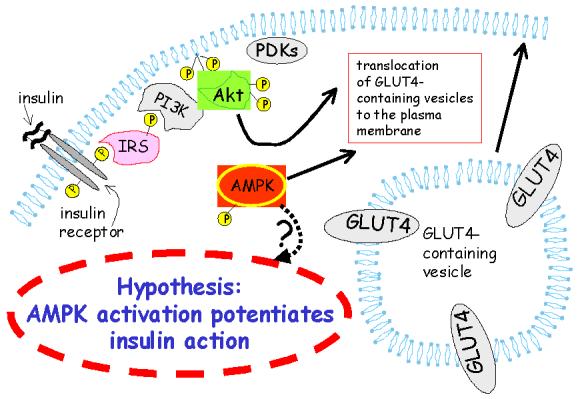
Is AMPK involved in potentiation of insulin action? Because several cellular stresses, chemical agents, and adipokines activate AMPK and also normalize blood glucose concentrations and/or improve insulin action, it seems possible that AMPK (shown activated by phosphorylation) could play a role in regulation of insulin action.
AS160
Phosphorylation and inactivation of AS160, identified as an 160 kDa Rab GTPase activating protein that is an Akt target (Kane et al., 2002), has been shown to be essential to stimulation of GLUT4 membrane localization by insulin in adipocytes (Sano et al., 2003). While the mechanistic work characterizing the role of AS160 in GLUT4 trafficking has been performed in adipocytes [e.g. (Eguez et al., 2005; Larance et al., 2005)], it now appears that AS160 is phosphorylated in skeletal muscle in response to insulin, exercise, and treatment with AICAR (Bruss, Arias, Lienhard, & Cartee, 2005; Karlsson et al., 2005; Plomgaard et al., 2005). AS160′s regulation by multiple pathways (i.e. insulin, muscle contractions, and AICAR) and its characterized role in regulation of glucose transport place it at the top of the list as a potential candidate in the control of insulin sensitivity.
Conclusion
Is AMPK indeed involved in regulation of insulin action? The evidence at this point is circumstantial. However, molecular tools such as siRNAs against AMPK, constitutively active AMPK, dominant negative forms of AMPK, and transgenic animals, will most certainly help determine whether AMPK activation is sufficient for or even necessary to the potentiated insulin action that is associated with cellular stress. If AMPK is found to regulate insulin-stimulated glucose transport, researchers in the field will have plenty of work ahead determining which of the multitude of downstream effectors of AMPK mediates the potentiation of insulin action by AMPK.
Non-technical summary
Insulin, a natural hormone secreted into the bloodstream after meals containing carbohydrates, causes a few tissues, including skeletal muscle (the muscles that make the body move), fat, and heart, to increase uptake of glucose (sugar). Of these tissues, skeletal muscle is by far the most massive, comprising approximately 40% of body weight. The majority of sugar cleared out of the bloodstream in response to insulin is stored in muscle. Therefore, muscle is of primary importance in the insulin-related control of blood sugar levels. Muscle that does not react properly to insulin often underlies the increased blood sugar concentration that is the hallmark of diabetes.
It has been known for over two decades that a short time after exercise insulin works much better at stimulating glucose transport into muscle. Recently, evidence has emerged that a protein called the “AMP-activated protein kinase” (AMPK) could be responsible for increasing the sensitivity of muscle to insulin. AMPK appears to be a cellular fuel gauge that senses metabolic stresses or nutritional deficiency and subsequently controls several cellular processes to muster fuel resources and conserve energy. For example, it appears that AMPK can increase glucose transport into muscle but also increase the rate of fat burning by muscle. AMPK appears to activate several signaling pathways in muscle that are also stimulated by insulin, so it is possible that AMPK and insulin together could have synergistic effects on the signaling events that lead to glucose transport. AMPK also seems to prevent the negative effects on insulin′s action of another cellular fuel gauge that senses nutrient sufficiency. Finally, it seems possible that the positive effect of AMPK on insulin′s ability to stimulate glucose transport may simply be a result of increased fat burning that clears fat molecules (that inhibit insulin action), out of muscle cells.
Exercise robustly potentiates insulin action. However, some people at risk of developing diabetes are unable to exercise. Understanding the cellular mechanisms for increased sugar uptake into muscle is an important first step in developing drug strategies for treating or preventing diabetes when exercise is not a viable option.
Acknowledgement
Jonathan S. Fisher is supported by NIH K01 DK066330.
References with links to PubMed entries
- Abu-Elheiga L, Oh W, Kordari P, Wakil SJ. Acetyl-CoA carboxylase 2 mutant mice are protected against obesity and diabetes induced by high-fat/high-carbohydrate diets. Proc.Natl.Acad.Sci.U.S.A. 2003;100:10207–10212. doi: 10.1073/pnas.1733877100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Al Khalili L, Krook A, Zierath JR, Cartee GD. Prior Serum and AICAR-Induced AMPK Activation in Primary Human Myocytes Does Not Lead to Subsequent Increase in Insulin-Stimulated Glucose Uptake. Am J.Physiol Endocrinol.Metab. 2004;287:E553–E557. doi: 10.1152/ajpendo.00161.2004. [DOI] [PubMed] [Google Scholar]
- Arciero PJ, Vukovich MD, Holloszy JO, Racette SB, Kohrt WM. Comparison of short-term diet and exercise on insulin action in individuals with abnormal glucose tolerance. J Appl Physiol. 1999;86:1930–1935. doi: 10.1152/jappl.1999.86.6.1930. [DOI] [PubMed] [Google Scholar]
- Bruss MD, Arias EB, Lienhard GE, Cartee GD. Increased Phosphorylation of Akt Substrate of 160 kDa (AS160) in Rat Skeletal Muscle in Response to Insulin or Contractile Activity. Diabetes. 2005;54:41–50. doi: 10.2337/diabetes.54.1.41. [DOI] [PubMed] [Google Scholar]
- Carling D. The AMP-activated protein kinase cascade--a unifying system for energy control. Trends Biochem.Sci. 2004;29:18–24. doi: 10.1016/j.tibs.2003.11.005. [DOI] [PubMed] [Google Scholar]
- Cartee GD, Young DA, Sleeper MD, Zierath J, Wallberg-Henriksson H, Holloszy JO. Prolonged increase in insulin-stimulated glucose transport in muscle after exercise. American Journal of Physiology. 1989;256:E494–E499. doi: 10.1152/ajpendo.1989.256.4.E494. [DOI] [PubMed] [Google Scholar]
- Chen ZP, Mitchelhill KI, Michell BJ, Stapleton D, Rodriguez-Crespo I, Witters LA, et al. AMP-activated protein kinase phosphorylation of endothelial NO synthase. FEBS Letters. 1999;443:285–289. doi: 10.1016/s0014-5793(98)01705-0. [DOI] [PubMed] [Google Scholar]
- Chen ZP, Stephens TJ, Murthy S, Canny BJ, Hargreaves M, Witters LA, et al. Effect of exercise intensity on skeletal muscle AMPK signaling in humans. Diabetes. 2003;52:2205–2212. doi: 10.2337/diabetes.52.9.2205. [DOI] [PubMed] [Google Scholar]
- Christ-Roberts CY, Mandarino LJ. Glycogen synthase: key effect of exercise on insulin action. Exerc.Sport Sci Rev. 2004;32:90–94. doi: 10.1097/00003677-200407000-00003. [DOI] [PubMed] [Google Scholar]
- Cline GW, Petersen KF, Krssak M, Shen J, Hundal RS, Trajanoski Z, et al. Impaired glucose transport as a cause of decreased insulin-stimulated muscle glycogen synthesis in type 2 diabetes. New England Journal of Medicine. 1999;341:240–246. doi: 10.1056/NEJM199907223410404. [DOI] [PubMed] [Google Scholar]
- Corton JM, Gillespie JG, Hawley SA, Hardie DG. 5-aminoimidazole-4-carboxamide ribonucleoside. A specific method for activating AMP-activated protein kinase in intact cells. European Journal of Biochemistry. 1995;229:558–565. doi: 10.1111/j.1432-1033.1995.tb20498.x. [DOI] [PubMed] [Google Scholar]
- Cushman SW, Goodyear LJ, Pilch PF, Ralston E, Galbo H, Ploug T, et al. Molecular mechanisms involved in GLUT4 translocation in muscle during insulin and contraction stimulation. Adv Exp Med Biol. 1998;441:63–71. doi: 10.1007/978-1-4899-1928-1_6. [DOI] [PubMed] [Google Scholar]
- Defronzo RA. Lilly lecture 1987. The triumvirate: beta-cell, muscle, liver. A collusion responsible for NIDDM. Diabetes. 1988;37:667–687. doi: 10.2337/diab.37.6.667. [DOI] [PubMed] [Google Scholar]
- Defronzo RA, Ferrannini E, Sato Y, Felig P, Wahren J. Synergistic interaction between exercise and insulin on peripheral glucose uptake. J Clin.Invest. 1981;68:1468–1474. doi: 10.1172/JCI110399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eguez L, Lee A, Chavez JA, Miinea CP, Kane S, Lienhard GE, et al. Full intracellular retention of GLUT4 requires AS160 Rab GTPase activating protein. Cell Metab. 2005;2:263–272. doi: 10.1016/j.cmet.2005.09.005. [DOI] [PubMed] [Google Scholar]
- Fisher JS, Gao J, Han D-H, Holloszy JO, Nolte LA. Activation of AMP kinase enhances sensitivity of muscle glucose transport to insulin. American Journal of Physiology: Endocrinology and Metabolism. 2002;282:E18–E23. doi: 10.1152/ajpendo.2002.282.1.E18. [DOI] [PubMed] [Google Scholar]
- Fisher J, Ju JS, Oppelt PJ, Smith JL, Suzuki A, Esumi H. Muscle contractions, AICAR, and insulin cause phosphorylation of an AMPK-related kinase. Am.J.Physiol Endocrinol.Metab. 2005;289:986–992. doi: 10.1152/ajpendo.00335.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fryer LG, Carling D. AMP-activated protein kinase and the metabolic syndrome. Biochem.Soc.Trans. 2005;33:362–366. doi: 10.1042/BST0330362. [DOI] [PubMed] [Google Scholar]
- Fryer LG, Hajduch E, Rencurel F, Salt IP, Hundal HS, Hardie DG, et al. Activation of glucose transport by AMP-activated protein kinase via stimulation of nitric oxide synthase. Diabetes. 2000;49:1978–1985. doi: 10.2337/diabetes.49.12.1978. [DOI] [PubMed] [Google Scholar]
- Fryer LG, Parbu-Patel A, Carling D. The Anti-diabetic drugs rosiglitazone and metformin stimulate AMP-activated protein kinase through distinct signaling pathways. J Biol Chem. 2002;277:25226–25232. doi: 10.1074/jbc.M202489200. [DOI] [PubMed] [Google Scholar]
- Fujii N, Aschenbach WG, Musi N, Hirshman MF, Goodyear LJ. Regulation of glucose transport by the AMP-activated protein kinase. Proc.Nutr.Soc. 2004;63:205–210. doi: 10.1079/PNS2004340. [DOI] [PubMed] [Google Scholar]
- Fujii N, Hirshman MF, Kane EM, Ho RC, Peter LE, Seifert MM, et al. AMP-activated protein kinase alpha2 activity is not essential for contraction-and hyperosmolarity-induced glucose transport in skeletal muscle. J Biol Chem. 2005 doi: 10.1074/jbc.M504208200. [DOI] [PubMed] [Google Scholar]
- Gao J, Gulve EA, Holloszy JO. Contraction-induced increase in muscle insulin sensitivity: requirement for a serum factor. American Journal of Physiology. 1994;266:E186–E192. doi: 10.1152/ajpendo.1994.266.2.E186. [DOI] [PubMed] [Google Scholar]
- Geiger PC, Wright DC, Han DH, Holloszy JO. Activation of p38 MAP kinase enhances sensitivity of muscle glucose transport to insulin. Am J Physiol Endocrinol.Metab. 2004;288:E782–E788. doi: 10.1152/ajpendo.00477.2004. [DOI] [PubMed] [Google Scholar]
- Gulve EA, Spina RJ. Effect of 7-10 days of cycle ergometer exercise on skeletal muscle GLUT-4 protein content. J Appl Physiol. 1995;79:1562–1566. doi: 10.1152/jappl.1995.79.5.1562. [DOI] [PubMed] [Google Scholar]
- Hardie DG. Minireview: the AMP-activated protein kinase cascade: the key sensor of cellular energy status. Endocrinology. 2003;144:5179–5183. doi: 10.1210/en.2003-0982. [DOI] [PubMed] [Google Scholar]
- Harwood HJ., Jr. Acetyl-CoA carboxylase inhibition for the treatment of metabolic syndrome. Curr.Opin.Investig.Drugs. 2004;5:283–289. [PubMed] [Google Scholar]
- Harwood HJ, Jr., Petras SF, Shelly LD, Zaccaro LM, Perry DA, Makowski MR, et al. Isozyme-nonselective N-substituted bipiperidylcarboxamide acetyl-CoA carboxylase inhibitors reduce tissue malonyl-CoA concentrations, inhibit fatty acid synthesis, and increase fatty acid oxidation in cultured cells and in experimental animals. J Biol Chem. 2003;278:37099–37111. doi: 10.1074/jbc.M304481200. [DOI] [PubMed] [Google Scholar]
- Hayashi T, Hirshman MF, Fujii N, Habinowski SA, Witters LA, Goodyear LJ. Metabolic stress and altered glucose transport: activation of AMP-activated protein kinase as a unifying coupling mechanism. Diabetes. 2000;49:527–531. doi: 10.2337/diabetes.49.4.527. [DOI] [PubMed] [Google Scholar]
- Holloszy JO. Exercise-induced increase in muscle insulin sensitivity. J Appl Physiol. 2005;99:338–343. doi: 10.1152/japplphysiol.00123.2005. [DOI] [PubMed] [Google Scholar]
- Holmes BF, Kurth-Kraczek EJ, Winder WW. Chronic activation of 5′-AMP-activated protein kinase increases GLUT-4, hexokinase, and glycogen in muscle. Journal of Applied Physiology. 1999;87:1990–1995. doi: 10.1152/jappl.1999.87.5.1990. [DOI] [PubMed] [Google Scholar]
- Horike N, Takemori H, Katoh Y, Doi J, Min L, Asano T, et al. Adipose-specific expression, phosphorylation of Ser794 in insulin receptor substrate-1, and activation in diabetic animals of salt-inducible kinase-2. J Biol Chem. 2003;278:18440–18447. doi: 10.1074/jbc.M211770200. [DOI] [PubMed] [Google Scholar]
- Hresko RC, Mueckler M. mTOR/RICTOR is the Ser473 kinase for Akt/PKB in 3T3-L1 adipocytes. J Biol Chem. 2005 doi: 10.1074/jbc.M508361200. in press.
- Iglesias MA, Ye JM, Frangioudakis G, Saha AK, Tomas E, Ruderman NB, et al. AICAR administration causes an apparent enhancement of muscle and liver insulin action in insulin-resistant high-fat-fed rats. Diabetes. 2002;51:2886–2894. doi: 10.2337/diabetes.51.10.2886. [DOI] [PubMed] [Google Scholar]
- Jakobsen SN, Hardie DG, Morrice N, Tornqvist HE. 5′-AMP-activated protein kinase phosphorylates IRS-1 on Ser-789 in mouse C2C12 myotubes in response to 5-aminoimidazole-4-carboxamide riboside. J.Biol Chem. 2001;276:46912–46916. doi: 10.1074/jbc.C100483200. [DOI] [PubMed] [Google Scholar]
- Jorgensen SB, Viollet B, Andreelli F, Frosig C, Birk JB, Schjerling P, et al. Knockout of the alpha2 but not alpha1 5′-AMP-activated protein kinase isoform abolishes 5-aminoimidazole-4-carboxamide-1-beta-4-ribofuranosidebut not contraction-induced glucose uptake in skeletal muscle. J Biol Chem. 2004;279:1070–1079. doi: 10.1074/jbc.M306205200. [DOI] [PubMed] [Google Scholar]
- Ju JS, Smith JL, Oppelt PJ, Fisher JS. Creatine feeding increases GLUT4 expression in rat skeletal muscle. Am J Physiol Endocrinol Metab. 2004;288:E347–E352. doi: 10.1152/ajpendo.00238.2004. [DOI] [PubMed] [Google Scholar]
- Kahn BB, Alquier T, Carling D, Hardie DG. AMP-activated protein kinase: ancient energy gauge provides clues to modern understanding of metabolism. Cell Metab. 2005;1:15–25. doi: 10.1016/j.cmet.2004.12.003. [DOI] [PubMed] [Google Scholar]
- Kane S, Sano H, Liu SC, Asara JM, Lane WS, Garner CC, et al. A method to identify serine kinase substrates. Akt phosphorylates a novel adipocyte protein with a Rab GTPase-activating protein (GAP) domain. J.Biol Chem. 2002;277:22115–22118. doi: 10.1074/jbc.C200198200. [DOI] [PubMed] [Google Scholar]
- Karlsson HKR, Zierath JR, Kane S, Krook A, Lienhard GE, Wallberg-Henriksson H. Insulin-stimulated phosphorylation of the Akt substrate AS160 is impaired in skeletal muscle of type 2 diabetic subjects. Diabetes. 2005;54:1692–1697. doi: 10.2337/diabetes.54.6.1692. [DOI] [PubMed] [Google Scholar]
- Kawanaka K, Han D-H, Gao J, Nolte LA, Holloszy JO. Development of glucose-induced insulin resistance in muscle requires protein synthesis. Journal of Biological Chemistry. 2001 doi: 10.1074/jbc.M010599200. in press.
- Kim DH, Sarbassov DD, Ali SM, King JE, Latek RR, Erdjument-Bromage H, et al. mTOR interacts with raptor to form a nutrient-sensitive complex that signals to the cell growth machinery. Cell. 2002;110:163–175. doi: 10.1016/s0092-8674(02)00808-5. [DOI] [PubMed] [Google Scholar]
- Kim J, Solis RS, Arias EB, Cartee GD. Post-Contraction Insulin Sensitivity: Relationship with Contraction Protocol, Glycogen Concentration, and 5′AMP-Activated Protein Kinase Phosphorylation. J.Appl.Physiol. 2003 doi: 10.1152/japplphysiol.00909.2003. [DOI] [PubMed] [Google Scholar]
- Konrad D, Rudich A, Bilan PJ, Patel N, Richardson C, Witters LA, et al. Troglitazone causes acute mitochondrial membrane depolarisation and an AMPK-mediated increase in glucose phosphorylation in muscle cells. Diabetologia. 2005;48:954–966. doi: 10.1007/s00125-005-1713-7. [DOI] [PubMed] [Google Scholar]
- Kraegen EW, Saha AK, Preston E, Wilks D, Hoy AJ, Cooney GJ, et al. Increased malonyl CoA and diacylglycerol content and reduced AMPK activity accompany insulin resistance induced by glucose infusion in muscle and liver of rats. Am J Physiol Endocrinol Metab. 2005 doi: 10.1152/ajpendo.00316.2005. in press.
- Krook A, Wallberg-Henriksson H, Zierath JR. Sending the signal: molecular mechanisms regulating glucose uptake. Med.Sci Sports Exerc. 2004;36:1212–1217. doi: 10.1249/01.mss.0000132387.25853.3b. [DOI] [PubMed] [Google Scholar]
- Larance M, Ramm G, Stockli J, van Dam EM, Winata S, Wasinger V, et al. Characterization of the role of the Rab GTPase-activating protein AS160 in insulin-regulated GLUT4 trafficking. J Biol Chem. 2005;280:37803–37813. doi: 10.1074/jbc.M503897200. [DOI] [PubMed] [Google Scholar]
- Lee WJ, Song KH, Koh EH, Won JC, Kim HS, Park HS, et al. Alpha-lipoic acid increases insulin sensitivity by activating AMPK in skeletal muscle. Biochem.Biophys.Res Commun. 2005;332:885–891. doi: 10.1016/j.bbrc.2005.05.035. [DOI] [PubMed] [Google Scholar]
- Lefebvre DL, Bai Y, Shahmolky N, Sharma M, Poon R, Drucker DJ, et al. Identification and characterization of a novel sucrose-non-fermenting protein kinase/AMP-activated protein kinase-related protein kinase, SNARK. Biochem.J. 2001;355:297–305. doi: 10.1042/0264-6021:3550297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lefebvre DL, Rosen CF. Regulation of SNARK activity in response to cellular stresses. Biochimica et Biophysica Acta. 2005;1724:71–85. doi: 10.1016/j.bbagen.2005.03.015. [DOI] [PubMed] [Google Scholar]
- Lemieux K, Konrad D, Klip A, Marette A. The AMP-activated protein kinase activator AICAR does not induce GLUT4 translocation to transverse tubules but stimulates glucose uptake and p38 mitogen-activated protein kinases alpha and beta in skeletal muscle. FASEB J. 2003;17:1658–1665. doi: 10.1096/fj.02-1125com. [DOI] [PubMed] [Google Scholar]
- Li J, DeFea K, Roth RA. Modulation of insulin receptor substrate-1 tyrosine phosphorylation by an Akt/phosphatidylinositol 3-kinase pathway. J Biol Chem. 1999;274:9351–9356. doi: 10.1074/jbc.274.14.9351. [DOI] [PubMed] [Google Scholar]
- Lizcano JM, Goransson O, Toth R, Deak M, Morrice NA, Boudeau J, et al. LKB1 is a master kinase that activates 13 kinases of the AMPK subfamily, including MARK/PAR-1. EMBO J. 2004;23:833–843. doi: 10.1038/sj.emboj.7600110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Merrill GF, Kurth EJ, Hardie DG, Winder WW. AICA riboside increases AMP-activated protein kinase, fatty acid oxidation, and glucose uptake in rat muscle. American Journal of Physiology. 1997;273:E1107–E1112. doi: 10.1152/ajpendo.1997.273.6.E1107. [DOI] [PubMed] [Google Scholar]
- Michelle FL, Poon V, Klip A. GLUT4 activation: thoughts on possible mechanisms. Acta Physiol Scand. 2003;178:287–296. doi: 10.1046/j.1365-201X.2003.01160.x. [DOI] [PubMed] [Google Scholar]
- Minokoshi Y, Kim YB, Peroni OD, Fryer LG, Muller C, Carling D, et al. Leptin stimulates fatty-acid oxidation by activating AMP-activated protein kinase. Nature. 2002;415:339–343. doi: 10.1038/415339a. [DOI] [PubMed] [Google Scholar]
- Mu J, Barton ER, Birnbaum MJ. Selective suppression of AMP-activated protein kinase in skeletal muscle: update on ′lazy mice′. Biochem.Soc.Trans. 2003;31:236–241. doi: 10.1042/bst0310236. [DOI] [PubMed] [Google Scholar]
- Ojuka EO, Nolte LA, Holloszy JO. Increased expression of GLUT-4 and hexokinase in rat epitrochlearis muscles exposed to AICAR in vitro. Journal of Applied Physiology. 2000;88:1072–1075. doi: 10.1152/jappl.2000.88.3.1072. [DOI] [PubMed] [Google Scholar]
- Ozes ON, Akca H, Mayo LD, Gustin JA, Maehama T, Dixon JE, et al. A phosphatidylinositol 3-kinase/Akt/mTOR pathway mediates and PTEN antagonizes tumor necrosis factor inhibition of insulin signaling through insulin receptor substrate-1. Proc.Natl.Acad.Sci.U.S.A. 2001;98:4640–4645. doi: 10.1073/pnas.051042298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Petersen KF, Hendler R, Price T, Perseghin G, Rothman DL, Held N, et al. 13C/31P NMR studies on the mechanism of insulin resistance in obesity. Diabetes. 1998;47:381–386. doi: 10.2337/diabetes.47.3.381. [DOI] [PubMed] [Google Scholar]
- Plomgaard P, Bouzakri K, Krogh-Madsen R, Mittendorfer B, Zierath JR, Pedersen BK. Tumor necrosis factoralpha induces skeletal muscle insulin resistance in healthy human subjects via inhibition of Akt substrate 160 phosphorylation. Diabetes. 2005;54:2939–2945. doi: 10.2337/diabetes.54.10.2939. [DOI] [PubMed] [Google Scholar]
- Qiao LY, Zhande R, Jetton TL, Zhou G, Sun XJ. In vivo phosphorylation of insulin receptor substrate 1 at serine 789 by a novel serine kinase in insulin-resistant rodents. J Biol Chem. 2002;277:26530–26539. doi: 10.1074/jbc.M201494200. [DOI] [PubMed] [Google Scholar]
- Richter EA, Garetto LP, Goodman MN, Ruderman NB. Muscle glucose metabolism following exercise in the rat: increased sensitivity to insulin. Journal of Clinical Investigation. 1982;69:785–793. doi: 10.1172/JCI110517. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roy D, Perreault M, Marette A. Insulin stimulation of glucose uptake in skeletal muscles and adipose tissues in vivo is NO dependent. Am J.Physiol. 1998;274:E692–E699. doi: 10.1152/ajpendo.1998.274.4.E692. [DOI] [PubMed] [Google Scholar]
- Ruderman N, Prentki M. AMP kinase and malonyl-CoA: targets for therapy of the metabolic syndrome. Nat.Rev.Drug Discov. 2004;3:340–351. doi: 10.1038/nrd1344. [DOI] [PubMed] [Google Scholar]
- Ruderman NB, Dean D. Malonyl CoA, long chain fatty acyl CoA and insulin resistance in skeletal muscle. J Basic Clin.Physiol Pharmacol. 1998;9:295–308. doi: 10.1515/jbcpp.1998.9.2-4.295. [DOI] [PubMed] [Google Scholar]
- Ruderman NB, Saha AK, Vavvas D, Witters LA. Malonyl-CoA, fuel sensing, and insulin resistance. Am.J.Physiol. 1999;276:E1–E18. doi: 10.1152/ajpendo.1999.276.1.E1. [DOI] [PubMed] [Google Scholar]
- Saha AK, Vavvas D, Kurowski TG, Apazidis A, Witters LA, Shafrir E, et al. Malonyl-CoA regulation in skeletal muscle: its link to cell citrate and the glucose-fatty acid cycle. Am.J.Physiol. 1997;272:E641–E648. doi: 10.1152/ajpendo.1997.272.4.E641. [DOI] [PubMed] [Google Scholar]
- Sakamoto K, Goransson O, Hardie DG, Alessi DR. Activity of LKB1 and AMPK-related kinases in skeletal muscle; effects of contraction, phenformin and AICAR. Am J.Physiol Endocrinol.Metab. 2004 doi: 10.1152/ajpendo.00074.2004. [DOI] [PubMed] [Google Scholar]
- Sano H, Kane S, Sano E, Miinea CP, Asara JM, Lane WS, et al. Insulin-stimulated phosphorylation of a Rab GTPase-activating protein regulates GLUT4 translocation. J Biol Chem. 2003;278:14599–14602. doi: 10.1074/jbc.C300063200. [DOI] [PubMed] [Google Scholar]
- Sarbassov DD, Ali SM, Kim DH, Guertin DA, Latek RR, Erdjument-Bromage H, et al. Rictor, a novel binding partner of mTOR, defines a rapamycin-insensitive and raptor-independent pathway that regulates the cytoskeleton. Curr.Biol. 2004;14:1296–1302. doi: 10.1016/j.cub.2004.06.054. [DOI] [PubMed] [Google Scholar]
- Sarbassov DD, Guertin DA, Ali SM, Sabatini DM. Phosphorylation and regulation of Akt/PKB by the rictormTOR complex. Science. 2005;307:1098–1101. doi: 10.1126/science.1106148. [DOI] [PubMed] [Google Scholar]
- Shearer J, Fueger PT, Vorndick B, Bracy DP, Rottman JN, Clanton JA, et al. AMP kinase-induced skeletal muscle glucose but not long-chain fatty acid uptake is dependent on nitric oxide. Diabetes. 2004;53:1429–1435. doi: 10.2337/diabetes.53.6.1429. [DOI] [PubMed] [Google Scholar]
- Smith JL, Patil PB, Fisher JS. AICAR and hyperosmotic stress increase insulin-stimulated glucose transport. Journal of Applied Physiology. 2005;99:877–883. doi: 10.1152/japplphysiol.01297.2004. [DOI] [PubMed] [Google Scholar]
- Smith JL, Patil PB, Minteer SD, Lipsitz JR, Fisher JS. Possibility of autocrine β-adrenergic signaling in C2C12 myotubes. Experimental Biology and Medicine. 2005;230:845–852. doi: 10.1177/153537020523001109. Abstract
- Somwar R, Kim DY, Sweeney G, Huang C, Niu W, Lador C, et al. GLUT4 translocation precedes the stimulation of glucose uptake by insulin in muscle cells: potential activation of GLUT4 via p38 mitogen-activated protein kinase. Biochem.J. 2001;359:639–649. doi: 10.1042/0264-6021:3590639. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Somwar R, Koterski S, Sweeney G, Sciotti R, Djuric S, Berg C, et al. A dominant-negative p38 MAPK mutant and novel selective inhibitors of p38 MAPK reduce insulin-stimulated glucose uptake in 3T3-L1 adipocytes without affecting GLUT4 translocation. J.Biol Chem. 2002;277:50386–50395. doi: 10.1074/jbc.M205277200. [DOI] [PubMed] [Google Scholar]
- Suzuki A, Kusakai GI, Kishimoto A, Lu J, Ogura T, Lavin MF, et al. Identification of a novel protein kinase mediating Akt survival signaling to ATM. Journal of Biological Chemistry. 2003;278:48–53. doi: 10.1074/jbc.M206025200. [DOI] [PubMed] [Google Scholar]
- Suzuki A, Lu J, Kusakai G, Kishimoto A, Ogura T, Esumi H. ARK5 is a tumor invasion-associated factor downstream of Akt signaling. Mol Cell Biol. 2004;24:3526–3535. doi: 10.1128/MCB.24.8.3526-3535.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Taha C, Tsakiridis T, McCall A, Klip A. Glucose transporter expression in L6 muscle cells: regulation through insulin-and stress-activated pathways. Am J.Physiol. 1997;273:E68–E76. doi: 10.1152/ajpendo.1997.273.1.E68. [DOI] [PubMed] [Google Scholar]
- Thong FS, Dugani CB, Klip A. Turning signals on and off: GLUT4 traffic in the insulin-signaling highway. Physiology (Bethesda.) 2005;20:271–284. doi: 10.1152/physiol.00017.2005. [DOI] [PubMed] [Google Scholar]
- Tortorella LL, Pilch PF. C2C12 myocytes lack an insulin-responsive vesicular compartment despite dexamethasone-induced GLUT4 expression. Am J Physiol Endocrinol Metab. 2002;283:E514–E524. doi: 10.1152/ajpendo.00092.2002. [DOI] [PubMed] [Google Scholar]
- Turban S, Beardmore VA, Carr JM, Sakamoto K, Hajduch E, Arthur JS, et al. Insulin-Stimulated Glucose Uptake Does Not Require p38 Mitogen-Activated Protein Kinase in Adipose Tissue or Skeletal Muscle. Diabetes. 2005;54:3161–3168. doi: 10.2337/diabetes.54.11.3161. [DOI] [PubMed] [Google Scholar]
- Um SH, Frigerio F, Watanabe M, Picard F, Joaquin M, Sticker M, et al. Absence of S6K1 protects against age- and diet-induced obesity while enhancing insulin sensitivity. Nature. 2004;431:200–205. doi: 10.1038/nature02866. [DOI] [PubMed] [Google Scholar]
- Winder WW, Hardie DG. AMP-activated protein kinase, a metabolic master switch: possible roles in type 2 diabetes. American Journal of Physiology. 1999;277:E1–E10. doi: 10.1152/ajpendo.1999.277.1.E1. [DOI] [PubMed] [Google Scholar]
- Wojtaszewski JF, Nielsen JN, Richter EA. Invited review: effect of acute exercise on insulin signaling and action in humans. J Appl.Physiol. 2002;93:384–392. doi: 10.1152/japplphysiol.00043.2002. [DOI] [PubMed] [Google Scholar]
- Xi X, Han J, Zhang JZ. Stimulation of glucose transport by AMP-activated protein kinase via activation of p38 mitogen-activated protein kinase. J.Biol Chem. 2001;276:41029–41034. doi: 10.1074/jbc.M102824200. [DOI] [PubMed] [Google Scholar]
- Yamauchi T, Kamon J, Minokoshi Y, Ito Y, Waki H, Uchida S, et al. Adiponectin stimulates glucose utilization and fatty-acid oxidation by activating AMP-activated protein kinase. Nat.Med. 2002;8:1288–1295. doi: 10.1038/nm788. [DOI] [PubMed] [Google Scholar]
- Zhou G, Myers R, Li Y, Chen Y, Shen X, Fenyk-Melody J, et al. Role of AMP-activated protein kinase in mechanism of metformin action. J.Clin.Invest. 2001;108:1167–1174. doi: 10.1172/JCI13505. [DOI] [PMC free article] [PubMed] [Google Scholar]