

Summary
The neuroaxonal dystrophies (NADs) in human beings are fatal, inherited, neurodegenerative diseases with distinctive pathological features. This report describes a new mouse model of NAD that was identified as a spontaneous mutation in a BALB/c congenic mouse strain. The affected animals developed clinical signs of a sensory axonopathy consisting of hindlimb spasticity and ataxia as early as 3 weeks of age, with progression to paraparesis and severe morbidity by 6 months of age. Hallmark histological lesions consisted of spheroids (swollen axons), in the grey and white matter of the midbrain, brain stem, and all levels of the spinal cord. Ultrastructural analysis of the spheroids revealed accumulations of layered stacks of membranes and tubulovesicular elements, strongly resembling the ultrastructural changes seen in the axons of human patients with endogenous forms of NAD. Mouse NAD would therefore seem a potentially valuable model of human NADs.
Keywords: man, mouse model, neuroaxonal dystrophy, spheroids
Introduction
Inherited, primary or endogenous neuroaxonal dystrophies (NADs) form a group of debilitating and ultimately fatal neurodegenerative diseases affecting human beings (Vuia, 1977; Williamson et al., 1982; Kimura et al., 1987; Taylor et al., 1996) and animals, including horses, cats, rats, several breeds of dog (Cork et al., 1983; Schmidt et al., 1983; Chrisman et al., 1984; Carmichael et al., 1993; Sacre et al., 1993; Franklin et al., 1995; Adams et al., 1996) and several inbred and genetically engineered mouse strains (Yamazaki et al., 1988; Dal Canto and Gurney, 1994; Schmidt et al., 1998; Dragatsis et al., 2000; Liedtke et al., 2002; Matsushima et al., 2005). The characteristic pathological feature of NAD in all species is the swollen axon or “spheroid”, often found throughout the central and peripheral nervous systems (Summers et al., 1995). The term “spheroid” refers non-specifically to swollen axons seen by light microscopy and does not describe the nature of the underlying axonal damage or distinguish between potential causative mechanisms. Although spheroids develop in all variants of endogenous NADs, differences in age of onset, clinical manifestations, and distribution of lesions separate one form from another (Vuia, 1977; Williamson et al., 1982; Kimura et al., 1987; Taylor et al., 1996). A number of neuropathological conditions including infectious, demyelinating, toxic or traumatic injuries can damage normal axons and produce spheroids. At the ultrastructural level, however, these “secondary” spheroids are seen to differ greatly from the spheroids found in primary axonopathies or endogenous NAD; thus, degenerate axons predominate in secondary axonopathies and consist of electron-dense bodies, whereas the spheroids in primary axonopathies contain organelles and structural elements that accumulate as a result of defects in axonal transport (Lampert, 1967; Koestner and Norton, 1991; Summers et al., 1995). Although similar lesions are seen in various mammalian species with spontaneously occurring NAD, appropriate animal models for studying the natural progression, biochemical composition and possible underlying mechanisms of spheroid formation are limited.
A new and potentially valuable mouse model of endogenous NAD arose from a spontaneous mutation in a congenic mouse strain, C.D2 Es-Hba (HBA). This strain contained <5% of the DBA/2 genome on the BALB/c mouse strain background and was originally bred to map susceptibility to lymphomagenesis, and subsequently utilized to study immune responses to antigen stimulation in models of atopy and asthma (Ruscetti et al., 1985; McIntire et al., 2001). In the course of these studies, which utilized thousands of HBA-derived progeny for positional cloning of a T-cell and airway hyperreactivity locus, mice were identified with clinical signs of hindlimb spasticity, mild ataxia and progressive paraparesis. The trait appeared to be hereditary, as it segregated within certain HBA sublines. This interesting neurological phenotype was investigated in the present study. Characterization of the neurological disease progression with ultrastructural and immunohistochemical analyses was performed to document similarities between the disease in these mice and human NAD, of which infantile neuroaxonal dystrophy (INAD) is one variant.
Materials and Methods
Animals
The congenic mouse strain, C.D2 Es-Hba, which contains <5% of the DBA/2 mouse strain genome, was originally produced by crossing BALB/c with DBA/2 mice, followed by backcrossing for 10 generations on to the BALB/c mouse strain. Mice were bred and representative progeny with the clinical phenotype were examined histologically, immunohistochemically and ultrastructurally at various stages of disease progression. All studies were approved by Stanford’s Institutional Animal Care and Use Committee (IACUC).
Necropsy and Histopathology
Approximately 100 mice aged 3 weeks to 6 months were submitted for necropsy and histopathological examinations. Mice with NAD and unaffected littermates were humanely killed with CO2, a terminal blood sample being taken from the heart to obtain a complete blood count (CBC) and standard clinical biochemical values. These were then compared with normal reference ranges for BALB/c mice, already established by the veterinary diagnostic laboratory at Stanford University. Complete necropsies were performed on most clinically affected mice and all tissues were fixed in 10% buffered neutral formalin (BNF). In addition, the brain and spinal column with spinal cord were harvested from other affected and unaffected mice and these tissues were fixed in 10% BNF or Cal ExII (Fisher Scientific, Bridgewater, NY, USA), for routine paraffin wax embedding and staining with haematoxylin and eosin (HE). Sections (4 μm) taken throughout the central nervous system (CNS) were examined to determine the overall distribution of spheroids. The following special stains were used: Bielschowsky’s silver stain, periodic acid-Schiff (PAS), Congo red, and Prussian blue.
Immunohistochemistry (IHC)
Paraffin wax-embedded sections of brain and spinal cord were treated with the following primary antibodies: mouse monoclonal antibody (mAb) anti-Tau (Sigma, St Louis, MO, USA), mouse mAb anti-neurofilament (Dako, Carpinteria, CA, USA), rabbit polyclonal anti-glial fibrillary acidic protein (GFAP) (Dako), and rabbit polyclonal anti-ubiquitin (Biomeda, Foster City, CA, USA). Primary antibodies were detected with biotinylated secondary antibodies directed against the species from which the primary antibody was obtained, and streptavidin—horseradish peroxidase (HRP) (Jackson Immunologicals, West Grove, PA, USA) was used for the third amplification step with stable 3,3′-diaminobenzidine tetrahydrochloride (DAB) (Research Genetics, Huntsville, AL, USA) serving as the chromogen.
Transmission Electron Microscopy (TEM)
Pairs of mice aged 0.75, 2, 4 or 6 months were examined. The animals were deeply anaesthetized with pentobarbital (90 mg/kg, intraperitoneally), and the brain and spinal cord were removed after intra-cardiac perfusion with chilled physiological saline followed by a chilled solution of glutaraldehyde 2% in cacodylate buffer (pH 7, 0.1 M). Brain and proximal spinal cord were processed for TEM as previously described (Bouley et al., 2001). Briefly, cubes (1 mm3) of tissue from the dorsal brain stem and spinal cord were post-fixed in glutaraldehyde 2% in cacodylate buffer, transferred overnight to 0.1 M cacodylate buffer, post-fixed in 1% osmium tetroxide, dehydrated, embedded in Epon, sectioned, mounted and stained with toluidine blue. Thin sections were stained with 1% uranyl acetate and lead citrate, mounted on grids and examined with a Philips CM12 electron microscope.
Results
Animals
The NAD trait was originally identified in <2% of the HBA offspring, but it was observed that affected litters usually contained more than one affected animal. Healthy siblings of affected animals were selected from the HBA colony, and subsequent breeding of these HBA sublines produced approximately equal numbers of males and females. In crosses that produced mice with NAD, approximately 25% of the offspring (representing over 100 litters) were clinically affected, suggesting an autosomal recessive mode of Mendelian inheritance. Affected NAD males were unable to breed, probably due to hind limb weakness or paraparesis, although a mild defect in spermatogenesis was observed in many of the adult male NAD mice. Affected females were not placed in breeding pairs.
Clinical Manifestations
At <3 weeks of age, i.e., very early in the course of disease, affected littermates were smaller than unaffected sibs but initially did not appear less mobile. Clinical features, which became clearly visible at 4 weeks of age and progressed thereafter, included hunched posture, unthrifty pelage, crouched stance, stilted gait, and atrophy of the lumbar and hind limb musculature (Fig. 1a), resulting in a 10–30% reduction in body weight. When lifted by the tail, mice would immediately adduct their hind legs and clasp their hind paws together (Fig. 1b). This non-specific clinical sign of sensory neuropathy was noted as early as 3–4 weeks of age. It was consistently seen in the smaller affected mice, but varied in severity. The clasping reflex is not unique to mice with NAD and can be seen with other diseases of the CNS (Tesseur et al., 2000) and in normal mice aged less than 3 weeks (personal observation). Nonetheless, the clasping reflex combined with slightly lower body weights at weaning could be relied upon to identify affected mice when debility and gait abnormalities were not yet detectable. CBC and clinical biochemistry values were unremarkable and no significant differences were detected between affected and unaffected mice.
Fig. 1.
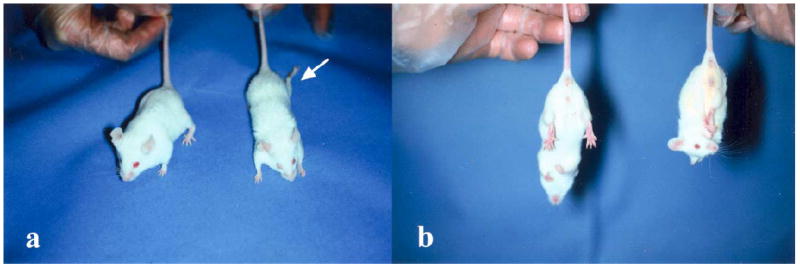
Clinical appearance of the NAD mouse. (a) The NAD mouse on the right has moderate muscle wasting and is smaller than its littermate on the left. Note the left hind limb, splayed caudolaterally (arrow). (b) The mouse on the right, when suspended by its tail, displays a strong clasping reflex, one of the identifying features of NAD mice.
Histopathology
Multiple sections of brain and spinal cord revealed numerous, variably sized, round to irregularly shaped eosinophilic bodies consistent with swollen axons or spheroids (Fig. 2a). The spheroids ranged in size from 2 to 100 μm at their largest dimension and had either a homogeneous appearance or contained pale crescent-shaped structures (Fig. 2b). Others had a “bull’s-eye” appearance with dark–staining centres. Spheroids were seen in both grey and white matter of the spinal cord, with all levels of the cord (cervical, thoracic and lumbar) being equally affected. They were present throughout the midbrain, thalamus, and cerebellar peduncles. The highest density of spheroids, however, was consistently seen in the medulla, predominantly in the sensory nuclei (gracilis, cuneatus and accessory cuneatus), and in the dorsal horns and dorsal funiculi of the spinal cord (Fig. 2c). Sparse, small spheroids were seen throughout the brain and spinal cord in a small proportion of very young (3 weeks) animals showing a pronounced clasp reflex. Spheroids were not detected in peripheral nerves, distal axon terminals of the skin, optic nerve, retina, dorsal root ganglia, trigeminal ganglia, or in the cerebral hemispheres rostral to the anterior commissure.
Fig. 2.
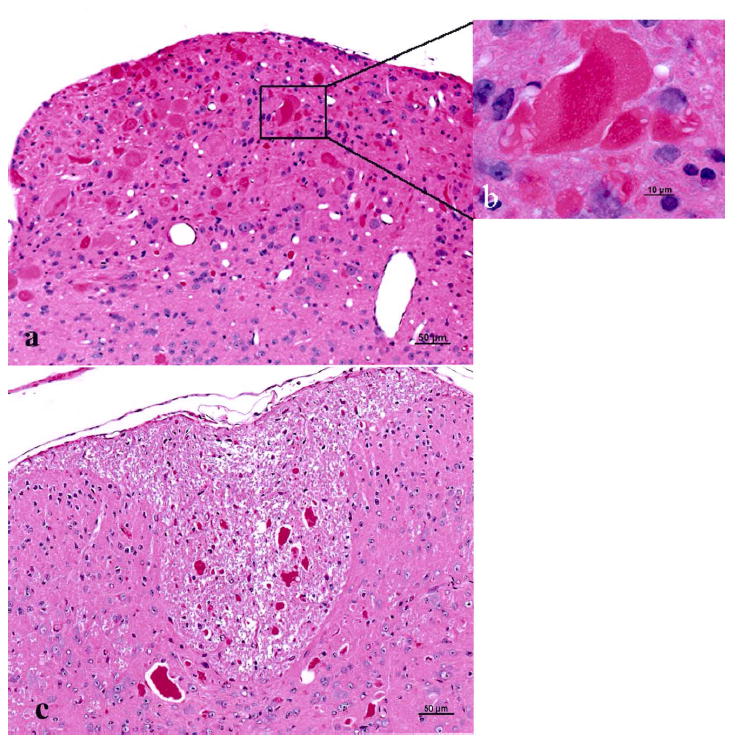
Spheroids in brain stem and spinal cord of a 6-month-old NAD mouse. (a) A sagittal section through the medulla shows a focus of intense spheroid formation. Bar, 50 μm. (b) Inset is a higher magnification of 2a. Note the clear clefts in the large centrally located spheroid. Bar, 10 μm. (c) Dorsal funiculi in the spinal cord contain numerous, variably sized spheroids. Bar, 50 μm. HE.
Other lesions noted in approximately 95% of NAD mice included discrete nodular accumulations of lymphocytes and plasma cells in the lung. Hyperplastic lymphoid nodules were present in typical bronchus-associated lymphoid tissue (BALT) locations, but were more frequently seen in sub-pleural sites, distant from conducting airways. Lesions such as conjunctivitis and periocular dermatitis, variably associated with common bacterial pathogens, were sometimes present but considered to be due to the general debility produced by NAD, rather than to NAD itself.
Special Staining and Immunohistochemistry
Spheroids reacted strongly with PAS and were strongly argyrophilic with Bielschowsky’s silver stain (Fig. 3a) but were not stained by Congo red, and no iron deposits were detected with Prussian blue. Spheroids were immunolabelled faintly and inconsistently with mAb to neurofilament (Fig. 3b) but not with tau antibodies. Most spheroids were labelled strongly with anti-ubiquitin antibody (Fig. 3c), and reactive gliosis was seen to be pronounced in sections treated with anti-GFAP (Fig. 3d).
Fig. 3.
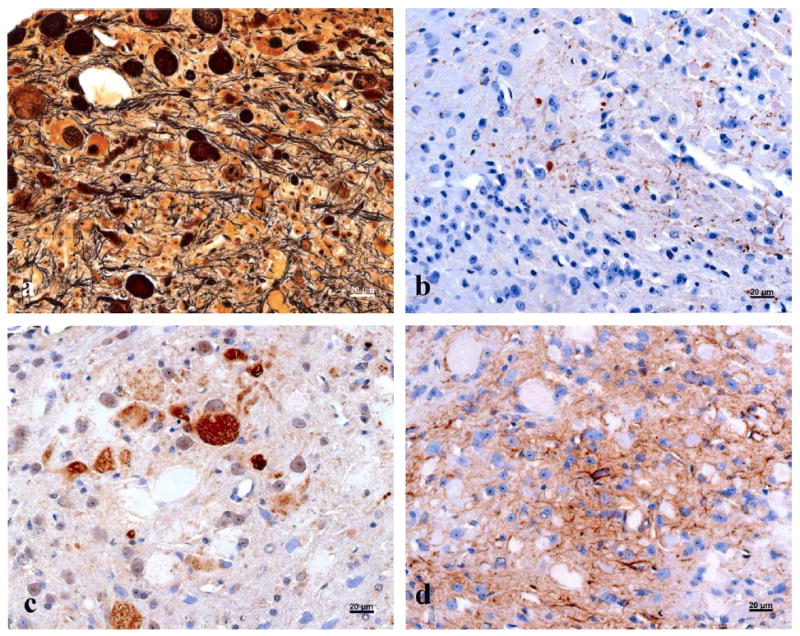
Spheroids were (a) stained with Bielschowsky’s silver stain, (b) labelled by IHC with anti-NF monoclonal antibody, (c) immunolabelled with anti-ubiquitin polyclonal antibody, and (d) immunolabelled with anti-GFAP polyclonal antibody. Bars, 20 μm.
TEM
Perfused brain stem and spinal cord from older mice were examined, focusing on the most heavily affected areas, i.e., the nuclei gracilis and cuneatus from the medulla and dorsal funiculi from the cervical spinal cord. Ultrastructural changes in all locations were similar. The lesions identified as spheroids by light microscopy consisted of distinctly swollen axons, many of which were ruptured, their contents spilling into the surrounding white matter. Two morphologically different types of accumulation were noted in the spheroids. The most prevalent form was composed of stacks of membranes, tubulovesicular profiles, and synaptic vesicles (Fig. 4a,b). Less frequently, a second type of spheroid was noted, consisting of accumulations of degenerate mitochondria, myelin figures, neurofilaments, and membranous dense bodies (Fig. 4c).
Fig. 4.
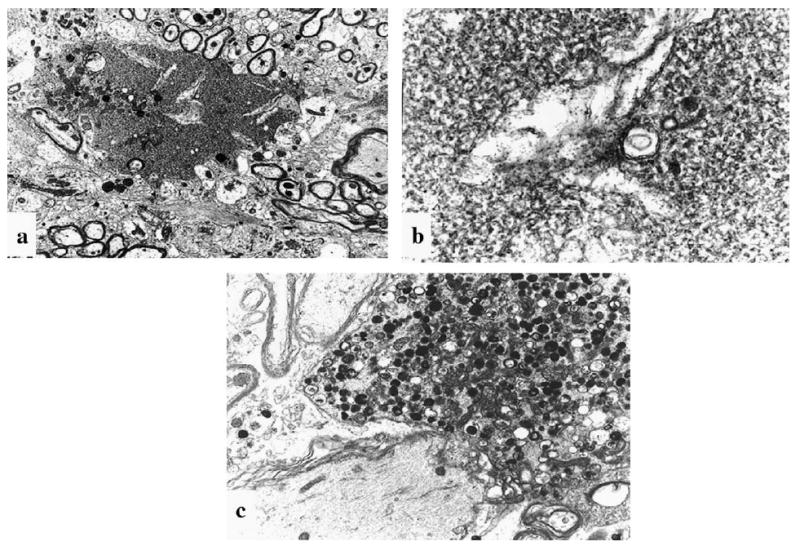
Ultrastructural features in a 6-month-old NAD mouse. (a) Axoplasm spills into the surrounding tissue. Clear clefts are present within densely stacked membranous structures. ×6000. (b) Higher magnification of 4a shows characteristic membranous stacks and tubulovesicular profiles, consistently observed in NAD spheroids. ×35,000. (c) A degenerate axon contains electron-dense bodies and degenerate organelles, suggestive of secondary axonal damage. The latter kind of axon was observed only rarely in the NAD mice. TEM. ×8000.
Discussion
The NAD mouse provides a potentially useful model for the study of endogenous human NAD, particularly in view of the parallels in ultrastructural pathology. The advantages of such an animal model over the use of human autopsy specimens are numerous. It enables nervous tissue to be examined before the onset of severe clinical signs, thus providing insight into pathogenesis. Moreover, it enables both primary disease progression and secondary lesion development to be investigated.
Many previous descriptions of animal models of NAD took the form of single case reports or reports of small kindred groups of animals with NAD. For example, spontaneously occurring mouse models (generalized neuroaxonal dystrophy, gnd/gnd, vestibulo-motor degeneration mouse, vmd/vmd, gracial axonal dystrophy mouse, gad/gad, iNAD) (Yamazaki et al., 1988; Bronson et al., 1992; Matsushima et al., 2005) and various genetically engineered mice (Stathmin −/−, SOD tg, tau tg, and APOE tg mice) (Dal Canto and Gurney, 1994; Spittaels et al., 1999; Tesseur et al., 2000; Liedtke et al., 2002), shared common clinical features with the NAD mouse yet differed in such manifestations as age of onset, lesion distribution and spheroid composition. The gad/gad is the only mouse model in which a specific mutation, thought to be responsible for dystrophic axon formation, has been identified (MacDonald, 1999; Saigoh et al., 1999).
Endogenous forms of human NAD vary in age of onset, clinical signs, and character and distribution of lesions. In INAD, the onset of disease occurs at less than 1 or 2 years of age and death usually occurs before puberty (Malandrini et al., 1995). Hallervorden-Spatz disease (HSS), a variant of INAD, also has an early onset but differs in lesion distribution and characteristics (Malandrini et al., 1995). Other forms do not become manifest until adolescence or even adulthood (Vuia, 1977). NAD mice develop clinical neurological signs (clasping reflex) and detectable spheroids in the brain stem and spinal cord at 3–4 weeks of age. The mice do not become severely debilitated until at least 4 months of age, i.e., well beyond sexual maturity, and often survive until 6 months of age. The histological lesions in the CNS and spinal cord, particularly at the ultrastructural level, closely resemble those seen at autopsy in patients with INAD (Lampert, 1967; Shimono et al., 1976; Malandrini et al., 1995). Large swollen axons in both the grey and white matter of the brain and spinal cord are hallmarks of the disease in human patients and NAD mice. However, two major differences occur, namely, the apparent sparing of the peripheral nervous system in NAD mice, and the lack of iron deposition in neurons in the brain; these are common in some forms of human NAD, such as HSS (Taylor et al., 1996). Despite examination of countless skin sections and sciatic and optic nerves, spheroids were not detected in NAD mice, and no neuronal iron deposits were seen in the brain. As with many diseases, slight differences in underlying pathogenesis may slightly affect the distribution of lesions.
Reactive astrocytosis is a common feature of many neurodegenerative diseases, but as yet the role of glial cells in lesion development remains unclear. GFAP, a marker for differentiated astrocytes, has been used to demonstrate areas of reactive gliosis in various CNS diseases, including INAD (Williamson et al., 1982). GFAP immunoreactivity was substantially increased in the NAD mice.
Abnormal accumulations of structural or functional proteins have been identified by IHC in spheroids and other lesions in patients with various neurodegenerative diseases, including the NADs (Goldman and Yen, 1986; Moretto et al., 1993; Bacci et al., 1994; Hardy and Gwinn-Hardy, 1998; Dunnett and Bjorklund, 1999; Newell et al., 1999; Duda et al., 2000). Neurofilaments (neuron-specific intermediate filaments) accumulate in proximal axons in diseases affecting fast anterograde axonal transport and in the spheroids of amyotrophic lateral sclerosis (ALS) (Goldman and Yen, 1986; Schlaepfer, 1987; Price et al., 1998; Warita et al., 1999). Ubiquitin, a protein that plays a role in intracellular protein degradation, accumulates in many neurodegenerative diseases and is co-localized with neurofilaments in small spheroids in some cases of INAD (Moretto et al., 1993). In the gad mouse, a truncated form of ubiquitin carboxy-terminal hydrolase (Uch-11), encoded by a mis-sense mutation on Chr 5, is believed to contribute to spheroid formation (Saigoh et al., 1999). Reactivity to tau, a 60-kD, phosphoprotein with a role in cross-linking of microtubules, is seen in neurofibrillary tangles, a prominent lesion in Alzheimer’s disease (Grundke-Iqbal et al., 1986; Wiche et al., 1991); moreover, both over-expression and deletion of tau have resulted in neuropathological effects in tg mice (Harada et al., 1994; Spittaels et al., 1999). The spheroids in the NAD mice were not immunolabelled with tau antibodies, and were labelled only minimally with neurofilament antibodies, suggesting that other substances may contribute more substantially to spheroid composition in NAD mice.
Interpretation of the ultrastructural composition of spheroids has been paramount in classifying human NADs, despite speculation that different variants all manifest a single disorder of retrograde axonal transport (Williamson et al., 1982; Malandrini et al., 1995). The ultrastructural morphology of spheroids in NAD mice closely resembles that previously described in primary distal axonopathies of human patients and some animals (Williamson et al., 1982; Cork et al., 1983; Schmidt et al., 1983; Malandrini et al., 1995). In canine NAD, described in the Rottweiler dog as a distal axonopathy, characterization of the spheroids by TEM revealed swollen axons and synaptic terminals distended by accumulations of tubulovesicular profiles and membrane stacks. These types of accumulation are generally described in dystrophic axons with primary defects in transport (Lampert, 1967), and this aspect of the neuropathology in canine NAD closely resembles that seen in the NAD mouse. In the latter, however, the less frequently observed second type of spheroid, consisting of accumulations of degenerate mitochondria, myelin figures, neurofilaments, and membranous dense bodies, are considered more typical of secondary axonopathies (Lampert, 1967).
Perturbations of axonal transport are generally believed to play a role in the pathogenesis of endogenous forms of NAD. Spheroids forming near the neuronal cell body characterize defects in slow anterograde transport and contain structural proteins, such as neurofilaments (Goldman and Yen, 1986; Dahlstrom et al., 1992; Summers et al., 1995). In INAD and HSS, the defect is thought to disrupt retrograde axonal transport, and spheroid formation occurs in the distal axon or at synaptic terminals, where substances such as synaptic vesicles or recycled membranes and organelles accumulate (Griffin and Yen, 1988; Malandrini et al., 1995). As in most distal axonopathies, the neurons in the NAD mice were normal, and spheroids were composed of tubulovesicular profiles and layered stacks of membranous material, consistent with those described in INAD. This suggests that some disturbance in retrograde axonal transport is responsible for the neuropathological changes seen in NAD mice.
In conclusion, the HBA-derived NAD mouse provides a potentially valuable model of endogenous human NAD, and mapping of the genetic defect in the NAD mouse is currently under investigation.
Acknowledgments
The authors thank Pauline Chu for expert technical assistance in histology and immunohistochemistry, Nafisa Ghori for electron microscopy, Elana Hadar for care and management of the NAD mouse colony, Dr Linda Cork for critical neuropathological advice, and the Department of Comparative Medicine for support of the mouse colony. This work was funded in part by the R.J. Murdock Foundation.
Contributor Information
D. M. Bouley, Department of Comparative Medicine, Stanford University School of Medicine, Stanford, CA
J. J. McIntire, Division of Immunology and Allergy (Department of Pediatrics), Stanford University School of Medicine, Stanford, CA
B. T. Harris, Department of Pathology, Dartmouth Medical School, Hanover, NH
R. J. Tolwani, Department of Comparative Medicine, Stanford University School of Medicine, Stanford, CA
G. M. Otto, Department of Comparative Medicine, Stanford University School of Medicine, Stanford, CA
R. H. DeKruyff, Division of Immunology and Allergy (Department of Pediatrics), Stanford University School of Medicine, Stanford, CA
S. J. Hayflick, Department of Genetics, Oregon Health Science Institute, Portland, OR, USA
References
- Adams AP, Collatos C, Fuentealba C, Illanes O, Blanchard R. Neuroaxonal dystrophy in a two-year-old quarter horse filly. Canadian Veterinary Journal. 1996;37:43–44. doi: 10.4141/cjas57-006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bacci B, Cochran E, Nunzi MG, Izeki E, Mizutani T, Patton A, Hite S, Sayre LM, Autilio-Gambetti L, Gambetti P. Amyloid beta precursor protein and ubiquitin epitopes in human and experimental dystrophic axons. Ultrastructural localization. American Journal of Pathology. 1994;144:702–710. [PMC free article] [PubMed] [Google Scholar]
- Bouley DM, Ghori N, Mercer KL, Falkow S, Ramakrishnan L. Dynamic nature of host-pathogen interactions in Mycobacterium marinum granulomas. Infection and Immunity. 2001;69:7820–7831. doi: 10.1128/IAI.69.12.7820-7831.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bronson RT, Sweet HO, Spencer CA, Davisson MT. Genetic and age related models of neurodegeneration in mice: dystrophic axons. Journal of Neurogenetics. 1992;8:71–83. doi: 10.3109/01677069209084153. [DOI] [PubMed] [Google Scholar]
- Carmichael KP, Howerth EW, Oliver JE, Klappenbach K. Neuroaxonal dystrophy in a group of related cats. Journal of Veterinary Diagnostic Investigation. 1993;5:585–590. doi: 10.1177/104063879300500414. [DOI] [PubMed] [Google Scholar]
- Chrisman CL, Cork LC, Gamble DA. Neuroaxonal dystrophy of Rottweiler dogs. Journal of the American Veterinary Medical Association. 1984;184:464–467. [PubMed] [Google Scholar]
- Cork LC, Troncoso JC, Price DL, Stanley EF, Griffin JW. Canine neuroaxonal dystrophy. Journal of Neuropathology and Experimental Neurology. 1983;42:286–296. doi: 10.1097/00005072-198305000-00006. [DOI] [PubMed] [Google Scholar]
- Dahlstrom AB, Czernik AJ, Li JY. Organelles in fast axonal transport. What molecules do they carry in anterograde vs retrograde directions, as observed in mammalian systems? Molecular Neurobiology. 1992;6:157–177. doi: 10.1007/BF02780550. [DOI] [PubMed] [Google Scholar]
- Dal Canto MC, Gurney ME. Development of central nervous system pathology in a murine transgenic model of human amyotrophic lateral sclerosis. American Journal of Pathology. 1994;145:1271–1279. [PMC free article] [PubMed] [Google Scholar]
- Dragatsis I, Levine MS, Zeitlin S. Inactivation of Hdh in the brain and testis results in progressive neurodegeneration and sterility in mice. Nature Genetics. 2000;26:300–306. doi: 10.1038/81593. [DOI] [PubMed] [Google Scholar]
- Duda JE, Giasson BI, Chen Q, Gur TL, Hurtig HI, Stern MB, Gollomp SM, Ischiropoulos H, Lee VM, Trojanowski JQ. Widespread nitration of pathological inclusions in neurodegenerative synucleinopathies. American Journal of Pathology. 2000;157:1439–1445. doi: 10.1016/S0002-9440(10)64781-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dunnett SB, Bjorklund A. Prospects for new restorative and neuroprotective treatments in Parkinson’s disease. Nature. 1999;399:A32–A39. doi: 10.1038/399a032. [DOI] [PubMed] [Google Scholar]
- Franklin RJ, Jeffery ND, Ramsey IK. Neuroaxonal dystrophy in a litter of papillon pups. Journal of Small Animal Practice. 1995;36:441–444. doi: 10.1111/j.1748-5827.1995.tb02774.x. [DOI] [PubMed] [Google Scholar]
- Goldman JE, Yen SH. Cytoskeletal protein abnormalities in neurodegenerative diseases. Annals of Neurology. 1986;19:209–223. doi: 10.1002/ana.410190302. [DOI] [PubMed] [Google Scholar]
- Griffin JE, Yen SH. Axonal transport in neurological disease. Annals of Neurology. 1988;23:3–13. doi: 10.1002/ana.410230103. [DOI] [PubMed] [Google Scholar]
- Grundke-Iqbal I, Iqbal K, Tung YC, Quinlan M, Wisniewski HM, Binder LI. Abnormal phosphorylation of the microtubule-associated protein tau (tau) in Alzheimer cytoskeletal pathology. Proceedings of the National Academy of Sciences of the USA. 1986;83:4913–4917. doi: 10.1073/pnas.83.13.4913. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harada A, Oguchi K, Okabe S, Kuno J, Terada S, Ohshima T, Sato-Yoshitake R, Takei Y, Noda T, Hirokawa N. Altered microtubule organization in small-calibre axons of mice lacking tau protein. Nature. 1994;369:488–491. doi: 10.1038/369488a0. [DOI] [PubMed] [Google Scholar]
- Hardy J, Gwinn-Hardy K. Genetic classification of primary neurodegenerative disease. Science. 1998;282:1075–1079. doi: 10.1126/science.282.5391.1075. [DOI] [PubMed] [Google Scholar]
- Kimura S, Sasaki Y, Warlo I, Goebel HH. Axonal pathology of the skin in infantile neuroaxonal dystrophy. Acta Neuropathologica. 1987;75:212–215. doi: 10.1007/BF00687084. [DOI] [PubMed] [Google Scholar]
- Koestner A, Norton S. Nervous system. In: Haschek WM, Rousseaux CG, editors. Handbook of Toxicology Pathology. Academic Press; San Diego, New York, Boston, London, Sydney, Tokyo, Toronto: 1991. pp. 625–674. [Google Scholar]
- Lampert PW. A comparative electron microscopic study of reactive, degenerating, regenerating, and dystrophic axons. Journal of Neuropathology and Experimental Neurology. 1967;26:345–368. doi: 10.1097/00005072-196707000-00001. [DOI] [PubMed] [Google Scholar]
- Liedtke W, Leman EE, Fyffe RE, Raine CS, Schubart UK. Stathmin-deficient mice develop an age-dependent axonopathy of the central and peripheral nervous systems. American Journal of Pathology. 2002;160:469–480. doi: 10.1016/S0002-9440(10)64866-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- MacDonald ME. Gadzooks! (news; comment) Nature Genetics. 1999;23:10–11. doi: 10.1038/12602. [DOI] [PubMed] [Google Scholar]
- Malandrini A, Cavallaro T, Fabrizi GM, Berti G, Salvestroni R, Salvadori C, Guazzi GC. Ultrastructure and immunoreactivity of dystrophic axons indicate a different pathogenesis of Hallervorden–Spatz disease and infantile neuroaxonal dystrophy. Virchows Archiv. 1995;427:415–421. doi: 10.1007/BF00199391. [DOI] [PubMed] [Google Scholar]
- Matsushima Y, Kikuchi T, Kikuchi H, Ichihara N, Ishikawa A, Ishijima Y, Tachibana M. A new mouse model for infantile neuroaxonal dystrophy, inad mouse, maps to mouse chromosome 1. Mammalian Genome. 2005;16:73–78. doi: 10.1007/s00335-004-3017-5. [DOI] [PubMed] [Google Scholar]
- McIntire JJ, Umetsu SE, Akbari O, Potter M, Kuchroo VK, Barsh GS, Freeman GJ, Umetsu DT, DeKruyff RH. Identification of Tapr (an airway hyperreactivity regulatory locus) and the linked Tim gene family. Nature Immunology. 2001;2:1109–1116. doi: 10.1038/ni739. [DOI] [PubMed] [Google Scholar]
- Moretto G, Sparaco M, Monaco S, Bonetti B, Rizzuto N. Cytoskeletal changes and ubiquitin expression in dystrophic axons of Seitelberger’s disease. Clinical Neuropathology. 1993;12:34–37. [PubMed] [Google Scholar]
- Newell KL, Boyer P, Gomez-Tortosa E, Hobbs W, Hedley-Whyte ET, Vonsattel JP, Hyman BT. Alpha-synuclein immunoreactivity is present in axonal swellings in neuroaxonal dystrophy and acute traumatic brain injury. Journal of Neuropathology and Experimental Neurology. 1999;58:1263–1268. doi: 10.1097/00005072-199912000-00007. [DOI] [PubMed] [Google Scholar]
- Price DL, Sisodia SS, Borchelt DR. Genetic neurodegenerative diseases: the human illness and transgenic models. Science. 1998;282:1079–1083. doi: 10.1126/science.282.5391.1079. [DOI] [PubMed] [Google Scholar]
- Ruscetti S, Matthai R, Potter M. Susceptibility of BALB/c mice carrying various DBA/2 genes to development of Friend murine leukemia virus-induced erythroleukemia. Journal of Experimental Medicine. 1985;162:1579–1587. doi: 10.1084/jem.162.5.1579. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sacre BJ, Cummings JF, De Lahunta A. Neuroaxonal dystrophy in a Jack Russell terrier pup resembling human infantile neuroaxonal dystrophy. Cornell Veterinarian. 1993;83:133–142. [PubMed] [Google Scholar]
- Saigoh K, Wang YL, Suh JG, Yamanishi T, Sakai Y, Kiyosawa H, Harada T, Ichihara N, Wakana S, Kikuchi T, Wada K. Intragenic deletion in the gene encoding ubiquitin carboxy-terminal hydrolase in gad mice. Nature Genetics. 1999;23:47–51. doi: 10.1038/12647. [DOI] [PubMed] [Google Scholar]
- Schlaepfer WW. Neurofilaments: structure, metabolism and implications in disease. Journal of Neuropathology and Experimental Neurology. 1987;46:117–129. [PubMed] [Google Scholar]
- Schmidt RE, Dorsey DA, Beaudet LN, Plurad SB, Parvin CA, Bruch LA. Vacuolar neuritic dystrophy in aged mouse superior cervical sympathetic ganglia is strain-specific. Brain Research. 1998;806:141–151. doi: 10.1016/s0006-8993(98)00678-7. [DOI] [PubMed] [Google Scholar]
- Schmidt RE, Plurad SB, Modert CW. Neuroaxonal dystrophy in the autonomic ganglia of aged rats. Journal of Neuropathology and Experimental Neurology. 1983;42:376–390. doi: 10.1097/00005072-198307000-00002. [DOI] [PubMed] [Google Scholar]
- Shimono M, Ohta M, Asada M, Kuroiwa Y. Infantile neuroaxonal dystrophy. Ultrastructural study of peripheral nerve. Acta Neuropathologica. 1976;36:71–79. doi: 10.1007/BF00685149. [DOI] [PubMed] [Google Scholar]
- Spittaels K, Van den Haut C, Van Dorpe J, Bruynseels K, Vandezande K, Laenen I, Geerts H, Mercken M, Sciot R, Van Lommel A, Loos R, Van Leuven F. Prominent axonopathy in the brain and spinal cord of transgenic mice overexpressing four-repeat human tau protein. American Journal of Pathology. 1999;155:2153–2165. doi: 10.1016/S0002-9440(10)65533-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Summers BA, Cummings JF, De Lahunta A. Principles of neuropathology. In: Summers BA, Cummings JF, De Lahunta A, editors. Veterinary Neuropathology. Mosby; St Louis, MO: 1995. pp. 1–67. [Google Scholar]
- Taylor TD, Litt M, Kramer P, Pandolfo M, Angelini L, Nardocci N, Davis S, Pineda M, Hattori H, Flett PJ, Cilio MR, Bertini E, Hayflick SJ. Homozygosity mapping of Hallervorden-Spatz syndrome to chromosome 20p12.3-p13. Nature Genetics. 1996;14:479–481. doi: 10.1038/ng1296-479. published erratum appears in Vol. 16 [1997], p. 109. [DOI] [PubMed] [Google Scholar]
- Tesseur I, Van Dorpe J, Bruynseels K, Bronfman F, Sciot R, Van Lommel A, Van Leuven F. Prominent axonopathy and disruption of axonal transport in transgenic mice expressing human apolipoprotein E4 in neurons of brain and spinal cord. American Journal of Pathology. 2000;157:1495–1510. doi: 10.1016/S0002-9440(10)64788-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vuia O. Neuroaxonal dystrophy, a juvenile-adult form. Clinical Neurology and Neurosurgery. 1977;79:305–315. [PubMed] [Google Scholar]
- Warita H, Itoyama Y, Abe K. Selective impairment of fast anterograde axonal transport in the peripheral nerves of asymptomatic transgenic mice with a G93A mutant SOD1 gene. Brain Research. 1999;819:120–131. doi: 10.1016/s0006-8993(98)01351-1. [DOI] [PubMed] [Google Scholar]
- Wiche G, Oberkanins C, Himmler A. Molecular structure and function of microtubule-associated proteins. International Review of Cytology—A Survey of Cell Biology. 1991;124:217–273. doi: 10.1016/s0074-7696(08)61528-4. [DOI] [PubMed] [Google Scholar]
- Williamson K, Sima AA, Curry B, Ludwin SK. Neuroaxonal dystrophy in young adults: a clinicopathological study of two unrelated cases. Annals of Neurology. 1982;11:335–343. doi: 10.1002/ana.410110403. [DOI] [PubMed] [Google Scholar]
- Yamazaki K, Wakasugi N, Tomita T, Kikuchi T, Mukoyama M, Ando K. Gracile axonal dystrophy (GAD), a new neurological mutant in the mouse. Proceedings of the Society of Experimental Biology and Medicine. 1988;187:209–215. doi: 10.3181/00379727-187-42656. [DOI] [PubMed] [Google Scholar]