

Abstract
DNA mismatch repair is thought to act through two subpathways involving the recognition of base-base and insertion/deletion mispairs by the Msh2-Msh6 heterodimer and the recognition of insertion/deletion mispairs by the Msh2-Msh3 heterodimer. Here, through genetic and biochemical approaches, we describe a previously unidentified role of the Msh2-Msh3 heterodimer in the recognition of base-base mispairs and the suppression of homology-mediated duplication and deletion mutations. Saccharomyces cerevisiae msh3 mutants did not show an increase in the rate of base substitution mutations by the CAN1 forward mutation assay compared to the rate for the wild type but did show an altered spectrum of base substitution mutations, including an increased accumulation of base pair changes from GC to CG and from AT to TA; msh3 mutants also accumulated homology-mediated duplication and deletion mutations. The mutation spectrum of mlh3 mutants paralleled that of msh3 mutants, suggesting that the Mlh1-Mlh3 heterodimer may also play a role in the repair of base-base mispairs and in the suppression of homology-mediated duplication and deletion mutations. Mispair binding analysis with purified Msh2-Msh3 and DNA substrates derived from CAN1 sequences found to be mutated in vivo demonstrated that Msh2-Msh3 exhibited robust binding to specific base-base mispairs that was consistent with functional mispair binding.
For a cell to survive and grow normally, it must maintain the fidelity of its genome. To do this, the cell utilizes multiple mechanisms to minimize the rate at which mutations occur. DNA mismatch repair is one such highly conserved mechanism that recognizes and repairs mispaired bases in DNA caused by replication errors, recombination, or chemical damage to DNA and DNA precursors (26, 37). The importance of mismatch repair is evidenced by the fact that inherited mutations in two human mismatch repair genes, MSH2 and MLH1, are responsible for most cases of hereditary nonpolyposis colorectal cancer and that epigenetic silencing of MLH1 underlies most cases of sporadic mismatch repair-defective cancer (31, 42).
The mechanism of mismatch repair is best understood for Escherichia coli, where mismatch repair has been reconstituted in vitro with purified proteins and defined DNA substrates (20, 37, 39). In this reaction, the MutS protein homodimer recognizes the abnormal DNA structure of base-base or insertion/deletion mispairs (24, 46, 49). The MutL homodimer binds to the MutS-DNA complex and activates MutH endonuclease, which nicks the unmethylated DNA strand at a hemimethylated GATC sequence, targeting repair to newly synthesized DNA strands (1, 5, 15, 17, 19, 47, 53). The nicked DNA strand is unwound by UvrD helicase and degraded by a number of exonucleases, resulting in the excision of the mispaired base; repair is completed by resynthesis of the excised strand (6, 27, 34). The detailed molecular mechanisms of many aspects of this reaction are still under investigation (1, 3, 25, 47).
The mismatch repair system in eukaryotes is more complex than, while conserved with, that of bacteria. Nonetheless, many of the proteins involved have been identified, their general biochemical properties have been determined, and at least partial repair reactions resembling those of bacteria have been reconstituted in vitro with purified proteins (10, 55). In eukaryotes, the dimeric MutS mismatch recognition protein has been replaced by two different heterodimers of MutS homologue proteins, the Msh2-Msh6 and Msh2-Msh3 complexes (26). Similarly, the MutL dimer has been replaced by two different heterodimers of MutL homologue proteins, the Mlh1-Pms1 (Pms2 in humans) and Mlh1-Mlh3 complexes (7, 14, 43, 52). In addition, it has been suggested that a third MutL homologue complex, Mlh1-Mlh2 (Pms1 in humans), may play a minor role in mismatch repair, although biochemical studies do not support this suggestion (21, 44). DNA polymerase δ, RPA, PCNA, RFC, and Exo1 have been shown to act in eukaryotic mismatch repair, although evidence suggests that additional proteins may be involved (38).
Current models of eukaryotic mismatch repair suggest that the Msh2-Msh6 complex is the major mismatch recognition complex and functions in the repair of base-base and insertion/deletion mispairs (20, 26, 37-39). The Msh2-Msh3 complex is redundant with the Msh2-Msh6 complex with respect to the repair of small insertion/deletion mispairs and is also able to recognize larger insertion/deletion mispairs (32, 33, 48). A number of genetic results are consistent with this scenario: null mutations in MSH2 result in a strong mutator phenotype characterized by the accumulation of base substitution and frameshift mutations; MSH6 defects result in a strong mutator phenotype with respect to base substitutions, but only a small increase in frameshift mutations; MSH3 defects cause weak mutator phenotypes characterized by the accumulation of frameshift mutations (however, in assays where larger frameshift mutations are analyzed, stronger mutator phenotypes are observed); and lastly, an msh3 msh6 double mutant recapitulates the mutator phenotype of an msh2 single mutant (32, 48).
Similar studies have led to the view that the Mlh1-Pms1 complex is the major MutL homologue complex that functions in eukaryotic mismatch repair, whereas the Mlh1-Mlh3 complex plays a minor role in mismatch repair and is partially redundant with the Mlh1-Pms1 complex (14, 43, 52). Genetic results supporting this view are as follows: null mutations in MLH1 and PMS1 result in a strong mutator phenotype characterized by the accumulation of base substitution and frameshift mutations; MLH3 defects result in a weak mutator phenotype primarily characterized by the accumulation of frameshift mutations; and the deletion of both MLH3 and PMS1 (PMS2 in human and mouse) is required to recapitulate the mutator phenotypes (and cancer prone phenotype in mice) caused by a defect in MLH1 (9, 23). Genetic analysis has also suggested that the Mlh1-Mlh3 complex primarily functions in conjunction with the Msh2-Msh3 complex (7, 14, 43, 44, 52). Biochemical studies are consistent, with the Mlh1-Pms1 complex playing the major role in mismatch repair, whereas the Mlh1-Mlh3 complex, which has been much less studied, has only weak in vitro mismatch repair activity (7, 10).
While the studies establishing the roles of the eukaryotic MutS and MutL homologue complexes in mismatch repair seem quite definitive, it is important to note that they have some limitations. First, the genetic results are based on a few types of assays. Reversion assays can detect only a limited number of mutation types. Forward mutation assays are less biased, but prior mutation spectrum analysis was performed at a time when it was not feasible to sequence large numbers of mutations in large unbiased forward mutation targets like the CAN1 gene. Even with analysis of small forward mutation targets, where large numbers of mutations can be analyzed, it is difficult to control biological variation within mutation spectrum analysis experiments. Second, the mutations observed in a given mutant background are the result of a complex process involving misincorporation errors at individual sites combined with how efficiently other competing pathways, including editing exonucleases, bypass DNA polymerases and the different mismatch repair pathways act on mispairs and mispair-producing errors. Third, because of the low mutation rates caused by defects in MSH3 and MLH3, it has been difficult to genetically characterize the roles of Msh2-Msh3 and Mlh1-Mlh3 in vivo, which is further complicated by the fact that these defects are masked by the activity of the Msh2-Msh6 and Mlh1-Pms1 complexes, respectively (14, 32, 48). Lastly, biochemical studies have used a limited diversity of substrates and mispairs predicted to occur in vivo have generally not been used as substrates, in vitro. Here we have used a genetic approach to identify mutations that arose in the absence of the Saccharomyces cerevisiae protein Msh6 or Msh3 in vivo and then used DNA substrates derived from the mutated sequences to analyze Msh2-Msh3 and Msh2-Msh6 binding affinities in vitro. Our results indicate that Msh2-Msh3 plays a previously unrecognized role in the repair of specific base-base mispairs and imply that the Mlh1-Mlh3 complex may also function in similar repair reactions. Additionally, we demonstrated that Msh2-Msh3 and Mlh1-Mlh3 play a previously unrecognized role in the suppression of homology-mediated duplication and deletion mutations.
MATERIALS AND METHODS
General methods and strains.
All media, including dropout medium and canavanine-containing dropout medium, have been previously described (2, 4, 45). All strains used in this study were derivatives of the S288c strain RDKY3686 MATα ura3-52 leu2-1 trp1-63 hom3-10 his3-200 lys2-10A (4). The relevant genotypes of these strains are as follows: msh3::hisG for RDKY4149; msh6::hisG for RDKY4151, mlh3::HIS3 for RDKY5295, and mlh1::hisG for RDKY4237. The protease-deficient strain RDKY2418 MATα ura3-52 leu2-1 his3-200 pep4::HIS3 prb1-1.6R can1 msh2::hisG msh6::hisG was used to overexpress proteins for purification (22). Genetic complementation of MSH3 derivatives was measured in S. cerevisiae strain RDKY4234 MATα ura3-52 leu2-1 trp1-63 hom3-10 his3-200 lys2-10A msh3::hisG msh6::hisG. Mutation rates were determined by fluctuation analysis using at least 14 independent colonies from each strain as previously described (2, 4, 11, 45).
Genetic complementation.
Site-directed mutagenesis of a wild-type MSH3 low-copy-number LEU2 plasmid was performed to mutate the Met codon at position 1 to Ala (M1A) or the Met codon at position 30 to Ala (M30A). Primers to create the msh3-M1A allele were JH67 5′-AATTTTGACAAAGCCAATTTGAACTCCAAAGCTGCCCCAGCTACCCCTAAACTTCTAAGACT and JH68 5′-AGTCTTAGAAGTTTTAGGGGTAGCTGGGGCAGCTTTGGAGTTCAAATTGGCTTTGTCAAAATT. Primers to create the msh3-M30A allele were JH69 5′-GAAAATGGCTCCACATCTTCTCAAAAGAAAGCTAAGCAATCGAGTTTGTTATCTTTTTTCTCA and JH70 5′-TGAGAAAAAAGATAACAAACTCGATTGCTTAGCTTTCTTTTGAGAAGATGTGGAGCCATTTTC. The msh3 mutant plasmids were sequenced to confirm that only the desired mutation was present. Plasmids were then transformed into strain RDKY4234, and transformants were patched onto media lacking Leu to maintain plasmid selection. Patches were then replica plated onto plates lacking Lys and Thr and grown at 30°C for 2 days to select for lys2-10A and hom3-10 revertants so as to visualize the mutator phenotype of the different plasmid-containing strains.
Mutation analysis.
Strains of interest were first streaked for single colonies on yeast extract-peptone-dextrose plates, and then individual colonies were patched onto yeast extract-peptone-dextrose plates. The patches were replica plated onto selective media without Arg/with Can, and canavanine-resistant mutants were allowed to grow at 30°C for 2 days. Mutation spectra were analyzed by isolating chromosomal DNA from one Canr mutant per patch, amplifying the CAN1 gene by PCR and sequencing to determine the inactivating mutation in the CAN1 gene (11, 14, 32). The PCR primer pair used for amplification of CAN1 was CAN1FX (5′-GTTGGATCCAGTTTTTAATCTGTCGTC) and CAN1RX (5′-TTCGGTGTATGACTTATGAGGGTG). The three primers used for sequencing CAN1 were CAN1G (5′-CAGTGGAACTTTGTACGTCC), CANSEQ3 (5′-TTCTGTCACGCAGTCCTTGG), and CANSEQ5 (5′-AACTAGTTGGTATCACTGCT). All DNA sequencing was performed by using an Applied Biosystems 3730XL DNA sequencer and standard chemistry. Sequence analysis was performed using Sequencher 4.2.2 (Gene Codes, Ann Arbor, MI).
Statistical analysis.
The significance of the observed overlap between the CAN1 base substitution mutation spectra in different strains was calculated with a Monte Carlo technique. Since the observed base substitution mutations were unlikely to be saturating, the total number of readily mutable CAN1-inactivating mutation sites was estimated by fitting the observed distribution of singly and multiply observed base substitution mutations to a theoretical Poisson distribution. We minimized the root-mean-square error between the expected and observed number of singly and multiply observed mutations using the equation
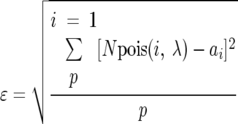 |
where p is the maximum number of events for any single mutation, ai is the number of mutations observed i times, and pois(i,λ) is the probability with the parameter λ = C/N, with C being the number of observed events and N being the total number of possible mutation sites, defined by both the position and the base substitution at that position. Minimization of ɛ by varying N in the range (1,600) gives the total number of mutations, including those not observed in experimental sampling. By using the Poisson distribution, we assumed that all observed base substitution mutations within a strain occur with equal efficiencies and that mutations in multiple isolates are independent of each other. For the wild-type and msh3 strains, the best fitted Poisson curves used values of N = 159 and N = 259, respectively.
Using the total number of inactivating mutations, the results for two different models were calculated. In model 1, the readily mutated CAN1-inactivating mutations are identical in both strains and each strain was allowed to accumulate mutations at any of the 259 or 159 mutation sites (the predicted number of mutations in the msh3 and wild-type strains, respectively). In model 2, the mutational spectra were treated as overlapping but distinct in that the wild-type strain was allowed to accumulate mutations at only 159 of the 259 mutation sites for the msh3 strain. Mutations were then randomly selected for both strains by using each of the two models. The total number of randomly chosen mutations was equal to the number of observed mutations in each strain. The overlap of these theoretical distributions of mutation sites was then calculated. We repeated this process 50,000 times and used a z score test to calculate the significance of the observed overlaps by using the null hypothesis that differences in overlap in base substitution mutation spectra between wild-type and msh3 strains were due to sampling and not due to differences in the specificity of mutation accumulation.
The two-tailed Mann-Whitney test, the chi square “goodness of fit” test, and the Fisher exact probability test were performed on the VassarStats website (http://faculty.vassar.edu/lowry/VassarStats.html).
Overexpression and purification of Msh2-Msh3 complex.
The S. cerevisiae Msh2-Msh3 heterodimer was coexpressed from GAL10 promoter plasmids in the protease-deficient yeast strain RDKY2418. The Msh2 expression vector contains a GAL10 promoter fused to the MSH2 gene on a 2μm URA3 Ampr plasmid. The Msh3 expression vector contains a GAL10 promoter fused to the MSH3 FLAG-tagged gene on a 2μm LEU2 Ampr plasmid. The Msh3 expression vector fuses the GAL10 promoter to the methionine at amino acid position 30 according to the Saccharomyces genome database (http://www.yeastgenome.org/) coding sequence using the leader sequence AAGGAGATATACATatg and contains a C-terminal FLAG tag sequence, cacGACTACAAGGACGACGATGACAAGtga, where the last codon of MSH3 (cac 1047) is shown in lowercase and the FLAG codons are shown in uppercase, followed by the stop codon; genetic complementation studies showed that this FLAG tag did not affect the biological function of MSH3 (data not shown). A fermentor was used to grow 10 liters of cells in synthetic dropout medium lacking uracil and leucine and containing 2% raffinose to an optical density of 0.8 at 30°C. The expression of Msh2 and Msh3 was then induced by adding galactose to a final concentration of 2% for 8 h. The cells were harvested by centrifugation, and the resulting cell pellet was resuspended in lysis buffer (500 mM NaCl, 50 mM Tris-HCl, pH 8.0, 1 mM EDTA, 5 mM dithiothreitol, 10% glycerol, 1 mM phenylmethylsulfonyl fluoride, leupeptin, benzamidine, pepstatin) and lysed with glass beads (Sigma) in a bead beater (Biospec Products, Inc., Bartlesville, OK). The Msh2-Msh3 heterodimer was purified by sequential chromatography on 30-ml Polybuffer Exchanger 94 resin, 10-ml High Trap Q, 5-ml heparin, 10-ml DNA cellulose, 1-ml SP-Sepharose, and 1-ml DEAE columns. Fractions either were frozen directly in liquid N2 and stored at −80°C or were concentrated by centrifugation in a Centricon YM30 (Millipore, Billerica, MA) and then frozen. Protein concentrations were determined by comparison to known protein concentrations on a Coomassie blue-stained gel. The yield from 10 liters of cells was 15 μg of Msh2-Msh3 protein. Additionally, the purified protein was digested with trypsin and subjected to mass spectrometry to confirm its identity. Msh2-Msh6 was provided by Dan Mazur (35).
DNA substrates.
Oligonucleotides were synthesized by Midland Certified Reagent Company (Midland, TX). Double-stranded DNA substrates were constructed by annealing 38-bp complementary oligonucleotides at 95°C for 5 min in annealing buffer (0.5 M NaCl, 10 mM Tris-HCl, pH 7.5, 1 mM EDTA), followed by slowly cooling over 2 h. DNA duplexes were purified by high-pressure liquid chromatography using a Waters GEN-PAK FAX column (33). The sequences of the different oligonucleotides and double-stranded DNA substrates are presented in Table S2 in the supplemental material.
In vitro DNA binding experiments.
Purified DNA substrates were 5′ end labeled using [γ-32P]ATP and T4 polynucleotide kinase and purified by centrifugation through mini Quick Spin oligonucleotide columns (Roche, Indianapolis, IN). DNA binding assays were performed by combining 16 nM protein (Msh2-Msh6 or Msh2-Msh3 heterodimer) with 14 nM 32P-labeled substrate in a final volume of 10 μl binding buffer (20 mM Tris-HCl, pH 8.0, 110 mM NaCl, 5 mM MgCl2, 1 mM dithiothreitol, 10 μM ADP, 5% glycerol, 70 nM unlabeled GC homoduplex, 100 mM bovine serum albumin). Reaction mixtures were incubated on ice for 15 min and then 500 μM ATP was added as indicated in individual experiments for 15 min before loading dye was added. Gel electrophoresis of the samples was performed on a 4 to 20% gradient TBE (45 mM Tris borate, 1 mM EDTA, pH 8.0) Criterion gel (Bio-Rad, Hercules, CA) run in 0.5× TBE, 5% glycerol for 3 h at 150 V at 4°C. Gels were then soaked for 1 h in 40% MeOH, 10% acetic acid, 5% glycerol before being dried and analyzed using a PhosphorImager and ImageQuant software (Molecular Dynamics, Sunnyvale, CA).
RESULTS
Mutation spectrum analysis of mismatch repair-deficient strains.
The roles of Msh2, Msh3, and Msh6 in mismatch repair were initially inferred by determining the rates and spectra of reversion of specific frameshift mutations as well as forward mutation of the CAN1 gene in S. cerevisiae strains lacking different combinations of Msh2, Msh3, and Msh6 (20, 32, 48). However, these studies analyzed only small numbers of forward mutations, potentially limiting the conclusions of these early experiments. To reinvestigate the role of Msh3 in mismatch repair, we analyzed the spectrum of mutations that accumulate in an msh3 strain (Table 1) in a larger number of independent mutants than previously analyzed. The msh3 strain displayed an increase in the proportion of frameshift and previously undescribed homology-mediated duplication and deletion mutations (see Table S1 in the supplemental material) and a decrease in base substitution mutations compared to the wild-type strain (P = 0.0001, chi square “goodness of fit” test). The duplicated and deleted sequences were flanked by direct repeats that varied in length from 4 to 8 nucleotides; these duplication and deletion mutations were of the same type as previously seen in rad27 mutants (51), and many of the duplication and deletion mutations found in the msh3 mutant were identical to those found in a rad27 mutant (data not shown). In contrast, the mutation spectrum of the msh6 strain consisted almost exclusively of base substitutions as previously reported (32) and showed no homology-mediated duplication and deletion mutations. The mutation spectrum of the mlh3 strain was intermediate between that of the msh3 and wild-type strains. For purposes of reference, data for a mlh1 mutant strain, expected to be null for mismatch repair, are presented in Tables 1 through 3.
TABLE 1.
Mutation spectrum analysis of mismatch repair-deficient strains
| Genotype | Strain | No. (%) of indicated mutation types
|
nd | |||
|---|---|---|---|---|---|---|
| Base substitution | Frameshift (±1 or 2) | Homologous duplication/deletionb | Otherc | |||
| WTa | RDKY3686 | 68 (81) | 12 (14) | 2 (2) | 2 (2) | 84 |
| msh3 | RDKY4149 | 61 (46) | 45 (34) | 23 (18) | 2 (2) | 132 |
| msh6 | RDKY4151 | 90 (89) | 10 (10) | 0 | 1 (1) | 101 |
| mlh3 | RDKY5295 | 50 (69) | 17 (24) | 4 (5) | 1 (2) | 72 |
| mlh1 | RDKY4237 | 23 (41) | 32 (57) | 0 | 1 (1) | 56 |
WT, wild type.
Homologous duplication/deletions are defined as the addition or removal of a sequence flanked by blocks of 4 to 8 nucleotides of homology (51); also see Table S1 in the supplemental material.
Other mutation types include single isolates that contain more than one mutation.
n, number of mutations.
TABLE 3.
Classes of base substitutions found in mismatch repair-deficient strainsa
| Base pair mutation class | % Substitutions for indicated genotypeb
|
||||
|---|---|---|---|---|---|
| WT | msh6 | msh3 | mlh3 | mlh1 | |
| GC to CG | 12 (8) | 0 | 18 (11) | 10 (5) | 9 (2) |
| GC to AT | 38 (26) | 42 (38) | 27 (17) | 30 (15) | 39 (9) |
| GC to TA | 26 (18) | 38 (35) | 30 (18) | 32 (16) | 26 (6) |
| AT to TA | 4 (3) | 2 (2) | 13 (8) | 10 (5) | 13 (3) |
| AT to GC | 6 (4) | 10 (9) | 7 (4) | 8 (4) | 4 (1) |
| AT to CG | 13 (9) | 8 (7) | 5 (3) | 10 (5) | 9 (2) |
n was 68 for the wild type (WT), 91 for msh6, 61 for msh3, 50 for mlh3, and 23 for mlh1.
The observed number of mutations of each type is shown in parentheses. For the GC to CG base pair class, msh6 significantly differed from the wild type (the Fisher exact test, P = 6.8 × 10−5). For the AT bp to TA bp class, msh3 significantly differed from the wild type (the Fisher exact test, P = 0.04). Both of these analyses used wild-type mutation spectrum data combined with that of Tishkoff et al. (51). Two other comparisons were of borderline significance: the GC to CG base pair class in msh3 compared to that in the wild type and the GC bp to TA bp class in msh6 compared to that in the wild type. For these latter comparisons to reach statistical significance, three-times- and two-times-larger sample sizes would be required, respectively.
To further characterize the mutator phenotype of the mismatch repair-defective strains, the overall mutation rate of each strain was determined by fluctuation analysis and the rate of each class of mutation was then calculated (Table 2). The overall mutation rates of the msh3 and mlh3 strains (P = 0.0001 and 0.0117, respectively, two-tailed Mann-Whitney test) were significantly higher than that of the wild-type strain and were indistinguishable from each other (P = 0.3735). The rate of base substitutions in the msh3 strain was not different from that in the wild-type strain (P = 0.7263), whereas the msh3 strain had significantly increased rates of both frameshift mutations (P = 0.0001) and homology-mediated duplication and deletion mutations (P = 0.0001). Compared to the wild-type strain, the mlh3 mutant showed a slight increase in base substitutions (P = 0.0308) and an increase in both frameshift (P = 0.0002) and homology-mediated deletion and duplication mutations (P = 0.0002), albeit not as large as that seen in the msh3 mutant. This result is consistent with previous models showing that Mlh1-Mlh3 is a MutL homologue protein complex that acts in a subset of Msh2-Msh3-dependent mismatch repair events (14, 20). These mutation spectra were different from those of the msh6 and mlh1 strains, which showed an increase in only frameshift and base substitution mutations. It should be noted that an mlh1 mutant is completely mismatch repair defective, resulting in sufficiently high rates of base substitution and frameshift mutations that, given our sample size, homology-mediated duplication and deletion mutations would not be detected had they occurred at the same rate as those in msh3 or mlh3 mutants.
TABLE 2.
Mutation rate analysis of mismatch repair-deficient strains
| Genotype | Strain | Rate (fold increase) for indicated mutationa
|
|||
|---|---|---|---|---|---|
| Overall Canr | Base substitution | Frameshift (±1 or 2) | Homologous duplication/deletion | ||
| WTb | RDKY3686 | 7.5 [3.8-8.9] × 10−8 (1) | 6.1 × 10−8 (1) | 1.1 × 10−8 (1) | 1.5 × 10−9 (1) |
| msh3 | RDKY4149 | 1.3 [1.1-1.4] × 10−7 (1.6) | 5.8 × 10−8 (0.94) | 4.4 × 10−8 (4) | 2.3 × 10−8 (15) |
| msh6 | RDKY4151 | 6.7 [5.9-10.1] × 10−7 (8.9) | 6.0 × 10−7 (9) | 6.7 × 10−8 (6) | 0 |
| mlh3 | RDKY5295 | 1.1 [0.7-1.6] × 10−7 (1.4) | 7.6 × 10−8 (1.3) | 2.6 × 10−8 (2.5) | 5.5 × 10−9 (3.7) |
| mlh1 | RDKY4237 | 3.2 [2.3-4.1] × 10−6 (43) | 1.3 × 10−6 (21) | 1.8 × 10−6 (164) | 0 |
The increase relative to the value for the wild type is shown in parentheses. Ninety-five percent confidence intervals are shown in brackets. The overall mutation rate was used to calculate the rate of each class of mutations.
WT, wild type.
To further study mispair recognition by the Msh2-Msh6 and Msh2-Msh3 heterodimers, we analyzed the spectra and classes of base substitutions that accumulate in mismatch repair-defective strains in detail (Table 3 and see Fig. S1 through S4 in the supplemental material). We found that although the overall rate of base substitutions in the msh3 mutant was not significantly different from that of the wild-type strain, the spectrum of base substitutions observed was different and showed very little overlap between the positions of either single or recurrent mutations between the two spectra. Of the 68 and 61 base substitutions observed in wild-type and msh3 strains, respectively, only 8 of the mutations in the wild-type spectrum were in common with those of the msh3 spectrum and only 5 of the mutations in the msh3 spectrum were in common with those of the wild-type spectrum; in addition, none of the six hotspots from the msh3 spectrum overlapped with the wild-type mutation spectrum. A particularly interesting example was seen at the mutation hotspot CAN1 codon 399 that was mutated from CGT (Arg) to CCT (Pro) in msh3 two times and mutated to CAT (His) three times in the wild type but not mutated in the other strains analyzed. Similarly, there was also little or no overlap between the mlh3 or msh6 mutation spectra and the wild-type mutation spectrum (see Fig. S1 through S4 in the supplemental material). These data raise the possibility that msh3 and mlh3 mutations, like msh6 mutations, alter the repair of base-base mispairs, although there are limitations to this analysis because it is difficult to ensure that the different mutation spectra have reached saturation. We did not make direct comparisons using the mlh1 mutation spectrum because the mlh1 mutator phenotype is dominated by frameshift mutations, and hence the base substitution mutation sample size was small.
Although saturation of the mutation target did not occur, we could analyze the overlap of the mutation spectra for the various strains. By fitting the base substitution mutation spectrum data (specifically the number of unique and recurrent mutations) to Poisson distributions, we estimated that the wild-type strain contains 159 sites available for mutation and the msh3 strain contains 259 sites available for mutation; the larger number of mutable sites in the msh3 mutant is consistent with a defect in repair leading to base substitution mutations. Next we examined the relationship between the 159 wild-type and 259 msh3 mutation sites. We found that (i) it is unlikely that the wild-type and msh3 strains explicitly share the same mutation sites (P was 0.0215, assuming 259 sites available to both strains; P was 7.992 × 10−5, assuming 159 sites available to both strains) and (ii) it is unlikely that the deletion of MSH3 simply added additional mutation sites to a wild-type strain (P = 0.032). Thus, our data suggests that while the spectra of base substitution mutations in both the wild-type and msh3 strains overlap to some degree, they are likely to be different from each other.
To further analyze the spectrum of base substitution mutations seen in the msh3, msh6, and mlh3 mutants, the classes of observed base substitution mutations were compared (Table 3). The msh3, msh6, and mlh3 strains showed statistically different spectra of mutation classes compared to that of the wild type (P = 0.0001, 0.0003, and 0.0147, respectively, two-tailed chi square “goodness of fit” test). Compared to the wild type, the msh3 strain showed increases in base pair mutations of GC to CG and AT to TA and decreases in base pair mutations of GC to AT and AT to CG. Consistent with the known role of Msh6 in the repair of base-base mispairs, the msh6 strain showed distinct differences from the wild-type strain, including a striking absence of base substitutions of GC to CG base pairs. One hypothesis that could explain these data is that the Msh2-Msh3 complex is able to specifically recognize one of the base-base mispairs involved in these two classes of base substitutions seen to increase either a GG or a CC mispair involved in a base substitution of GC to CG base pairs and either an AA or a TT mispair involved in a base substitution of AT to TA base pairs.
Expression of Msh3.
We initially attempted to overproduce the Msh2-Msh3 complex by fusing the GAL10 promoter and an optimal Kozak consensus sequence to codon 1 of MSH3 and coexpress it with Msh2 but were unable to detect significant amounts of Msh3-FLAG by Western blotting. Similarly, a reconstructed version of a previously published Msh3 expression vector containing the GAL10 promoter upstream of codon 1 (18) only resulted in low-level Msh3 expression. This led us to consider whether Met codon 1 was in fact the correct translational start codon. By aligning the conserved PCNA binding motif present in various Saccharomyces Msh3 proteins, we found that the equivalent of S. cerevisiae Met codon 30 was conserved among all of the Msh3 proteins analyzed, whereas only S. cerevisiae and Saccharomyces paradoxus MSH3 contained 29 upstream codons, including Met codon 1, suggesting that the start site for translation might be at codon 30 (Fig. 1A). Plasmids were then constructed that contained the native MSH3 promoter and gene to analyze Ala substitution mutations at each position, M1A and M30A. The msh3-M1A allele was able to complement the mutator phenotype of the msh3 msh6 strain to the same level as did wild-type MSH3 (Fig. 1B), but neither the msh3-M30A allele nor the vector control was able to complement. These results indicate that Met codon 30 is the initiation codon for in vivo translation of the S. cerevisiae MSH3 gene. Consistent with this result, fusion of the GAL10 promoter and an optimal Kozak consensus sequence to codon 30 of MSH3 resulted in approximately 10-times-higher levels of Msh3 expression when Msh2 was coexpressed compared to that observed with the longer MSH3 allele (18).
FIG. 1.

Identification of MSH3 translation start site. (A) Alignment of predicted fungal Msh3 protein sequences. The protein sequence, according to the Saccharomyces genome database, of Msh3 from various organisms is aligned based on the conserved PCNA-binding motif. (B) MSH3 complementation of a msh3 msh6 strain. The msh3 alleles were expressed on a low-copy-number plasmid bearing a marker allowing growth on media lacking Leu. Plasmids were transformed into the msh3 msh6 strain, and isolates were patched onto plates lacking Leu and then replica plated onto plates lacking Lys and Thr as shown.
Biochemical characterization of the Msh2-Msh3 complex.
The S. cerevisiae Msh2-Msh3 complex was overexpressed and purified to near homogeneity as described in Materials and Methods. The mispair recognition properties of the Msh2-Msh3 heterodimer were investigated using electrophoretic mobility shift assays and compared to Msh2-Msh6 (Fig. 2). As a positive control for Msh2-Msh3 mispair binding, we used a 38-bp oligonucleotide duplex based on a previously described backbone and containing a 6-nucleotide insertion (18, 22). In addition, we created a series of 27 different control and mispair-containing 38-bp oligonucleotide duplexes derived from the sequence of the CAN1 gene (see Table S2 in the supplemental material). The mispaired DNA substrates generated were from sites found to be mutated in the msh3, msh6, or wild-type mutation spectra.
FIG. 2.

Binding of Msh2-Msh3 to mispaired DNA substrates in vitro. (A) Sixteen nanomolar of purified heterodimer (white bars, Msh2-Msh3; black bars, Msh2-Msh6) was incubated with 14 nM 32P-labeled substrates as described in Materials and Methods and analyzed by gel shift assay. The percentage of substrate shifted from the total signal per lane was calculated by densitometry. Error bars indicate one standard deviation. (B) The addition of ATP promotes dissociation of Msh2-Msh3 from mispaired DNA. Binding reactions were performed, and then 500 μM ATP was added on ice for 15 min (black bars). −, absence of; +, presence of. (C) Gel shift analysis of Msh2-Msh3 specifically bound to a CC mispair.
We initially examined binding of Msh2-Msh3 and Msh2-Msh6 to a GC base pair and GG, CC, AC and GT mispairs at CAN1 coding nucleotide 1196 (Fig. 2A), which is the position in codon 399 where a mutation of base pairs GC to CG was found two times in the msh3 mutation spectrum exclusively. As predicted, Msh2-Msh3 bound most robustly to the +6 insertion mispair and high level binding to the CC mispair was also found. Msh2-Msh3 also bound to the GG, AC, and GT mispairs at a level which was weak but above the GC binding background. The Msh2-Msh6 complex bound the GG, AC, GT, and +6 insertion substrates weakly and did not appear to significantly bind the CC mispair. Furthermore, the addition of ATP caused a decrease in binding of Msh2-Msh3 to CC and +6 insertion mispairs, consistent with rapid release via sliding (Fig. 2B and C) (36, 54). This mispair binding specificity is consistent with the formation of a CC mispair at nucleotide 1196 of CAN1 that subsequently escapes repair in an msh3 mutant.
To extend the above results, binding of Msh2-Msh3 to a greater diversity of CAN1 derived base-base mispairs was analyzed (Table 4); three were at sites found to be mutated in the msh3 spectrum, and two each were at sites found to be mutated in the msh6 or wild-type spectra. The substrates analyzed included sites that were and were not mutated in msh3 mutants and also included mispairs that were and were not predicted to underlie the classes of mutations that were preferentially found in the msh3 mutation spectrum. In these experiments, very strong binding (greater than fourfold over the GC control) was observed for four mispairs (CC 1196, CT 1196, AA 1193, and AC 1193) and binding that was at least twofold above the control binding was observed for an additional seven mispairs (CC 413, AA 1196, AC 1196, GG 1196, AA 1628, AC 807, and AG 1196). Weak or no binding was observed for an additional 14 mispairs. ATP promoted dissociation from the mispair in each case (data not shown). Of the 11 mispairs showing the strongest binding, 6 were of the classes suggested by mutation spectra analysis to undergo Msh2-Msh3-dependent repair, GG or CC and AA or TT. Of the seven total sites analyzed, Msh2-Msh3 did not show high-level mispair binding at the following two sites: 955, found to be mutated in an msh6 mutant and 314 found to be mutated in an msh3 mutant. However, Msh2-Msh3 did show binding at five other sites: 1193 and 1196, found to be mutated in an msh3 mutant; 413 and 1628, found to be mutated in the wild-type strain; and 807, found to be mutated in an msh6 mutant.
TABLE 4.
Binding of Msh2-Msh3 to mispairs in different sequence contextsa
| Origin | Mispair | Position in CAN1 | % Bindingb | Scorec | Expectedd |
|---|---|---|---|---|---|
| msh3 | GC | 1196 | 7.1 | − | No |
| GG | 1196 | 18.5 | + | Yes | |
| CC | 1196 | 45.4 | ++ | Yes | |
| AA | 1196 | 20.2 | + | Yes | |
| TT | 1196 | 9.5 | − | No | |
| GT | 1196 | 13.5 | − | No | |
| AC | 1196 | 18.6 | + | No | |
| CT | 1196 | 37 | ++ | No | |
| AG | 1196 | 25.5 | + | No | |
| +6 | NA | 75.7 | ++ | NA | |
| msh3 | GC | 1193 | 8.4 | − | No |
| GG | 1193 | 8.7 | − | No | |
| AA | 1193 | 33.3 | ++ | Yes | |
| TT | 1193 | 13.4 | − | No | |
| GT | 1193 | 11 | − | No | |
| AC | 1193 | 47.9 | ++ | No | |
| CT | 1193 | 9.6 | − | No | |
| AG | 1193 | 12 | − | No | |
| +6 | NA | 85.4 | ++ | NA | |
| WT | GG | 413 | 8.4 | − | No |
| CC | 413 | 17.3 | + | Yes | |
| msh6 | AA | 955 | 9.9 | − | No |
| TT | 955 | 8.7 | − | No | |
| WT | AA | 1628 | 16.2 | + | Yes |
| TT | 1628 | 12.6 | − | No | |
| msh3 | GT | 314 | 11.4 | − | No |
| AC | 314 | 9.7 | − | No | |
| msh6 | GT | 807 | 11.6 | − | No |
| AC | 807 | 22.3 | + | No | |
| +6 | NA | 44 | ++ | NA |
NA, not applicable; WT, wild type.
The percentage of substrate shifted from the total signal per lane was calculated by densitometry and is shown as “% Binding” for the base pairs/mispairs at the indicated CAN1 positions.
The three degrees of binding are noted under “Score.” ++, increased binding of more than fourfold compared to GC; +, increased binding of two- to fourfold compared to GC; −, increased binding of less than twofold compared to GC.
“Expected” indicates whether a mispair was predicted to be recognized based on the mutations seen in the CAN1 mutation spectra.
DISCUSSION
In the present study, we have used a combined genetic and biochemical approach to investigate the role of the Msh2-Msh3 complex in mismatch repair. Mutation spectrum analysis showed that msh3 mutants, while having low overall rates of base substitution mutations nonetheless accumulated a spectrum of such mutations that appeared distinct from those that accumulated either in msh6 mutants or in the wild-type strain, suggesting a role for the Msh2-Msh3 complex in the repair of some base-base mispairs. Mutation spectrum analysis similarly suggested that the Mlh1-Mlh3 heterodimer might also function in the repair of base-base mispairs. In addition, msh3 and mlh3, but not msh6 or mlh1, mutants accumulated homology-mediated duplication and deletion mutations of the type only previously seen in rad27 mutants (51). The parallels between the mutation spectra of mlh3 and msh3 mutants are consistent with previous observations showing that the Mlh1-Mlh3 complex functions in conjunction with the Msh2-Msh3 complex (14). Mispair binding analysis with DNA substrates derived from CAN1 sequences found to be mutated in vivo demonstrated that Msh2-Msh3 had robust binding to specific base-base mispairs that was reduced upon the addition of ATP. Overall, the results presented here are consistent with an unexpected role of the Msh2-Msh3 complex and the Mlh1-Mlh3 complex in the repair of base-base mispairs as well as in the suppression of homology-mediated duplication and deletion mutations. This result could explain how, in one genetic study, Msh3 could partially suppress homeologous recombination between substrates containing four single-base differences (but note that the effects of individual single base differences were not examined in the study) (41). Additionally, another study examining the repair of defined mismatches on transformed plasmids detected possible repair defects of some individual base-base mismatches caused by an msh3 mutant in one strain background but not in another strain background (30). The observation presented here, that Msh2-Msh3 is able to bind to specific mispairs in vitro that were initially identified as potential mutation intermediates in vivo, demonstrates a specific mechanism that explains some of these earlier studies.
Previous studies have analyzed the effect of different mismatch repair defects in a number of mutator assays sometimes combined with sequencing of mutation spectra to infer the role of different proteins in mismatch repair. As noted above, these types of studies have a number of limitations. In the current study, we sequenced larger numbers of independent mutations in a large, relatively unbiased forward mutation target than the numbers in prior studies and found that a msh3 strain appeared to accumulate a spectrum of base substitutions that differed from that of wild-type or msh6 strains; differences in the spectrum of frameshift mutations were also observed, although we did not further analyze these mutations as it is well accepted that Msh2-Msh3 and Msh2-Msh6 both function in the repair of insertion/deletion mispairs (20, 26, 37-39). It is probably difficult to completely saturate the CAN1 mutation spectrum in the strains tested; nonetheless, there was very little overlap in the mutation spectra and there were significant differences between the overall mutation spectra observed as well as in the classes of base substitutions seen. These data support the hypothesis that the Msh2-Msh3 complex functions in the repair of base-base mispairs. However, it is difficult to determine how efficiently the Msh2-Msh3 complex can act in such repair events in vivo because competition by Msh2-Msh6-dependent repair clearly obscures the msh3 mutator phenotype. In mammalian cells, the Msh6 mismatch repair pathway dominates mismatch repair because the Msh2-Msh6 complex is found at 6-fold-higher to 10-fold-higher levels than the Msh2-Msh3 complex is (13, 16). The ratio of the two complexes is not known in S. cerevisiae due to the lack of good Msh3 antibodies. However, the observations that msh3 mutants have detectable mutator phenotypes (32, 48) and that a single-copy MSH3 plasmid can suppress dominant msh6 mutations (11) suggest that the Msh3 pathway plays a significant role in mismatch repair in wild-type S. cerevisiae.
By performing mispair binding studies with oligonucleotide duplexes based on the sequence of the CAN1 gene that contained mispairs that were or were not found at sites mutated in the msh3 mutation spectrum and that were or were not the mispairs predicted to underlie the mutation seen, we were able to demonstrate that the Msh2-Msh3 complex could robustly bind specific base-base mispairs, including ones that were not well recognized by Msh2-Msh6; base-base mispairs that were not bound by Msh2-Msh3 were also found. The Msh2-Msh3 base-base mispair binding was sensitive to ATP addition, upon which Msh2-Msh3 quickly dissociated from the DNA substrate, as predicted for bona-fide mispair binding (36, 54). Significant binding of base-base mispairs by Msh2-Msh3 has not previously been observed; our use of mispairs based on in vivo mutation sites is likely what made it possible to observe binding of base-base mispairs by Msh2-Msh3. These results support the hypothesis that Msh2-Msh3 can function in the repair of base-base mispairs and suggest that such repair augments Msh2-Msh6-dependent repair of base-base mispairs. A considerable amount of structural information is available on how MutS recognizes mispairs, and Msh6 shares the key MutS mispair recognition structural determinants, including the Phe residue that stacks on the mispaired base and other residues that contact the DNA backbone (12, 28, 40). Msh3 lacks this key Phe residue but retains six residues that contact the DNA backbone in MutS and are present in Msh6 as well (29). However, four of these residues have been mutated in Msh3 and only one was found to be important for Msh3-dependent mismatch repair (12, 29). Given our analysis indicating that the Msh2-Msh6 and Msh2-Msh3 complexes can recognize the same classes of mispairs (i.e., base-base and insertion/deletion mispairs), the lack of the mispair-contacting Phe residue and the lack of a requirement for many of the other predicted critical DNA-contacting residues suggest that Msh3 may utilize a distinct structural mechanism for mispair recognition.
Additionally, we found that msh3 and mlh3 mutants exhibit a significant increase in the rate of accumulation of homology-mediated duplication and deletion mutations. This type of mutation has been suggested to arise in rad27 mutants due to errors in processing the ends of Okazaki fragments, leading to double-strand breaks and aberrant repair of the double-strand breaks (51) possibly by single-strand annealing recombination (SSA). However, the deletion and duplication mutations seen in an msh3 mutant (and probably in an mlh3) probably cannot be mediated by SSA because Msh3 is required for SSA and, in particular, for those events that occur by SSA between short homologous DNA sequences like those implicated in the deletion and duplication mutations seen here (50). It seems unlikely that loss of Msh3 and Mlh3 causes the same type of defects as does loss of Rad27 since an msh3 mutation did not cause an increase in the rate of gross chromosomal rearrangements as seen for rad27 mutants (data not shown) (8). It is also unlikely that all errors arising in the absence of Rad27, which result in duplication and deletion events, are normally repaired by mismatch repair since mismatch repair defects do not result in a synergistic increase in mutation rate when combined with a rad27 mutation (unpublished results) (51). More likely possibilities are either that Rad27, Msh2-Msh3, and Mlh1-Mlh3 function together in a subclass of repair events or that only a proportion of the errors induced by the absence of Rad27 are repaired by Msh2-Msh3- and Mlh1-Mlh3-dependent mismatch repair. For example, Msh2-Msh3 and possibly Rad27 could interact with aberrant branched structures that form at stalled or damaged replication forks; additionally, Mlh1-Mlh3 could act in subsequent repair events (50). Additional studies will be required to elucidate the exact mechanisms involved.
In summary, the results presented here indicate a need for modification of the current models of mismatch repair, such that in the early step of mismatch repair, both Msh2-Msh6 and Msh2-Msh3 recognize base-base and insertion/deletion mispairs; this redundancy likely increases the overall efficiency of mismatch repair. In addition, our results have implicated the Msh2-Msh3 and Mlh1-Mlh3 complexes in the suppression of homology-mediated duplication and deletion mutations like those that occur in rad27 mutants, thus expanding current views of the role of mismatch repair in suppressing mutations.
Supplementary Material
Acknowledgments
We acknowledge Dan Mazur for supplying purified Msh2-Msh6 heterodimer for biochemical studies, Christopher Putnam for help with mutation analysis, and Jason Chan for help with statistical analysis. We also thank Scarlet Shell for supplying the +6 DNA substrate and the msh6 mutation spectra and fluctuation rates. Finally, we thank Scarlet Shell and Marc Mendillo for critical reading of the manuscript.
This work was funded by NIH grant GM50006.
We declare that we have no competing financial interests.
Footnotes
Published ahead of print on 16 July 2007.
Supplemental material for this article may be found at http://mcb.asm.org/.
REFERENCES
- 1.Acharya, S., P. L. Foster, P. Brooks, and R. Fishel. 2003. The coordinated functions of the E. coli MutS and MutL proteins in mismatch repair. Mol. Cell 12:233-246. [DOI] [PubMed] [Google Scholar]
- 2.Alani, E., R. A. Reenan, and R. D. Kolodner. 1994. Interaction between mismatch repair and genetic recombination in Saccharomyces cerevisiae. Genetics 137:19-39. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Allen, D. J., A. Makhov, M. Grilley, J. Taylor, R. Thresher, P. Modrich, and J. D. Griffith. 1997. MutS mediates heteroduplex loop formation by a translocation mechanism. EMBO J. 16:4467-4476. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Amin, N. S., M. N. Nguyen, S. Oh, and R. D. Kolodner. 2001. exo1-dependent mutator mutations: model system for studying functional interactions in mismatch repair. Mol. Cell. Biol. 21:5142-5155. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Au, K. G., K. Welsh, and P. Modrich. 1992. Initiation of methyl-directed mismatch repair. J. Biol. Chem. 267:12142-12148. [PubMed] [Google Scholar]
- 6.Burdett, V., C. Baitinger, M. Viswanathan, S. T. Lovett, and P. Modrich. 2001. In vivo requirement for RecJ, ExoVII, ExoI, and ExoX in methyl-directed mismatch repair. Proc. Natl. Acad. Sci. USA 98:6765-6770. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Cannavo, E., G. Marra, J. Sabates-Bellver, M. Menigatti, S. M. Lipkin, F. Fischer, P. Cejka, and J. Jiricny. 2005. Expression of the MutL homologue hMLH3 in human cells and its role in DNA mismatch repair. Cancer Res. 65:10759-10766. [DOI] [PubMed] [Google Scholar]
- 8.Chen, C., and R. D. Kolodner. 1999. Gross chromosomal rearrangements in Saccharomyces cerevisiae replication and recombination defective mutants. Nat. Genet. 23:81-85. [DOI] [PubMed] [Google Scholar]
- 9.Chen, P. C., S. Dudley, W. Hagen, D. Dizon, L. Paxton, D. Reichow, S. R. Yoon, K. Yang, N. Arnheim, R. M. Liskay, and S. M. Lipkin. 2005. Contributions by MutL homologues Mlh3 and Pms2 to DNA mismatch repair and tumor suppression in the mouse. Cancer Res. 65:8662-8670. [DOI] [PubMed] [Google Scholar]
- 10.Constantin, N., L. Dzantiev, F. A. Kadyrov, and P. Modrich. 2005. Human mismatch repair: reconstitution of a nick-directed bidirectional reaction. J. Biol. Chem. 280:39752-39761. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Das Gupta, R., and R. D. Kolodner. 2000. Novel dominant mutations in Saccharomyces cerevisiae MSH6. Nat. Genet. 24:53-56. [DOI] [PubMed] [Google Scholar]
- 12.Drotschmann, K., W. Yang, F. E. Brownewell, E. T. Kool, and T. A. Kunkel. 2001. Asymmetric recognition of DNA local distortion. Structure-based functional studies of eukaryotic Msh2-Msh6. J. Biol. Chem. 276:46225-46229. [DOI] [PubMed] [Google Scholar]
- 13.Drummond, J. T., J. Genschel, E. Wolf, and P. Modrich. 1997. DHFR/MSH3 amplification in methotrexate-resistant cells alters the hMutSalpha/hMutSbeta ratio and reduces the efficiency of base-base mismatch repair. Proc. Natl. Acad. Sci. USA 94:10144-10149. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Flores-Rozas, H., and R. D. Kolodner. 1998. The Saccharomyces cerevisiae MLH3 gene functions in MSH3-dependent suppression of frameshift mutations. Proc. Natl. Acad. Sci. USA 95:12404-12409. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Galio, L., C. Bouquet, and P. Brooks. 1999. ATP hydrolysis-dependent formation of a dynamic ternary nucleoprotein complex with MutS and MutL. Nucleic Acids Res. 27:2325-2331. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Genschel, J., S. J. Littman, J. T. Drummond, and P. Modrich. 1998. Isolation of MutSbeta from human cells and comparison of the mismatch repair specificities of MutSbeta and MutSalpha. J. Biol. Chem. 273:19895-19901. [DOI] [PubMed] [Google Scholar]
- 17.Grilley, M., K. M. Welsh, S. S. Su, and P. Modrich. 1989. Isolation and characterization of the Escherichia coli mutL gene product. J. Biol. Chem. 264:1000-1004. [PubMed] [Google Scholar]
- 18.Habraken, Y., P. Sung, L. Prakash, and S. Prakash. 1996. Binding of insertion/deletion DNA mismatches by the heterodimer of yeast mismatch repair proteins MSH2 and MSH3. Curr. Biol. 6:1185-1187. [DOI] [PubMed] [Google Scholar]
- 19.Hall, M. C., and S. W. Matson. 1999. The Escherichia coli MutL protein physically interacts with MutH and stimulates the MutH-associated endonuclease activity. J. Biol. Chem. 274:1306-1312. [DOI] [PubMed] [Google Scholar]
- 20.Harfe, B. D., and S. Jinks-Robertson. 2000. DNA mismatch repair and genetic instability. Annu. Rev. Genet. 34:359-399. [DOI] [PubMed] [Google Scholar]
- 21.Harfe, B. D., B. K. Minesinger, and S. Jinks-Robertson. 2000. Discrete in vivo roles for the MutL homologs Mlh2p and Mlh3p in the removal of frameshift intermediates in budding yeast. Curr. Biol. 10:145-148. [DOI] [PubMed] [Google Scholar]
- 22.Hess, M. T., R. D. Gupta, and R. D. Kolodner. 2002. Dominant Saccharomyces cerevisiae msh6 mutations cause increased mispair binding and decreased dissociation from mispairs by Msh2-Msh6 in the presence of ATP. J. Biol. Chem. 277:25545-25553. [DOI] [PubMed] [Google Scholar]
- 23.Heyer, J., K. Yang, M. Lipkin, W. Edelmann, and R. Kucherlapati. 1999. Mouse models for colorectal cancer. Oncogene 18:5325-5333. [DOI] [PubMed] [Google Scholar]
- 24.Joshi, A., S. Sen, and B. J. Rao. 2000. ATP-hydrolysis-dependent conformational switch modulates the stability of MutS-mismatch complexes. Nucleic Acids Res. 28:853-861. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Junop, M. S., G. Obmolova, K. Rausch, P. Hsieh, and W. Yang. 2001. Composite active site of an ABC ATPase: MutS uses ATP to verify mismatch recognition and authorize DNA repair. Mol. Cell 7:1-12. [DOI] [PubMed] [Google Scholar]
- 26.Kolodner, R. D., and G. T. Marsischky. 1999. Eukaryotic DNA mismatch repair. Curr. Opin. Genet. Dev. 9:89-96. [DOI] [PubMed] [Google Scholar]
- 27.Lahue, R. S., K. G. Au, and P. Modrich. 1989. DNA mismatch correction in a defined system. Science 245:160-164. [DOI] [PubMed] [Google Scholar]
- 28.Lamers, M. H., A. Perrakis, J. H. Enzlin, H. H. Winterwerp, N. de Wind, and T. K. Sixma. 2000. The crystal structure of DNA mismatch repair protein MutS binding to a G × T mismatch. Nature 407:711-717. [DOI] [PubMed] [Google Scholar]
- 29.Lee, S. D., J. A. Surtees, and E. Alani. 2007. Saccharomyces cerevisiae MSH2-MSH3 and MSH2-MSH6 complexes display distinct requirements for DNA binding domain I in mismatch recognition. J. Mol. Biol. 366:53-66. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Lühr, B., J. Scheller, P. Meyer, and W. Kramer. 1998. Analysis of in vivo correction of defined mismatches in the DNA mismatch repair mutants msh2, msh3 and msh6 of Saccharomyces cerevisiae. Mol. Gen. Genet. 257:362-367. [DOI] [PubMed] [Google Scholar]
- 31.Lynch, H. T., and A. de la Chapelle. 2003. Hereditary colorectal cancer. N. Engl. J. Med. 348:919-932. [DOI] [PubMed] [Google Scholar]
- 32.Marsischky, G. T., N. Filosi, M. F. Kane, and R. Kolodner. 1996. Redundancy of Saccharomyces cerevisiae MSH3 and MSH6 in MSH2-dependent mismatch repair. Genes Dev. 10:407-420. [DOI] [PubMed] [Google Scholar]
- 33.Marsischky, G. T., and R. D. Kolodner. 1999. Biochemical characterization of the interaction between the Saccharomyces cerevisiae MSH2-MSH6 complex and mispaired bases in DNA. J. Biol. Chem. 274:26668-26682. [DOI] [PubMed] [Google Scholar]
- 34.Matson, S. W., and A. B. Robertson. 2006. The UvrD helicase and its modulation by the mismatch repair protein MutL. Nucleic Acids Res. 34:4089-4097. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Mazur, D. J., M. L. Mendillo, and R. D. Kolodner. 2006. Inhibition of Msh6 ATPase activity by mispaired DNA induces a Msh2(ATP)-Msh6(ATP) state capable of hydrolysis-independent movement along DNA. Mol. Cell 22:39-49. [DOI] [PubMed] [Google Scholar]
- 36.Mendillo, M. L., D. J. Mazur, and R. D. Kolodner. 2005. Analysis of the interaction between the Saccharomyces cerevisiae MSH2-MSH6 and MLH1-PMS1 complexes with DNA using a reversible DNA end-blocking system. J. Biol. Chem. 280:22245-22257. [DOI] [PubMed] [Google Scholar]
- 37.Modrich, P. 1991. Mechanisms and biological effects of mismatch repair. Annu. Rev. Genet. 25:229-253. [DOI] [PubMed] [Google Scholar]
- 38.Modrich, P. 2006. Mechanisms in eukaryotic mismatch repair. J. Biol. Chem. 281:30305-30309. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Modrich, P., and R. Lahue. 1996. Mismatch repair in replication fidelity, genetic recombination, and cancer biology. Annu. Rev. Biochem. 65:101-133. [DOI] [PubMed] [Google Scholar]
- 40.Natrajan, G., M. H. Lamers, J. H. Enzlin, H. H. Winterwerp, A. Perrakis, and T. K. Sixma. 2003. Structures of Escherichia coli DNA mismatch repair enzyme MutS in complex with different mismatches: a common recognition mode for diverse substrates. Nucleic Acids Res. 31:4814-4821. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Nicholson, A., M. Hendrix, S. Jinks-Robertson, and G. F. Crouse. 2000. Regulation of mitotic homeologous recombination in yeast. Functions of mismatch repair and nucleotide excision repair genes. Genetics 154:133-146. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Peltomäki, P. 2003. Role of DNA mismatch repair defects in the pathogenesis of human cancer. J. Clin. Oncol. 21:1174-1179. [DOI] [PubMed] [Google Scholar]
- 43.Prolla, T. A., Q. Pang, E. Alani, R. D. Kolodner, and R. M. Liskay. 1994. MLH1, PMS1, and MSH2 interactions during the initiation of DNA mismatch repair in yeast. Science 265:1091-1093. [DOI] [PubMed] [Google Scholar]
- 44.Räschle, M., G. Marra, M. Nystrom-Lahti, P. Schar, and J. Jiricny. 1999. Identification of hMutLbeta, a heterodimer of hMLH1 and hPMS1. J. Biol. Chem. 274:32368-32375. [DOI] [PubMed] [Google Scholar]
- 45.Reenan, R. A., and R. D. Kolodner. 1992. Characterization of insertion mutations in the Saccharomyces cerevisiae MSH1 and MSH2 genes: evidence for separate mitochondrial and nuclear functions. Genetics 132:975-985. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Schofield, M. J., S. Nayak, T. H. Scott, C. Du, and P. Hsieh. 2001. Interaction of Escherichia coli MutS and MutL at a DNA mismatch. J. Biol. Chem. 276:28291-28299. [DOI] [PubMed] [Google Scholar]
- 47.Selmane, T., M. J. Schofield, S. Nayak, C. Du, and P. Hsieh. 2003. Formation of a DNA mismatch repair complex mediated by ATP. J. Mol. Biol. 334:949-965. [DOI] [PubMed] [Google Scholar]
- 48.Sia, E. A., M. Dominska, L. Stefanovic, and T. D. Petes. 2001. Isolation and characterization of point mutations in mismatch repair genes that destabilize microsatellites in yeast. Mol. Cell. Biol. 21:8157-8167. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Su, S. S., R. S. Lahue, K. G. Au, and P. Modrich. 1988. Mispair specificity of methyl-directed DNA mismatch correction in vitro. J. Biol. Chem. 263:6829-6835. [PubMed] [Google Scholar]
- 50.Surtees, J. A., and E. Alani. 2006. Mismatch repair factor MSH2-MSH3 binds and alters the conformation of branched DNA structures predicted to form during genetic recombination. J. Mol. Biol. 360:523-536. [DOI] [PubMed] [Google Scholar]
- 51.Tishkoff, D. X., N. Filosi, G. M. Gaida, and R. D. Kolodner. 1997. A novel mutation avoidance mechanism dependent on S. cerevisiae RAD27 is distinct from DNA mismatch repair. Cell 88:253-263. [DOI] [PubMed] [Google Scholar]
- 52.Wang, T. F., N. Kleckner, and N. Hunter. 1999. Functional specificity of MutL homologs in yeast: evidence for three Mlh1-based heterocomplexes with distinct roles during meiosis in recombination and mismatch correction. Proc. Natl. Acad. Sci. USA 96:13914-13919. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Welsh, K. M., A. L. Lu, S. Clark, and P. Modrich. 1987. Isolation and characterization of the Escherichia coli mutH gene product. J. Biol. Chem. 262:15624-15629. [PubMed] [Google Scholar]
- 54.Wilson, T., S. Guerrette, and R. Fishel. 1999. Dissociation of mismatch recognition and ATPase activity by hMSH2-hMSH3. J. Biol. Chem. 274:21659-21664. [DOI] [PubMed] [Google Scholar]
- 55.Zhang, Y., F. Yuan, S. R. Presnell, K. Tian, Y. Gao, A. E. Tomkinson, L. Gu, and G. M. Li. 2005. Reconstitution of 5′-directed human mismatch repair in a purified system. Cell 122:693-705. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.