

Summary
We asked whether a movement disorder could be elicited by deprivation of pantothenic acid (PA; vitamin B5), the substrate for the enzyme pantothenate kinase 2 (PANK2), which is deficient in the inherited neurological disorder PKAN (pantothenate kinase-associated neurodegeneration formerly called Hallervorden–Spatz syndrome). This study was undertaken because mice made null for Pank2 failed to show the neurological manifestations of the human disease. Wild-type and Pank2 mutant mice were fed pantothenic acid-deficient diets and were monitored for general health, fertility and movement compared with animals on control diets over time. Mice of both genotypes on PA-deficient diets exhibited poor grooming, greying of fur and decreased body weight. With PA deprivation, wild-type mice manifested azoospermia (a phenotype also seen in Pank2 mice) as well as a movement disorder with a low-lying pelvis and slow steps. Rear limbs appeared to drag and occasionally extended into unnatural postures for 16–17 s duration, possibly indicative of dystonia. Movement disruption probably also occurs in PA-deprived Pank2 mutant mice, but they died precipitously before undergoing detailed analysis. Remarkably, restoration of dietary PA led to recovery of general health and grooming, weight gain, reversal of the movement disorder, and reappearance of mature sperm within 4 weeks. This study confirms the primacy of PA metabolism in the mechanism of disease in PKAN. PA deprivation provides a useful phenocopy for PKAN and allows us to test pharmacological and other interventional strategies in the treatment of this devastating disease.
Introduction
Pantothenate kinase-associated neurodegeneration (PKAN, formerly Hallervorden–Spatz syndrome, OMIM #234200 and #606157) is an autosomal recessively inherited, neurodegenerative disease typically characterized by childhood onset of disturbance in gait, dystonia, dysarthria and dysphagia (Dooling et al 1974). A primary site of degeneration is the globus pallidus, in which cellular tissue is gradually replaced by iron, culminating in a characteristic image referred to as ‘eye of the tiger’ on T2-weighted magnetic resonance imaging. Retinitis pigmentosa is also a common feature of the disease.
PKAN results from a large collection of mutations in PANK2, one of four genes discovered to encode multiple isoforms of pantothenate kinases (NM_153638; Hayflick et al 2003; Zhou et al 2001). Pantothenate kinase (EC 2.7.1.33) metabolizes pantothenic acid (vitamin B5) in the first of five steps in the enzymatic production of coenzyme A (CoA), a ubiquitous metabolic substrate. CoA is an essential ingredient in the tricarboxylic acid cycle, in fatty acid metabolism, and in the synthesis of some amino acids. These findings point to a defect in CoA metabolism as the basis for PKAN, yet leave open the question of which pathway is primary in its pathogenesis. Of the several isoforms of pantothenate kinase, PANK2 appears to be uniquely targeted to the mitochondria in both human and the mouse, highlighting a potentially special niche for this particular enzyme (Hortnagel et al 2003; Johnson et al 2004). Recent biochemical experiments on PANK2 also document its key regulatory role in CoA biosynthesis in the mitochondria, as it is extremely sensitive to inhibition by CoA esters but is activated by palmitoylcarnitine (Leonardi et al 2007; Zhang et al 2006).
To dissect mechanistic questions in vivo, as well as to provide a mouse model for testing therapeutic strategies for PKAN, we previously generated a mouse lacking the murine orthologue (Pank2) of pantothenate kinase 2 (NM_153501; Kuo et al 2005). We demonstrated that a defect in Pank2 causes growth retardation, azoospermia and retinal degeneration; however, the mutant failed to exhibit the movement disorder and brain pathology characteristic of the human disease, even when followed over 18 months of age. We pursued several strategies to provoke a neurological phenotype in the mutant animals by taking advantage of their predicted vulnerabilities, including both chemical and genetic perturbations, but each of these failed (Y. M. Kuo, unpublished data).
In this study we asked whether a deficit in the substrate, pantothenic acid, in wild-type and mutant mice could produce a murine phenocopy of the human movement disorder. We found that, in addition to the growth retardation and azoospermia seen in the Pank2 mutant animals, deprivation of pantothenic acid indeed manifests in a movement disorder reminiscent of the human disease.
Materials and methods
Animals, care and diets
Wild-type C57BL/6J and 129X1/SvJ (formerly 129/SvJ) mice were purchased commercially from Jackson Laboratories. Pank2 (−/−) mice and their wild-type (+/+) littermates were obtained from our breeding colony of heterozygous (+/−) animals (Kuo et al 2005). Confirmation of genotypes was assessed routinely by PCR amplification of genomic DNA prepared from mouse tails using nested pairs of primers and a primer specific for neomycin, yielding 446 bp and 344 bp products for the wild-type and mutant alleles, respectively (Kuo et al 2005).
Mice were fed ad libitum various diets that were formulated and prepared by Teklad Laboratories, Madison, WI, USA. Diets were made up of alcohol-extracted casein and a vitamin mix with and without pantothenic acid (PA) corresponding to control (TD 99366), PA-depleted (TD 03346), 2 mg/kg PA (TD 04361), and 5 mg/kg PA (TD 04362). For the control amino acid diet the vitamin mix supplies approximately 20.8 mg/kg Ca-pantothenic acid (about 19 mg/kg pantothenic acid). The estimated requirement is about 9.1 mg/kg of pantothenic acid per se, and the alcohol-extracted casein added only about 0.6–1.2 mg/kg. Mice were given free access to diet and drinking water. All animals were maintained at 24°C with 55% relative humidity on a 12 h light cycle (0700–1900). All protocols were approved by the University of California, San Francisco Institutional Animal Care and Use Committee.
Motor test
For preliminary assessment of general motor activity, animals were tested in an open field (10-minute test sessions) and video-taped to evaluate any gross abnormalities in locomotion (Crawley 2000). Locomotion was videotaped and assessed using the open-field test (Pekhletski et al 1996), which measures exploratory behaviour in an open field consisting of a 40 cm×60 cm clear-walled Plexiglas box with a clear Plexiglas bottom in standard room conditions of light and temperature. Data were collected weekly over 10 min time intervals. Mice walking on a metal screen were observed for missteps. Missteps were defined as placing any paw between the grids of the metal screen. Gait abnormalities were tested from the footprint pattern of the mouse (Barlow et al 1999; Carter et al 1999; Crawley and Paylor 1997). This footprint test is performed by labelling hind paws with nontoxic paint and placing mice at one end of an enclosed, dark tunnel on a piece of white paper (10 cm wide×50 cm long×10 cm high). Mice walked three times along a 50 cm long, 10 cm wide strip (with 15 cm high walls). Stride length and width (between left and right footprints) were measured from seven consecutive steps on a paper covering the floor of their walking path.
Histological analysis
Tissues were isolated and fixed for 12–14 h in Bouin's solution (Sigma). Fixed tissues were dehydrated, embedded in paraffin and sectioned (5 μm) on the microtome as previously described (Kuo et al 1997). For histological analysis of the eyes, mice were euthanized by carbon dioxide inhalation and immediately perfused intracardially with 2% paraformaldehyde and 2.5% glutaraldehyde as previously described (Kuo et al 2005). Sections were examined using a Nikon E800 microscope and images were captured using a Spot II digital camera.
Results
Effect of PA deficiency on growth and appearance
We monitored two strains of wild-type male mice, 129X1/SvJ and C57BL/6J, on control and PA-deficient diets starting at 4 weeks of age. A reduction in the size of both 129X1/SvJ and C57BL/6J mice on PA-deficient diets was readily apparent, with weekly body-weight measurements indicating differences from 7 weeks through approximately 29 weeks of age (Fig. 1A; C57BL/6J similar result, not shown). The weight of the animals on PA-deficient diets was roughly 74% that of their counterparts on control diets. Although body length was not routinely measured, mice fed on PA-deficient diets were obviously shorter (Fig. 1A, inset).
Fig. 1.
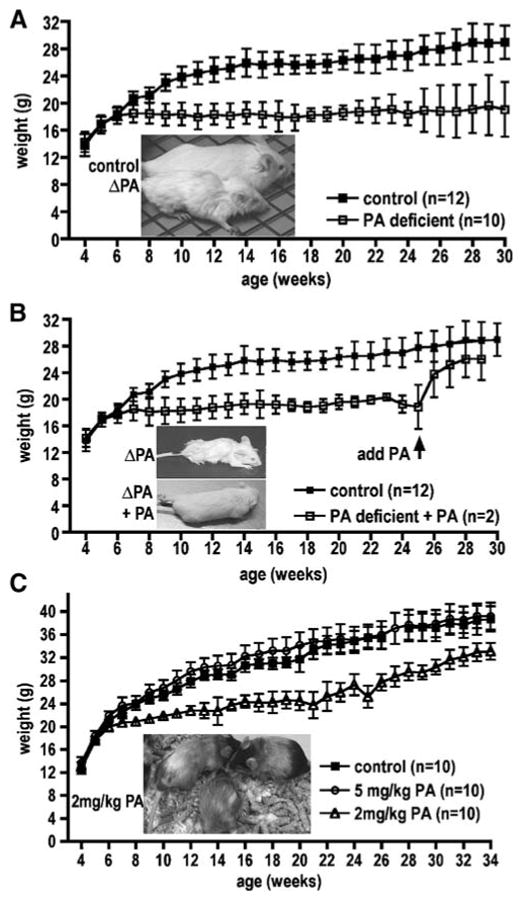
Growth in response to PA deficiency. (A) Growth reduction in 129X1/SvJ mice on pantothenic acid-deficient diets. Body weight (in grams) of the mice on PA-deficient diets (open squares) is compared with that of mice on control diets (solid squares) as a function of age from weeks 4 to 29. Graph shows mean±SEM of body weight. (Inset) Representative photograph of mice on either control or PA-deficient diet at week 20. Weight and size differences in the C57BL/6J in response PA-deficient diets were similar (data not shown). (B) Body weight (in grams) of mice on PA-deficient diets (open squares) is compared to that of mice on control diets (solid squares) as a function of age from weeks 4 to 29. PA supplementation after 25 weeks on a PA-deficient diet results in body weight increase. Graph shows mean±SEM of body weight. (Inset) Photograph of representative mouse on PA-deficient diet at week 25 and the same mouse 4 weeks later given PA supplementation control diet at week 29. (C) Mice on 2 mg/kg pantothenic acid diets show growth reduction. Body weight (in grams) of the mice on 2 mg/kg (open triangles) and 5 mg/kg (open circles) is compared with that of mice on control diets (solid squares) as a function of age from weeks 4 to 34. Graph shows mean±SEM of body weight. (Inset) Representative photograph of mice on 2 mg/kg pantothenic acid diet shows greying of the fur at week 24
In addition to the growth impairment, deprivation of PA led to a number of other physical characteristics, as shown in Fig. 2. Wild-type animals on PA-deficient diets showed poorly groomed fur and hunched over postures compared to those on control diets (Fig. 2A). Greying patches in the coats can be seen in the darkly coloured C57BL/6J mice (Fig. 2B). On PA-deficient diets, over half the mice were more susceptible to eye lesions (Fig. 2C) and facial sores (Fig. 2D), two defects that were similarly observed in a proportion of older (16 months and older) mutant Pank2 mice (Kuo et al 2005; unpublished observation). On control diets, mice do not develop eye or facial lesions (Fig. 2E).
Fig. 2.
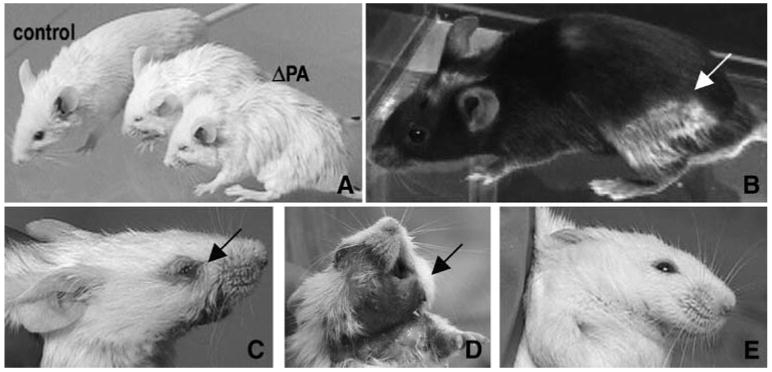
Effect of PA-deficient diets in wild-type mice. (A) 129X1/SvJ mice on PA-deficient diets are poorly groomed and hunched over versus mouse on control diet. (B) C57BL/6J mouse on PA-deficient diet shows greying of the fur. (C, D) Example of a mouse on a PA-deficient diet showing an eye lesion (C) and skin dermatitis (D). (E) Example of a mouse on control diet with no eye or skin lesions
Neurological assessment of wild-type mice on PA-deficient diets
The animals on PA-deficient diets were continually monitored for evidence of movement abnormalities. Wild-type mice on control diets were active, continually moving and exploring their environment, whereas mice on PA-deficient diets were mostly inactive. A substantial reduction of spontaneous movement, which ensued at about 18–20 weeks, was an initial sign of a progressive neurological degeneration.
Video monitoring documented that PA-deficient mice developed a movement disorder with their belly low because of a low-lying pelvis and slow steps (mostly rear limbs) and overall weakness (video clips available upon request from authors). The rear limbs in particular seemed to drag, and they were occasionally extended into abnormal postures for approximately 16–17 s duration. Occasional signs of spasticity and seizure were also apparent. Figure 3A shows an example of a 129X1/SvJ mouse on control diet with normal posture, whereas mice on PA-deficient diets could be distinguished by bizarre limb positioning (Fig. 3B–D). These disabilities were reflected further in footprint tests that recorded compromised motor functions and gait disturbance of the mice on PA-deficient diets at the phenotypic stage of 20 weeks (Fig. 3E, right column).
Fig. 3.
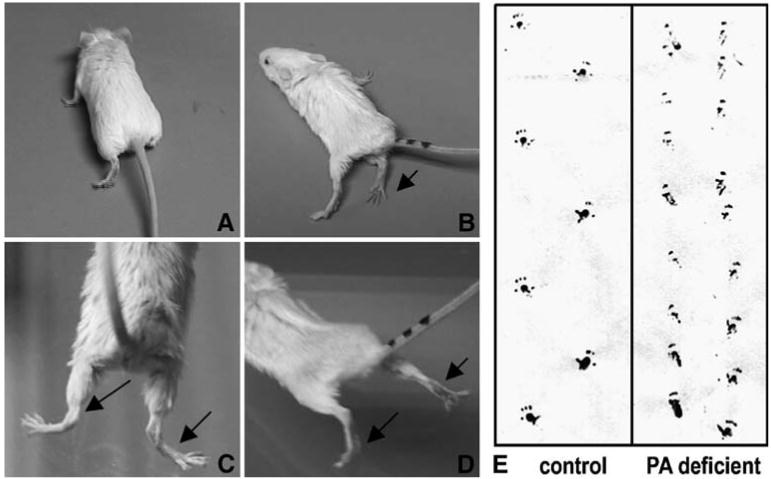
Effect of diet on motor function in wild-type mice. Compared to the mice on control diets (A) the 129X1/SvJ mice on PA-deficient diets show limb weakness and bizarre positioning (B–D). Footprint analysis (E) indicates compromised motor function in the 129X1/SvJ mice on PA-deficient diets. The hind paw prints of mice were recorded while walking on white paper. Footprint analyses indicated that mice on a PA-deficient diet showed decreased average stride length and decreased distance between the feet during walking compared to mice on control diets
Despite their neurological presentation, the wild-type mice fed PA-deficient diets showed no evidence of brain iron accumulation by Perls–DAB staining (methods and data not shown). This finding is concordant with our earlier failure to detect any iron accumulation in the Pank2 mutant mice by either histology or MRI (Kuo et al 2005). Whether the PA-deficient mice would ultimately have shown iron accumulation in the globus pallidus remains an open question; PA was reintroduced into the diet at 25 weeks because of animal morbidity. The PA-deficient mice also did not show any histological evidence for retinal degeneration (data not shown); however, in Pank2 mutant animals, retinal degeneration is observed anatomically only after one year of age (Kuo et al 2005).
Azoospermia in the PA-deprived mice
Like the Pank2 mice, wild-type mice on PA-deficient diets failed to produce progeny. Histological analyses of testes taken from mice on PA-deficient diets revealed a complete absence of elongated spermatids in the seminiferous tubules. As shown in Fig. 4A, mature elongated spermatozoa were seen in the lumen of animals on control diets, but were completely absent in the 129X1/SvJ mice on PA-deficient diets (Fig. 4B). Only round spermatids were present near the luminal border of the mutant tubule. These failed to elongate, and instead appeared to accumulate vacuoles. Compared to normal spermatogenesis, in which maturation into haploid spermatids occurs in a synchronous and spatially orderly process, the spermatocytes and spermatids were somewhat disordered in the testes tubule of mice on PA-deficient diets. This phenotype of arrested spermatid development in PA-deficiency in wild-type animals is identical to that seen in Pank2 animals.
Fig. 4.
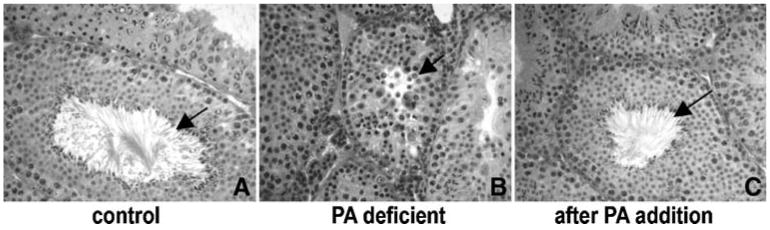
Histological analysis of testis sections taken from mice and stained with haematoxylin and eosin. Sections at 29 weeks of age from a mouse on control diet (A) show mature spermatozoa that are absent in sections from a mouse on PA-deficient diet (B). A mouse that was on a PA-deficient diet then given 4 weeks of PA supplementation starting at 25 weeks of age shows mature spermatozoa at age 29 weeks old (C). Images were taken at ×40 magnification
Effect of PA supplementation
At 25 weeks, introduction of PA into the diets of PA-depleted mice yielded a significant improvement in every aspect of their health within 4 weeks. Figure 1B documents the rapid weight gain in the test mice, although they failed to reach the full weight of their counterparts on control diets. They also exhibited remarkable restoration in general health and grooming (Fig. 1B inset). Mature sperm were also produced in the testes within 4 weeks of PA addition to the diet (Fig. 4C). Deficits in movement also appeared to be fully reversible; video monitoring showed that the animals moved with no difficulty and were active and alert with spontaneous movement, indistinguishable from mice on control diets (video clips available upon request from authors).
Effect of PA deficiency in Pank2 mutant mice
PA deficiency also led to impairment of growth in the Pank2 mutant animals by ∼25%; however, on both control and PA-deficient diets, the Pank2 animals were ∼20% lighter than their wild-type littermates. These Pank2 animals also showed fur discoloration and poor grooming. Most Pank2 mice fed PA-deficient diets died precipitously without our being able to discern a neurological problem or observe any eye or skin lesions; however, we have observed a few who, prior to their death, exhibited mild neurological abnormalities (absence of spontaneous activity and slow movement). Pank2 mice fed control diets exhibited no ill effects within the period of this experiment other than growth retardation and azoospermia, reported previously (Kuo et al 2005).
Effect of intermediate diets on wild-type and mutant mice
We tested 2 mg/kg and 5 mg/kg PA diets in an attempt to elicit a phenotype less severe than the sudden death exhibited by Pank2 mice on the PA-deficient diets. Growth (Fig. 1C), appearance, and behaviour of wild-type mice on 5 mg/kg diets were similar to those on control diets. On 2 mg/kg diets the wild-type had an intermediate phenotype, including weight loss (Fig. 1C), greying of fur and poor grooming (Fig. 1C inset). Pank2 mutant mice similarly showed growth reduction on 2 mg/kg PA, but not on 5 mg/kg, compared with the control diet. No eye or skin disease or movement disorder was elicited in either mutant or wild-type mice, even after 1 year of observation on 2 mg/kg diet. Curiously, fur discoloration and grooming deficits were not as pronounced in the Pank2 mutant animals on 2 mg/kg diets compared with the wild-type mice on the same diet. A 2 mg/kg diet was sufficient to maintain spermatogenesis in the wild-type animals and the generation of surviving offspring.
Discussion
We report that mice raised on pantothenic acid-deficient diets (0 mg/kg) exhibit impaired growth, poorly groomed fur with discoloured patches, azoospermia, eye and facial lesions, and, most significantly, a movement disorder developing around 18 weeks. Specifically, the wild-type mice developed an abnormal gait with abdominal dragging and hind limb dyskinesia. Rear limbs in particular seemed to drag, demonstrating occasional action-induced dystonia with foot extension during ambulation. Remarkably, all of these disease symptoms could be reversed by the addition of PA back into the diet, although body weight appeared not to be fully restored.
Our data support previously published studies on PA deficiency in animals, including dogs, calves, monkeys and rodents, that show high incidence of spontaneous infection and morbidity, fur depigmentation, hair loss and hind leg muscle weakness (McCall et al 1946; Plesofsky-Vig 1996; Sheppard and Johnson 1957). In addition, pigs deficient in PA develop a movement disorder described as ‘goose stepping’ and raise their hind legs rapidly after stepping down hard (Wintrobe et al 1943). PA-deficient animals also have altered reproductive systems, as determined by various studies in pigs, chickens and turkeys, and rats (Nelson and Evans 1946; Ullrey et al 1955), due to gestational growth or to unspecified fertility problems. Our results suggest that at least some of the latter may be attributed to impairment of spermatogenesis.
Comparison of the PA nutritional deficiency with the phenotype of the Pank2 mutant mice allows us to dissect which features of the nutritional deficiency can be ascribed to lack of coenzyme A production by the enzyme Pank2. As summarized in Table 1, features seen in the nutritional deficiency that are recapitulated in the genetic deficiency include poor growth and eye and skin lesions, although the lesions present at a later age (18 months) in the mutant animals. In addition, the wild-type mice on PA-deficient diets become azoospermic, histologically indistinguishable from the Pank2 mice. These comparisons suggest that the above aspects of PA-deficiency syndrome are likely to directly involve the particular mitochondrial-specific coenzyme A synthetic pathway that utilizes Pank2.
Table 1.
Comparison of phenotypes in PA-deficient mice, Pank2-deficient mice, and human patients with PKAN
| Phenotype | PA-deficient diet | Pank2 mice | PKAN patients |
|---|---|---|---|
| Movement disorder | + | − | + |
| Retinal degeneration | N/A | + | + |
| Azoospermia | + | + | ? |
| Eye and skin lesions | + | − | − |
| Premature greying | + | − | − |
| Growth retardation | + | + | − |
In contrast, the patchy fur discoloration and poor grooming caused by PA deficiency is not observed in the Pank2 mutant, possibly implicating a different pantothenate kinase isoform in the mechanism of these manifestations. Further support for this hypothesis lies in the curious observation that the mutant animals on 2 mg/kg diet do not show as pronounced fur discoloration or grooming deficits as their wild-type littermates on the same diet. This may suggest that a knockout of Pank2 leaves more PA substrate available for the pantothenate kinase 1, 3 and 4 isoforms, or that synthesis of the other isoforms is augmented in the mutant animal. These possibilities can potentially be addressed when knockout mutants of the other genes become available. Moreover, quantitative measurement of both RNA (by real-time RT PCR) and protein (using isoform-specific antibodies) of the various isoforms of wild-type, mutant and pantothenate-deficient animals will also allow metabolic relationships between the isoforms to be dissected.
The movement disorder seen in the nutritional deficiency was anticipated but not observed in the Pank2 mutant mice, even though a movement disorder is the primary presentation of the human disease PKAN. A possible explanation for this difference could lie in the variability in CoA concentrations in different tissues, the variability in sequestration of CoA between the cytoplasmic and mitochondrial compartments, and the variability in the relative expression levels of the four PANK genes (Zhou et al 2001; Hayflick, unpublished observations). We hypothesize that CoA levels produced by Pank 1, 3 and 4 in the rodent brain may be sufficient to compensate for the mitochondrial CoA deficit in Pank2-null animals, but insufficient in the brains of human patients with PKAN. In the nutritional model, total brain CoA is probably depleted below a protective threshold level, leading to manifestation of a movement disorder.
Iron accumulation in the globus pallidus, a hallmark of the human disease, was not observed in either the PA-deficient wild-type or Pank2 mutant mice on either replete or PA-deficient diets. Although the mechanism underlying iron deposition is unknown, we might speculate on several factors that could contribute to the difference observed between mice and humans. First, the iron accumulation seen in the human condition, as evidence by the ‘eye of the tiger’ impression on MRI, is gradual, manifesting after onset of disease, typically beyond the lifespan of the average mouse, and certainly well beyond the 26 weeks when the PA-deficient mice are so compromised that PA must be introduced back into the diet. Second, the human basal ganglia of are normally rich in iron, probably contributing to the pathological finding; iron may not be as abundant in the corresponding structures in mice. Third, along similar lines, we previously speculated that cysteine accumulation could be a contributing factor to iron accumulation in the globus pallidus in PKAN (Zhou et al 2001); if this is true, lower free cysteine in the mouse brain could also offer protection.
The contrast between the neurology of the PA-deprived and Pank2 mutant animals also causes us to question whether an additional genetic element in the mutant animal might protect the mice from manifesting the movement disorder. If so, understanding the nature of this modifier might provide insights into the human pathophysiology of disease. We are currently testing this hypothesis by back-crossing Pank2 in a variety of backgrounds, including AKR and SWR strains, which have high endogenous levels of iron (Clothier et al 2000; Fleming et al 2001), as strain backgrounds can influence manifestation of disease (Buchner et al 2003).
Nutritional pantothenate deficiency provides a useful phenocopy for the human disorder PKAN and may allow us to test pharmacological and other interventional strategies as therapy for of this devastating disease. The rapid reversibility of symptoms with addition of pantothenic acid in the mouse model also suggests that pantothenate supplementation could be useful in treating PKAN. Even in patients with null mutations in PANK2, it may be possible to stimulate production of sufficient levels of CoA through PANK1, 3 and 4 by providing excess substrate to compensate for the loss of CoA through PANK2 deficiency. Indeed, some patients anecdotally report improvement in symptoms with pantothenic acid supplementation, but no formal clinical trial has yet addressed this question.
Acknowledgments
We are very grateful to Dr Hyder A. Jinnah for reviewing videotapes of the mice. We appreciate the excellent technical assistance Hernan Consengco and Angela Yu and members of our laboratories for helpful discussion and support. Research was funded by grants to S. J. H from the NEI and Oregon Center for Complementary and Alternative Medicine in Neurological Disorders, P50 AT00066. Y. M. K and J. G were also supported by the Howard Hughes Medical Institute during much of this work.
Abbreviations
- PA
pantothenic acid
- PANK2
pantothenate kinase 2
- PKAN
pantothenate kinase-associated neurodegeneration
Footnotes
Contributor Information
Y. M. Kuo, Departments of Medicine and Pediatrics, University of California, San Francisco, CA, USA
S. J. Hayflick, Department of Molecular and Medical Genetics, Oregon Health and Science University, Portland, OR, USA
J. Gitschier, Departments of Medicine and Pediatrics, University of California, San Francisco, CA, USA.
References
- Barlow C, Hirotsune S, Paylor R, et al. Atm-deficient mice: a paradigm of ataxia telangiectasia. Cell. 1999;86(1):159–171. doi: 10.1016/s0092-8674(00)80086-0. [DOI] [PubMed] [Google Scholar]
- Buchner DA, Trudeau M, Meisler MH. SCNM1, a putative RNA splicing factor that modifies disease severity in mice. Science. 2003;301(5635):967–969. doi: 10.1126/science.1086187. [DOI] [PubMed] [Google Scholar]
- Carter RJ, Lione LA, Humby T, et al. Characterization of progressive motor deficits in mice transgenic for the human Huntington's disease mutation. J Neurosci. 1999;19(8):3248–3257. doi: 10.1523/JNEUROSCI.19-08-03248.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Clothier B, Robinson S, Akhtar RA, et al. Genetic variation of basal iron status, ferritin and iron regulatory protein in mice: potential for modulation of oxidative stress. Biochem Pharmacol. 2000;59(2):115–122. doi: 10.1016/s0006-2952(99)00306-8. [DOI] [PubMed] [Google Scholar]
- Crawley JN. Behavioral Phenotyping of Transgenic and Knockout Mice. New York: Wiley-Liss; 2000. What's Wrong With My Mouse? pp. 31–45.pp. 47–63. [Google Scholar]
- Crawley JN, Paylor R. A proposed test battery and constellations of specific behavioral paradigms to investigate the behavioral phenotypes of transgenic and knockout mice. Horm Behav. 1997;31(3):197–211. doi: 10.1006/hbeh.1997.1382. [DOI] [PubMed] [Google Scholar]
- Dooling EC, Schoene WC, Richardson EP., Jr Hallervorden–Spatz syndrome. Arch Neurol. 1974;30:70–83. doi: 10.1001/archneur.1974.00490310072012. [DOI] [PubMed] [Google Scholar]
- Fleming RE, Holden CC, Tomatsu S, et al. Mouse strain differences determine severity of iron accumulation in Hfe knockout model of hereditary hemochromatosis. Proc Natl Acad Sci USA. 2001;98(5):2707–2711. doi: 10.1073/pnas.051630898. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hayflick SJ, Westaway SK, Levinson B, et al. Genetic, clinical, and radiographic delineation of Hallervorden–Spatz syndrome. N Engl J Med. 2003;348:33–40. doi: 10.1056/NEJMoa020817. [DOI] [PubMed] [Google Scholar]
- Hortnagel K, Prokisch H, Meitinger T. An isoform of hPANK2, deficient in pantothenate kinase-associated neurodegeneration, localizes to mitochondria. Hum Mol Genet. 2003;12:321–327. doi: 10.1093/hmg/ddg026. [DOI] [PubMed] [Google Scholar]
- Johnson MA, Kuo YM, Westaway SK, et al. Mitochondrial localization of human PANK2 and hypotheses of secondary iron accumulation in pantothenate kinase-associated neurodegeneration. Ann NY Acad Sci. 2004;1012:282–298. doi: 10.1196/annals.1306.023. [DOI] [PubMed] [Google Scholar]
- Kuo YM, Gitschier J, Packman S. Developmental expression of the mouse mottled and toxic milk genes suggests distinct functions for the Menkes and Wilson disease copper transporters. Hum Mol Genet. 1997;6:1043–1049. doi: 10.1093/hmg/6.7.1043. [DOI] [PubMed] [Google Scholar]
- Kuo YM, Duncan JL, Westaway SK, et al. Deficiency of pantothenate kinase 2 (Pank2) in mice leads to retinal degeneration and azoospermia. Hum Mol Genet. 2005;14:49–57. doi: 10.1093/hmg/ddi005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leonardi R, Rock CO, Jackowski S, Zhang YM. Activation of human mitochondrial pantothenate kinase 2 by palmitoylcarnitine. Proc Natl Acad Sci USA. 2007;104:1494–1499. doi: 10.1073/pnas.0607621104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McCall KB, Waisman HA, Elvehjem CA, et al. A study of pyridoxine and pantothenic deficiencies in the monkey (Macaca mulatta) J Nutr. 1946;31:685–697. doi: 10.1093/jn/31.6.685. [DOI] [PubMed] [Google Scholar]
- Nelson MM, Evans HM. Pantothenic acid deficiency and reporoduction in the rat. J Nutr. 1946;31:497–450. doi: 10.1093/jn/31.4.497. [DOI] [PubMed] [Google Scholar]
- Pekhletski R, Gerlai R, Overstreet LS, et al. Impaired cerebellar synaptic plasticity and motor performance in mice lacking the mGluR4 subtype of metabotropic glutamate receptor. J Neurosci. 1996;16:6364–6473. doi: 10.1523/JNEUROSCI.16-20-06364.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Plesofsky-Vig N. Pantothenic acid. In: Ziegler EE, Filer LJ, editors. Present Knowledge in Nutrition. 7th. Washington DC: ILSI Press; 1996. pp. 237–244. [Google Scholar]
- Sheppard AJ, Johnson BC. Pantothenic acid deficiency in the growing calf. J Nutr. 1957;61:195–205. doi: 10.1093/jn/61.2.195. [DOI] [PubMed] [Google Scholar]
- Ullrey DE, Becker DE, Terrill SW, et al. Dietary levels of pantothenic acid and reproductive performance of female swine. J Nutr. 1955;57:410–414. doi: 10.1093/jn/57.3.401. [DOI] [PubMed] [Google Scholar]
- Wintrobe MM, Follis RH, Jr, Alcayaga R, et al. Pantothenic acid deficiency in swine. Bull John Hopkins Hosp. 1943;73:313–342. [Google Scholar]
- Zhang YM, Rock CO, Jackowski S. Biochemical properties of human pantothenate kinase 2 isoforms and mutations linked to pantothenate kinase-associated neurodegeneration. J Biol Chem. 2006;281:107–114. doi: 10.1074/jbc.M508825200. [DOI] [PubMed] [Google Scholar]
- Zhou B, Westaway SK, Levinson B, et al. A novel pantothenate kinase gene (PANK2) is defective in Hallervorden–Spatz syndrome. Nat Genet. 2001;28:345–349. doi: 10.1038/ng572. [DOI] [PubMed] [Google Scholar]