

Abstract
A complete and accurate set of human protein-coding gene annotations is perhaps the single most important resource for genomic research after the human-genome sequence itself, yet the major gene catalogs remain incomplete and imperfect. Here we describe a genome-wide effort, carried out as part of the Mammalian Gene Collection (MGC) project, to identify human genes not yet in the gene catalogs. Our approach was to produce gene predictions by algorithms that rely on comparative sequence data but do not require direct cDNA evidence, then to test predicted novel genes by RT–PCR. We have identified 734 novel gene fragments (NGFs) containing 2188 exons with, at most, weak prior cDNA support. These NGFs correspond to an estimated 563 distinct genes, of which >160 are completely absent from the major gene catalogs, while hundreds of others represent significant extensions of known genes. The NGFs appear to be predominantly protein-coding genes rather than noncoding RNAs, unlike novel transcribed sequences identified by technologies such as tiling arrays and CAGE. They tend to be expressed at low levels and in a tissue-specific manner, and they are enriched for roles in motor activity, cell adhesion, connective tissue, and central nervous system development. Our results demonstrate that many important genes and gene fragments have been missed by traditional approaches to gene discovery but can be identified by their evolutionary signatures using comparative sequence data. However, they suggest that hundreds—not thousands—of protein-coding genes are completely missing from the current gene catalogs.
In the excitement about new noncoding elements in mammalian genomes—including enhancers (Bejerano et al. 2006; Pennacchio et al. 2006), insulators (Xie et al. 2007), and various species of noncoding RNAs (Mattick and Makunin 2006; Pollard et al. 2006)—it is easy to lose sight of the central importance of protein-coding genes. A complete and accurate protein-coding gene set for each sequenced genome is still perhaps the single most important resource for genomic research after the genome sequence itself. Good gene sets are critical for microarray design, association studies, the identification of drug targets, evolutionary analyses, systems biology, and many other endeavors. Even most noncoding elements must be examined in relation to nearby or interacting genes. Nevertheless, gene annotation in many ways has not kept pace with genome sequencing. Six years after the draft sequence of the human genome first became available (International Human Genome Sequencing Consortium 2001; Venter et al. 2001), not only is no complete human gene set available, but the number of human genes is still not precisely known, with estimates ranging from 20,000 to 25,000 (International Human Genome Sequencing Consortium 2004). Furthermore, many genes are erroneously, incompletely, or inconsistently annotated in the major human gene catalogs—RefSeq (Pruitt et al. 2005), Vega (Ashurst et al. 2005), and Ensembl (Hubbard et al. 2007) (see Clamp et al. 2007; http://www.ncbi.nlm.nih.gov/CCDS/).
A steady stream of new discoveries has made a complete human gene catalog a moving target. Studies of the mammalian transcriptome have revealed pervasive transcription, thousands of noncoding RNAs, extensive antisense transcription, tandem chimerisms, and widespread alternative splicing and alternative promoters (Bertone et al. 2004; Cheng et al. 2005; Carninci et al. 2006; Parra et al. 2006; Kapranov et al. 2007). Recently, many of these observations were corroborated in a close examination of 1% of the human genome (Harrow et al. 2006; Denoeud et al. 2007; ENCODE Project Consortium 2007). Other studies have revealed extensive and elaborate regulation and modification at post-transcriptional stages (Bass 2002; Bartel 2004). These discoveries point to an unanticipated level of complexity in the way the genome encodes functional molecules, and they call into question our working definition of the gene (Gerstein et al. 2007).
Nevertheless, for its day-to-day work, the research community depends on sets of gene annotations that are as complete and accurate as possible, by some reasonable working definition of the gene. Also needed are essential reagents related to genes, such as cDNA clones. The Mammalian Gene Collection (MGC) project was created to provide the community with a representative high-quality full-length cDNA clone for every human and mouse gene, as well as for a large subset of rat genes (Strausberg et al. 1999). Roughly three of four human and mouse genes, and thousands of rat genes, are now represented in the MGC (http://mgc.nci.nih.gov). However, the goal of a full-length clone for every gene has proven difficult to achieve. In response to declining yields from methods based on random expressed sequence tag (EST) sequencing (Gerhard et al. 2004), several years ago the MGC adopted a more directed strategy, by which candidate genes not in the collection were amplified by RT–PCR, then were cloned and validated by full-length sequencing (Baross et al. 2004; Wu et al. 2004a). A component of this strategy was to use ab initio computational gene prediction to identify candidates missing from catalogs of known genes and poorly supported by ESTs, yet still detectable from subtle signatures in the genome sequence. In this way, the goals of completing the MGC and of obtaining a complete set of gene annotations have become intertwined.
Until recently it would have been impractical to test otherwise unsupported computational predictions by RT–PCR at the scale of an entire mammalian genome. However, improvements in gene prediction accuracy, the completeness of gene catalogs, and the cost effectiveness of RT–PCR have helped to make a project of this kind feasible (Guigó et al. 2003; Wu et al. 2004b; Eyras et al. 2005; Brzoska et al. 2006; Harrow et al. 2006). Perhaps the most important development has been the dramatic decrease in the false-positive rates of ab initio predictions, owing to the incorporation of comparative sequence data in gene finders (Korf et al. 2001; Parra et al. 2003; Siepel and Haussler 2004; Gross and Brent 2006). Comparative gene-finding programs have reduced false-positive rates by roughly half (at the nucleotide and exon levels) with little or no cost in sensitivity, by making use of the patterns of nucleotide substitutions and insertions/deletions that are characteristic of protein-coding genes (Flicek et al. 2003; Siepel and Haussler 2004; Gross and Brent 2006). Improved methods to filter out pseudogenes and make use of EST evidence where available have further improved accuracy (Arumugam et al. 2006; van Baren and Brent 2006). These improvements are especially important in novel gene discovery, because predictions outside of known genes are strongly enriched for false positives.
Here we describe the results of a genome-wide effort to identify novel human genes by computational gene prediction followed by RT–PCR validation. Because success rates for RT–PCR tend to decline with product length, our approach was first to target short, intron-spanning fragments of predicted genes for validation, then—if sufficient support for expression and splicing was found—to submit larger predictions to the MGC pipeline for full-length cloning (see Fig. 1). This initial phase of gene prediction and fragment validation produced relatively short EST-like sequences—here called RT–PCR amplified sequence tags (RSTs)—that provide evidence of transcription and splicing, but do not define full-length transcripts. Thus, this approach can be thought of as a kind of directed EST sequencing, which targets likely protein-coding exons that have been undersampled by ordinary EST-sequencing methods. We refer to this method as computational exon discovery (CED). In this article, we present an analysis of more than 2000 novel human exons identified by CED.
Figure 1.

(A) Flowchart for computational exon discovery (CED). Beginning with three sets of gene predictions, candidate novel genes are tested for evidence of expression and splicing in several rounds of candidate selection, RT–PCR amplification, and sequencing. The result is a large set of EST-like sequences, called RSTs, that provided supporting evidence for novel protein-coding exons, but do not define full-length transcripts. (B) Illustration of CED. Gene 1 is known and well-supported by public cDNA sequences, so overlapping gene predictions are ignored. Predicted gene 2 appears to be novel and is selected for RT–PCR validation, but the validation experiment fails. Predicted gene 3 also appears to be novel and is tested by two RT–PCR experiments, both of which produce valid RSTs (“hits”). The first experiment validates the TRANSMAP prediction, and the second validates the N-SCAN prediction and one of two Exoniphy predictions. A cDNA cluster is constructed to summarize each set of overlapping cDNAs (including RSTs), and a novel gene fragment (NGF) is constructed by merging the two RSTs that support novel exons (NEs; in red).
Results
Selection of targets and RT–PCR validation
For the initial gene predictions, we used three programs that have high-prediction accuracy, yet do not depend on direct cDNA evidence: N-SCAN, Exoniphy, and TRANSMAP. All of these methods make use of comparative sequence data, but in different ways (Table 1). They were expected to complement one another by identifying somewhat different sets of novel genes. We selected intron-containing predictions that did not overlap known genes and had little or no support from publicly available human EST or mRNA sequences, and tested them for expression and splicing in pooled mRNA sources by RT–PCR (see Methods and Supplemental material). RT–PCR experiments that produced sequences with high-quality spliced alignments to the genome were considered “hits,” while other experiments were considered “misses” (Fig. 1B; Methods). Stringent filters ensured that the targeted genes, and not paralogs, had been amplified and sequenced. Notably, a hit implies that a targeted region is expressed and spliced, but does not prove that it encodes a functional protein. In addition, misses may arise for reasons other than false-positive predictions, such as failures of reverse transcription, PCR amplification, or sequencing, or incomplete sampling of tissues in the mRNA pool.
Table 1.
Computational gene finders used in this study

A total of 12,164 RT–PCR experiments were performed, excluding ones that produced RSTs with ambiguous mappings to the genome. Of these, 2767 (22.7%) were hits (Table 2). We also evaluated hits at the level of “prediction clusters,” or maximal sets of co-tested predictions (Methods), to account for multiple sets of overlapping predictions and multiple experiments per prediction. A total of 4140 prediction clusters were tested, of which 1090 yielded at least one hit, for a hit rate of 26.3% (Table 2). While targets were, in practice, identified from individual predictions (a large majority being based on N-SCAN), each experiment was retrospectively considered to be a test of all compatible predictions (see Fig. 1B and Methods).
Table 2.
RT–PCR hit rates by gene prediction source

aClusters of co-tested predictions (see Methods).
b(NS) N-SCAN, (EX) Exoniphy, (TM) TRANSMAP.
cTotal number of hits and misses (excludes ambiguous mappings to genome; see Methods).
dNumber of hits divided by number of hits and misses ×100.
ePercentage of all hits that this source contributed (e.g., 2602/2767 = 94% for experiments in first row).
The three prediction sources displayed quite different hit rates, ranging from 26.0% at the cluster level (22.4% at the experiment level) for N-SCAN to 72.8% (49.8%) for Exoniphy and 71.7% (54.2%) for TRANSMAP (Table 2). At the same time, far more hits were accounted for by N-SCAN than by the other two sources. Predictions supported by multiple sources were validated at a significantly higher rate than those that were supported by a single predictor, with a hit rate of >80% for predictions supported by all three sources. Predictions in segmental duplications were validated at a much lower rate (8.2%) than those outside segmental duplications (23.9%; P < 2.2 × 10−16, Fisher’s exact test), probably owing to an enrichment for pseudogenes in duplicated regions (data not shown). The differences in hit rates between predictors primarily result from different strategies in candidate selection rather than differences in the native false-positive rates of the gene predictors. For example, the Exoniphy predictions were passed through stringent filters before they were submitted for validation, while a more inclusive strategy was used with N-SCAN predictions (see Supplemental material). The goal of this work was not to perform an unbiased evaluation of gene prediction accuracy, but to identify as many novel genes (exons) as possible. However, these results do demonstrate that an inclusive strategy for candidate selection in CED can identify fairly large numbers of novel genes, even in a well-annotated genome, but hit rates may be fairly low (∼25%), while stringent filtering can improve hit rates considerably (to >70%), but will reduce yields.
Novel exons and novel gene fragments
We sought to quantify how much novel evidence of transcription the 2767 hits provided beyond what was already available from public cDNA sequence data. Because most cDNAs are fragments, we devised a system for measuring evidence of transcription at the level of individual exons. Based on alignments to the genome sequence of the RSTs and all other public (human) cDNAs, we defined a set of benchmark exons (BMEs), representing our current best estimate of the true genomic boundaries of all cDNA-supported exons (Supplemental Fig. S1). Each BME was then classified as having complete support (spanning both splice sites of an internal exon or the single splice site of an initial/terminal exon), partial support (spanning one splice site of an internal exon), or no significant support (no coverage of splice sites), from either the RSTs or from prior cDNA evidence (Supplemental Fig. S2; Methods). BMEs that had complete support from the RSTs and, at most, partial support from prior cDNA evidence were designated as novel exons (NEs) (Supplemental Fig. S2).
The cDNA database is constantly expanding, so the set of NEs is a function of the cut-off date used to define prior cDNA evidence. However, the NEs turn out not to be highly sensitive to the choice of date. A cut-off date of January 1, 2005 (when the first RSTs were sequenced) defined a set of 2188 NEs, most of which (91%) had no significant previous support (Supplemental Table S1). A cut-off date of June 1, 2007 decreased the number of NEs by only 14% to 1892, and left the proportion with no significant previous support essentially unchanged. Thus, while the number of public cDNAs has nearly doubled since early 2005, the NEs identified by our methods appear to be relatively impervious to other methods for exon discovery. For simplicity, we assume a cut-off date of January 1, 2005 for the remainder of this article.
We define a novel gene fragment (NGF) to be a set of n connected exons supported by RSTs that contain NEs. (If there are multiple overlapping NE-containing RSTs with consistent splice junctions, they are merged to create one NGF; see Fig. 1B.) NGFs provide partial information about the transcripts to which NEs belong. The 2767 hits yielded 734 NGFs. Nearly half of the NGFs are completely novel, in the sense that they are isolated gene fragments that do not overlap prior cDNA evidence (as were given priority in target selection). About a third represent 5′ or 3′ extensions of prior cDNA clusters (with slightly more 5′ than 3′ extensions), another 12% contribute single internal exons, and the remainder represent other combinations of internal exons and transcript extensions (Supplemental Table S2).
To assess the degree to which the NGFs represent independent genes, we built clusters based on several combined sources of evidence—including the RSTs, other cDNAs, the predictions, known human genes, and homologous genes from other species—and conservatively assumed that NGFs in the same cluster represented the same gene. This procedure produced 563 distinct NGF clusters (NGFCs). Compared with the latest curated, cDNA-supported gene sets (RefSeq and Vega), 327 (58%) of these NGFCs are completely novel, 99 (18%) are 5′ or 3′ extensions, and 43 (8%) augment genes by contributing novel internal exons (Table 3). A total of 94 (17%) of the NGFCs are no longer novel with respect to these gene sets, in many cases because they have already been used in defining new genes. Comparisons with more inclusive sets of known genes result in fewer completely novel NGFCs and more no-longer-novel, extending, and augmenting NGFCs. For example, adding the Ensembl gene predictions (which were not considered in candidate selection; see Supplemental material) reduces the completely novel set to 178 (32% of NGFCs), and extending known genes by overlapping cDNA clusters further reduces it to 164 (29%). Thus, the NGFCs are estimated to represent between 164 and 327 novel genes, depending on how the known genes are defined. In all cases, hundreds of known genes are found to be extended or otherwise augmented by NGFCs.
Table 3.
NGF clusters by relationship with current known genes

aBackground gene set: (R) RefSeq; (V) Vega; (E) Ensembl; (ext) extended by overlapping cDNA clusters.
bDoes not overlap known genes.
cOverlaps gene(s) and extends in 5′ direction.
dOverlaps gene(s) and extends in 3′ direction.
eOverlaps gene(s), does not extend, but contributes novel internal exons.
fAll exons now represented in gene set.
Protein-coding potential of novel gene fragments
To address the possibility that many NGFs might be noncoding RNAs (ncRNAs) falsely predicted as protein-coding genes (Mattick and Makunin 2006), we searched for homologs of the NGFs in a large database of proteins. For comparison, we performed the same search with 509 sequences annotated as ncRNAs in the RefSeq database. Most NGFs (86%) had at least one significant homolog, compared with only about 15% of ncRNAs. This difference is significant even after correcting for differences in query sequence length (see Supplemental materials). Similarly, 70% of NGFs and only 11% of ncRNAs had at least one significant match to a conserved domain. At the same time, only about 5% of NGFs had high-scoring matches to ncRNAs from the Rfam database (Griffiths-Jones et al. 2003), compared with 12% of RefSeq protein-coding genes and 75% of RefSeq ncRNAs.
We also compared the NGFs with annotated coding sequences (CDSs), untranslated regions (UTRs), and noncoding RNAs (ncRNAs) from RefSeq in terms of two signatures of protein-coding potential: the length distribution of indels and the distribution of distances between mismatches in human/mouse alignments. Both of these measures show a pronounced periodicity (with period three) in CDSs, and both show an absence of periodicity in UTRs and ncRNAs (Fig. 2; Supplemental Fig. S3). The NGFs display pronounced periodicity by both measures, although it is somewhat dampened in comparison with CDSs. This dampening may result from some ncRNAs among the NGFs, but it may also reflect increased sequencing and alignment error, as the fragmentary RSTs map to the genome less precisely than do the full-length mRNAs in RefSeq. It also appears to reflect a reduction in the overall level of conservation of the NGFs.
Figure 2.

Distributions of distances between nearest mismatches in human–mouse alignments for NGFs vs. CDSs, UTRs, and ncRNAs from RefSeq.
Another possibility is that the NGFs include transcribed pseudogenes (Zheng et al. 2007). However, these pseudogenes would have to be spliced as well as transcribed, and if they were identified by N-SCAN or Exoniphy (as were all but a few; see Table 2), they would have to have been pseudogenized recently enough so that their substitution and indel patterns still strongly resembled those of functional genes. They would also have to have eluded our pseudogene filters. It is therefore unlikely that a large number of pseudogenes are included.
Taken together, these results strongly suggest that, while the NGFs may contain some ncRNAs and pseudogenes, they consist predominantly of genuine protein-coding sequences.
Historical exon discovery
As a side benefit, our database of BMEs allows the discovery of novel human exons to be tracked over time. Our data show that the number of BMEs completely supported by public cDNA data began to grow rapidly in about 1993 and experienced a dramatic acceleration of growth through the mid and late 1990s (Fig. 3). The growth rate reached a peak between 2000 and 2001, and has steadily declined since—except for a pronounced spike in 2006 from 5′-end sequencing (Kimura et al. 2006). The decline in growth around 2001, when about two-thirds of BMEs had been identified, primarily reflect “saturation” in cDNA sequencing, with new sequences becoming less likely to identify new exons and more likely to provide additional support for known exons (Supplemental Fig. S4). By 2004, exon discovery had declined to its 1993 level. Coding exons appear to have reached saturation somewhat earlier than noncoding exons. Notably, most novel exons since 2004 are apparent noncoding exons, contributed by methods designed to enrich for the 5′ ends of transcripts.
Figure 3.

Number of benchmark exons completely supported by at least one cDNA sequence in GenBank as a function of time, and the rate of growth of this number (computed in a 12-mo sliding window). Separate curves are shown for all exons and for exons that overlap annotated CDSs of known genes. Four spikes in growth can be traced to major EST submissions by (1) Adams et al. (1993a, b), (2) Hillier et al. (1996), (3) Adams et al. (1995) and L.D. Hillier and colleagues (“The WashU-Merck EST Project,” unpubl.), and (4) Kimura et al. (2006). The largest spike, between (3) and (4), comes from various sources.
Our contribution of ∼2000 NEs is not on the scale of the largest contributions from EST-sequencing projects, some of which numbered in the tens of thousands (Fig. 3). Nevertheless, despite saturation in exon discovery, the NEs are equal in number to ∼1% of all annotated coding exons, and they represent >0.5% of all cDNA-supported exons.
Functional categories of novel gene fragments
To obtain information about the possible functions of the NGFs, we translated them into peptide sequences, searched for homologous vertebrate genes, and assigned the NGFs to the Gene Ontology (GO) (Ashburner et al. 2000) categories of their closest homologs. We also identified conserved protein domains within the NGFs. To avoid overcounting the categories or domains of especially long or fragmented genes, we analyzed the NGF clusters (NGFCs) instead of the individual NGFs.
Compared with a background set of RefSeq genes, several GO categories were significantly over-represented among the NGFCs (Supplemental Table S3). If these categories are clustered by the NGFs assigned to them (Fig. 4), two main groups emerge: (A) “motor activity” and related categories such as “ciliary or flagellar motility” and “response to mechanical stimulus;” and (B) “extracellular region” and related categories such as “extracellular matrix,” “collagen binding,” and “cell adhesion.” There were fewer over-represented protein domains than GO categories (Supplemental Table S3), and they generally corresponded closely to the enriched GO categories.
Figure 4.

Hierarchical clustering of over-represented GO categories, based on the NGFs assigned to each category. This dendrogram is derived from a dissimilarity matrix defined such that any two GO categories, X and Y, have dissimilarity 0 when all NGFs assigned to X are also assigned to Y (or vice-versa), and dissimilarity 1 when the sets of NGFs assigned to X and Y do not overlap. (Specifically, X and Y have dissimilarity dXY = 1 − [|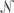 (X)∩(Y)|/ min{|(X)|,|(Y)|}], where (C) denotes the (nonempty) set of NGFs assigned to GO category C.) As a result, GO categories associated with similar sets of NGFs group together in the dendrogram, even if these categories are not closely related in the GO hierarchy (such as “liver development” and “cell adhesion”). Here, two major groups of related categories are evident, broadly related to motor activity (Group A) and the extracellular region (Group B). (Dendrogram produced by the hclust function in R with method = “average.”)
(X)∩(Y)|/ min{|(X)|,|(Y)|}], where (C) denotes the (nonempty) set of NGFs assigned to GO category C.) As a result, GO categories associated with similar sets of NGFs group together in the dendrogram, even if these categories are not closely related in the GO hierarchy (such as “liver development” and “cell adhesion”). Here, two major groups of related categories are evident, broadly related to motor activity (Group A) and the extracellular region (Group B). (Dendrogram produced by the hclust function in R with method = “average.”)
The enrichments for “motor activity” and related categories came primarily from more than a dozen NGFCs homologous to dynein and myosin heavy-chain polypeptides (HCPs). In particular, several NGFCs showed strong homology with the HCPs of axonemal dyneins, large protein complexes that are responsible for the movement of cilia and flagella. Other clusters were homologous to HCPs of cytoplasmic dynein 2, which plays a role in intraflagellar transport. Some of these NGFCs were extensions of well-studied genes, such as ngf338–ngf339, which extend DNAH17 by 14 exons in the 5′ direction (Supplemental Table S5). Others appear to be essentially novel. For example, ngf51–ngf55 contain 24 novel exons that apparently belong to a new axonemal dynein HCP gene of ∼66 exons (Fig. 5). With myosins, as with dyneins, the NGFCs included both novel genes (e.g., ngf634–ngf638) and extensions of known genes (e.g., ngf408–ngf409).
Figure 5.

Gene predictions, cDNA evidence, and novel gene fragments in the region on chromosome 1 that includes ngf51–ngf55. Gene predictions are shown in green, prior cDNA evidence is in black, RSTs (which are represented in GenBank as ESTs) are in gold, and NGFs are in blue, with novel exons colored red. cDNA sequences recently deposited in GenBank (post 1/1/05) and ignored in evaluating novelty are shown in purple. This cluster of NGFs contributes 24 novel exons to a gene that spans >450 kb and consists of an estimated 66 exons. This gene appears to code for a novel axonemal dynein heavy-chain polypeptide.
The dynein and myosin HCP gene families are both diverse, with large numbers of functionally specialized—and often quite divergent—members. At the same time, orthologs in these families are generally well conserved across long evolutionary distances (Weiss and Leinwand 1996; Pfister et al. 2006). In addition, many of these genes are known to exhibit tissue and cell-specific expression. For example, DYNC2H1 (Supplemental Table S5), is specifically expressed in ciliated cell species of the mammalian brain, the olfactory epithelium, and the retina (Mikami et al. 2002). Moreover, many of these genes are quite large, so EST coverage is likely to be incomplete, and attempts at acquiring full-length mRNAs are likely to have failed. Combined, these factors could have caused these genes to be missed by conventional methods for gene discovery, yet allowed them to be readily detectable by CED.
Many of the NGFs assigned to the “extracellular region” group of categories showed strong homology with cell-adhesion molecules such as mucin-like proteins, integrins, cadherins, and von Willebrand factors. Thus, these genes may function as components of biofilms, in blood coagulation, in epithelial tissues, or in other extracellular capacities. Others were homologous to structural proteins such as collagens, or to extracellular enzymes such as the serine proteases trypsin, neurotrypsin, and neutrophil. Several of the NGFCs in this group were nearly or completely novel, such as ngf167–ngf171, which contribute 24 NEs to a homolog of von Willebrand factors and mucins, and ngf510–ngf513, which cover most of a novel collagen homolog. Others contributed major extensions to known genes, such as ngf101–ngf103, which extends the otogelin (OTOG) gene in both the 5′ and 3′ directions (Supplemental Fig. S5). OTOG is an example of a well-studied gene that has been slow to make its way into the human gene catalogs, probably because tissue-specific expression has resulted in poor cDNA coverage (Cohen-Salmon et al. 1997; El-Amraoui et al. 2001). Similar examples include MUC19, COL28A1, and HMCN2. Notably, several of the NGFs in this group—such as ones overlapping SSPO, CNTN3, and SDK2—appear to function in central nervous system development and/or synaptic transmission.
Despite their over-representation, these groups of categories account for only about one-fourth of all NGFs, and the remaining NGFs have diverse functional roles. Thus, the deficiencies of the current gene catalogs cannot be attributed to any particular class of genes.
In situ hybridizations to zebrafish embryos
To test the possibility that some NGFs might be specifically expressed in embryonic development, we identified 23 that had little or no other cDNA support and that could be mapped, via whole-genome syntenic alignments, to the zebrafish genome. We then synthesized probes for the zebrafish orthologs of these NGFs, and used them for whole-mount in situ hybridizations to zebrafish embryos.
Clear expression was observed for three NGFs, of 19 for which probe synthesis was successful. The first case, ngf136, consists of two of the three exons of the brain-specific homeobox (BSX) gene, which has recently been added to RefSeq. We observed specific expression of this gene in the hypothalamus during embryonic development, consistent with other findings (Cremona et al. 2004). The second case, ngf674, now corresponds to a minimally annotated three-exon kelch-like gene (RefSeq NM 001081675). This gene was found to be highly expressed in the zebrafish embryo’s branchial arches (precursors of gills) and pronephric duct (precursor of the kidney). The third case, ngf60, is a nine-exon NGF consisting completely of novel exons (Supplemental Table S5). This gene has no known vertebrate orthologs, and its predicted product shows only weak homology with several kinesin-like proteins. In zebrafish embryos, it displays an expression pattern in the telencephelon and hindbrain similar to the transcription factor OTP, a homeobox transcription factor that is essential for the development of the hypothalamus (Fig. 6). Therefore, ngf60 may play a critical role in development. These examples show that at least some NGFs exhibit tissue-specific expression during embryonic development in zebrafish, and probably in human as well.
Figure 6.

Whole-mount in situ hybridization for a zebrafish sequence orthologous to ngf60, showing its expression pattern in the brain 48 h past fertilization (hpf). For comparison, the expression pattern is also shown for OTP, a homeobox transcription factor that was used as a positive control because of its highly specific and well described expression profile (Eaton and Glasgow 2007). The expression patterns of the two genes remain generally similar at 72 hpf (Supplemental material).
Expression levels of novel exons
We examined the expression levels of NEs and NGFs using publicly available data from the Affymetrix Human Exon 1.0 ST Array, which, in addition to probes for known genes, has probes for a large number of ab initio gene predictions—including 75% of our NEs and 95% of our NGFs. In all 11 tissues for which data was available, the NEs showed significantly less detectable expression than exons of known genes, with the fraction of NEs displaying significant expression above background ranging from 17% to 63% (median 27%) compared with 63% to 86% (median 70%) for RefSeq exons (P < 1 × 10−103, one-sided Fisher’s exact test; Supplemental Fig. S7A). Furthermore, among exons showing detectable expression, the NEs showed a significant decrease in estimated expression levels compared with RefSeq exons, with median expression levels 25%–39% lower (Supplemental Fig. S7B). The NEs also showed significantly greater variation across tissues in expression levels, with a median coefficient of variation of 0.21 compared with 0.16 for RefSeq exons (Mann Whitney P < 1 × 10−15). About 3.5% of novel exons that were expressed in at least one tissue displayed tissue-specific expression compared with only 0.8% of RefSeq exons (P = 5 × 10−15, one-sided Fisher’s exact test). Thus, the NEs and NGFs on average are expressed at lower levels, and in a more tissue-specific manner, than are known exons.
Discussion
There have been several previous attempts at gene or exon discovery in vertebrate genomes by combined computational prediction and experimental validation (CED). Methods similar to ours have been applied in human and mouse (Guigó et al. 2003), rat (Wu et al. 2004b), chicken (Eyras et al. 2005), and, most recently, in the 1% of the human genome targeted by the ENCODE project (Harrow et al. 2006). However, these efforts were done at substantially smaller scales, and generally either for genomes that lacked mature gene sets (Wu et al. 2004b; Eyras et al. 2005), making it relatively easy to identify novel genes, or for regions that were already so well annotated that essentially no new genes (and only a few new exons) could be found (Harrow et al. 2006). Even since the work of Guigó et al. (2003), the human genome has had 4 yr of close scrutiny by manual annotators and computational algorithms, and it is considerably more difficult to find new genes now than it was in 2003. Despite these challenges, we have found evidence for thousands of novel exons corresponding to hundreds of genes.
The previous work most similar to ours was a project by Brzoska et al. (2006), in which over 7000 human ab initio gene predictions were tested in a high-throughput RT–PCR pipeline. Brzoska and colleagues were able to validate 796 predictions, 163–296 of which were entirely novel and 505–574 of which included novel exons, with the exact numbers depending on the choice of reference set. Thus, their yields were roughly comparable to ours. However, the sequence data from this project has not been made public, and therefore it is not reflected in the public gene catalogs. Brzoska et al. (2006) did not use the newest and most accurate comparative gene predictors, and partly for this reason, they had a validation rate of only ∼12% (approximately half our rate). They also did not attempt to evaluate the protein-coding potential of their novel genes. On the other hand, they performed a large number of 5′ and 3′ rapid amplification of cDNA ends (RACE) reactions, and were able to augment their RT–PCR validated gene fragments (the equivalent of our NGFs) with nearly 400 complete transcripts. Their project and ours, despite different strengths and weaknesses, both demonstrate that large-scale CED projects can produce significant numbers of novel genes.
As a method for discovering novel exons, CED can be seen as an alternative not only to EST sequencing, but to transcription tiling arrays (Bertone et al. 2004; Cheng et al. 2005; Kapranov et al. 2007) and techniques for identifying transcript ends such as cap analysis of gene expression (CAGE) and oligo-capping (Carninci et al. 2005; Kimura et al. 2006). In comparison with these approaches, CED is generally more sensitive to rare transcripts because of its targeted PCR amplification step. Indeed, only ∼10% of our novel exons are reasonably well covered (≥50% of bases) by Affymetrix-transcribed fragments (transfrags) (Kapranov et al. 2007) from a typical cell line, and only ∼30% by a merged set of transfrags from all cell lines (Supplemental Table S6). In addition, because the targeted exons are selected by computational gene finders, CED enriches strongly for protein-coding exons. Unlike tiling arrays, it also captures splice junctions, allowing for more precise definitions of exon boundaries and some information about splice patterns. In addition, with careful primer design and analysis of sequenced products, it is far more specific than arrays, which suffer from the problem of cross-hybridization. On the other hand, at least if used with comparative gene-finding methods, CED may miss some lineage-specific or fast-evolving genes. Also, unlike methods such as CAGE, it has no built-in capacity for identifying transcript ends.
An important question that still remains to be answered is how far CED can be pushed in detecting novel genes. As shown here, CED is most effective at identifying conserved genes that have a strong evolutionary footprint, but a weak expression footprint (at least when mRNA samples are pooled across tissues and/or developmental stages). In contrast, most cDNA-based methods require strong expression, but do not require evolutionary conservation. There are almost certainly genes that are essentially invisible to both types of methods—for example, lineage-specific, fast-evolving, very short, or single-exon genes that are expressed at low levels. Some of these genes may be detectable by more sophisticated computational gene finders that can effectively combine weak comparative and weak expression-based signals, perhaps along with chromatin state or other information. Comparative gene finders would also benefit from richer evolutionary models that allow for gene duplication, lineage-specific gains and losses of genes, or changes across species in gene structure. Better gene predictors would allow our synteny and duplication filters to be relaxed, opening up heavily duplicated and rearranged regions of the genome to CED. New computational tools of this kind and/or new high-throughput methods for detecting low-abundance transcripts will be needed before the remaining “dark matter” of the proteome can be characterized.
The number of human genes has been estimated to be about 20,000–25,000 (International Human Genome Sequencing Consortium 2004). The major gene catalogs together currently contain ∼24,500 genes, near the upper limit of this range, but a recent comparative analysis of mammalian genomes (Clamp et al. 2007) suggests that the number of well-supported genes is only ∼20,500, closer to the lower limit of the estimated range (see also Goodstadt and Ponting 2006). Our identification of 164–327 additional genes does not dramatically change the number of human genes, and assuming Clamp et al.’s estimates are accurate, it leaves the total number well below 21,000. Similarly, Brzoska et al. (2006) provide evidence for, at most, hundreds of new human genes (see also Lee et al. 2006). However, current high-throughput approaches to gene discovery all have fundamental limitations that cause whole classes of genes to be invisible to them. In addition, no method is completely efficient at detecting genes in its target class. For example, some well-conserved genes were undoubtedly missed by our methods because of errors in gene prediction, primer design, PCR, reverse transcription, sequencing, or alignment (although a precise estimate of the overall false-negative rate is quite difficult to obtain). Consequently, while improved gene sets are giving more confident lower bounds on the number of human genes, a tight upper bound is much more difficult to establish.
Identifying novel genes tends to be the main focus of efforts in gene discovery, but obtaining a complete representation of each gene is equally important. Our results show that many genes are only partially represented in the gene catalogs, by being truncated at the 5′ or 3′ ends, by missing one or more internal exons, or by being represented as separate genes despite strong evidence that they are joined. Some of these genes are missing dozens of exons. This evidence is consistent with recent studies identifying many novel 5′ extensions of known genes (Harrow et al. 2006; Kimura et al. 2006; Denoeud et al. 2007). As full-length clones, based in part on our NGFs, are produced by the MGC pipeline, additional exons and exon boundaries will be identified, and it will become clearer which NGFs belong to the same transcript. Standard protocols will allow the RefSeq and Ensembl gene catalogs to be updated based on the MGC clones.
If gene completeness is followed to its logical conclusion, however, alternative splicing and alternative promoters must be considered, as well as more exotic phenomena such as tandem chimerisms. At present, these issues are largely ignored by the MGC, and are addressed in a simplified way by the major gene catalogs. Even the GENCODE Consortium, which made an effort to capture as many alternative transcripts as possible in its detailed annotation of 1% of the human genome, simply enumerated transcripts (and associated open reading frames) (Harrow et al. 2006). Ideally, these resources would also have information about tissue-, cell-, and developmental stage-specific distributions over transcripts, and perhaps would even have information about the joint distributions for multiple transcripts.
Attempts to enrich the representation of genes will inevitably bump up against the thorny question of what a gene is (Gerstein et al. 2007). However, it may be that the definition of the gene will become less, rather than more important as more information becomes available about alternative transcripts. With complete information about expression patterns, protein products, and functions at the transcript level, a “gene” becomes just a label for a set of transcripts; the real information is in the transcripts themselves. Similarly, as more becomes known about transcript diversity, counts of genes become less interesting. Therefore, it may be that “a complete representation of functional transcripts” would be a more appropriate long-term goal than a “complete gene set.” In any case, it is clear that much work remains to be done.
Methods
Selection of targets
Gene predictions were based on BLASTZ (Schwartz et al. 2003) and MULTIZ (Blanchette et al. 2004) alignments of the July 2003 (hg16) and May 2004 (hg17) assemblies of the human genome with the mouse (mm3/mm5), rat (rn3), and/or chicken (galGal2) genome assemblies. Target selection occurred over a 2-yr period, using various versions of the alignments and prediction programs and using post-processing filters that differed somewhat by prediction source. However, in all cases, candidate genes were required, at the time of selection, not to overlap the RefSeq (Pruitt et al. 2005) or Vega (Ashurst et al. 2005) gene sets, genes either already in the MGC, or genes in the MGC pipeline for full-length cloning. In addition, preference was given to candidates with little or no cDNA support, as defined by overlap in genomic coordinates with alignments of public EST or mRNA sequences. In some cases, additional filters were used to eliminate likely pseudogenes, to avoid recent duplications, and to require conserved synteny between species. Predictions that did not contain at least one intron between coding exons were removed, and any predicted UTRs were ignored. The procedure for target selection was designed to maximize the number of validated novel genes (exons), not to evaluate the (absolute or relative) performance of the gene predictors. See the Supplemental material for further details.
RT–PCR and sequencing
PCR primers were designed for each candidate gene, such that predicted amplicons would span at least one intron and would have lengths of ∼500–800 bases. The number of exons spanned by the amplicons ranged from two to 13, with a median of four. Equal amounts of total RNA were pooled from 20 human tissues, including adrenal gland, bone marrow, cerebellum, brain (whole), fetal brain, fetal liver, heart, kidney, liver, lung, placenta, prostate, salivary gland, skeletal muscle, spleen, testis, thymus, thyroid gland, trachea, and uterus (Human Total RNA master panel II, BD Biosciences Clontech). Pooled total RNA was reverse transcribed using Superscript III reverse transcriptase with Oligo dT primer according to the manufacturer’s instructions (Invitrogen). Reverse transcription was followed by ‘touchdown’ PCR amplification (Don et al. 1991) using Phusion high-fidelity DNA polymerase (New England Biolabs). PCR products were directly sequenced, and forward and reverse reads were assembled into contigs, if possible, using Phrap (P. Green and B. Ewing, unpubl.).
The resulting sequences (either assembled or unassembled) were then aligned to the genome sequence using BLAT (Kent 2002) or Pairagon (Arumugam et al. 2006). For sequences aligned with BLAT, est2genome was used to re-align cDNAs to BLAT-extracted regions of the genome. Any sequence that formed a high-quality alignment (>75% identity and >80% identity within 10 bases of splice sites) and revealed at least one intron with canonical (GT-AG) donor and acceptor splice sites was considered a valid RST. Failure to produce a valid RST could occur for various reasons, including failures of PCR amplification, sequencing, or alignment. Positive controls succeed at an average rate of 93%. All valid RSTs were submitted to GenBank as ESTs. Due to mispriming, the best alignment of an RST to the genome occasionally did not match the original targeted gene prediction.
Alignment of cDNAs to genome sequence
EST and mRNA sequences available in GenBank as of June 1, 2007—including the RSTs—were aligned to the human genome sequence (hg17) using BLAT. Each cDNA sequence with at least one high-quality alignment (≥25% coverage and ≥95% identity) was assigned its best-matching position in the genome, plus any secondary positions having high-quality alignments within 1% identity of the best match. Any cDNAs without high-quality alignments were discarded. RSTs assigned multiple genomic positions (usually because of a recent genomic duplication) were excluded from subsequent analyses. (See Supplemental material)
Evaluation of hit rates
Each RT–PCR experiment was associated with one or more gene predictions by mapping the PCR primer pair used in the experiment to the genome with isPcr (J. Kent, unpubl.; http://hgdownload.cse.ucsc.edu/downloads.html) and identifying overlapping predictions. Success rates were evaluated for prediction clusters as well as for individual predictions, because predictions tend to overlap and some (such as those from Exoniphy) are more fragmented than others. Prediction clusters correspond to the connected components of a graph in which nodes represent predictions, and an edge is present between two nodes if, and only if, the corresponding predictions are both associated with the same experiment. An experiment was considered a “hit” if it produced a valid RST that had an unambiguous mapping to the genome, a “miss” if it did not produce a valid RST, and otherwise was ignored. A prediction cluster was considered a “hit” if any associated experiment was a “hit,” and a “miss” if it had no associated “hits” and at least one “miss.” Hit rates were calculated as the number of hits divided by the number of hits and misses. (See Supplemental material)
Definition of benchmark exons
Benchmark exons (BMEs) were derived from cDNAs aligned to the genome with canonical (GT-AG) flanking introns and an unambiguous direction of transcription. Any internal cDNA exon with canonical flanking introns defined an internal BME. An initial exon with a flanking canonical intron defined an initial BME, provided no overlapping cDNA suggested additional exons in the 5′ direction, and provided no other initial exon with the same 3′ boundary extended farther in the 5′ direction (Supplemental Fig. S1). Terminal BMEs were defined in a symmetric manner. Because of uncertainty in the alignment of cDNAs, two exon boundaries were considered “equal” if they were within 2 bp of one another in genomic coordinates. (See Supplemental material.)
Identification of novel exons and novel gene fragments
Novel exons were defined as BMEs having complete support from RSTs, but at most, partial support from prior cDNA evidence. RSTs that overlapped and had equal exon boundaries in their region of overlap were merged (Fig. 1B). Novel gene fragments (NGFs) were defined as merged RSTs that provided complete support for novel exons. To merge RSTs, overlapping exons were merged, then all exons were concatenated together. Note that this simple approach may inaccurately represent complex alternative splicing scenarios such as mutually exclusive exon incorporation.
To cluster together the NGFs that are likely to correspond to the same transcript, the NGFs were combined with the N-SCAN, Exoniphy, and TRANSMAP predictions, the latest human RefSeq genes, human mappings of non-human RefSeq genes (as defined by the “Non-Human RefSeq Genes” track in the UCSC Browser), and clusters of cDNAs from the PASA program (Haas et al. 2003). These features were then clustered by same-stranded exonic overlap using the UCSC clusterGenes program (http://hgdownload.cse.ucsc.edu/downloads.html). All non-NGF features were then discarded. There were 563 remaining non-empty NGF clusters.
Methods for the analyses of protein-coding potential, functional categories, and expression levels, and for the in situ hybridization experiments, are provided in the Supplemental material.
Acknowledgments
Funding was provided by National Cancer Institute subcontracts N01-CO-12400 (MBR) and 22XS013A (D.H., A.S.), a University of California Biotechnology Research and Education Program Graduate Research and Education in Adaptive Biotechnology fellowship (A.S.), and NSF Faculty Early Career Development grant DBI-0644111 (A.S.). We thank numerous colleagues for assistance, feedback, and advice, including R. Baertsch, A.G. Clark, R.A. Gibbs, D. Gordon, G. Lunter, J.S. Pedersen, C. Sugnet, T. Vinar, and two anonymous reviewers of an earlier version of the manuscript.
Footnotes
[Supplemental material is available online at www.genome.org.]
Article published online before print. Article and publication date are online at http://www.genome.org/cgi/doi/10.1101/gr.7128207
References
- Adams M.D., Kerlavage A.R., Fields C., Venter J.C., Kerlavage A.R., Fields C., Venter J.C., Fields C., Venter J.C., Venter J.C. 3,400 new expressed sequence tags identify diversity of transcripts in human brain. Nat. Genet. 1993a;4:256–267. doi: 10.1038/ng0793-256. [DOI] [PubMed] [Google Scholar]
- Adams M.D., Soares M.B., Kerlavage A.R., Fields C., Venter J.C., Soares M.B., Kerlavage A.R., Fields C., Venter J.C., Kerlavage A.R., Fields C., Venter J.C., Fields C., Venter J.C., Venter J.C. Rapid cDNA sequencing (expressed sequence tags) from a directionally cloned human infant brain cDNA library. Nat. Genet. 1993b;4:373–380. doi: 10.1038/ng0893-373. [DOI] [PubMed] [Google Scholar]
- Adams M.D., Kerlavage A.R., Fleischmann R.D., Fuldner R.A., Bult C.J., Lee N.H., Kirkness E.F., Weinstock K.G., Gocayne J.D., White O., Kerlavage A.R., Fleischmann R.D., Fuldner R.A., Bult C.J., Lee N.H., Kirkness E.F., Weinstock K.G., Gocayne J.D., White O., Fleischmann R.D., Fuldner R.A., Bult C.J., Lee N.H., Kirkness E.F., Weinstock K.G., Gocayne J.D., White O., Fuldner R.A., Bult C.J., Lee N.H., Kirkness E.F., Weinstock K.G., Gocayne J.D., White O., Bult C.J., Lee N.H., Kirkness E.F., Weinstock K.G., Gocayne J.D., White O., Lee N.H., Kirkness E.F., Weinstock K.G., Gocayne J.D., White O., Kirkness E.F., Weinstock K.G., Gocayne J.D., White O., Weinstock K.G., Gocayne J.D., White O., Gocayne J.D., White O., White O., et al. Initial assessment of human gene diversity and expression patterns based upon 83 million nucleotides of cDNA sequence. Nature. 1995;377 (Suppl):3–174. [PubMed] [Google Scholar]
- Arumugam M., Wei C., Brown R.H., Brent M.R., Wei C., Brown R.H., Brent M.R., Brown R.H., Brent M.R., Brent M.R. Pairagon+N-scan EST: A model-based gene annotation pipeline. Genome Biol. 2006;7 (Suppl 1):1–10. doi: 10.1186/gb-2006-7-s1-s5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ashburner M., Ball C.A., Blake J.A., Botstein D., Butler H., Cherry J.M., Davis A.P., Dolinski K., Dwight S.S., Eppig J.T., Ball C.A., Blake J.A., Botstein D., Butler H., Cherry J.M., Davis A.P., Dolinski K., Dwight S.S., Eppig J.T., Blake J.A., Botstein D., Butler H., Cherry J.M., Davis A.P., Dolinski K., Dwight S.S., Eppig J.T., Botstein D., Butler H., Cherry J.M., Davis A.P., Dolinski K., Dwight S.S., Eppig J.T., Butler H., Cherry J.M., Davis A.P., Dolinski K., Dwight S.S., Eppig J.T., Cherry J.M., Davis A.P., Dolinski K., Dwight S.S., Eppig J.T., Davis A.P., Dolinski K., Dwight S.S., Eppig J.T., Dolinski K., Dwight S.S., Eppig J.T., Dwight S.S., Eppig J.T., Eppig J.T., et al. Gene Ontology: Tool for the unification of biology. Nat. Genet. 2000;25:25–29. doi: 10.1038/75556. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ashurst J.L., Chen C.-K., Gilbert J.G.R., Jekosch K., Keenan S., Meidl P., Searle S.M., Stalker J., Storey R., Trevanion S., Chen C.-K., Gilbert J.G.R., Jekosch K., Keenan S., Meidl P., Searle S.M., Stalker J., Storey R., Trevanion S., Gilbert J.G.R., Jekosch K., Keenan S., Meidl P., Searle S.M., Stalker J., Storey R., Trevanion S., Jekosch K., Keenan S., Meidl P., Searle S.M., Stalker J., Storey R., Trevanion S., Keenan S., Meidl P., Searle S.M., Stalker J., Storey R., Trevanion S., Meidl P., Searle S.M., Stalker J., Storey R., Trevanion S., Searle S.M., Stalker J., Storey R., Trevanion S., Stalker J., Storey R., Trevanion S., Storey R., Trevanion S., Trevanion S., et al. The vertebrate genome annotation (Vega) database. Nucleic Acids Res. 2005;33:459–465. doi: 10.1093/nar/gki135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baross A., Butterfield Y.S.N., Coughlin S.M., Zeng T., Griffith M., Griffith O.L., Petrescu A.S., Smailus D.E., Khattra J., McDonald H.L., Butterfield Y.S.N., Coughlin S.M., Zeng T., Griffith M., Griffith O.L., Petrescu A.S., Smailus D.E., Khattra J., McDonald H.L., Coughlin S.M., Zeng T., Griffith M., Griffith O.L., Petrescu A.S., Smailus D.E., Khattra J., McDonald H.L., Zeng T., Griffith M., Griffith O.L., Petrescu A.S., Smailus D.E., Khattra J., McDonald H.L., Griffith M., Griffith O.L., Petrescu A.S., Smailus D.E., Khattra J., McDonald H.L., Griffith O.L., Petrescu A.S., Smailus D.E., Khattra J., McDonald H.L., Petrescu A.S., Smailus D.E., Khattra J., McDonald H.L., Smailus D.E., Khattra J., McDonald H.L., Khattra J., McDonald H.L., McDonald H.L., et al. Systematic recovery and analysis of full-ORF human cDNA clones. Genome Res. 2004;14:2083–2092. doi: 10.1101/gr.2473704. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bartel D.P. MicroRNAs: Genomics, biogenesis, mechanism, and function. Cell. 2004;116:281–297. doi: 10.1016/s0092-8674(04)00045-5. [DOI] [PubMed] [Google Scholar]
- Bass B. RNA editing by adenosine deaminases that act on RNA. Annu. Rev. Biochem. 2002;71:817–846. doi: 10.1146/annurev.biochem.71.110601.135501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bejerano G., Lowe C.B., Ahituv N., King B., Siepel A., Salama S.R., Rubin E.M., Kent W.J., Haussler D., Lowe C.B., Ahituv N., King B., Siepel A., Salama S.R., Rubin E.M., Kent W.J., Haussler D., Ahituv N., King B., Siepel A., Salama S.R., Rubin E.M., Kent W.J., Haussler D., King B., Siepel A., Salama S.R., Rubin E.M., Kent W.J., Haussler D., Siepel A., Salama S.R., Rubin E.M., Kent W.J., Haussler D., Salama S.R., Rubin E.M., Kent W.J., Haussler D., Rubin E.M., Kent W.J., Haussler D., Kent W.J., Haussler D., Haussler D. A distal enhancer and an ultraconserved exon are derived from a novel retroposon. Nature. 2006;441:87–90. doi: 10.1038/nature04696. [DOI] [PubMed] [Google Scholar]
- Bertone P., Stolc V., Royce T.E., Rozowsky J.S., Urban A.E., Zhu X., Rinn J.L., Tongprasit W., Samanta M., Weissman S., Stolc V., Royce T.E., Rozowsky J.S., Urban A.E., Zhu X., Rinn J.L., Tongprasit W., Samanta M., Weissman S., Royce T.E., Rozowsky J.S., Urban A.E., Zhu X., Rinn J.L., Tongprasit W., Samanta M., Weissman S., Rozowsky J.S., Urban A.E., Zhu X., Rinn J.L., Tongprasit W., Samanta M., Weissman S., Urban A.E., Zhu X., Rinn J.L., Tongprasit W., Samanta M., Weissman S., Zhu X., Rinn J.L., Tongprasit W., Samanta M., Weissman S., Rinn J.L., Tongprasit W., Samanta M., Weissman S., Tongprasit W., Samanta M., Weissman S., Samanta M., Weissman S., Weissman S., et al. Global identification of human transcribed sequences with genome tiling arrays. Science. 2004;306:2242–2246. doi: 10.1126/science.1103388. [DOI] [PubMed] [Google Scholar]
- Blanchette M., Kent W.J., Riemer C., Elnitski L., Smit A.F.A., Roskin K.M., Baertsch R., Rosenbloom K., Clawson H., Green E.D., Kent W.J., Riemer C., Elnitski L., Smit A.F.A., Roskin K.M., Baertsch R., Rosenbloom K., Clawson H., Green E.D., Riemer C., Elnitski L., Smit A.F.A., Roskin K.M., Baertsch R., Rosenbloom K., Clawson H., Green E.D., Elnitski L., Smit A.F.A., Roskin K.M., Baertsch R., Rosenbloom K., Clawson H., Green E.D., Smit A.F.A., Roskin K.M., Baertsch R., Rosenbloom K., Clawson H., Green E.D., Roskin K.M., Baertsch R., Rosenbloom K., Clawson H., Green E.D., Baertsch R., Rosenbloom K., Clawson H., Green E.D., Rosenbloom K., Clawson H., Green E.D., Clawson H., Green E.D., Green E.D., et al. Aligning multiple genomic sequences with the threaded blockset aligner. Genome Res. 2004;14:708–715. doi: 10.1101/gr.1933104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brzoska P.M., Brown C., Cassel M., Ceccardi T., Di Francisco V., Dubman A., Evans J., Fang R., Harris M., Hoover J., Brown C., Cassel M., Ceccardi T., Di Francisco V., Dubman A., Evans J., Fang R., Harris M., Hoover J., Cassel M., Ceccardi T., Di Francisco V., Dubman A., Evans J., Fang R., Harris M., Hoover J., Ceccardi T., Di Francisco V., Dubman A., Evans J., Fang R., Harris M., Hoover J., Di Francisco V., Dubman A., Evans J., Fang R., Harris M., Hoover J., Dubman A., Evans J., Fang R., Harris M., Hoover J., Evans J., Fang R., Harris M., Hoover J., Fang R., Harris M., Hoover J., Harris M., Hoover J., Hoover J., et al. An efficient and high-throughput approach for experimental validation of novel human gene predictions. Genomics. 2006;87:437–445. doi: 10.1016/j.ygeno.2005.11.016. [DOI] [PubMed] [Google Scholar]
- Burge C., Karlin S., Karlin S. Prediction of complete gene structures in human genomic DNA. J. Mol. Biol. 1997;268:78–94. doi: 10.1006/jmbi.1997.0951. [DOI] [PubMed] [Google Scholar]
- Carninci P., Kasukawa T., Katayama S., Gough J., Frith M.C., Maeda N., Oyama R., Ravasi T., Lenhard B., Wells C., Kasukawa T., Katayama S., Gough J., Frith M.C., Maeda N., Oyama R., Ravasi T., Lenhard B., Wells C., Katayama S., Gough J., Frith M.C., Maeda N., Oyama R., Ravasi T., Lenhard B., Wells C., Gough J., Frith M.C., Maeda N., Oyama R., Ravasi T., Lenhard B., Wells C., Frith M.C., Maeda N., Oyama R., Ravasi T., Lenhard B., Wells C., Maeda N., Oyama R., Ravasi T., Lenhard B., Wells C., Oyama R., Ravasi T., Lenhard B., Wells C., Ravasi T., Lenhard B., Wells C., Lenhard B., Wells C., Wells C., et al. The transcriptional landscape of the mammalian genome. Science. 2005;309:1559–1563. doi: 10.1126/science.1112014. [DOI] [PubMed] [Google Scholar]
- Carninci P., Sandelin A., Lenhard B., Katayama S., Shimokawa K., Ponjavic J., Semple C.A.M., Taylor M.S., Engstrom P.G., Frith M.C., Sandelin A., Lenhard B., Katayama S., Shimokawa K., Ponjavic J., Semple C.A.M., Taylor M.S., Engstrom P.G., Frith M.C., Lenhard B., Katayama S., Shimokawa K., Ponjavic J., Semple C.A.M., Taylor M.S., Engstrom P.G., Frith M.C., Katayama S., Shimokawa K., Ponjavic J., Semple C.A.M., Taylor M.S., Engstrom P.G., Frith M.C., Shimokawa K., Ponjavic J., Semple C.A.M., Taylor M.S., Engstrom P.G., Frith M.C., Ponjavic J., Semple C.A.M., Taylor M.S., Engstrom P.G., Frith M.C., Semple C.A.M., Taylor M.S., Engstrom P.G., Frith M.C., Taylor M.S., Engstrom P.G., Frith M.C., Engstrom P.G., Frith M.C., Frith M.C., et al. Genome-wide analysis of mammalian promoter architecture and evolution. Nat. Genet. 2006;38:626–635. doi: 10.1038/ng1789. [DOI] [PubMed] [Google Scholar]
- Cheng J., Kapranov P., Drenkow J., Dike S., Brubaker S., Patel S., Long J., Stern D., Tammana H., Helt G., Kapranov P., Drenkow J., Dike S., Brubaker S., Patel S., Long J., Stern D., Tammana H., Helt G., Drenkow J., Dike S., Brubaker S., Patel S., Long J., Stern D., Tammana H., Helt G., Dike S., Brubaker S., Patel S., Long J., Stern D., Tammana H., Helt G., Brubaker S., Patel S., Long J., Stern D., Tammana H., Helt G., Patel S., Long J., Stern D., Tammana H., Helt G., Long J., Stern D., Tammana H., Helt G., Stern D., Tammana H., Helt G., Tammana H., Helt G., Helt G., et al. Transcriptional maps of 10 human chromosomes at 5-nucleotide resolution. Science. 2005;308:1149–1154. doi: 10.1126/science.1108625. [DOI] [PubMed] [Google Scholar]
- Clamp M., Fry B., Kamal M., Xie X., Cuff J., Lin M.F., Kellis M., Lindblad-Toh K., Lander E.S., Fry B., Kamal M., Xie X., Cuff J., Lin M.F., Kellis M., Lindblad-Toh K., Lander E.S., Kamal M., Xie X., Cuff J., Lin M.F., Kellis M., Lindblad-Toh K., Lander E.S., Xie X., Cuff J., Lin M.F., Kellis M., Lindblad-Toh K., Lander E.S., Cuff J., Lin M.F., Kellis M., Lindblad-Toh K., Lander E.S., Lin M.F., Kellis M., Lindblad-Toh K., Lander E.S., Kellis M., Lindblad-Toh K., Lander E.S., Lindblad-Toh K., Lander E.S., Lander E.S. Distinguishing protein-coding and non-coding genes in the human genome. Proc. Natl. Acad. Sci. 2007 doi: 10.1073/pnas.0709013104. (in press) [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cohen-Salmon M., El-Amraoui A., Leibovici M., Petit C., El-Amraoui A., Leibovici M., Petit C., Leibovici M., Petit C., Petit C. Otogelin: A glycoprotein specific to the acellular membranes of the inner ear. Proc. Natl. Acad. Sci. 1997;94:14450–14455. doi: 10.1073/pnas.94.26.14450. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cremona M., Colombo E., Andreazzoli M., Cossu G., Broccoli V., Colombo E., Andreazzoli M., Cossu G., Broccoli V., Andreazzoli M., Cossu G., Broccoli V., Cossu G., Broccoli V., Broccoli V. Bsx, an evolutionary conserved brain specific homeoboX gene expressed in the septum, epiphysis, mammillary bodies and arcuate nucleus. Brain Res. Gene Expr. Patterns. 2004;4:47–51. doi: 10.1016/s1567-133x(03)00151-0. [DOI] [PubMed] [Google Scholar]
- Denoeud F., Kapranov P., Ucla C., Frankish A., Castelo R., Drenkow J., Lagarde J., Alioto T., Manzano C., Chrast J., Kapranov P., Ucla C., Frankish A., Castelo R., Drenkow J., Lagarde J., Alioto T., Manzano C., Chrast J., Ucla C., Frankish A., Castelo R., Drenkow J., Lagarde J., Alioto T., Manzano C., Chrast J., Frankish A., Castelo R., Drenkow J., Lagarde J., Alioto T., Manzano C., Chrast J., Castelo R., Drenkow J., Lagarde J., Alioto T., Manzano C., Chrast J., Drenkow J., Lagarde J., Alioto T., Manzano C., Chrast J., Lagarde J., Alioto T., Manzano C., Chrast J., Alioto T., Manzano C., Chrast J., Manzano C., Chrast J., Chrast J., et al. Prominent use of distal 5′ transcription start sites and discovery of a large number of additional exons in ENCODE regions. Genome Res. 2007;17:746–759. doi: 10.1101/gr.5660607. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Don R.H., Cox P.T., Wainwright B.J., Baker K., Mattick J.S., Cox P.T., Wainwright B.J., Baker K., Mattick J.S., Wainwright B.J., Baker K., Mattick J.S., Baker K., Mattick J.S., Mattick J.S. ‘Touchdown’ PCR to circumvent spurious priming during gene amplification. Nucleic Acids Res. 1991;19:4008. doi: 10.1093/nar/19.14.4008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eaton J.L., Glasgow E., Glasgow E. Zebrafish orthopedia (otp) is required for isotocin cell development. Dev. Genes Evol. 2007;217:149–158. doi: 10.1007/s00427-006-0123-2. [DOI] [PubMed] [Google Scholar]
- El-Amraoui A., Cohen-Salmon M., Petit C., Simmler M.C., Cohen-Salmon M., Petit C., Simmler M.C., Petit C., Simmler M.C., Simmler M.C. Spatiotemporal expression of otogelin in the developing and adult mouse inner ear. Hear. Res. 2001;158:151–159. doi: 10.1016/s0378-5955(01)00312-4. [DOI] [PubMed] [Google Scholar]
- ENCODE Project Consortium Identification and analysis of functional elements in 1% of the human genome by the ENCODE pilot project. Nature. 2007;447:799–816. doi: 10.1038/nature05874. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eyras E., Reymond A., Castelo R., Bye J.M., Camara F., Flicek P., Huckle E.J., Parra G., Shteynberg D.D., Wyss C., Reymond A., Castelo R., Bye J.M., Camara F., Flicek P., Huckle E.J., Parra G., Shteynberg D.D., Wyss C., Castelo R., Bye J.M., Camara F., Flicek P., Huckle E.J., Parra G., Shteynberg D.D., Wyss C., Bye J.M., Camara F., Flicek P., Huckle E.J., Parra G., Shteynberg D.D., Wyss C., Camara F., Flicek P., Huckle E.J., Parra G., Shteynberg D.D., Wyss C., Flicek P., Huckle E.J., Parra G., Shteynberg D.D., Wyss C., Huckle E.J., Parra G., Shteynberg D.D., Wyss C., Parra G., Shteynberg D.D., Wyss C., Shteynberg D.D., Wyss C., Wyss C., et al. Gene finding in the chicken genome. BMC Bioinformatics. 2005;6:131. doi: 10.1186/1471-2105-6-131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Flicek P., Keibler E., Hu P., Korf I., Brent M.R., Keibler E., Hu P., Korf I., Brent M.R., Hu P., Korf I., Brent M.R., Korf I., Brent M.R., Brent M.R. Leveraging the mouse genome for gene prediction in human: From whole-genome shotgun reads to a global synteny map. Genome Res. 2003;13:46–54. doi: 10.1101/gr.830003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gerhard D.S., Wagner L., Feingold E.A., Shenmen C.M., Grouse L.H., Schuler G., Klein S.L., Old S., Rasooly R., Good P., Wagner L., Feingold E.A., Shenmen C.M., Grouse L.H., Schuler G., Klein S.L., Old S., Rasooly R., Good P., Feingold E.A., Shenmen C.M., Grouse L.H., Schuler G., Klein S.L., Old S., Rasooly R., Good P., Shenmen C.M., Grouse L.H., Schuler G., Klein S.L., Old S., Rasooly R., Good P., Grouse L.H., Schuler G., Klein S.L., Old S., Rasooly R., Good P., Schuler G., Klein S.L., Old S., Rasooly R., Good P., Klein S.L., Old S., Rasooly R., Good P., Old S., Rasooly R., Good P., Rasooly R., Good P., Good P., et al. The status, quality, and expansion of the NIH full-length cDNA project: The Mammalian Gene Collection (MGC) Genome Res. 2004;14:2121–2127. doi: 10.1101/gr.2596504. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gerstein M.B., Bruce C., Rozowsky J.S., Zheng D., Du J., Korbel J.O., Emanuelsson O., Zhang Z.D., Weissman S., Snyder M., Bruce C., Rozowsky J.S., Zheng D., Du J., Korbel J.O., Emanuelsson O., Zhang Z.D., Weissman S., Snyder M., Rozowsky J.S., Zheng D., Du J., Korbel J.O., Emanuelsson O., Zhang Z.D., Weissman S., Snyder M., Zheng D., Du J., Korbel J.O., Emanuelsson O., Zhang Z.D., Weissman S., Snyder M., Du J., Korbel J.O., Emanuelsson O., Zhang Z.D., Weissman S., Snyder M., Korbel J.O., Emanuelsson O., Zhang Z.D., Weissman S., Snyder M., Emanuelsson O., Zhang Z.D., Weissman S., Snyder M., Zhang Z.D., Weissman S., Snyder M., Weissman S., Snyder M., Snyder M., et al. What is a gene, post-ENCODE? History and updated definition. Genome Res. 2007;17:669–681. doi: 10.1101/gr.6339607. [DOI] [PubMed] [Google Scholar]
- Goodstadt L., Ponting C.P., Ponting C.P. Phylogenetic reconstruction of orthology, paralogy, and conserved synteny for dog and human. PLoS Comput. Biol. 2006;2:e133. doi: 10.1371/journal.pcbi.0020133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Griffiths-Jones S., Bateman A., Marshall M., Khanna A., Eddy S.R., Bateman A., Marshall M., Khanna A., Eddy S.R., Marshall M., Khanna A., Eddy S.R., Khanna A., Eddy S.R., Eddy S.R. Rfam: An RNA family database. Nucleic Acids Res. 2003;31:439–441. doi: 10.1093/nar/gkg006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gross S.S., Brent M.R., Brent M.R. Using multiple alignments to improve gene prediction. J. Comput. Biol. 2006;13:379–393. doi: 10.1089/cmb.2006.13.379. [DOI] [PubMed] [Google Scholar]
- Guigó R., Dermitzakis E.T., Agarwal P., Ponting C.P., Parra G., Reymond A., Abril J.F., Keibler E., Lyle R., Ucla C., Dermitzakis E.T., Agarwal P., Ponting C.P., Parra G., Reymond A., Abril J.F., Keibler E., Lyle R., Ucla C., Agarwal P., Ponting C.P., Parra G., Reymond A., Abril J.F., Keibler E., Lyle R., Ucla C., Ponting C.P., Parra G., Reymond A., Abril J.F., Keibler E., Lyle R., Ucla C., Parra G., Reymond A., Abril J.F., Keibler E., Lyle R., Ucla C., Reymond A., Abril J.F., Keibler E., Lyle R., Ucla C., Abril J.F., Keibler E., Lyle R., Ucla C., Keibler E., Lyle R., Ucla C., Lyle R., Ucla C., Ucla C., et al. Comparison of mouse and human genomes followed by experimental verification yields an estimated 1,019 additional genes. Proc. Natl. Acad. Sci. 2003;100:1140–1145. doi: 10.1073/pnas.0337561100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Haas B.J., Delcher A.L., Mount S.M., Wortman J.R., Smith R.K.J., Hannick L.I., Maiti R., Ronning C.M., Rusch D.B., Town C.D., Delcher A.L., Mount S.M., Wortman J.R., Smith R.K.J., Hannick L.I., Maiti R., Ronning C.M., Rusch D.B., Town C.D., Mount S.M., Wortman J.R., Smith R.K.J., Hannick L.I., Maiti R., Ronning C.M., Rusch D.B., Town C.D., Wortman J.R., Smith R.K.J., Hannick L.I., Maiti R., Ronning C.M., Rusch D.B., Town C.D., Smith R.K.J., Hannick L.I., Maiti R., Ronning C.M., Rusch D.B., Town C.D., Hannick L.I., Maiti R., Ronning C.M., Rusch D.B., Town C.D., Maiti R., Ronning C.M., Rusch D.B., Town C.D., Ronning C.M., Rusch D.B., Town C.D., Rusch D.B., Town C.D., Town C.D., et al. Improving the Arabidopsis genome annotation using maximal transcript alignment assemblies. Nucleic Acids Res. 2003;31:5654–5666. doi: 10.1093/nar/gkg770. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harrow J., Denoeud F., Frankish A., Reymond A., Chen C.-K., Chrast J., Lagarde J., Gilbert J.G.R., Storey R., Swarbreck D., Denoeud F., Frankish A., Reymond A., Chen C.-K., Chrast J., Lagarde J., Gilbert J.G.R., Storey R., Swarbreck D., Frankish A., Reymond A., Chen C.-K., Chrast J., Lagarde J., Gilbert J.G.R., Storey R., Swarbreck D., Reymond A., Chen C.-K., Chrast J., Lagarde J., Gilbert J.G.R., Storey R., Swarbreck D., Chen C.-K., Chrast J., Lagarde J., Gilbert J.G.R., Storey R., Swarbreck D., Chrast J., Lagarde J., Gilbert J.G.R., Storey R., Swarbreck D., Lagarde J., Gilbert J.G.R., Storey R., Swarbreck D., Gilbert J.G.R., Storey R., Swarbreck D., Storey R., Swarbreck D., Swarbreck D., et al. GENCODE: Producing a reference annotation for ENCODE. Genome Biol. 2006;7:S4. doi: 10.1186/gb-2006-7-s1-s4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hillier L.D., Lennon G., Becker M., Bonaldo M.F., Chiapelli B., Chissoe S., Dietrich N., DuBuque T., Favello A., Gish W., Lennon G., Becker M., Bonaldo M.F., Chiapelli B., Chissoe S., Dietrich N., DuBuque T., Favello A., Gish W., Becker M., Bonaldo M.F., Chiapelli B., Chissoe S., Dietrich N., DuBuque T., Favello A., Gish W., Bonaldo M.F., Chiapelli B., Chissoe S., Dietrich N., DuBuque T., Favello A., Gish W., Chiapelli B., Chissoe S., Dietrich N., DuBuque T., Favello A., Gish W., Chissoe S., Dietrich N., DuBuque T., Favello A., Gish W., Dietrich N., DuBuque T., Favello A., Gish W., DuBuque T., Favello A., Gish W., Favello A., Gish W., Gish W., et al. Generation and analysis of 280,000 human expressed sequence tags. Genome Res. 1996;6:807–828. doi: 10.1101/gr.6.9.807. [DOI] [PubMed] [Google Scholar]
- Hubbard T.J.P., Aken B.L., Beal K., Ballester B., Caccamo M., Chen Y., Clarke L., Coates G., Cunningham F., Cutts T., Aken B.L., Beal K., Ballester B., Caccamo M., Chen Y., Clarke L., Coates G., Cunningham F., Cutts T., Beal K., Ballester B., Caccamo M., Chen Y., Clarke L., Coates G., Cunningham F., Cutts T., Ballester B., Caccamo M., Chen Y., Clarke L., Coates G., Cunningham F., Cutts T., Caccamo M., Chen Y., Clarke L., Coates G., Cunningham F., Cutts T., Chen Y., Clarke L., Coates G., Cunningham F., Cutts T., Clarke L., Coates G., Cunningham F., Cutts T., Coates G., Cunningham F., Cutts T., Cunningham F., Cutts T., Cutts T., et al. Ensembl 2007. Nucleic Acids Res. 2007;35:D610–D617. doi: 10.1093/nar/gkl996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- International Human Genome Sequencing Consortium Initial sequencing and analysis of the human genome. Nature. 2001;409:860–921. doi: 10.1038/35057062. [DOI] [PubMed] [Google Scholar]
- International Human Genome Sequencing Consortium Finishing the euchromatic sequence of the human genome. Nature. 2004;431:931–945. doi: 10.1038/nature03001. [DOI] [PubMed] [Google Scholar]
- Kapranov P., Cheng J., Dike S., Nix D.A., Duttagupta R., Willingham A.T., Stadler P.F., Hertel J., Hackermuller J., Hofacker I.L., Cheng J., Dike S., Nix D.A., Duttagupta R., Willingham A.T., Stadler P.F., Hertel J., Hackermuller J., Hofacker I.L., Dike S., Nix D.A., Duttagupta R., Willingham A.T., Stadler P.F., Hertel J., Hackermuller J., Hofacker I.L., Nix D.A., Duttagupta R., Willingham A.T., Stadler P.F., Hertel J., Hackermuller J., Hofacker I.L., Duttagupta R., Willingham A.T., Stadler P.F., Hertel J., Hackermuller J., Hofacker I.L., Willingham A.T., Stadler P.F., Hertel J., Hackermuller J., Hofacker I.L., Stadler P.F., Hertel J., Hackermuller J., Hofacker I.L., Hertel J., Hackermuller J., Hofacker I.L., Hackermuller J., Hofacker I.L., Hofacker I.L., et al. RNA maps reveal new RNA classes and a possible function for pervasive transcription. Science. 2007;316:1484–1488. doi: 10.1126/science.1138341. [DOI] [PubMed] [Google Scholar]
- Kent W.J. BLAT: The BLAST-like alignment tool. Genome Res. 2002;12:656–664. doi: 10.1101/gr.229202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kimura K., Wakamatsu A., Suzuki Y., Ota T., Nishikawa T., Yamashita R., Yamamoto J.-i., Sekine M., Tsuritani K., Wakaguri H., Wakamatsu A., Suzuki Y., Ota T., Nishikawa T., Yamashita R., Yamamoto J.-i., Sekine M., Tsuritani K., Wakaguri H., Suzuki Y., Ota T., Nishikawa T., Yamashita R., Yamamoto J.-i., Sekine M., Tsuritani K., Wakaguri H., Ota T., Nishikawa T., Yamashita R., Yamamoto J.-i., Sekine M., Tsuritani K., Wakaguri H., Nishikawa T., Yamashita R., Yamamoto J.-i., Sekine M., Tsuritani K., Wakaguri H., Yamashita R., Yamamoto J.-i., Sekine M., Tsuritani K., Wakaguri H., Yamamoto J.-i., Sekine M., Tsuritani K., Wakaguri H., Sekine M., Tsuritani K., Wakaguri H., Tsuritani K., Wakaguri H., Wakaguri H., et al. Diversification of transcriptional modulation: Large-scale identification and characterization of putative alternative promoters of human genes. Genome Res. 2006;16:55–65. doi: 10.1101/gr.4039406. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Korf I., Flicek P., Duan D., Brent M.R., Flicek P., Duan D., Brent M.R., Duan D., Brent M.R., Brent M.R. Integrating genomic homology into gene structure prediction. Bioinformatics. 2001;17:S140–S148. doi: 10.1093/bioinformatics/17.suppl_1.s140. [DOI] [PubMed] [Google Scholar]
- Lee L., Hughes T., Frey B., Hughes T., Frey B., Frey B. How many new genes are there? Science. 2006;311:1709–1711. doi: 10.1126/science.311.5768.1709b. [DOI] [PubMed] [Google Scholar]
- Mattick J.S., Makunin I.V., Makunin I.V. Non-coding RNA. Hum. Mol. Genet. 2006;15:R17–R29. doi: 10.1093/hmg/ddl046. [DOI] [PubMed] [Google Scholar]
- Mikami A., Tynan S.H., Hama T., Luby-Phelps K., Saito T., Crandall J.E., Besharse J.C., Vallee R.B., Tynan S.H., Hama T., Luby-Phelps K., Saito T., Crandall J.E., Besharse J.C., Vallee R.B., Hama T., Luby-Phelps K., Saito T., Crandall J.E., Besharse J.C., Vallee R.B., Luby-Phelps K., Saito T., Crandall J.E., Besharse J.C., Vallee R.B., Saito T., Crandall J.E., Besharse J.C., Vallee R.B., Crandall J.E., Besharse J.C., Vallee R.B., Besharse J.C., Vallee R.B., Vallee R.B. Molecular structure of cytoplasmic dynein 2 and its distribution in neuronal and ciliated cells. J. Cell Sci. 2002;115:4801–4808. doi: 10.1242/jcs.00168. [DOI] [PubMed] [Google Scholar]
- Parra G., Agarwal P., Abril J.F., Wiehe T., Fickett J.W., Guigó R., Agarwal P., Abril J.F., Wiehe T., Fickett J.W., Guigó R., Abril J.F., Wiehe T., Fickett J.W., Guigó R., Wiehe T., Fickett J.W., Guigó R., Fickett J.W., Guigó R., Guigó R. Comparative gene prediction in human and mouse. Genome Res. 2003;13:108–117. doi: 10.1101/gr.871403. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Parra G., Reymond A., Dabbouseh N., Dermitzakis E.T., Castelo R., Thomson T.M., Antonarakis S.E., Guigo R., Reymond A., Dabbouseh N., Dermitzakis E.T., Castelo R., Thomson T.M., Antonarakis S.E., Guigo R., Dabbouseh N., Dermitzakis E.T., Castelo R., Thomson T.M., Antonarakis S.E., Guigo R., Dermitzakis E.T., Castelo R., Thomson T.M., Antonarakis S.E., Guigo R., Castelo R., Thomson T.M., Antonarakis S.E., Guigo R., Thomson T.M., Antonarakis S.E., Guigo R., Antonarakis S.E., Guigo R., Guigo R. Tandem chimerism as a means to increase protein complexity in the human genome. Genome Res. 2006;16:37–44. doi: 10.1101/gr.4145906. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pennacchio L.A., Ahituv N., Moses A.M., Prabhakar S., Nobrega M.A., Shoukry M., Minovitsky S., Dubchak I., Holt A., Lewis K.D., Ahituv N., Moses A.M., Prabhakar S., Nobrega M.A., Shoukry M., Minovitsky S., Dubchak I., Holt A., Lewis K.D., Moses A.M., Prabhakar S., Nobrega M.A., Shoukry M., Minovitsky S., Dubchak I., Holt A., Lewis K.D., Prabhakar S., Nobrega M.A., Shoukry M., Minovitsky S., Dubchak I., Holt A., Lewis K.D., Nobrega M.A., Shoukry M., Minovitsky S., Dubchak I., Holt A., Lewis K.D., Shoukry M., Minovitsky S., Dubchak I., Holt A., Lewis K.D., Minovitsky S., Dubchak I., Holt A., Lewis K.D., Dubchak I., Holt A., Lewis K.D., Holt A., Lewis K.D., Lewis K.D., et al. In vivo enhancer analysis of human conserved non-coding sequences. Nature. 2006;444:499–502. doi: 10.1038/nature05295. [DOI] [PubMed] [Google Scholar]
- Pfister K.K., Shah P.R., Hummerich H., Russ A., Cotton J., Annuar A.A., King S.M., Fisher E.M.C., Shah P.R., Hummerich H., Russ A., Cotton J., Annuar A.A., King S.M., Fisher E.M.C., Hummerich H., Russ A., Cotton J., Annuar A.A., King S.M., Fisher E.M.C., Russ A., Cotton J., Annuar A.A., King S.M., Fisher E.M.C., Cotton J., Annuar A.A., King S.M., Fisher E.M.C., Annuar A.A., King S.M., Fisher E.M.C., King S.M., Fisher E.M.C., Fisher E.M.C. Genetic analysis of the cytoplasmic dynein subunit families. PLoS Genet. 2006;2:e1. doi: 10.1371/journal.pgen.0020001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pollard K.S., Salama S.R., Lambert N., Lambot M.-A., Coppens S., Pedersen J.S., Katzman S., King B., Onodera C., Siepel A., Salama S.R., Lambert N., Lambot M.-A., Coppens S., Pedersen J.S., Katzman S., King B., Onodera C., Siepel A., Lambert N., Lambot M.-A., Coppens S., Pedersen J.S., Katzman S., King B., Onodera C., Siepel A., Lambot M.-A., Coppens S., Pedersen J.S., Katzman S., King B., Onodera C., Siepel A., Coppens S., Pedersen J.S., Katzman S., King B., Onodera C., Siepel A., Pedersen J.S., Katzman S., King B., Onodera C., Siepel A., Katzman S., King B., Onodera C., Siepel A., King B., Onodera C., Siepel A., Onodera C., Siepel A., Siepel A., et al. An RNA gene expressed during cortical development evolved rapidly in humans. Nature. 2006;443:167–172. doi: 10.1038/nature05113. [DOI] [PubMed] [Google Scholar]
- Pruitt K.D., Tatusova T., Maglott D.R., Tatusova T., Maglott D.R., Maglott D.R. NCBI Reference Sequence (RefSeq): A curated non-redundant sequence database of genomes, transcripts and proteins. Nucleic Acids Res. 2005;33:D501–D504. doi: 10.1093/nar/gki025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schwartz S., Kent W.J., Smit A., Zhang Z., Baertsch R., Hardison R.C., Haussler D., Miller W., Kent W.J., Smit A., Zhang Z., Baertsch R., Hardison R.C., Haussler D., Miller W., Smit A., Zhang Z., Baertsch R., Hardison R.C., Haussler D., Miller W., Zhang Z., Baertsch R., Hardison R.C., Haussler D., Miller W., Baertsch R., Hardison R.C., Haussler D., Miller W., Hardison R.C., Haussler D., Miller W., Haussler D., Miller W., Miller W. Human-mouse alignments with BLASTZ. Genome Res. 2003;13:103–107. doi: 10.1101/gr.809403. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Siepel A., Haussler D., Haussler D. Proceedings of the Eighth International Conference on Research in Computational Molecular Biology. ACM Press; New York: 2004. Computational identification of evolutionarily conserved exons; pp. 177–186. [Google Scholar]
- Strausberg R.L., Feingold E.A., Klausner R.D., Collins F.S., Feingold E.A., Klausner R.D., Collins F.S., Klausner R.D., Collins F.S., Collins F.S. The mammalian gene collection. Science. 1999;286:455–457. doi: 10.1126/science.286.5439.455. [DOI] [PubMed] [Google Scholar]
- van Baren M.J., Brent M.R., Brent M.R. Iterative gene prediction and pseudogene removal improves genome annotation. Genome Res. 2006;16:678–685. doi: 10.1101/gr.4766206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Venter J.C., Adams M.D., Myers E.W., Li P.W., Mural R.J., Sutton G.G., Smith H.O., Yandell M., Evans C.A., Holt R.A., Adams M.D., Myers E.W., Li P.W., Mural R.J., Sutton G.G., Smith H.O., Yandell M., Evans C.A., Holt R.A., Myers E.W., Li P.W., Mural R.J., Sutton G.G., Smith H.O., Yandell M., Evans C.A., Holt R.A., Li P.W., Mural R.J., Sutton G.G., Smith H.O., Yandell M., Evans C.A., Holt R.A., Mural R.J., Sutton G.G., Smith H.O., Yandell M., Evans C.A., Holt R.A., Sutton G.G., Smith H.O., Yandell M., Evans C.A., Holt R.A., Smith H.O., Yandell M., Evans C.A., Holt R.A., Yandell M., Evans C.A., Holt R.A., Evans C.A., Holt R.A., Holt R.A., et al. The sequence of the human genome. Science. 2001;291:1304–1351. doi: 10.1126/science.1058040. [DOI] [PubMed] [Google Scholar]
- Weiss A., Leinwand L.A., Leinwand L.A. The mammalian myosin heavy chain gene family. Annu. Rev. Cell Dev. Biol. 1996;12:417–439. doi: 10.1146/annurev.cellbio.12.1.417. [DOI] [PubMed] [Google Scholar]
- Wu J.Q., Garcia A.M., Hulyk S., Sneed A., Kowis C., Yuan Y., Steffen D., McPherson J.D., Gunaratne P.H., Gibbs R.A., Garcia A.M., Hulyk S., Sneed A., Kowis C., Yuan Y., Steffen D., McPherson J.D., Gunaratne P.H., Gibbs R.A., Hulyk S., Sneed A., Kowis C., Yuan Y., Steffen D., McPherson J.D., Gunaratne P.H., Gibbs R.A., Sneed A., Kowis C., Yuan Y., Steffen D., McPherson J.D., Gunaratne P.H., Gibbs R.A., Kowis C., Yuan Y., Steffen D., McPherson J.D., Gunaratne P.H., Gibbs R.A., Yuan Y., Steffen D., McPherson J.D., Gunaratne P.H., Gibbs R.A., Steffen D., McPherson J.D., Gunaratne P.H., Gibbs R.A., McPherson J.D., Gunaratne P.H., Gibbs R.A., Gunaratne P.H., Gibbs R.A., Gibbs R.A., et al. Large-scale RT-PCR recovery of full-length cDNA clones. Biotechniques. 2004a;36:690–696. doi: 10.2144/04364DD03. [DOI] [PubMed] [Google Scholar]
- Wu J.Q., Shteynberg D., Arumugam M., Gibbs R.A., Brent M.R., Shteynberg D., Arumugam M., Gibbs R.A., Brent M.R., Arumugam M., Gibbs R.A., Brent M.R., Gibbs R.A., Brent M.R., Brent M.R. Identification of rat genes by TWINSCAN gene prediction, RT-PCR, and direct sequencing. Genome Res. 2004b;14:665–671. doi: 10.1101/gr.1959604. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xie X., Mikkelsen T.S., Gnirke A., Lindblad-Toh K., Kellis M., Lander E.S., Mikkelsen T.S., Gnirke A., Lindblad-Toh K., Kellis M., Lander E.S., Gnirke A., Lindblad-Toh K., Kellis M., Lander E.S., Lindblad-Toh K., Kellis M., Lander E.S., Kellis M., Lander E.S., Lander E.S. Systematic discovery of regulatory motifs in conserved regions of the human genome, including thousands of CTCF insulator sites. Proc. Natl. Acad. Sci. 2007;104:7145–7150. doi: 10.1073/pnas.0701811104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zheng D., Frankish A., Baertsch R., Kapranov P., Reymond A., Choo S.W., Lu Y., Denoeud F., Antonarakis S.E., Snyder M., Frankish A., Baertsch R., Kapranov P., Reymond A., Choo S.W., Lu Y., Denoeud F., Antonarakis S.E., Snyder M., Baertsch R., Kapranov P., Reymond A., Choo S.W., Lu Y., Denoeud F., Antonarakis S.E., Snyder M., Kapranov P., Reymond A., Choo S.W., Lu Y., Denoeud F., Antonarakis S.E., Snyder M., Reymond A., Choo S.W., Lu Y., Denoeud F., Antonarakis S.E., Snyder M., Choo S.W., Lu Y., Denoeud F., Antonarakis S.E., Snyder M., Lu Y., Denoeud F., Antonarakis S.E., Snyder M., Denoeud F., Antonarakis S.E., Snyder M., Antonarakis S.E., Snyder M., Snyder M., et al. Pseudogenes in the ENCODE regions: Consensus annotation, analysis of transcription, and evolution. Genome Res. 2007;17:839–851. doi: 10.1101/gr.5586307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhu J., Sanborn J.Z., Diekhans M., Lowe C.B., Pringle T., Haussler D., Sanborn J.Z., Diekhans M., Lowe C.B., Pringle T., Haussler D., Diekhans M., Lowe C.B., Pringle T., Haussler D., Lowe C.B., Pringle T., Haussler D., Pringle T., Haussler D., Haussler D. Comparative genomics search for losses of long-established genes on the human lineage. PLoS Comput. Biol. 2007 doi: 10.1371/journal.pcbi.0030247.eor. (in press) [DOI] [PMC free article] [PubMed] [Google Scholar]