

Abstract
Compartmentation of cAMP is thought to generate the specificity of Gs-coupled receptors action in cardiac myocytes, with phosphodiesterases (PDEs) playing a major role in this process by preventing cAMP diffusion. We have tested this hypothesis in adult rat ventricular myocytes (ARVMs) by characterizing PDEs involved in the regulation of cAMP signals and L-type Ca2+ current (ICa,L) upon stimulation with β1-adrenergic receptors (β1-AR), β2-adrenergic receptors (β2-AR), glucagon receptors (Glu-R) and prostaglandin E1 receptors (PGE1-R). All receptors but PGE1-R increased total cAMP, and inhibition of PDEs with IBMX strongly potentiated these responses. When monitored in single cells by high affinity cyclic nucleotide-gated (CNG) channels, stimulation of β1-AR and Glu-R increased cAMP, whereas β2-AR and PGE1-R had no detectable effect. Selective inhibition of PDE3 by cilostamide and PDE4 by Ro 20-1724 potentiated β1-AR cAMP signals, whereas Glu-R cAMP was augmented only by PD4 inhibition. PGE1-R and β2-AR generated substantial cAMP increases only when PDE3 and PDE4 were blocked. For all receptors except PGE1-R, the measurements of ICa,L closely matched the ones obtained with CNG channels. Indeed, PDE3 and PDE4 controlled β1-AR and β2-AR regulation of ICa,L, while only PDE4 controlled Glu-R regulation of ICa,L thus demonstrating that receptor-PDE coupling has functional implications downstream of cAMP. PGE1 had no effect on ICa,L even after blockade of PDE3 or PDE4, suggesting that other mechanisms prevent cAMP produced by PGE1 to diffuse to L-type Ca2+ channels. These results identify specific functional coupling of individual PDE families to Gs-coupled receptors as a major mechanism enabling cardiac cells to generate heterogeneous cAMP signals in response to different hormones.
Keywords: Animals; Calcium; pharmacology; Calcium Channels, L-Type; physiology; Cyclic AMP; physiology; Heart Ventricles; cytology; Ion Channel Gating; physiology; Muscle Cells; drug effects; enzymology; physiology; Patch-Clamp Techniques; Phosphoric Diester Hydrolases; metabolism; Rats; Signal Transduction
Keywords: cAMP, heart, G protein-coupled receptor, phosphodiesterase
Introduction
Cardiac myocytes express a number of Gs-coupled receptors (GsPCRs) that raise intracellular cAMP levels and activate cAMP-dependent protein kinase (PKA) but exert different downstream effects. For instance, β1-AR stimulation produces a major and sustained increase in force of contraction, accelerates relaxation, and stimulates glycogen phosphorylase.1 β2-AR stimulation also increases contractile force but does not activate glycogen phosphorylase2 and does not accelerate relaxation1,3 (but see Bartel et al. (2003)2); Glu-R stimulation activates phosphorylase and exerts positive inotropic and lusitropic effects, but the contractile effects fade with time.4 Finally, PGE-1 has no effect on contractile activity or glycogen metabolism.5,6
Such observations led to the proposal that activation of different GsPCRs results in the accumulation of cAMP and phosphorylation of hormone target proteins in distinct compartments.7 The discovery of A-kinase anchoring proteins, responsible for the subcellular distribution of particulate PKA,8 and the development of new methodologies allowing to track local cAMP in living cells have provided additional support for this concept.9–15
Because cAMP is a small and readily diffusible molecule, cAMP compartments can only exist under a restrictive set of conditions for the localization and activity of the different components of the cAMP signaling cascade, namely GsPCRs, adenylyl cyclases, PKA and the cAMP phosphodiesterases (PDEs). Cardiac PDEs fall into four families: PDE1, which is activated by Ca2+/calmodulin; PDE2, which is stimulated by cGMP; PDE3, which is inhibited by cGMP; and PDE4. While PDE1 and PDE2 can hydrolyze both cAMP and cGMP, PDE3 preferentially hydrolyzes cAMP and PDE4 is specific for cAMP. Until now, only a limited number of studies have addressed directly the participation of PDEs to cAMP compartmentation and hormonal specificity in cardiac cells.9,10,12,15,16 In a recent study, we used recombinant cyclic nucleotide-gated (CNG) channels as cAMP biosensors in adult rat ventricular myocytes (ARVMs) to show that β-adrenergic cAMP signals are localized by PKA-mediated activation of PDE3 and/or PDE4.13 However, PDE regulation of other hormonal cAMP signals is presently unknown. In the following, we address this point by comparing the cAMP signals generated by β1-AR, β2-AR, Glu-R and PGE1-R, their regulation by individual PDE families, and the functional consequences on L-type Ca2+ channels.
Materials and Methods
Detailed methods are included in the online-only Data Supplement to this article, which is available at http://circres.ahajournals.org.
Total cAMP determination
cAMP was measured by RIA after acetylation of the samples.20,21
PDE Assay
PDE activity was measured according to a modification of the two-step assay procedure method described by Thompson & Appleman17 with 1 μM cAMP and 105 cpm [3H]-cAMP, as detailed previously.18
Electrophysiological Experiments
The whole-cell configuration of the patch-clamp technique was used to record the L-type calcium current (ICa,L) and the CNG current (ICNG). The extracellular Cs+-Ringer solution contained 1.8 mM [Ca2+] and 1.8 mM [Mg2+] for ICa,L recordings and nominal [Ca2+] and [Mg2+] plus 1μmol/L nifedipine for ICNG recordings. Patch pipettes were filled with a Cs+-solution.
Data Analysis
ICa,L amplitude was measured as the difference between the peak inward current and the current at the end of the 400-ms duration pulse.19 ICNG amplitude was measured at the end of the 200 ms pulse. Capacitance and leak currents were not compensated. In 141 ARVMs, mean capacitance was 160.5 ± 3.9 pF. ICNG density was calculated for each experiment as the ratio of current amplitude to cell capacitance. All the data are expressed as mean ± S.E.M. When appropriate, the Student’s t-test was used for statistical evaluation.
Results
PDE activities in ARVMs
Figure 1 summarizes experiments where the relative activity of PDE2, PDE3 and PDE4 in quiescent ARVMs was determined. Total cAMP hydrolytic activity was on average 109.8±16.6 pmoles/min/mg protein (n=4) and was mainly due to PDE3 and PDE4, which represented 31% and 38% of the total, respectively. Under these assay conditions, PDE2 activity was minimal (Fig. 1). Immunoprecipitation experiments showed that PDE4 activity is contributed by PDE4A, PDE4B and PDE4D variants, while the PDE3A form is the predominant PDE3 expressed (data not shown). The relative activity of individual PDE families did not vary significantly after 24 h and 48 h of culture (data not shown). Moreover, the ratio of PDE3/PDE4 was similar to that measured in extracts of adult rat ventricle (data not shown). Thus, cultured ARVMs closely resemble the in vivo pattern of PDE expression.
Figure 1.
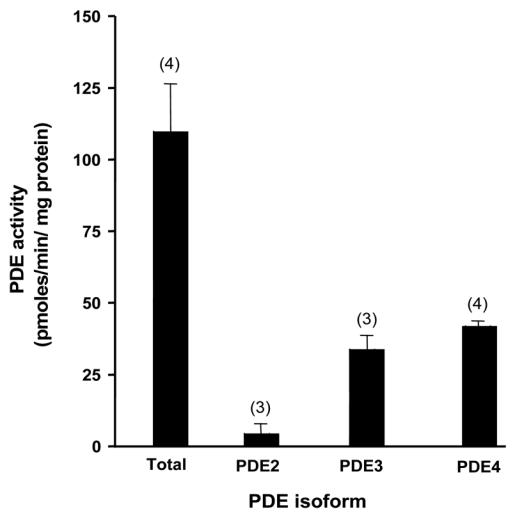
Relative PDE activities in ARVMs. PDE2 activity was determined as the fraction of total hydrolytic activity inhibited by 10 μmol/L EHNA; PDE3 as the fraction inhibited by 1 μmol/L cilostamide; and PDE4 activity as the fraction inhibited by 1 μmol/L rolipram. To some instances, rolipram was replaced by RS25344 (1 μmol/L) or piclamilast (1 μmol/L) with identical results. The bars represent the mean±s.e.m of the number of independent experiments indicated.
Total cAMP production in response to different GsPCRs in ARVMs
We next determined whether stimulation of different GsPCRs increased total cAMP in ARVMs (Fig. 2). We used a combination of isoprenaline (ISO, 5 μmol/L) and the β2-AR antagonist ICI 118551 (ICI, 1 μmol/L) to stimulate β1-AR, a combination of ISO (5 μmol/L) in the presence of the β1-AR antagonist CGP 20712A (CGP, 1 μmol/L) to stimulate the β2-AR, glucagon (Glu, 1 μmol/L) to stimulate Glu-R, and PGE1 (1 μmol/L) to stimulate PGE1-R. Basal cAMP was more than doubled by ISO + ICI and Glu, and increased by ≈70% by ISO + CGP. PGE1 had no effect on cAMP content under these experimental conditions. A 15 min incubation with the broad spectrum PDE inhibitor IBMX (100 μmol/L) increased the total cAMP content by about two-fold and dramatically potentiated the response to all agonists except PGE1. The effect of ISO + ICI and Glu were multiplied by 8 and 7, respectively, while that of ISO + CGP was increased 3-fold. Cyclic AMP content after PGE1 + IBMX was slightly increased when compared to IBMX alone although it did not reach statistical significance.
Figure 2.
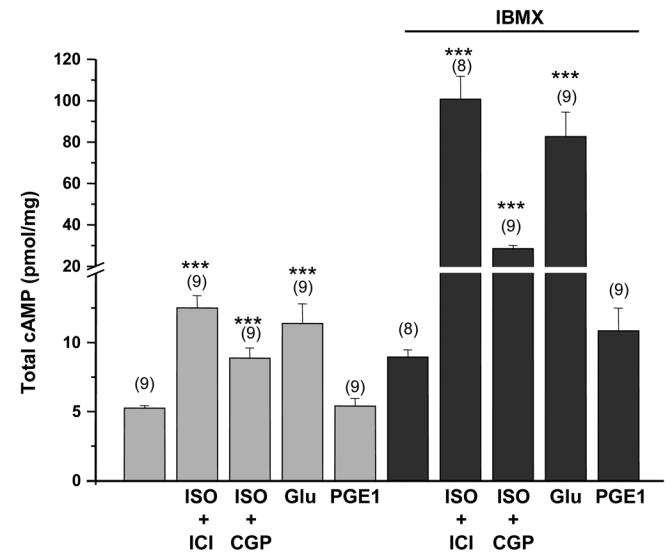
Effect of various stimuli linked to adenylyl cyclase activation on total cAMP content in freshly isolated ARVMs. Where necessary, the β1-AR antagonist CGP 20712A (CGP, 1 μmol/L), the β2-AR antagonist ICI 118551 (ICI, 1 μmol/L) and IBMX (100 μmol/L) were added 15 min before the 3 min stimulation with ISO (5 μmol/L), glucagon (Glu, 1 μmol/L) or PGE1 (1 μmol/L). The bars show the means±s.e.m. of the number of experiments indicated near the bars. Statistically significant difference in cAMP content between basal and agonists alones or between IBMX and agonists with IBMX are indicated as ***, p<0.001.
Real-time monitoring of subsarcolemmal cAMP signals elicited by different GsPCRs in ARVMs
To further compare cAMP signals elicited by the different receptors, we used CNG channels that allow direct monitoring of cAMP variations beneath the plasma membrane in intact ARVMs.13 Figure 3 summarizes the results obtained with two CNGA2 mutants which differ in their sensitivity to cAMP, E583M (EC50 for cAMP ≈ 10 μmol/L) and C460W/E583M (EC50 for cAMP ≈1 μmol/L).20 As depicted in Fig. 3A, activation of β1-AR by a combination of ISO (5 μmol/L) and ICI (1 μmol/L) increased basal ICNG density ≈3-fold, whereas specific activation of β2-AR by ISO (5 μmol/L) plus CGP (1 μmol/L) had no effect. Similarly, activation of Glu-R by Glu (1 μmol/L) or PGE1-R by PGE1 (1 μmol/L) failed to activate ICNG (Fig. 3A). These results indicate that cAMP concentration was below the activation threshold of E583M CNGA2 upon stimulation of β2-AR, Glu-R and PGE1-R. When using the C460W/E583M mutant (Fig. 3B), activation of β1-AR increased basal ICNG density 6-fold. This effect was significantly higher (p<0.01) than that obtained in cells expressing E583M CNGA2, as expected from the relative affinities of the two channels toward cAMP. Interestingly, application of Glu now activated ICNG by approximately 5-fold, but specific activation of β2-AR or PGE1-R still had no effect. Thus, the increased sensitivity of C460W/E583M CNGA2 allowed the detection of subsarcolemmal cAMP generated by activation of Glu-R but not by activation of β2-AR or PGE1-R.
Figure 3.
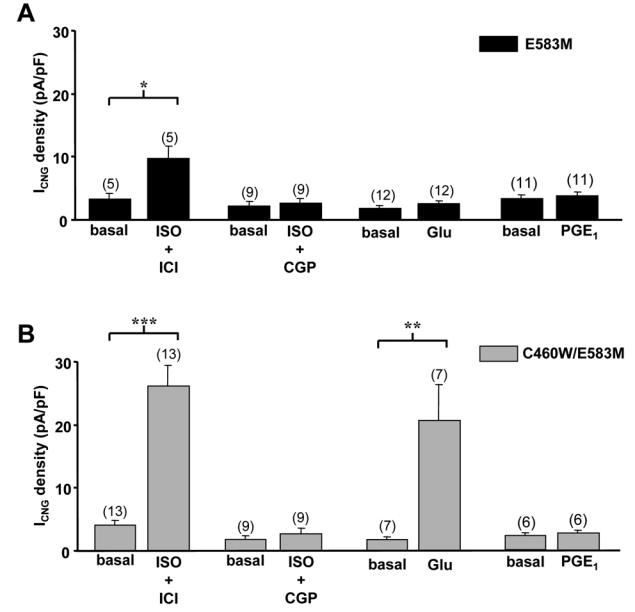
Subsarcolemmal cAMP signals reported by CNG channels upon activation of distinct GsPCRs in ARVMs. CNG current (ICNG) density from recombinant E583M (A) and C460W/E583M (B) CNGA2 channels was measured by the whole-cell patch-clamp technique in rat ventricular myocytes 24 h after isolation. Activation of β1-AR was achieved by application of isoprenaline (ISO, 5 μmol/L) in the presence of the ICI 118551 (ICI, 1 μmol/L). Activation of β2-AR was done by application of ISO (5 μmol/L) in combination with CGP 20712A (CGP, 1 μmol/L). Activation of Glu-R (C) and PGE1-R (D) was done by application of glucagon (Glu, 1 μmol/L) and PGE1 (1 μmol/L). Statistically significant differences are indicated as *, p<0.05; **, p<0.01 and ***, p<0.001.
Regulation of subsarcolemmal cAMP signals by PDEs
The above results suggest that cAMP signals elicited by the four different GsPCRs at the plasma membrane are not identical. We next investigated the contribution of PDEs to such specificity. Both CNGA2 mutants were used with essentially similar results, therefore the low affinity E583M channel data are presented as supplementary Figures I and II. Preliminary experiments showed that IBMX (100 μmol/L) had no significant effect on ICNG from myocytes expressing E583M/C460W (n=6, data not shown). Figure 4A shows a typical experiment in which the amplitude of ICNG is depicted as a function of time, thus providing an instant readout of subsarcolemmal cAMP level in a single cardiac myocyte. β1-AR stimulation with ISO (5 μmol/L) + ICI (1 μmol/L) induced a substantial increase of ICNG, which was strongly potentiated by selective PDE3 inhibition with cilostamide (Cil, 1 μmol/L) or selective PDE4 inhibition with Ro 20-1724 (Ro, 10 μmol/L). Concomitant inhibition of all PDEs with IBMX (100 μmol/L) resulted in a similar potentiation of β1-AR stimulation to that obtained with selective PDE4 inhibition. At the end of each experiment, the cell was challenged with a saturating concentration (100 μmol/L) of the forskolin analog L-858051 (L-85) to determine the maximal ICNG response. In Figures 4C and 4D, we investigated the role of PDEs in shaping the β2-AR cAMP response. As seen before, application of ISO (5 μmol/L) + CGP (1 μmol/L) had no effect on ICNG. Selective inhibition of PDE3 during β2-AR stimulation with ISO + CGP induced a small and transient increase of ICNG. A somewhat stronger effect was observed upon PDE4 inhibition, although it was also transient (Fig. 4C). In contrast, concomitant inhibition of PDE3 and PDE4 unmasked a robust and sustained cAMP increase during β2-AR stimulation. These results indicate that PDE3 and PDE4 control cAMP elicited by β1-AR and β2-AR. Both PDEs are necessary to circumvent the cAMP rise elicited by β1-AR, while either PDE3 or PDE4 individually are sufficient to prevent cAMP to rise beneath membrane upon β2-AR activation.
Figure 4.
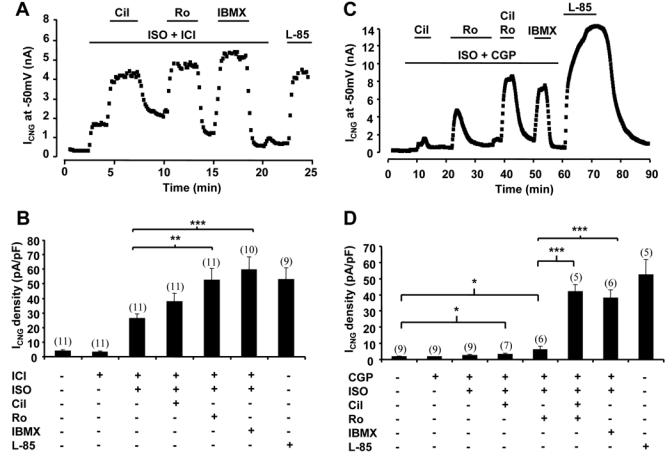
PDE regulation of cAMP signals from β1-AR and β2-AR. A and C, Time course of ICNG in ARVMs expressing C460W/E583M CNGA2. The cells were first superfused with control external Ringer and then challenged with the drugs during the periods indicated by the solid lines. B and D, Summary of the results obtained in a series of experiments as in A and C, respectively. Specific activation of β1-AR and β2-AR as in Fig. 1. Selective PDE3 inhibition by cilostamide (Cil, 1 μmol/L) or selective PDE4 inhibition by Ro 20-1724 (Ro, 10 μmol/L) strongly potentiated cAMP triggered by β1-AR. Upon activation of β2-AR, robust cAMP accumulation necessitated concomitant PDE3 and PDE4 blockade. Experiment was ended with application of 100 μmol/L L-85. .Statistically significant differences as in figure 3.
Figure 5A shows that inhibition of PDE3 had little effect on ICNG augmented by 1 μmol/L Glu while a selective inhibition of PDE4 by Ro produced a strong stimulation of the current. Concomitant inhibition of both PDE3 and PDE4 by Cil + Ro, or of all PDEs by IBMX, resulted in a comparable effect as inhibition of PDE4 alone (Fig. 5A and 5B). These results indicate that Glu-R cAMP signals at the membrane are exclusively controlled by PDE4. Although no increase in cAMP with PGE1 could be detected before, we tested whether PDE inhibition could reveal a PGE1 effect on ICNG. As shown in Fig. 5C, application of 1 μmol/L PGE1 did not affect ICNG. However, in the continuous presence of PGE1, simultaneous inhibition of PDE3 and PDE4 (by Cil + Ro) clearly activated the CNG current. IBMX was more efficient than Cil + Ro, indicating that other PDEs besides PDE3 and PDE4 might play a role under these conditions (Fig. 5C). Thus, similarly to β2-AR, cAMP mobilization by PGE1-R needs concomitant inhibition of PDE3 and PDE4 to be detected at the membrane. However, in contrast to β2-AR, other PDEs besides PDE3 and PDE4 regulate PGE1-R cAMP signals in cardiac cells.
Figure 5.
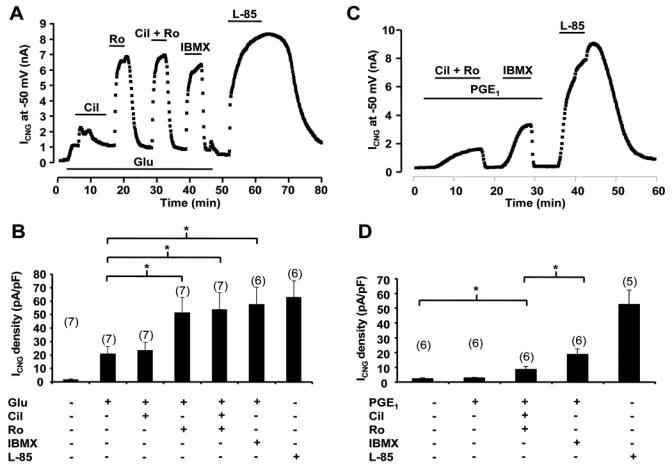
PDE regulation of cAMP signals from Glu-R and PGE1-R. A and C, Time course of ICNG in ARVMs expressing C460W/E583M CNGA2. B and D, Summary of the results obtained in a series of experiments as in A and C, respectively. A and B, Cil (1 μmol/L) exerted a transient potentiation of ICNG previously enhanced by glucagon (Glu, 1 μmol/L), while Ro (10 μmol/L) exerted a major and sustained cAMP accumulation. C and D, The cAMP response to PGE1 was revealed at the membrane by simultaneous blockade of PDE3 and PDE4 and further increased by application of IBMX. Statistically significant differences are indicated as *, p<0.05.
Regulation of the L-type Ca2+ current by GsPCRs and PDEs
Collectively, the above results show that different PDEs are involved in the regulation of cAMP generated by distinct GsPCRs in cardiac myocytes. In order to evaluate the functional implication of these findings, we examined the regulation of a major sarcolemmal cAMP target, the L-type Ca2+ current (ICa,L), by GsPCRs and PDEs. The experiments were performed at 24 h, as with CNG channels experiments. Selective inhibition of PDE3 by Cil (1 μmol/L) or PDE4 by Ro (10 μmol/L) had no effect on basal ICa,L (data not shown). As the concentration of ISO (5 μmol/L) used in the β1-AR stimulation of ICNG produced a maximal stimulation of ICa,L (121.3±22.2% over control, n=9, data not shown), a lower dose of ISO (1 nmol/L, with 1 μmol/L ICI) was used in subsequent experiments in order to assess the contribution of PDE isoforms to the β1-AR regulation of ICa,L. In these conditions, selective inhibition of PDE3 by Cil or PDE4 by Ro resulted in a marked potentiation of the prestimulated ICa,L (Fig. 6A and 6B). As shown in Fig. 6C and 6D, β2-AR stimulation with ISO (5 μmol/L) and CGP (1 μmol/L) produced a small but significant stimulation of ICa,L. This effect could be further potentiated by PDE3 inhibition with Cil and more markedly by PDE4 blockade with Ro. These results indicate that PDE3, and more prominently PDE4, are integral components of ICa,L regulation by β1-AR and β2-AR. In another series of experiments, the regulation of ICa,L by Glu was investigated. At high (1 μmol/L) concentration, Glu maximally stimulated ICa,L and addition of PDE inhibitors had no effect (data not shown). At the concentration of 10 nmol/L, Glu slightly decreased ICa,L and PDE3 inhibition by Cil had no effect. However, application of Ro to block PDE4 clearly increased ICa,L (Fig. 7A and 7B). Finally, as shown in Fig. 7C and 7D, PGE1 had no effect on ICa,L, even when either PDE3 or PDE4 was blocked. However, concomitant inhibition of PDE3 and PDE4 increased ICa,L in the presence of PGE1, but this effect was not different from the effect of the inhibitors on the basal ICa,L (Fig. 7D). These results confirm the specific and functional coupling of Glu-R to PDE4, and show that the cAMP compartment generated by activation of PGE1-R is not in contact with L-type Ca2+ channels.
Figure 6.
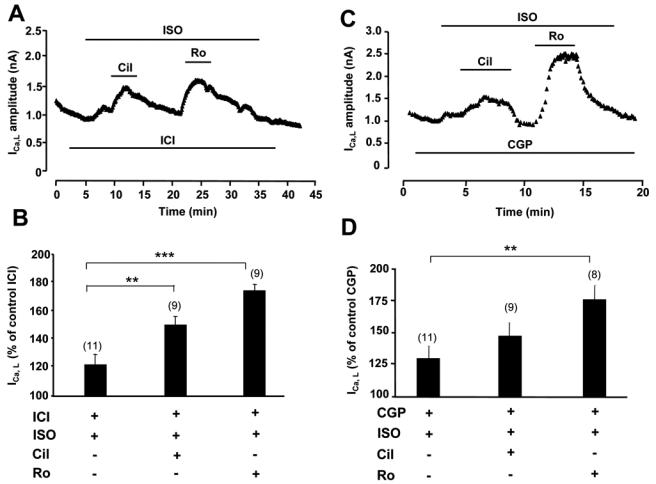
Regulation of ICa,L by β-AR and PDEs. A and C, Time course of ICa,L in ARVMs cultured for 24h. The cells were first superfused with control external Ringer and then challenged with the drugs during the periods indicated by the solid lines. ISO was used at 1 nmol/L in A and B, and at 5 μmol/L in C and D; all other drugs as in previous figures. B and D, Summary of the results obtained in a series of experiments as in A and C, respectively. Statistically significant differences indicated as in previous figures.
Figure 7.
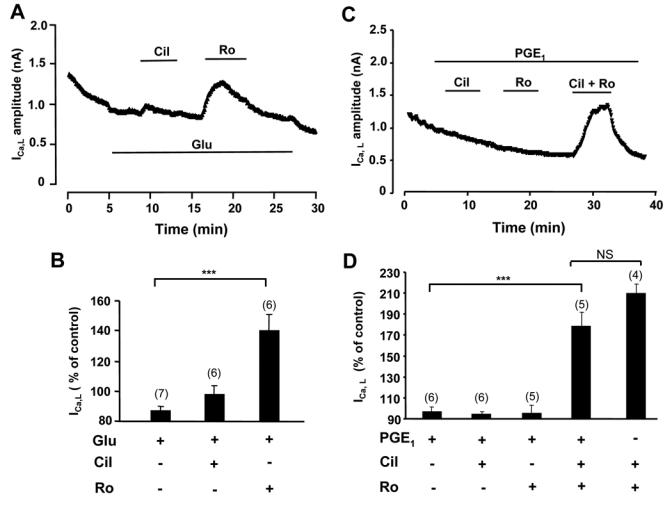
Regulation of ICa,L by Glu-R, PGE1-R and PDEs. A and C, Time course of ICa,L in ARVMs cultured for 24h. Glucagon (Glu) was used at 1 nmol/L in A and B; all other drugs as in previous figures. B and D, Summary of the results obtained in a series of experiments as in A and C, respectively. Statistically significant differences indicated as in previous figures. NS, non significant.
Discussion
While a number of recent studies have underscored the role of PDEs in tailoring hormonal cAMP signals,9–13,21–23 none has addressed directly whether distinct hormonal cAMP signals are controlled by different PDEs. To gain some insight into this question, we used complementary biochemical and electrophysiological techniques to resolve cAMP at the cellular, subsarcolemmal and local levels. We show that four canonical GsPCRs expressed in ARVMs generate different cAMP signals due to a specific contribution of individual PDE families. For all receptors but PGE1-R, the nature of this functional coupling determines the regulation of a major downstream cAMP target, the L-type Ca2+ channels.
In agreement with previous studies, we found that PDE3 and PDE4 represent the major cAMP hydrolytic activities in rodent cardiomyocytes.12,13,24 While there is a general agreement that β1-AR increases cAMP in adult rat cardiac preparations,3,25–27 the ability of β2-AR to do so is a matter of debate.3,25,26 Although glucagon increases cAMP in various cardiac preparations,4 recent studies failed to detect it.27,28. Thus, we checked whether stimulation of the different receptors led to detectable increases in total cAMP in ARVMs. We found that stimulation of β1-AR, Glu-R, and to a lesser extent β2-AR, increased total cAMP content, and that IBMX greatly accentuated these effects. In contrast, PGE1 alone did not increase cAMP, although a small effect could be observed in the presence of IBMX. This result is at variance with those from Buxton & Brunton29 in rabbit cardiomyocytes, where ISO and PGE1 similarly increased cAMP. However, in addition to species differences, the experimental conditions used here are different, as PGE1 was used at a 10-fold lower concentration (1 vs. 10 μmol/L) and application lasted 5-fold less (3 vs. 15 min).
We next used CNG channels to monitor cAMP signals triggered by distinct routes near the plasma membrane.11,13 We found that activation of β1-AR and Glu-R elicit readily detectable cAMP at the membrane, whereas β2-AR and PGE1-R do not. In heterologous expression systems and in rat olfactory epithelium, CNG channels associate preferentially with non-caveolar lipid rafts.30 If this holds true in ARVMs, failure to detect cAMP elicited by β2-AR and PGE1-R could be due to localization of these receptors in caveolae.31–34 However, this localization does not prevent cAMP diffusion to CNG channels when PDEs are inhibited Indeed, we found that IBMX, at 100 μmol/L concentration, increased the response of ICNG not only to β1-AR and Glu-R stimulation, but also revealed cAMP generated by β2-AR and PGE1-R stimulation. Given the Ki of IBMX for the different PDEs,35 100 μmol/L of this inhibitor should block most of the PDE activity, PDE1, PDE2, PDE3 and PDE4 being inhibited by ≈98%, ≈93%, ≈97% and ≈87%, respectively. To delineate the specific contribution of PDE4, we used Ro 20-1724, which at 10 μmol/L should inhibit ≈71% of its activity without affecting the other PDEs.35 We found that PDE4 hydrolyzes cAMP produced by all the receptors tested, thus limiting PKA activation and Ca2+ channel phosphorylation upon β1-AR, β2-AR and Glu-R activation. These results are consistent with previous studies showing that PDE4 is critically involved in the regulation of ICa,L and contractility by catecholamines and glucagon.21,28,36 PDE4 isoforms are likely to play this ubiquitous role due to their molecular diversity and specific subcellular localization.37–39 However, whether specific PDE4 isoforms are associated with different receptors remains unclear. Recent data in neonatal cardiomyocytes reported an important role for PDE4B2 in controlling norepinephrine-induced cAMP increase.12 However, in the same model, other studies suggest that PDE4D isoforms are preferentially associated with the β2-AR. For instance, PDE4D5 and to a lesser extent PDE4D3 are recruited to the β2-AR by β-arrestin upon ISO challenge.22,40 Moreover, Xiang et al. (2005)16 showed that PDE4D selectively affects β2-AR vs. β1-AR positive chronotropism. Whether these findings apply to ARVMs remains to be established.
Nearly complete (≈95%) and selective PDE3 inhibition should be achieved by 1 μmol/L cilostamide35. Using this inhibitor, we found that PDE3 controls the cAMP signals generated by β1-AR, β2-AR and PGE1-R, but not Glu-R. However, on ICa,L response, PDE3 controlled the effects of β1-AR and β2-AR, but had no impact on Glu-R or PGE1-R stimulation. We showed previously that PDE3 controlled cAMP diffusion and consequently ICa,L activation in cardiac myocytes subjected to β–adrenergic stimulation.13,21,36,41,42 Our findings contrast with results obtained in neonatal rat ventricular myocytes where PDE3 was found to have a moderate effect on β1-AR (with norepinephrine) induced cAMP.12 Moreover, in right ventricular strips from adult rat, PDE3 inhibition had no effect on the contractile response to the β2-AR agonist dobutamine but potentiated that of glucagon.27 In parallel cAMP assays, these authors found that PDE3 inhibition had also no effect on the response to dobutamine but revealed a substantial cAMP increase caused by glucagon. While obvious differences in developmental stage, cardiac territory or rat strains might account for some of the discrepancies, one should emphasize that CNG channels provide a readout of subsarcolemmal cAMP, while other methods measure cytosolic or total cAMP. Thus, one could speculate that upon β1-AR stimulation, PDE3 controls the membrane cAMP pool but not a cytosolic one, possibly involved in contractility (sarcoplasmic reticulum-associated PDE3 for instance43). In the case of Glu-R, the opposite would be true: PDE3 would not control subsarcolemmal cAMP generated by Glu-R, but would prevent the access of the second messenger to other, non sarcolemmal inotropic targets. For such a hypothesis to be valid, one must further speculate either that totally segregated cAMP pathways are triggered by the different receptors (including PKA targets), or that differential regulation of PDE3 is triggered by receptor activation. The latter mechanisms was found in frog heart but not in rat.44
While inhibition of either PDE3 or PDE4 potentiated the β1-AR response, and selective inhibition of PDE4 only potentiated the response to Glu, concomitant inhibition of both low Km cAMP PDEs was necessary to observe a substantial cAMP accumulation upon β2-AR and PGE1-R stimulations. This indicates that both PDE3 and PDE4 are functionally associated with β2-AR and PGE1-R, but that one is sufficient to lower cAMP below the detection threshold of CNG channels. When examining the regulation of ICa,L by β2-AR, single inhibition of PDE3 or PDE4 potentiated the effect of ISO+CGP. This may reflect the ≈3–10-fold higher sensitivity of PKA vs. CNG channels to cAMP. In contrast, single inhibition of PDE3 or PDE4 failed to reveal any effect of PGE1 on ICa,L. Although concomitant inhibition of both PDEs readily stimulate ICa,L in these cells and may mask a PGE1 effect (Fig. 7D), the data suggest that PGE1-R lie in a distinct compartment from L-type Ca2+ channels.
Upon stimulation of β1-AR, β2-AR, and Glu-R, inhibition of PDE1-4 by IBMX had a similar effect on ICNG as did concomitant PDE3 and PDE4 inhibition. This suggests that no other PDE, besides PDE3 and PDE4, played a significant role under our experimental conditions. However, a very different situation occurred with PGE1: in this case, inhibition of PDE 1-4 by IBMX had a greater effect than concomitant inhibition of PDE3 and PDE4. Thus, other PDEs, most likely PDE1 or PDE2, may also regulate PGE1-R-mediated intracellular cAMP signals.
Although this study was performed on healthy cardiac myocytes, it is tempting to speculate that changes in the organization of the cAMP compartments might take place in pathological conditions, such as heart failure. Cardiac hypertrophy and failure are associated with a profound remodeling of the cAMP signaling pathway in cardiomyocytes45 As to changes in PDE expression or activity, the results published so far are limited and contradictory. In rapid pacing-induced heart failure in dog, PDE3 mRNA and activity were decreased while PDE4D was unchanged. In the same model, a modest reduction in total PDE activity was found in the left ventricular subendocardium but not in the epicardium, suggesting regional differences.47 In human, sarcoplasmic reticulum-associated PDE3 activity was shown to be unchanged in heart failure,43 although total PDE3 activity appears reduced and PDE3A protein expression down-regulated in end-stage failing human heart.48 In rat, the expression and activity of PDE3 and PDE4 are strongly augmented in hypertension-induced hypertrophy.49 Finally, a recent study shows that PDE4D deficiency favours ryanodine receptors aberrant phosphorylation and promotes heart failure in mice.50 Although more work is needed to obtain a clear picture of the PDE rearrangement occurring in hypertrophy and heart failure, this study suggests that PDE alteration may affect cAMP compartmentation, leading to untargeted hormonal cAMP signals, aberrant phosphorylation of targets proteins and, in fine, contribute to cardiac dysfunction.
Supplementary Material
Acknowledgments
We thank Patrick Lechêne (INSERM U769) for skilful technical assistance and Dr. Frank Lezoualc’h for critical reading of the manuscript. This work was supported by an INSERM fellowship (to M.C.), Fondation de France (to G.V.), French Ministry of Education and Research (F.R. and A.A.-G.), Association Française contre les Myopathies (F.R.), National Institutes of Health (RO1 HD 20788) (to M.C. and K.H.) and by European Union Contract n°LSHM-CT-2005-018833/EUGeneHeart (to R.F.).
Footnotes
Conflict of interest statement
NONE
References
- 1.Xiao RP, Lakatta EG. Beta 1-adrenoceptor stimulation and beta 2-adrenoceptor stimulation differ in their effects on contraction, cytosolic Ca2+, and Ca2+ current in single rat ventricular cells. Circ Res. 1993;73:286–300. doi: 10.1161/01.res.73.2.286. [DOI] [PubMed] [Google Scholar]
- 2.Bartel S, Krause EG, Wallukat G, Karczewski P. New insights into beta2-adrenoceptor signaling in the adult rat heart. Cardiovasc Res. 2003;57:694–703. doi: 10.1016/s0008-6363(02)00720-4. [DOI] [PubMed] [Google Scholar]
- 3.Kuznetsov V, Pak E, Robinson RB, Steinberg SF. Beta 2-adrenergic receptor actions in neonatal and adult rat ventricular myocytes. Circ Res. 1995;76:40–52. doi: 10.1161/01.res.76.1.40. [DOI] [PubMed] [Google Scholar]
- 4.Farah AE. Glucagon and the circulation. Pharmacol Rev. 1983;35:181–217. [PubMed] [Google Scholar]
- 5.Hayes JS, Brunton LL, Brown JH, Reese JB, Mayer SE. Hormonally specific expression of cardiac protein kinase activity. Proc Natl Acad Sci U S A. 1979;76:1570–1574. doi: 10.1073/pnas.76.4.1570. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Brunton LL, Hayes JS, Mayer SE. Hormonally specific phosphorylation of cardiac troponin I and activation of glycogen phosphorylase. Nature. 1979;280:78–80. doi: 10.1038/280078a0. [DOI] [PubMed] [Google Scholar]
- 7.Steinberg SF, Brunton LL. Compartmentation of G protein-coupled signaling pathways in cardiac myocytes. Annu Rev Pharmacol Toxicol. 2001;41:751–773. doi: 10.1146/annurev.pharmtox.41.1.751. [DOI] [PubMed] [Google Scholar]
- 8.Wong W, Scott JD. AKAP signalling complexes: focal points in space and time. Nat Rev Mol Cell Biol. 2004;5:959–970. doi: 10.1038/nrm1527. [DOI] [PubMed] [Google Scholar]
- 9.Jurevicius J, Fischmeister R. cAMP compartmentation is responsible for a local activation of cardiac Ca2+ channels by beta-adrenergic agonists. Proc Natl Acad Sci U S A. 1996;93:295–299. doi: 10.1073/pnas.93.1.295. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Zaccolo M, Pozzan T. Discrete microdomains with high concentration of cAMP in stimulated rat neonatal cardiac myocytes. Science. 2002;295:1711–1715. doi: 10.1126/science.1069982. [DOI] [PubMed] [Google Scholar]
- 11.Rich TC, Fagan KA, Tse TE, Schaack J, Cooper DM, Karpen JW. A uniform extracellular stimulus triggers distinct cAMP signals in different compartments of a simple cell. Proc Natl Acad Sci U S A. 2001;98:13049–13054. doi: 10.1073/pnas.221381398. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Mongillo M, McSorley T, Evellin S, Sood A, Lissandron V, Terrin A, Huston E, Hannawacker A, Lohse MJ, Pozzan T, Houslay MD, Zaccolo M. Fluorescence resonance energy transfer-based analysis of cAMP dynamics in live neonatal rat cardiac myocytes reveals distinct functions of compartmentalized phosphodiesterases. Circ Res. 2004;95:67–75. doi: 10.1161/01.RES.0000134629.84732.11. [DOI] [PubMed] [Google Scholar]
- 13.Rochais F, Vandecasteele G, Lefebvre F, Lugnier C, Lum H, Mazet JL, Cooper DM, Fischmeister R. Negative feedback exerted by cAMP-dependent protein kinase and cAMP phosphodiesterase on subsarcolemmal cAMP signals in intact cardiac myocytes: an in vivo study using adenovirus-mediated expression of CNG channels. J Biol Chem. 2004;279:52095–52105. doi: 10.1074/jbc.M405697200. [DOI] [PubMed] [Google Scholar]
- 14.Warrier S, Belevych AE, Ruse M, Eckert RL, Zaccolo M, Pozzan T, Harvey RD. Beta-adrenergic- and muscarinic receptor-induced changes in cAMP activity in adult cardiac myocytes detected with FRET-based biosensor. Am J Physiol Cell Physiol. 2005;289:C455–461. doi: 10.1152/ajpcell.00058.2005. [DOI] [PubMed] [Google Scholar]
- 15.Mongillo M, Tocchetti CG, Terrin A, Lissandron V, Cheung YF, Dostmann WR, Pozzan T, Kass DA, Paolocci N, Houslay MD, Zaccolo M. Compartmentalized Phosphodiesterase-2 Activity Blunts {beta}-Adrenergic Cardiac Inotropy via an NO/cGMP-Dependent Pathway. Circ Res. 2006;98:226–234. doi: 10.1161/01.RES.0000200178.34179.93. [DOI] [PubMed] [Google Scholar]
- 16.Xiang Y, Naro F, Zoudilova M, Jin SL, Conti M, Kobilka B. Phosphodiesterase 4D is required for {beta} 2 adrenoceptor subtype-specific signaling in cardiac myocytes. Proc Natl Acad Sci U S A. 2005;102:909–914. doi: 10.1073/pnas.0405263102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Thompson WJ, Appleman MM. Multiple cyclic nucleotide phosphodiesterase activities from rat brain. Biochemistry. 1971;10:311–316. [PubMed] [Google Scholar]
- 18.Oki N, Takahashi SI, Hidaka H, Conti M. Short term feedback regulation of cAMP in FRTL-5 thyroid cells. Role of PDE4D3 phosphodiesterase activation. J Biol Chem. 2000;275:10831–10837. doi: 10.1074/jbc.275.15.10831. [DOI] [PubMed] [Google Scholar]
- 19.Kirstein M, Rivet-Bastide M, Hatem S, Benardeau A, Mercadier JJ, Fischmeister R. Nitric oxide regulates the calcium current in isolated human atrial myocytes. J Clin Invest. 1995;95:794–802. doi: 10.1172/JCI117729. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Rich TC, Tse TE, Rohan JG, Schaack J, Karpen JW. In vivo assessment of local phosphodiesterase activity using tailored cyclic nucleotide-gated channels as cAMP sensors. J Gen Physiol. 2001;118:63–78. doi: 10.1085/jgp.118.1.63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Jurevicius J, Skeberdis VA, Fischmeister R. Role of cyclic nucleotide phosphodiesterase isoforms in cAMP compartmentation following beta2-adrenergic stimulation of ICa,L in frog ventricular myocytes. J Physiol. 2003;551:239–252. doi: 10.1113/jphysiol.2003.045211. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Baillie GS, Sood A, McPhee I, Gall I, Perry SJ, Lefkowitz RJ, Houslay MD. beta-Arrestin-mediated PDE4 cAMP phosphodiesterase recruitment regulates beta-adrenoceptor switching from Gs to Gi. Proc Natl Acad Sci U S A. 2003;100:940–945. doi: 10.1073/pnas.262787199. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 23.Barnes AP, Livera G, Huang P, Sun C, O’neal WK, Conti M, Stutts MJ, Milgram SL. Phosphodiesterase 4D Forms a cAMP diffusion barrier at the apical membrane of the airway epithelium. J Biol Chem. 2004;280:7997–8003. doi: 10.1074/jbc.M407521200. [DOI] [PubMed] [Google Scholar]
- 24.Georget M, Mateo P, Vandecasteele G, Lipskaia L, Defer N, Hanoune J, Hoerter J, Lugnier C, Fischmeister R. Cyclic AMP compartmentation due to increased cAMP-phosphodiesterase activity in transgenic mice with a cardiac-directed expression of the human adenylyl cyclase type 8 (ACS) FASEB J. 2003;17:1380–1391. doi: 10.1096/fj.02-0784com. [DOI] [PubMed] [Google Scholar]
- 25.Xiao RP, Hohl C, Altschuld R, Jones L, Livingston B, Ziman B, Tantini B, Lakatta EG. Beta 2-adrenergic receptor-stimulated increase in cAMP in rat heart cells is not coupled to changes in Ca2+ dynamics, contractility, or phospholamban phosphorylation. J Biol Chem. 1994;269:19151–19156. [PubMed] [Google Scholar]
- 26.Laflamme MA, Becker PL. Do beta 2-adrenergic receptors modulate Ca2+ in adult rat ventricular myocytes? Am J Physiol. 1998;274:H1308–H1314. doi: 10.1152/ajpheart.1998.274.4.H1308. [DOI] [PubMed] [Google Scholar]
- 27.Juan-Fita MJ, Vargas ML, Hernandez J. The phosphodiesterase 3 inhibitor cilostamide enhances inotropic responses to glucagon but not to dobutamine in rat ventricular myocardium. Eur J Pharmacol. 2005;512:207–213. doi: 10.1016/j.ejphar.2005.01.056. [DOI] [PubMed] [Google Scholar]
- 28.Juan-Fita MJ, Vargas ML, Kaumann AJ, Hernandez Cascales J. Rolipram reduces the inotropic tachyphylaxis of glucagon in rat ventricular myocardium. Naunyn Schmiedebergs Arch Pharmacol. 2004;370:324–329. doi: 10.1007/s00210-004-0978-6. [DOI] [PubMed] [Google Scholar]
- 29.Buxton IL, Brunton LL. Compartments of cyclic AMP and protein kinase in mammalian cardiomyocytes. J Biol Chem. 1983;258:10233–10239. [PubMed] [Google Scholar]
- 30.Brady JD, Rich TC, Le X, Stafford K, Fowler CJ, Lynch L, Karpen JW, Brown RL, Martens JR. Functional role of lipid raft microdomains in cyclic nucleotide-gated channel activation. Mol Pharmacol. 2004;65:503–511. doi: 10.1124/mol.65.3.503. [DOI] [PubMed] [Google Scholar]
- 31.Rybin VO, Pak E, Alcott S, Steinberg SF. Developmental changes in beta2-adrenergic receptor signaling in ventricular myocytes: the role of Gi proteins and caveolae microdomains. Mol Pharmacol. 2003;63:1338–1348. doi: 10.1124/mol.63.6.1338. [DOI] [PubMed] [Google Scholar]
- 32.Head BP, Patel HH, Roth DM, Lai NC, Niesman IR, Farquhar MG, Insel PA. G-protein-coupled receptor signaling components localize in both sarcolemmal and intracellular caveolin-3-associated microdomains in adult cardiac myocytes. J Biol Chem. 2005;280:31036–31044. doi: 10.1074/jbc.M502540200. [DOI] [PubMed] [Google Scholar]
- 33.Calaghan S, White E. Caveolae modulate excitation-contraction coupling and beta(2)-adrenergic signalling in adult rat ventricular myocytes. Cardiovasc Res. 2006;69:816–824. doi: 10.1016/j.cardiores.2005.10.006. [DOI] [PubMed] [Google Scholar]
- 34.Ostrom RS, Gregorian C, Drenan RM, Xiang Y, Regan JW, Insel PA. Receptor number and caveolar co-localization determine receptor coupling efficiency to adenylyl cyclase. J Biol Chem. 2001;276:42063–42069. doi: 10.1074/jbc.M105348200. [DOI] [PubMed] [Google Scholar]
- 35.Stoclet J-C, Keravis T, Komas N, Lugnier C. Cyclic nucleotide phosphodiesterases as therapeutic targets in cardiovascular diseases. Expert Opin Invest Drugs. 1995;4:1081–1100. [Google Scholar]
- 36.Verde I, Vandecasteele G, Lezoualc’h F, Fischmeister R. Characterization of the cyclic nucleotide phosphodiesterase subtypes involved in the regulation of the L-type Ca2+ current in rat ventricular myocytes. Br J Pharmacol. 1999;127:65–74. doi: 10.1038/sj.bjp.0702506. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Conti M, Richter W, Mehats C, Livera G, Park JY, Jin C. Cyclic AMP-specific PDE4 phosphodiesterases as critical components of cyclic AMP signaling. J Biol Chem. 2003;278:5493–5496. doi: 10.1074/jbc.R200029200. [DOI] [PubMed] [Google Scholar]
- 38.Houslay MD, Adams DR. PDE4 cAMP phosphodiesterases: modular enzymes that orchestrate signalling cross-talk, desensitization and compartmentalization. Biochem J. 2003;370:1–18. doi: 10.1042/BJ20021698. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Richter W, Jin SL, Conti M. Splice variants of the cyclic nucleotide phosphodiesterase PDE4D are differentially expressed and regulated in rat tissue. Biochem J. 2005;388:803–811. doi: 10.1042/BJ20050030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Bolger GB, McCahill A, Huston E, Cheung YF, McSorley T, Baillie GS, Houslay MD. The unique amino-terminal region of the PDE4D5 cAMP phosphodiesterase isoform confers preferential interaction with beta-arrestins. J Biol Chem. 2003;278:49230–49238. doi: 10.1074/jbc.M303772200. [DOI] [PubMed] [Google Scholar]
- 41.Méry PF, Lohmann SM, Walter U, Fischmeister R. Ca2+ current is regulated by cyclic GMP-dependent protein kinase in mammalian cardiac myocytes. Proc Natl Acad Sci U S A. 1991;88:1197–1201. doi: 10.1073/pnas.88.4.1197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Vandecasteele G, Verde I, Rucker-Martin C, Donzeau-Gouge P, Fischmeister R. Cyclic GMP regulation of the L-type Ca(2+) channel current in human atrial myocytes. J Physiol. 2001;533:329–340. doi: 10.1111/j.1469-7793.2001.0329a.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Movsesian MA, Smith CJ, Krall J, Bristow MR, Manganiello VC. Sarcoplasmic reticulum-associated cyclic adenosine 5′-monophosphate phosphodiesterase activity in normal and failing human hearts. J Clin Invest. 1991;88:15–19. doi: 10.1172/JCI115272. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Méry PF, Brechler V, Pavoine C, Pecker F, Fischmeister R. Glucagon stimulates the cardiac Ca2+ current by activation of adenylyl cyclase and inhibition of phosphodiesterase. Nature. 1990;345:158–161. doi: 10.1038/345158a0. [DOI] [PubMed] [Google Scholar]
- 45.Lohse MJ, Engelhardt S, Eschenhagen T. What is the role of beta-adrenergic signaling in heart failure? Circ Res. 2003;93:896–906. doi: 10.1161/01.RES.0000102042.83024.CA. [DOI] [PubMed] [Google Scholar]
- 46.Smith CJ, Huang R, Sun D, Ricketts S, Hoegler C, Ding JZ, Moggio RA, Hintze TH. Development of decompensated dilated cardiomyopathy is associated with decreased gene expression and activity of the milrinone-sensitive cAMP phosphodiesterase PDE3A. Circulation. 1997;96:3116–3123. doi: 10.1161/01.cir.96.9.3116. [DOI] [PubMed] [Google Scholar]
- 47.Sato N, Asai K, Okumura S, Takagi G, Shannon RP, Fujita-Yamaguchi Y, Ishikawa Y, Vatner SF, Vatner DE. Mechanisms of desensitization to a PDE inhibitor (milrinone) in conscious dogs with heart failure. Am J Physiol. 1999;276:H1699–H1705. doi: 10.1152/ajpheart.1999.276.5.H1699. [DOI] [PubMed] [Google Scholar]
- 48.Ding B, Abe J, Wei H, Huang Q, Walsh RA, Molina CA, Zhao A, Sadoshima J, Blaxall BC, Berk BC, Yan C. Functional role of phosphodiesterase 3 in cardiomyocyte apoptosis: implication in heart failure. Circulation. 2005;111:2469–2476. doi: 10.1161/01.CIR.0000165128.39715.87. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Takahashi K, Osanai T, Nakano T, Wakui M, Okumura K. Enhanced activities and gene expression of phosphodiesterase types 3 and 4 in pressure-induced congestive heart failure. Heart Vessels. 2002;16:249–256. doi: 10.1007/s003800200032. [DOI] [PubMed] [Google Scholar]
- 50.Lehnart SE, Wehrens XH, Reiken S, Warrier S, Belevych AE, Harvey RD, Richter W, Jin SL, Conti M, Marks AR. Phosphodiesterase 4D deficiency in the ryanodine-receptor complex promotes heart failure and arrhythmias. Cell. 2005;123:25–35. doi: 10.1016/j.cell.2005.07.030. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.