

Abstract
The pleckstrin homology domain-interacting protein (PHIP) was originally identified as a 902-amino-acid (aa) protein that regulates insulin receptor-stimulated GLUT4 translocation in skeletal-muscle cells. Immunoblotting and immunohistological analyses of pancreatic β-cells reveal prominent expression of a 206-kDa PHIP isoform restricted to the nucleus. Herein, we report the cloning of this larger, 1,821-aa isoform of PHIP (PHIP1), which represents a novel WD40 repeat-containing protein. We demonstrate that PHIP1 overexpression stimulates insulin-like growth factor 1-dependent and -independent proliferation of β-cells, an event which correlates with transcriptional upregulation of the cyclin D2 promoter and the accumulation of cyclin D2 protein. RNA interference knockdown of PHIP1 in INS-1 cells abrogates insulin receptor substrate 2 (IRS2)-mediated DNA synthesis, providing for a specific role for PHIP1 in the enhancement of IRS2-dependent signaling responses leading to β-cell growth. Finally, we provide evidence that PHIP1 overexpression blocks free fatty acid-induced apoptosis in INS-1 cells, which is accompanied by marked activation of phosphoprotein kinase B (PKB)/AKT and the concomitant inhibition of caspase-9 and caspase-3 cleavage. Our finding that the restorative effect of PHIP1 on β-cell lipotoxicity can be attenuated by the overexpression of dominant-negative PKB suggests a key role for PKB in PHIP1-mediated cytoprotection. Taken together, these findings provide strong support for PHIP1 as a novel positive regulator of β-cell function. We suggest that PHIP1 may be involved in the induction of long-term gene expression programs to promote β-cell mitogenesis and survival.
Type 1 and type 2 are the two principal forms of diabetes and account for more than 99% of all known forms of this metabolic disorder (29). Type 1 diabetes results from autoimmune destruction of the pancreatic β cells and manifests itself when less than 10 to 20% of functional β cells remain in the islets (13). Type 2 diabetes is characterized by the development of β-cell dysfunction and the progressive reduction of β-cell mass via reduced proliferation and increased apoptosis, although, unlike type 1 diabetes, it is the result of peripheral insulin resistance and chronic exposure to high levels of glucose and long-chain free fatty acids (FFA), known as glucotoxicity and lipotoxicity, respectively (28, 31).
Biochemical and genetic evidence strongly implicate insulin and insulin-like growth factor 1 (IGF-1) as critical factors in β-cell function (21). The activated receptors engage and phosphorylate various cellular proteins, including insulin receptor substrate (IRS) protein family members. Recent studies, both in vitro and in transgenic animals, have revealed an important role for IRS2 in the control of β-cell growth and survival (15, 17, 25). There are two signaling cascades downstream of IRS2 in β cells that have been characterized extensively: the phosphatidylinositol 3′-kinase (PI3-kinase)/protein kinase B (PKB) pathway and the Ras/Raf/MEK/ERK pathway, with the former being the major regulator of β-cell survival. PKB is also a central player in pancreatic β-cell growth downstream of IRS2. Several independent studies have demonstrated that chronic FFA exposure inhibits β-cell mitogenesis and promotes β-cell apoptosis via inhibition of PKB activity (6). Importantly, FFA-induced apoptosis can be prevented by the expression of constitutively active PKB or the overexpression of IRS2 (34).
Efficient targeting of IRS proteins to the plasma membrane is mediated via their pleckstrin homology (PH) domain (15). The PH domain is a conserved 100- to 120-amino-acid (aa) domain, first identified in pleckstrin, with a demonstrated affinity for lipophilic molecules. This domain is found in a variety of proteins, including protein kinases, GTPases and their regulators, and phospholipases, as well as adaptor proteins (16). PH domain-containing proteins are further classified into four categories based on their affinities for different phosphoinositides. The majority of these proteins have an affinity for either PI(3,4,5)P3, PI(4,5)P2, or PI(3,4)P2, with a few proteins showing no clear specificity (16).
A PH domain-interacting protein (PHIP) was first identified in our laboratory as a 902-aa protein that interacts specifically with the PH domain of IRS1 and is shown to bind to IRS2 (10). In addition, we have shown that a dominant-negative N-terminal truncation mutant of PHIP inhibits transcriptional and proliferative responses downstream of the insulin receptor and attenuates the signal transduction pathway linking the insulin receptor to GLUT4 translocation in muscle cells (9). Given the apparent role of PHIP in IRS-mediated signaling pathways and the importance of IRS proteins in the proliferation and survival of insulin-producing β cells (33), we decided to investigate the functional role of PHIP in β-cells. Immunoblotting and immunohistological analyses of pancreatic β cells revealed a 206-kDa isoform of PHIP (PHIP1), which was localized exclusively in the nucleus.
In the present study, we describe the cloning and characterization of this novel WD40 repeat-containing isoform of PHIP. We demonstrate that PHIP1 overexpression promotes the growth of INS-1 β cells, an event which correlates with the transactivation of the cyclin D2 promoter and an increase in the accumulation of cyclin D2 protein. Importantly, we show that small interfering RNA (siRNA) knockdown of PHIP1 markedly inhibits IRS2-mediated DNA synthesis and cyclin D2 protein accumulation independently of phospho-PKB activation. Finally, PHIP1 overexpression blocks FFA-induced apoptosis in INS-1 cells, which is mediated via the activation of phospho-PKB and the inhibition of caspase-9 and caspase-3 activity. Collectively, these findings provide strong support for PHIP1 as a novel positive regulator of β-cell growth and survival.
MATERIALS AND METHODS
Materials.
Oleic acid (OA), fatty acid-free bovine serum albumin (BSA), propidium iodide, and collagenase type V were purchased from Sigma Chemical Co. (St. Louis, MO). Cell culture media, glutamine, and antibiotics were from Invitrogen/GIBCO (Grand Island, NY). Antibodies to total and phosphorylated (Ser473 and Thr308) PKB as well as cleaved caspase-9/caspase-3 were purchased from Cell Signaling Technology (Beverly, MA). The IRS2 antibody was from Upstate Biotechnology, Inc. (Lake Placid, NY), and the Bax antibody was from Upstate (Charlottesville, VA). The insulin, glucagon, and actin antibodies were from Sigma Chemical Co. (St. Louis, MO). The human recombinant IGF-1 was purchased from Calbiochem (San Diego, CA). The reagents for sodium dodecyl sulfate-polyacrylamide gel electrophoresis were from Bio-Rad Laboratories, Inc. (Hercules, CA).
Cell cultures.
The glucose-sensitive pancreatic β-cell line INS-1 (a kind gift from H. Y. Gaisano, University of Toronto) was maintained in RPMI 1640 medium (11 mM glucose) containing 10% (vol/vol) fetal bovine serum (FBS) (Wisent, Canada), 50 μM β-mercaptoethanol, 10 mM HEPES, 2 mM glutamine, 1 mM sodium pyruvate, 100 units/ml penicillin, 100 μg/ml streptomycin and incubated at 37°C, 5% CO2. MIN6 cells (passages, 26 to 30) were kindly provided by R. N. Kulkarni at Joshlin Diabetes Center and were grown in Dulbecco's modified Eagle's medium (DMEM) (25 mM glucose) containing 15% FBS, 100 units/ml penicillin, 100 μg/ml streptomycin and incubated at 5% CO2, 37°C.
Pancreatic islets.
Islets were isolated from male C57BL/6 mice. Briefly, the mice were anesthetized by using Avertin and the pancreatic duct was perfused with Hanks balanced salt solution (HBSS) (molecular composition in mM: 1.23 CaCl2, 0.8 MgSO4, 5 KCl, 0.44 KH2PO4, 4.1 NaHCO3, 0.138 NaCl, 0.3 Na2HPO4, 5 d-glucose) containing type V collagenase (0.7 mg/ml) and HEPES (2.5%). The pancreas was then removed and digested at 37°C. Islets were subsequently purified on a Ficoll gradient, transferred to fresh media, and maintained at 37°C. The islets were cultured in RPMI 1640 supplemented with 10% FBS, 11 mM glucose, 100 units/ml penicillin, 100 μg/ml streptomycin, 10 mM HEPES, pH 7.4. Following collagenase digestion, the islets were allowed to recover for at least 6 h prior to subsequent experiments.
Dispersed islet cells.
Isolated islets were washed twice in Ca2+-free phosphate-buffered solution (2 mM EGTA, 3 mM glucose, 100 units/ml penicillin, and 100 μg/ml streptomycin). Subsequently, the islets were centrifuged and incubated with dispase II (Roche Diagnostics) for 10 min at 37°C for digestion, followed by the addition of RPMI 1640 medium containing 11.1 mM glucose, 10% FBS, 100 units/ml penicillin, 100 μg/ml streptomycin, and 10 mM HEPES, pH 7.4. The suspension was centrifuged in order to remove the dispase II, and the cell pellet was resuspended in the same medium. The cells were plated on plastic coverslips coated with poly(l-lysine) and maintained in culture overnight prior to fixation in 4% paraformaldehyde for immunostaining.
Cloning of PHIP1 cDNA.
Full-length human PHIP1 (hPHIP1) cDNA was amplified from a human MCF7 cDNA library using high-fidelity Taq DNA polymerase (Invitrogen, Canada). The amplicon (size, 5.53 kb), spanning the entire 1,821-aa open reading frame (ORF), was digested with ClaI and BamHI and subcloned in frame with a hemagglutinin antigen (HA) epitope into compatible restriction enzyme sites in the pcDNA3 mammalian expression vector. The pcDNA3-HA-hPHIP1 construct was purified by CsCl gradient centrifugation and completely sequenced to confirm the absence of mutations.
Ad constructs.
Adenovirus (Ad) constructs were generated by using an AdEasy kit from Stratagene (La Jolla, CA). Briefly, HA-tagged hPHIP1 was subcloned into the multiple cloning site of the pShuttle/IRES-GFP-1 vector. Viral amplification of the recombinant HA-hPHIP1 Ad (AdPHIP1) was performed according to the manufacturer's instructions (Stratagene, La Jolla, CA). IRS2-expressing recombinant Ad (AdIRS2) and “kinase-dead” PKB-expressing recombinant Ad (AdKD) were a generous gift from C. J. Rhodes. Virus titers were measured for optical density at 260 nm and dialyzed immediately before usage, as described elsewhere (12).
Cell fractionation.
The extraction of the two subcellular protein fractions (cytosol and nucleus) was achieved by using the method of Wadman et al. (32).
Murine PHIP1 (mPHIP1) expression in tissue.
Total RNA was isolated from MIN6 cells and mouse tissues using Trizol reagent (Invitrogen, Canada) according to the manufacturer's instructions. First-strand synthesis of cDNA was carried out using Superscript II RNase H reverse transcriptase (Invitrogen, Canada). Briefly, 1 μg of total RNA was reverse transcribed using OligodT primers (Sigma Chemical Co, St. Louis, MO) following the manufacturer's instructions. The resulting cDNA was used for amplification in quantitative real-time PCR (qPCR). qPCR was performed in an ABI Prism 7900 HT sequence detection system (Applied Biosystems) with the following primers: mPHIP1 5′-GGACACTTATCATCGGTGAC-3′ and 3′-TTTCAGCAGCATGTCCTCTT-5′ (amplicon size, 141 bp) and mouse β-actin 5′-CTGAATGGCCCAGGTCTGA-3′ and 3′-CCCTGGCTGCCTCAACAC-5′ (amplicon size, 104 bp). Values were normalized to the expression of mouse β-actin mRNA and represent the averages of three independent experiments. Calculations were performed using the 2−ΔΔCT method.
Ad infection.
The optimal titers for each recombinant Ad were determined experimentally by infecting INS-1 cells grown in six-well plates (∼70% confluent; 2 × 106 cells) with various dilutions to achieve a multiplicity of infection (MOI) ranging from 100 to 400 as determined by the assay measuring optical density at 260 nm. For INS-1 infections, cells were infected for 2 h in FBS-free medium, and subsequently, the virus was removed and the medium replaced with complete medium. The efficacy of infection was 80 to 100% as determined by immunostaining with anti-HA antibodies and green fluorescent protein (GFP) fluorescence.
RNA interference.
INS-1 cells were transfected with an siRNA pool (50 or 100 nM; Dharmacon) using Lipofectamine 2000 (Invitrogen, Canada) according to the manufacturer's instructions.
Cell proliferation assay.
INS-1 cells were plated in 96-well plates (20,000 cells/well). Twenty-four hours after being plated, the cells were infected with appropriate doses of Ad. The 3-(4,5-dimethylthiazol-2-yl)-5-(3-carboxymethoxyphenyl)-2-(4-sulfophenyl)-2H-tetrazolium (MTS) assay was performed using a CellTiter 96 AQueous nonradioactive cell proliferation assay kit (Promega) at 24- to 96-h time intervals after viral infection, according to the manufacturer's instructions.
BrdU incorporation.
INS-1 cells were seeded at a density of 20,000 cells/well in 96-well plates. The cells were infected with Ad, and 16 h postinfection, the cells were washed once with 10 mM phosphate-buffered saline (PBS) (pH 7.4) and subsequently starved in 1 mM glucose-0.5% BSA RPMI medium for 24 h. They were then incubated for 24 h in RPMI medium with 15 mM glucose-FBS in the presence/absence of 10 nM of IGF-1. During the last 8 h of stimulation, 20 μl of bromodeoxyuridine (BrdU) solution was added, and an enzyme-linked immunosorbent assay (BrdU cell proliferation assay kit; Calbiochem) was performed according to the manufacturer's protocol.
[3H]thymidine incorporation.
INS-1 cells were plated in 24-well plates at a density of 100,000 cells/well and transfected with siRNAs (100 nM) using Lipofectamine 2000. Twelve hours after transfection, the cells were infected with an Ad that expressed GFP (AdGFP) or AdIRS2 (MOI, 200) and starved in the presence of 0.5% BSA-1 mM RPMI 1640 for 24 h. Subsequently, the cells were stimulated with 10 nM of IGF-1 in the presence of 15 mM glucose-RPMI. During the last 4 h of the incubation period, 3 μCi of [3H]thymidine/ml was added. After this final incubation, the cells were washed twice with PBS and lysed and the [3H]thymidine incorporation into INS-1 cell DNA was measured by liquid scintillation counting.
Apoptosis assay.
Apoptotic measurements were performed by using a fluorimetric method as described previously (1). The experiments were performed at least three times.
FFA treatment.
OA was complexed to fatty acid-free BSA by stirring 4 mM OA with 5% (wt/vol) BSA in Krebs-Ringer HEPES buffer (KRBH) (135 mM NaCl, 3.6 mM KCl, 5 mM NaHCO3, 0.5 mM NaH2PO4, 0.5 mM MgCl2, 1.5 mM CaCl2, 10 mM HEPES, pH 7.4) for 30 min at 50°C. FFA quantification in the resulting solution was achieved by the use of a NEFA C test kit assay (VWR International, Canada). The OA-BSA complex stock solution was then cooled to 25°C, filter sterilized, and stored at −20°C until further use. INS-1 cells were cultured on six-well dishes (60 to 70% confluence) with recombinant Ad for 16 h. The INS-1 cells were incubated in modified INS-1 cell RPMI medium containing 15 mM glucose with or without 10 ng/ml IGF-1 with 0.5% (wt/vol) BSA alone or 0.4 mM OA for 15 h. At the end of the experiments, both floating and attached cells were collected, combined, and subsequently used for apoptosis and Western blotting assays.
Luciferase assay.
INS-1 cells with 50% confluence on 24-well dishes were transfected into serum-free DMEM using Lipofectamine 2000 (Invitrogen) with 1 μg/well of a luciferase reporter construct containing 3.0 kb of the mouse cyclin D2 promoter. Twelve hours after transfection, the cells were infected with AdGFP or AdPHIP1 and subsequently starved in the presence of 0.5% BSA-1 mM RPMI 1640 for 24 h, and luciferase activity was measured 24 h after stimulation with 15 mM glucose with or without 10 nM of IGF-1. Measurements were performed using a firefly luciferase assay system (Promega) and were normalized to the protein concentration.
Generation of PHIP antibodies.
The bacterially expressed glutathione S-transferase (GST)-PHIP fusion protein corresponding to residues 1548 to 1821 of mPHIP1 was injected into rabbits to raise polyclonal PHIP antibodies. Anti-GST antibodies were removed by the passage of serum over a GST affinity column. Subsequently, the antibodies were affinity purified on nitrocellulose strips blotted with GST-PHIP as previously described (10). Anti-PHIP antibodies were used at a 1:500 ratio for Western blotting.
Immunofluorescence.
Mouse pancreases were dissected, washed with 0.1 M phosphate buffer, pH 7.4, embedded in OCT compound (Miles, Elkhart, IN), and then frozen by immersion in liquid nitrogen. Five-micrometer frozen sections were cut with a cryostat, transferred to poly(l-lysine)-coated thin slides, and stored at −80°C. For insulin, glucagon, and PDX-1 immunostaining, sections were fixed in 4% paraformaldehyde (10 min) after rehydration (0.1 M PBS; 30 min) and permeabilized (1% Triton X-100; 20 min). For PHIP staining, the fixation step was omitted because it provided significant nonspecific background. After the permeabilization, all sections were blocked with 2% BSA in PBS for 30 min. Subsequently, sections were incubated with primary antibodies (dilution, 1:200) in PBS containing 2% BSA (2 h at room temperature), followed by 1 h of incubation with secondary antibodies (goat anti-mouse immunoglobulin G [IgG] conjugated to fluorescein isothiocyanate or goat anti-rabbit IgG conjugated to Cy3 [dilution, 1:300; Molecular Probes, Eugene, OR]). MIN6 and INS-1 cells were fixed with ice-cold 100% methanol for 20 min and were permeabilized with 1% Triton X-100 (10 min). The cells were incubated with the primary antibodies in PBS for 2 h at room temperature, followed by the addition of secondary antibodies (goat anti-mouse IgG conjugated to fluorescein isothiocyanate or goat anti-rabbit IgG conjugated to Cy3 [dilution, 1:300; Molecular Probes, Eugene, OR]) for 1 h at room temperature. The immunofluorescent images were recorded with a Nikon epifluorescence microscope (Nikon, Tokyo, Japan) equipped with a PXL 1400 cooled charge-coupled-device camera system (Photometrics, Tucson, AZ) and SimplePCI software (C Imaging Systems, PA).
Statistical analysis.
Results are expressed as the means ± standard deviations (SD) unless otherwise specified. The t test and Mann-Whitney U test were used for comparisons between two groups for parametric and nonparametric data, respectively. P < 0.05 is considered statistically significant.
RESULTS
Expression, cloning, and characterization of PHIP1 in mouse tissues and insulin-producing cell lines.
The NCBI mouse database (NCBI Build 36; mm8) predicts at least four alternatively spliced variants of mPHIP, with multiple translation initiation start sites that are predicted to encode proteins with molecular masses ranging from 105 to 206 kDa (Fig. 1A). To detect which isoform is predominantly expressed in pancreatic β cells, whole-cell lysates (WCL) from mouse islets and the insulinoma cell lines MIN6 and INS-1 were subjected to immunoblot analysis with anti-PHIP antibodies. As shown in Fig. 1B, Western blots of cellular lysates revealed a prominent immunoreactive species at a molecular mass of approximately 206 kDa. We also observed the presence of additional, weaker immunoreactive bands in these lysates, migrating at approximately 105 kDa and 165 kDa. These lower-molecular-mass species may represent alternatively spliced or translational variants of mPHIP, such as those predicted in the mouse genome database.
FIG. 1.

Expression analysis of mPHIP in pancreatic β cells and insulin-producing β-cell lines. (A) Schematic representation of PHIP1 translation products illustrates the presence of eight WD40 motifs spanning residues 172 to 498 (black boxes), two bromodomains (BD) located between residues 1158 and 1423 (gray boxes), and two NLS at positions 912 to 924 and 1438 to 1465 (thick bar). Alternative splicing of mPHIP predicts multiple transcript variants that can be recognized by raised anti-PHIP antibodies. mPHIP variant 2 (GenBank accession number XP_900529) and variant 11 (GenBank accession number XP_900632) produce proteins that retain six and one of the eight WD40 repeats, respectively, whereas mouse variant 9 (GenBank accession number XP_900614.1), which we previously cloned, is lacking WD40 repeats. (B) Pancreatic β cells express a prominent 206-kDa form of PHIP. Total proteins (50 μg) from freshly extracted mouse islets and insulin-producing cell lines INS-1 and MIN6 were fractionated on a 6% sodium dodecyl sulfate-polyacrylamide gel and immunoblotted with anti-PHIP antibodies. (C) qPCR analysis of PHIP1 mRNA expression in various mouse tissues and MIN6 cells was performed with primers specific for mPHIP1. Results are expressed as the means ± standard errors of the means for three independent experiments done in triplicate. WAT, white adipose tissue; BAT, brown adipose tissue. (D) Anti-PHIP antibodies specifically detect two recombinant isoforms of PHIP. MIN6 cells were transiently transfected with either pcDNA3 control or the pcDNA3-HA-hPHIP1 or pcDNA3-HA-mPHIP vectors encoding the 1,821-aa and 902-aa PHIP isoforms, respectively. Cellular lysates were collected 48 h posttransfection and immunoblotted with anti-PHIP antibodies. Exogenously expressed HA-hPHIP1 and HA-mPHIP are denoted by arrowheads. Endogenous mPHIP1 is shown by an arrow (→). (E) siRNA knockdown of PHIP in INS-1 cells. INS-1 cells were transfected with 50 nM of siRNA pool to rat PHIP1 or nonsilencing scrambled (Scr) siRNAs. Immunoblot analysis shows knockdown of 206- and ∼145-kDa immunoreactive signals representing possible transcript variants of PHIP1. Actin was used as a loading control. IB, immunoblot.
The NCBI human and mouse reference sequence database currently predicts the existence of a long, 1,821-aa isoform of PHIP (PHIP1), with a predicted molecular mass of 206 kDa, which likely encodes the major immunoreactive species observed in β cells. Using the genome annotation as a guideline, we proceeded to clone full-length hPHIP1 from MCF-7 cells by reverse-transcriptase PCR, yielding a 5.53-kb cDNA containing the complete ORF of the predicted PHIP1 isoform (GenBank accession number NM_017934). Homology analysis revealed that hPHIP1 shares very high (96%) sequence identity with the predicted mPHIP1 isoform (Fig. 2). The hPHIP1 and mPHIP1 protein sequences were examined for conserved regions that could underline possible functional similarities. Sequence analyses employing the PFAM database of protein motifs (http://pfam.janelia.org/) indicate that hPHIP1 and mPHIP1 contain eight WD40 repeats (residues 171 to 211, 214 to 253, 256 to 299, 310 to 349, 354 to 393, 408 to 452, 455 to 495, and 498 to 542) and two bromodomains (residues 1158 to 1261 and 1318 to 1423) (Fig. 1A). In addition, the PredictNLS program (4) identified the presence of two putative nuclear localization signals (NLS).
FIG. 2.

Alignment of deduced aa sequences of cloned hPHIP1 (GenBank accession number DQ924532) and predicted mPHIP1 (GenBank accession number XP_358384) isoforms reveals 96% aa identity (highlighted in black).
In order to examine the expression pattern of PHIP1 in different mouse tissues, we used qPCR with primers designed to amplify the 5′ end of mPHIP1, as depicted in Fig. 1A. We observed that the mPHIP1 transcript is ubiquitously expressed in all tissues examined, although the level of expression varied in different tissues, with the most abundant expression in pancreatic islets, the brain, and skeletal muscle (Fig. 1C).
To confirm that the recombinant full-length PHIP1 cDNA we cloned encoded the 206-kDa isoform expressed in β cells, we transiently transfected an epitope-tagged HA-hPHIP1 (1,821-aa) plasmid in MIN6 cells. As shown in Fig. 1D, immunoblotting with anti-PHIP antibodies revealed that the exogenously expressed PHIP1 plasmid gave rise to a prominent band that comigrated with endogenous mPHIP1 in MIN6 cells. From a parallel experiment, exogenous expression of the smaller mPHIP variant 9 (902 aa) is shown for comparison. The 105-kDa protein encoded by this cDNA is not readily observed in MIN6 cell lysates.
We next attempted to silence PHIP1 expression in INS-1 cells by using RNA interference. A SMARTpool of siRNA oligonucleotides targeting the PHIP1 transcript was selected for transient knockdown. Using Alexa Fluor 488-labeled negative siRNA as a reporter, we could measure the transfection efficiency of the fluorescent oligonucleotides following transfection. The transfection efficiency was greater than 85% (data not shown). Gene silencing efficiency as measured by Western blot analysis was greater than 70% in all experiments performed, as shown in Fig. 1E. Transfection with PHIP siRNAs significantly reduced the intensity of the prominent 206-kDa protein corresponding to PHIP1, as well as the weaker immunoreactive band at 165 kDa which most likely represents a translational variant of mPHIP, confirming the specificity of the anti-PHIP antibodies employed in this study.
Given that PHIP1 has two predicted NLS, we next explored its subcellular localization in insulin-producing cells. PHIP1 was visualized in mouse pancreatic sections and MIN6 and INS-1 cells by immunofluorescence microscopy. Immunostaining of the mouse pancreas showed that PHIP1 is localized to the nucleus of islet cells and that these cells have much higher levels of PHIP1 than the surrounding acinar cells (Fig. 3A), which is consistent with the relative abundance of PHIP1 transcript levels assessed by qPCR analysis (Fig. 1C). As PHIP antibodies require unique fixation protocols that preserve their antigenicity, double immunostaining with insulin or glucagon was not feasible. Immunostaining of consecutive pancreatic sections for PHIP1, glucagon, and insulin demonstrated that PHIP1 was found distributed in both α and β cells (Fig. 3B). Moreover, immunostaining of dispersed islets from MIP-GFP transgenic mice (transgenic mice that express GFP under the control of the mouse insulin I gene promoter) (14) confirmed that PHIP1 was not restricted to GFP-positive β-cells only (Fig. 3C).
FIG. 3.

PHIP is localized to the nucleus of pancreatic β cells. (A) Section of mouse pancreas stained with anti-PHIP (red) and 4′,6′-diamidino-2-phenylindole (DAPI; blue) demonstrates predominant PHIP expression in nuclei of islet cells. Original magnification, ×100. (B) Five-micrometer consecutive sections of mouse pancreas were stained with anti-PHIP (green), anti-insulin (red), antiglucagon (green), and anti-PDX-1 (red) antibodies as described in Materials and Methods and counterstained with hematoxylin and eosin (H&E). The specificity of staining was verified by the inclusion of appropriate preimmune antibodies as a negative control. Original magnification, ×200; scale bar, 20 μm. (C) Coimmunostaining of PHIP1 and glucagon in dispersed islets from MIP-GFP transgenic mice shows comparable levels of PHIP (blue) in β cells (green) and in glucagon-positive α cells (red). (D) Immunostaining of MIN6 cells with anti-PHIP (upper left panel, red) and anti-IRS2 (upper right panel, green) antibodies reveals differential subcellular localization for PHIP and IRS2. In the upper middle panel, MIN6 cells were infected with AdPHIP1 (MOI, 400) and localization of ectopically expressed PHIP1 (red) was detected by anti-HA antibodies 48 h postinfection. Note the exclusively nuclear anti-HA staining. DAPI counterstaining indicates localization of nuclei. Original magnification, ×400; scale bar, 20 μm. (E) Subcellular fractionation shows that endogenously and exogenously expressed PHIP1 localized in the nuclear fraction. INS-1 cells were infected at an MOI of 400 with AdPHIP1 and, 48 h afterwards, were collected, and 50 μg of WCL and cytosolic and nuclear fractions were loaded for Western blotting analysis. Levels of IRS2 and cyclin D2 were measured as a control for subcellular fractionation efficacy. (F) IGF-1 does not induce nuclear-cytoplasmic shuttling of PHIP1. After serum starvation (DMEM and 0.5% BSA) for 24 h, INS-1 and MIN6 cells were exposed for 16 h to 10 nM of IGF-1 in DMEM and 0.5% BSA (in the presence of IGF-1 [+IGF-1]) and immunostained for PHIP1 (red). Cells from control groups were exposed to medium containing 0.5% BSA only (in the absence of IGF-1 [−IGF-1]). DAPI counterstaining indicates localization of nuclei. Original magnification, ×400.
To further confirm the nuclear localization of PHIP1 in insulin-producing cells, the distribution patterns of both endogenous and ectopically expressed PHIP1 were visualized in MIN6 and INS-1 cells by immunofluorescence staining and Western blot analysis. Indeed, both endogenous and exogenous PHIP1 showed nuclear localization with immunostaining with anti-PHIP and anti-HA antibodies (Fig. 3D and F). Cell fractionation experiments showed that exogenous PHIP1 paralleled the distribution pattern of endogenous PHIP1, in that it was localized selectively in the nuclear fraction of INS-1 cell lysates (Fig. 3E). In addition, the treatment of INS-1 and MIN6 cells with IGF-1 did not result in changes in the localization of PHIP1, which remained exclusively nuclear (Fig. 3F).
We have previously demonstrated that the short isoform of PHIP (variant 9) was able to interact with IRS2 (10). Taking into account that both the short and long isoforms of PHIP share the same structural domains, we next sought to investigate whether PHIP1 and IRS2 physically interact with each other. Due to inherent difficulties associated with solubilization of the larger PHIP1 isoform, attempts at immunoprecipitation of this species were unsuccessful and thus prevented any direct examination of a possible association with IRS2 or other IRS family members. Using immunofluorescence microscopy and Western blotting, we compared the subcellular localization of IRS2 and PHIP1 in pancreatic β cells. As shown in Fig. 3D and E, IRS2 is localized in the cytosolic compartment, whereas PHIP1 is nuclear, an observation that reduces the probability of a possible interaction between the two proteins, given their disparate subcellular localizations.
PHIP1 promotes proliferation and potentiates IGF-1-stimulated mitogenesis in β cells.
As we had previously observed that the short isoform of PHIP (variant 9) regulates insulin-stimulated mitogenic responses in fibroblasts, we wanted to determine next whether overexpression of PHIP1 had any modulatory effect on β-cell proliferation. To this end, we used AdPHIP1 to transduce INS-1 cells. Western blot analysis of INS-1 cells harvested 96 h postinfection with AdPHIP1 (Fig. 4A) showed a 3- to 10-fold increase in PHIP1 protein levels with increasing virus titer (MOI, 100 to 400). AdGFP was used as a control for these experiments at the same dose range and demonstrated 90 to 100% gene transfer efficacy (data not shown). Importantly, adenoviral infection with the control AdGFP did not exert any inhibitory effect on endogenous PHIP1 protein expression (Fig. 4A). Since IRS2 is well established as a positive regulator of β-cell proliferation, we employed AdIRS2 at the same dose range as AdPHIP1 and AdGFP. As shown in Fig. 4B, PHIP1 and IRS2 overexpression in INS-1 cells promoted cell proliferation in a time-dependent manner. The proliferation rates of AdPHIP1- and AdIRS2-infected cells, assessed 96 h after infection, were increased 1.3- ± 0.1- and 1.4- ± 0.13-fold, respectively, compared to that of AdGFP-infected INS-1 cells (P < 0.05).
FIG. 4.

Ad-mediated overexpression of PHIP1 promotes proliferation and potentiates IGF-1-stimulated mitogenesis of β cells. (A) Immunoblot analysis of PHIP1 expression in INS-1 cells infected with increasing doses of AdGFP and AdPHIP1 (MOI, 100 to 400, as indicated) indicates dose-dependent increase of exogenous PHIP1 expression (left). The IRS2 immunoblot demonstrates the level of IRS2 overexpression in AdIRS2-infected cells (right). Actin expression was used as a loading control. (B) Time course of INS-1 and NIH 3T3 proliferation assessed by MTS assay and counting of live cells, respectively, 24 to 96 h postinfection with AdGFP, AdPHIP1, and AdIRS2 (MOI, 200). Cells were incubated in the presence of 11 mM glucose-10% FBS. Experiments were performed three times in sextuplicate. Results are shown as means ± SD. *, P < 0.05 versus results for AdGFP-infected cells. OD450, optical density at 450 nm. (C) Ad-mediated overexpression of PHIP1 enhances IGF-1-dependent mitogenesis and promotes increase of cyclin D2 protein levels in INS-1 cells. Cells were infected with AdGFP, AdPHIP1, and AdIRS2 (MOI, 200) and, 16 h postinfection, were made quiescent for 24 h. Subsequently, cells were incubated in RPMI medium with 15 mM glucose in the presence (+) or absence (−) of 10 nM IGF-1. BrdU incorporation was measured as indicated in Materials and Methods. The data are expressed as the increases ± standard errors of the means compared to results for control cells (AdGFP-infected cells treated with 15 mM glucose in the absence of IGF-1 [GFP/−IGF group]). Experiments were performed three times in quadruplicate. *, P < 0.05. Bottom panel, cells were treated as described above and lysates were immunoblotted with cyclin D2 antibodies. Actin was used as a loading control. (D) Densitometry analysis from the three independent experiments described for panel C is summarized as a histogram. The data are expressed as the increases ± standard errors of the means. *, P < 0.05 versus results for AdGFP-infected cells treated in the absence of IGF-1; #, P < 0.05 versus results for AdGFP-infected cells treated in the presence of IGF-1. (E) Ad-mediated overexpression of PHIP1 enhances IGF-1-dependent cyclin D2 promoter activity in INS-1 cells. Cyclin D2 luciferase reporter was transfected into INS-1 cells. Subsequently, cells were infected with increasing MOIs, as indicated, of AdGFP or AdPHIP1 and incubated in the presence (+) or absence (−) of IGF-1 (10 nM). Luciferase assay was performed as indicated in Materials and Methods. Experiments were performed three times in triplicate. The data are expressed as the increases ± SD compared to results for control cells (AdGFP-infected cells treated with 15 mM glucose in the absence of IGF-1 [AdGFP/−IGF group]). *, P < 0.05 versus results for AdGFP group infected with same MOI in the absence of IGF-1; #, P < 0.05 versus results for AdPHIP1 group infected with same MOI in the presence of 10 nM of IGF-1. +, present; −, absent.
IGF-1 is a potent regulator of β-cell growth (21). INS-1 but not MIN6 cells have been reported to respond mitogenetically to IGF-1 treatment. We therefore sought to investigate whether overexpression of hPHIP1 can modulate IGF-1-dependent mitogenesis in INS-1 cells as measured by BrdU incorporation. As shown in Fig. 4C, AdPHIP1 increased BrdU incorporation by 1.3-fold (P < 0.05) compared to that in AdGFP-infected cells in the absence of IGF-1 treatment. More importantly, AdPHIP1 enhanced IGF-1-induced BrdU incorporation to the same extent as AdIRS2 (1.8-fold) compared to the incorporation in control AdGFP-infected cells (P < 0.05). To investigate the possible molecular mechanism(s) responsible for the observed increase in proliferation rate of β-cell lines overexpressing either PHIP1 or IRS2, we assessed the expression of cyclin D2, a key modulator of β-cell proliferation. As shown in Fig. 4C and D, PHIP1 and IRS2 overexpression induced a marked upregulation in the cyclin D2 protein levels in this cell line. There was no discernible effect on cyclin D1 expression. In order to determine whether PHIP1 is a general modulator of cell proliferation, we tested its ability to induce mitogenesis in non-β cells, namely, mouse fibroblasts (NIH 3T3). As shown in Fig. 4B, PHIP1 overexpression in NIH 3T3 cells mediates an increase in cellular proliferation comparable to that which we observed for INS-1 cells upon stimulation with IGF-1.
PHIP1 drives the transcriptional induction of the cyclin D2 gene promoter.
To address the role of PHIP1 in cyclin D2 transcriptional control, we performed promoter-reporter gene analysis. INS-1 cells were transiently transfected with the firefly luciferase gene under the control of a 3.0-kb promoter fragment of the mouse cyclin D2 promoter and subsequently transduced with increasing doses of AdPHIP1 or AdGFP control virus. As shown in Fig. 4E, PHIP1 was sufficient to drive transcriptional activation of the cyclin D2 promoter, which was further enhanced (2.3-fold) in response to IGF-1 treatment.
siRNA-mediated knockdown of PHIP1 inhibits IRS2-dependent mitogenic effects.
Having shown that PHIP1 potentiates IGF-1-mediated DNA synthesis in β cells, we next sought to determine whether PHIP1 serves as a downstream effector of IRS2 signaling pathways leading to mitogenic responses in INS-1 cells. To this end, we used siRNA interference oligonucleotides to knock down endogenous PHIP1 levels, and INS-1 cells were then incubated in parallel with AdGFP or AdIRS2 for 48 h and left untreated or stimulated with IGF-1. As shown in Fig. 5A, IGF-1-induced mitogenesis, as measured by [3H]thymidine incorporation assays, was increased twofold over the basal level in AdIRS2-infected cells. Silencing of PHIP1 in INS-1 cells abrogated the IRS2-mediated effect, which was accompanied by a reduction in the level of cyclin D2 protein, independent of any changes in phospho-PKB activation (Fig. 5B).
FIG. 5.
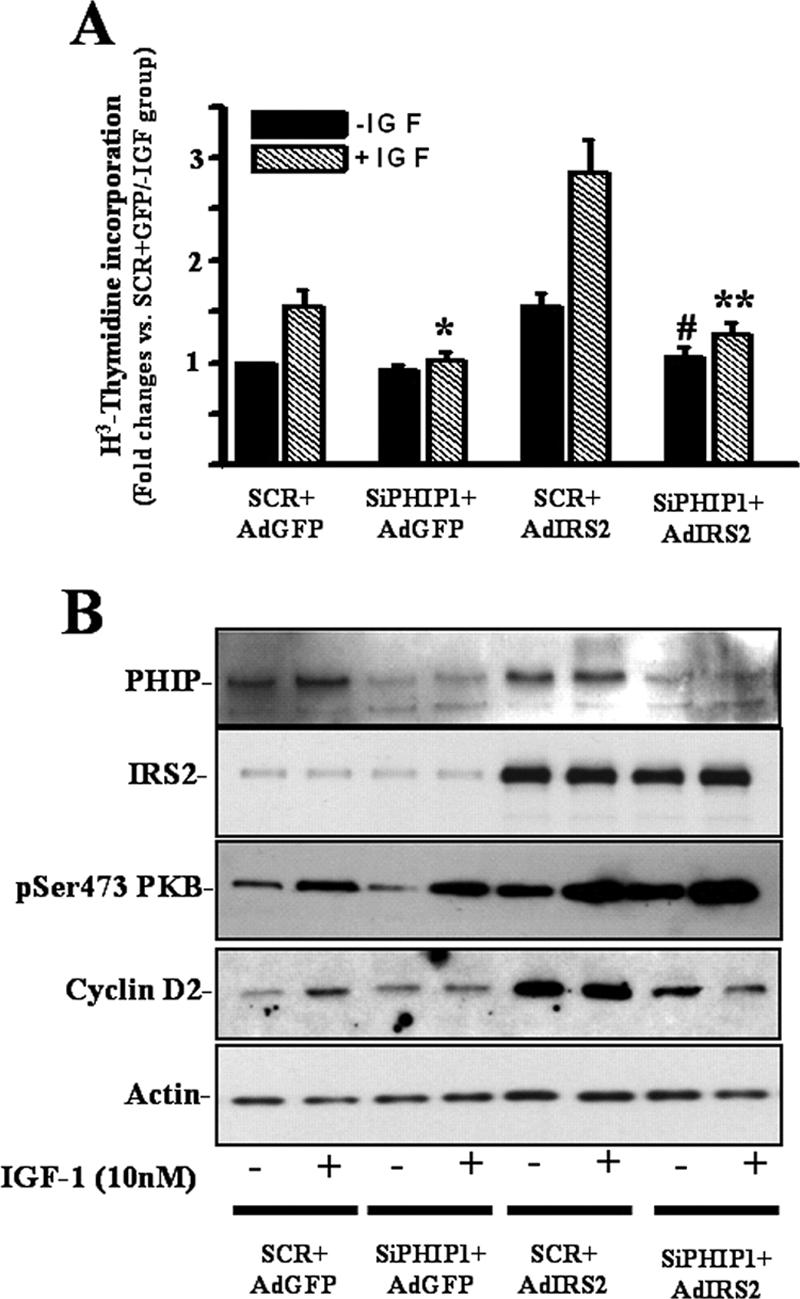
Inhibition of PHIP1 expression blocks IGF-1- and IRS2-induced mitogenesis. INS-1 cells were transiently transfected with nonsilencing scrambled (SCR) and anti-PHIP1 (SiPHIP1) siRNAs and infected with AdGFP or AdIRS2 at an MOI of 200. (A) [3H]thymidine incorporation was measured 24 h after IGF-1 stimulation as indicated in Materials and Methods. Experiments were performed three times in triplicate. Results are shown as means ± SD. *, P < 0.05 versus results for cells transfected with SCR siRNA, infected with AdGFP (SCR + AdGFP), and stimulated with IGF-1 (+IGF); #, P < 0.05 versus results for cells transfected with SCR, transfected with AdIRS2 (SCR + AdIRS2), and not stimulated with IGF-1 (−IGF); **, P < 0.05 versus results for cells transfected with SCR siRNA, infected with AdIRS2, and stimulated with IGF-1. (B) Immunoblot analyses for PHIP1, IRS2, phospho-Ser473 PKB, cyclin D2, and actin in cells treated as described for panel A. The immunoblots are representative of at least two independent experiments. +, present; −, absent.
Adenovirus-mediated expression of PHIP1 prevents FFA-induced apoptosis of INS-1 cells.
Having established that PHIP1 overexpression potentiates mitogenic responses in INS-1 cells, we next asked whether PHIP1 overexpression influences β-cell survival. OA has previously been shown to induce apoptosis, while having a minor effect on necrosis in β cells (34). Moreover, increased IRS2 expression or cellular exposure to IGF-1 (10 ng/ml) is capable of mitigating OA-induced apoptosis in INS-1 cells (34). To address whether PHIP1 can confer cytoprotective effects on OA-treated cells, INS-1 cells were infected with low and high levels of AdGFP and AdPHIP1 (MOI, 75 and 400, respectively) and subsequently incubated for 15 h at 15 mM glucose with either 0 or 10 ng/ml of IGF-1 in the presence of 0.4 mM OA-0.5% BSA or 0.5% BSA alone. Apoptosis was monitored by measuring the fraction of cells in the sub-G1 phase by using fluorescence-activated cell sorter analysis. As shown in Fig. 6A, upon exposure to 0.4 mM of OA, the incidence of apoptosis was significantly increased in cells infected with AdGFP at an MOI of either 75 or 400 and incubated in the absence of IGF-1 (MOI of 75, 24.2% ± 3.0% [P < 0.01], and MOI of 400, 17.8% ± 1.2% [P < 0.01]) compared to the incidence of apoptosis in AdGFP-infected cells incubated with BSA alone (7.1% ± 0.9%). IGF-1 exposure inhibited the induction of apoptosis in the population of AdGFP-infected cells treated with 0.4 mM of OA to near-basal levels.
FIG. 6.

Ad-mediated overexpression of PHIP1 prevents FFA-induced INS-1 cell apoptosis and induces PKB activation. (A) INS-1 cells were infected in six-well plates with AdGFP, AdPHIP1, and AdIRS2 (MOI, 75 and 400, as indicated). Sixteen hours postinfection, cells were incubated for 15 h with 15 mM glucose in the presence of 0.5% BSA alone or 0.4 mM of OA-0.5% BSA (OA). The incidence of apoptosis was assayed as described in Materials and Methods. Results are shown as the means ± SD for three independent experiments. *, P < 0.05 comparing results with AdPHIP1 to results with AdGFP in the absence of IGF-1 (−IGF-1); #, P < 0.05 comparing results with AdPHIP1 to results with AdGFP in the presence of IGF-1 (+IGF-1). (B) Ectopic PHIP1 overexpression induces PKB phosphorylation. WCL of AdGFP- and AdPHIP1-infected cells (MOI, 400) treated for 15 h with 15 mM glucose in the presence of 0.5% BSA and 10 ng/ml of IGF-1 were subjected to Western blot analysis for phospho-Ser473 (pSer473) and total PKB. Densitometry analysis from three independent experiments is summarized as a histogram. The data are expressed as n-fold increases ± standard errors of the means. *, P < 0.05 comparing results with AdPHIP1 to results with AdGFP in the absence of IGF-1; #, P < 0.05 comparing results with AdPHIP1 to results with AdGFP in the presence of IGF-1. +, present; −, absent. (C) Ectopic PHIP1 overexpression induces PKB phosphorylation and inhibits caspase-9 and -3 activation. WCL of AdGFP-, AdPHIP1-, and AdIRS2-infected (MOI, 400) INS-1 cells treated as described above were subjected to Western blot analysis for activated PKB (phospho-Ser473 [pSer473 PKB] and phospho-Thr308 [pThr308 PKB] PKB), total PKB, the proteolytically cleaved, activated forms of caspase-9 (Casp9) and -3 (Casp3), and Bax. Actin was used as a loading control. Representative immunoblots are shown. +, present; −, absent. (D) Densitometry analyses from the three independent experiments described for Fig. 6B are summarized as histograms. The data are expressed as n-fold increases ± standard errors of the means. Phospho-PKB levels were normalized to total PKB level, and cleaved procaspase-9 and -3 and Bax levels to actin level. *, P < 0.05 comparing results with AdPHIP1 to results with AdGFP in the absence of IGF-1; #, P < 0.05 comparing results with AdPHIP1 to results with AdGFP in the presence of IGF-1. +, present; −, absent.
It is noteworthy that Ad-mediated overexpression of PHIP1 in INS-1 cells elicited a more pronounced cytoprotective effect in OA-treated cells than IGF-1 alone (4.6-fold versus 2.2-fold, respectively; P < 0.05). Moreover, we observed that the incidence of apoptosis in AdPHIP1-infected cells treated with 0.4 mM of OA was similar in the absence (3.9% ± 0.2%) or presence of IGF-1 (3.4% ± 0.5%). Analogously, Ad-mediated overexpression of IRS2 had a marked inhibitory effect on FFA-induced apoptosis in INS-1 cells. The incidence of OA-induced apoptosis in AdIRS2-infected cells incubated in the absence of IGF-1 was significantly lower (2.9% ± 0.6%; P < 0.01) than that of AdGFP-infected cells incubated with IGF-1 in the presence of 0.4 mM of OA (7.9% ± 0.1%). However, there was no statistically significant difference in the incidence of apoptosis between OA-treated AdPHIP1- and AdIRS2-infected cells regardless of IGF-1 treatment. In a similar fashion, adenoviral infection of PHIP1 at a lower titer (MOI, 75) resulted in comparable cytoprotective effects against FFA-induced apoptosis (Fig. 6A). This observation is consistent with an essential role for PHIP1 in the signaling pathways that promote β-cell survival in the face of a lipotoxic stimulus.
Ectopic PHIP1 overexpression induces PKB phosphorylation and inhibits caspase-9 and -3 activation.
Genetic and biochemical studies have implicated PKB as a critical factor in maintaining β-cell survival. It is generally accepted that PKB activation is mediated by IGF-1-induced tyrosine phosphorylation of IRS2, followed by PI3-kinase activation and the subsequent PKB translocation to the plasma membrane, where 3-phosphoinositide-dependent kinase-1 (PDK1) phosphorylates PKB at Thr308 and mTORC2 at Ser473 (3). Activated PKB inhibits the activity of several proteins that induce apoptosis in β-cells, such as members of the Bcl-2 family (BAD), and the cysteine protease caspase-9. To investigate the possible molecular mechanisms involved in the observed antiapoptotic effect of PHIP1, we examined PKB expression and activity. As shown in Fig. 6B, overexpression of PHIP1 did not alter the levels of total PKB. However, under both basal (0.5% BSA) and IGF-1-stimulated conditions, we observed a significant increase in the phosphorylation status of PKB at Ser473 in AdPHIP1-infected cells compared to that in AdGFP-infected control cells. Overexpression of PHIP1 and IRS2 in OA-treated cells led to more than a 4.5-fold increase in the phosphorylation level of PKB (Ser473 and Thr308) (Fig. 6C and D). However, the extent of PKB phosphorylation was only marginally enhanced following IGF-1 stimulation with either Ad (Fig. 6C and D).
Caspase-9 is a substrate of PKB and plays an important role in the execution of apoptotic cell death in β-cells. This being the case, we proceeded to assess the level of cleaved (activated) caspase-9 in the context of the experimental conditions described above. We observed that OA treatment of AdGFP-infected cells led to a 1.7-fold increase in the level of activated caspase-9 compared to the level in control cells (P < 0.05), which was subsequently decreased by 25% upon the addition of IGF-1 (Fig. 6C and D). Importantly, Ad-mediated overexpression of either PHIP1 or IRS2 markedly suppressed caspase-9 cleavage in OA-treated INS-1 cells independent of IGF-1 treatment. Initiator caspases, such as caspase-9, can directly activate effector caspases, such as caspase-3, which cause cellular destruction. Caspase-3 levels were dramatically decreased in OA-treated cells infected with AdPHIP1 or IRS2 compared to the levels in AdGFP-infected cells (Fig. 6C and D). PKB signaling also leads to the down-regulation of bax gene transcription via the sequestration of Foxo1 from the nucleus (19). Furthermore, although OA treatment led to an increase in BAX expression, there was only a marginal decrease in the BAX levels in OA-treated cells infected with AdPHIP1 or IRS2 compared to the level in AdGPF-infected cells (Fig. 6C and D).
Inhibition of PKB activity blocks the protective effect of PHIP1 on FFA-induced apoptosis in INS-1 cells.
In order to investigate whether PHIP1 mediates its protective effect on FFA-induced apoptosis via the activation of PKB, we employed AdKD (created using a K179M mutation which eliminates the ATP-binding site and results in loss of kinase activity) (5). As shown in Fig. 7, overexpression of AdKD in INS-1 cells completely abrogated the protective effect of PHIP1 on FFA-induced apoptosis, coincident with a reversal of the PHIP1-induced activation of PKB/AKT. These findings are consistent with the idea that PKB/AKT has an essential role in the PHIP1 signaling pathway that promotes β-cell survival.
FIG. 7.
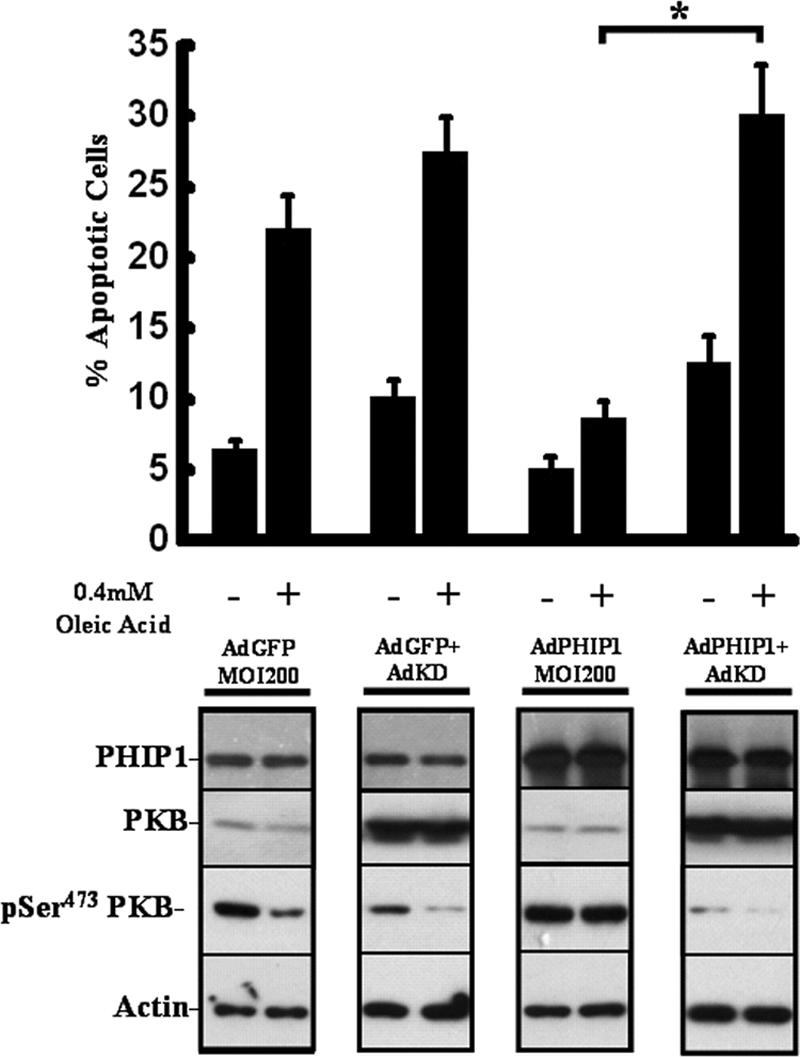
Ad-mediated overexpression of “kinase-dead” PKB blocks the protective effect of PHIP1 on FFA-induced INS-1 cell apoptosis. INS-1 cells were infected into six-well plates with AdGFP (MOI, 200), AdPHIP1 (MOI, 200), AdGFP (MOI, 200) plus AdKD (MOI, 600), or AdPHIP1 (MOI, 200) plus AdKD (MOI, 600). Sixteen hours postinfection, cells were incubated for 15 h with 15 mM glucose in the presence of 0.5% BSA alone or 0.4 mM OA-0.5% BSA. The incidence of apoptosis was assayed as described in Materials and Methods. Results are shown as the means ± SD of three independent experiments. *, P < 0.05 comparing results with AdPHIP1 to results with AdPHIP1 and AdKD in the absence of IGF-1. Lower panel, immunoblot analysis for PHIP1, phospho-Ser473 PKB (pSer473 PKB), and actin in cells treated as indicated above. The immunoblots are representative of two independent experiments. +, present; −, absent.
DISCUSSION
This study is the first to report and functionally characterize the long isoform of hPHIP (PHIP1). The ORF of PHIP1 is 5,530 bp and the conceptual translation predicts an 1,821-aa protein with a molecular mass of 206 kDa. The encoded protein contains a number of structural motifs, including eight WD40 repeats at the N terminus and two tandem bromodomains. WD40 repeats span a broad spectrum of important biological functions in eukaryotes. Proteins containing this motif often act as scaffolds facilitating protein-protein interactions. Such proteins serve as regulatory components of multiprotein complexes that participate in a wide variety of molecular and cellular processes, including mRNA splicing, transcriptional repression or activation, chromatin remodeling, pre-mRNA and pre-rRNA processing, cell cycle regulation, signal transduction, apoptosis, and ubiquitination (27, 30).
The characterization of bromodomains as acetyl-lysine binding motifs has led to their recognition as key modules involved in targeting the molecules that harbor them to areas of active chromatin (8, 11, 35). Proteins possessing bromodomains are known to function as transcriptional coactivators, transcription factors, histone acetyltransferases, and chromatin remodeling proteins. The observation that PHIP1 contains two bromodomains evokes the possibility that it too may be involved in similar cellular processes. Consistent with this idea, we demonstrate that PHIP1 is localized in the nucleus of insulinoma and primary β cells, where it functions as a transcriptional activator. It is noteworthy to mention that the distribution pattern of PHIP in skeletal-muscle cells differs from that in β cells in that PHIP is found in both cytosolic and nuclear compartments in the former, whereas it is only nuclear in the latter. As such, the PHIP protein interaction networks in these two cell types will be determined by differences in their intracellular distribution.
We have demonstrated that PHIP1 is capable of activating the cyclin D2 promoter in INS-1 cells both in the absence and presence of IGF-1 stimulation. Our results illustrate that the cell cycle regulator cyclin D2 is a direct target for PHIP1. D-type cyclins are critical modulators of β-cell growth (23). β cells primarily express three distinct isoforms of D-type cyclins (D1, D2, and D3), with cyclin D2 showing the highest expression levels in mouse islets. Targeted disruption of cyclin D2 in mice results in decreased levels of β-cell proliferation and islet mass and impaired glucose tolerance that progresses to frank diabetes by 12 months of age (23).
The transcriptional activation of cyclin D2 by the overexpression of PHIP1 suggests that PHIP1 may be involved in the induction of long-term gene expression programs to promote the growth and differentiation of β cells. Although the mechanism of cyclin D2 regulation in islets is largely unknown, evidence suggests that STAT5 regulates cyclin D2 expression via an interaction with its promoter (12). It is thus possible that PHIP1 also drives cellular proliferation via an indirect effect on the activation of STAT5, although further experimental work is required to address this hypothesis. It is worth mentioning that qPCR analysis showed that STAT5 levels of mRNA are increased following PHIP1 overexpression (data not shown), a fact that further supports this concept. Moreover, the pronounced increase in cyclin D2 protein levels may also reflect the direct stimulatory effect of PHIP1-induced phospho-PKB on protein translation or stability (7, 26).
We also provide evidence demonstrating that PHIP1 is essential for IGF-1- and IRS2-induced β-cell replication. Indeed, the significance of PHIP1 in particular for amplifying signaling events leading to β-cell mitogenesis was highlighted by our findings that Ad-mediated PHIP1 overexpression in INS-1 cells led to more than a twofold enhancement in glucose- and IGF-1-induced DNA synthesis. Moreover, using siRNA to decrease endogenous PHIP1 levels in INS-1 cells led to a complete blockade of IRS2-mediated DNA synthesis, providing for a specific role of PHIP1 in the enhancement of IRS2-dependent β-cell growth. The involvement of the IGF-1 receptor in the stimulation of β-cell proliferation largely stems from studies in pancreatic β-cell line models (6). In these studies, IGF-1 is crucial in the potentiation of glucose-dependent β-cell mitogenesis (34). However, a recent study by Kulkarni and coworkers (22) clearly demonstrates that β-cell-specific IGF-1 receptor knockout does not affect β-cell growth. Thus, it is now apparent that there exist IGF-1-independent pathways, such as placental lactogen and growth hormone, that contribute to pancreatic β-cell growth and development,. In this context, PHIP1 may promote β-cell proliferation in vivo by impinging on the signaling pathways of these growth factors independent of IGF-1.
It is well recognized that type 2 diabetes and the progressive deterioration of β-cell function are attributed to chronic hyperlipidemia and/or hyperglycemia. In particular, an elevation of the level of nonesterified FFA in models of chronic lipotoxicity leads to impaired insulin secretion at stimulatory glucose concentrations. In addition, FFAs inhibit insulin gene expression (18), induce oxidative stress (20), and increase β-cell death in vitro and in vivo (2). A central player in the glucose- and mitogen-induced effect on β-cell proliferation is PKB, the cellular homologue of the viral oncoprotein v-Akt (34). The inhibitory effect of FFAs on β-cell proliferation is primarily attributed to a significant inhibition of PKB activation, leading to a decrease in cell growth and an increase in apoptosis. In our study, overexpression of PHIP1 led to a marked protection against FFA-induced apoptosis. This effect was shown to be attributed to enhanced phosphorylation of PKB residues Ser473 and Thr308, with no changes in the total level of PKB. Notably, blocking PKB activity by overexpressing a “kinase dead” form led to abrogation of the PHIP1 protective effect on FFA-induced apoptosis in INS-1 cells, suggesting that PKB is a critical component of PHIP1's action in β-cell survival. Our findings further indicate that the cytoprotective effects of PHIP1 involve the inhibition of initiator caspase-9 and downstream effector caspase-3, likely mediated through the activation of PKB. Procaspase-9 has been shown to be phosphorylated by PKB on Ser196, an event that hinders its proteolytic activation to caspase-9. A general cytoprotective role for PHIP1 was also demonstrated by its ability to prevent cytotoxicity induced by serum starvation in the non-β-cell line NIH 3T3 (data not shown).
An important question arising from our study concerns the mechanism(s) by which PHIP1 mediates its antiapoptotic effects on β cells. IRS2, for instance, mediates many of its effects through activation of the PI3-kinase cascade, whereby the inositol phospholipids PI(3,4)P2 and PI(3,4,5)P3 recruit PDK1 and PKB to the plasma membrane and trigger downstream signaling cascades. The absence of any effect of siRNA-mediated knockdown of PHIP1 on IGF-1- or IRS2-induced activation of PKB suggests that PHIP1 may be acting downstream of IRS2 and PKB in β cells.
One possible mechanism by which PHIP1 mediates its effects on PKB activation that lead to enhanced protection from lipotoxic stress in pancreatic β cells may be via the transcriptional repression of negative regulators of PKB, such as the lipid phosphatase and tensin homologue on chromosome 10 (PTEN), and/or the SH2-domain-containing inositol 5-phosphatase 2 (SHIP2) (3). PTEN is a well-documented inhibitor of the PI3-kinase/PKB signaling cascade. Targeted inactivation of PTEN via antisense knockdown in db/db and ob/ob mice reverses the hyperglycemia and ameliorates insulin resistance in these mouse models of type 2 diabetes. Additionally, pancreas-specific deletion of PTEN in mouse islets leads to enhanced phosphorylation of PKB and increased islet mass (24). However, our data indicate that the overexpression of PHIP1 does not modulate PTEN or SHIP2 transcript levels (Fig. 8). Alternatively, we cannot exclude the possibility that PHIP1 overexpression leads to a transcriptional enhancement of positive regulators of PKB, such as the mTORC2 kinase complex, which was recently demonstrated to be required for the phosphorylation of PKB on Ser473 (3), although our initial investigation did not show statistically significant changes in mTOR steady-state mRNA levels following PHIP1 overexpression (Fig. 8). The precise mechanism by which PHIP1 signals in the nucleus converge on PKB survival pathways awaits further study of the transcriptional networks regulated by PHIP1 in pancreatic β cells.
FIG. 8.
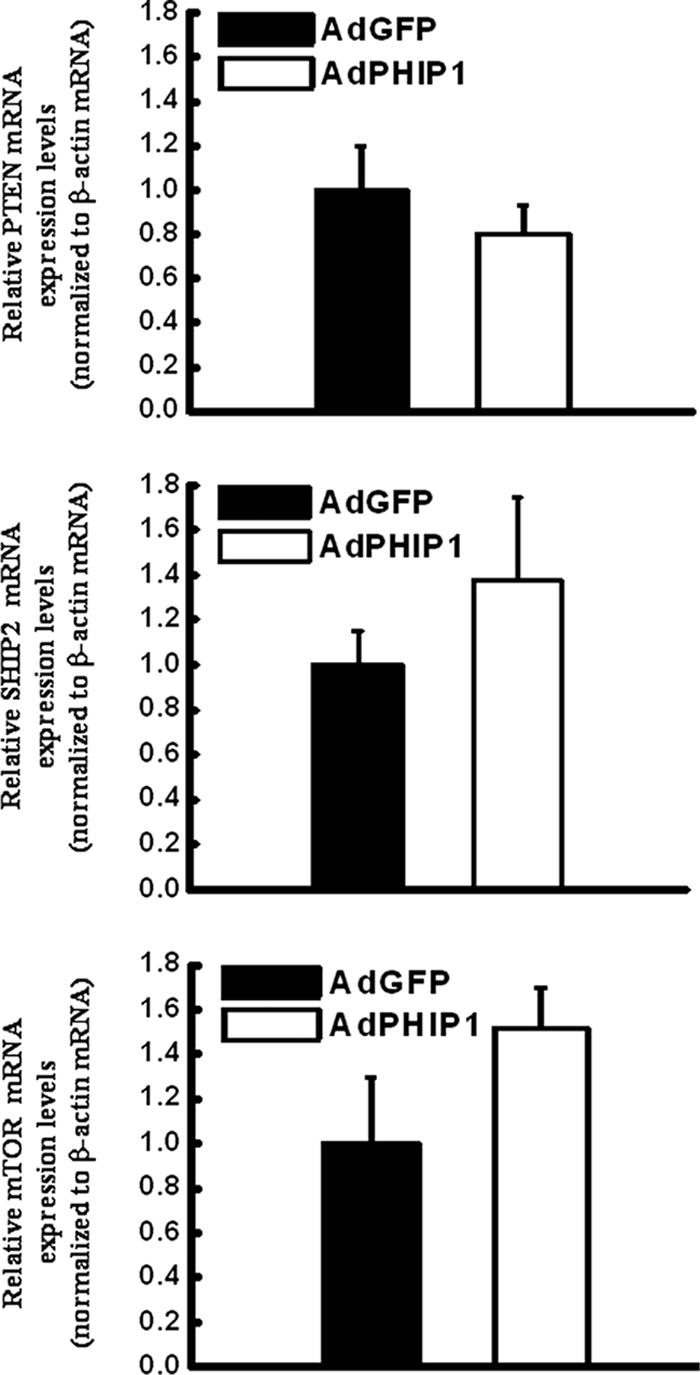
Effect of PHIP1 on mRNA levels of PTEN, SHIP2, and mTOR in INS-1 cells. INS-1 cells were infected into six-well plates with AdGFP and AdPHIP1 (MOI, 400). Sixteen hours postinfection, cells were incubated for 15 h with 15 mM glucose in the presence of 0.5% BSA and collected for qPCR analysis. Experiments were performed three times in duplicate. Results are shown as the means ± SD.
In summary, we have identified a WD40 repeat-containing isoform of PHIP as a novel regulator of β-cell growth and survival. The overexpression of PHIP1 in β cells results in enhanced levels of proliferation and decreased apoptosis. The PHIP signaling network seems to regulate survival and apoptosis through a variety of intracellular mediators, such as caspases, D-type cyclins, and the master regulator PKB. PHIP1 appears to be a new physiological regulator of IRS2 signaling in pancreatic β cells. This implies that a critical expression level of PHIP1 is required for general β-cell survival. Thus, approaches that promote PHIP1 expression in β cells could provide important treatments for β-cell failure and diabetes.
Acknowledgments
This work was supported by a grant from the National Cancer Institute of Canada to Maria Rozakis-Adcock. George Bikopoulos is the recipient of an NSERC postgraduate scholarship.
We thank Chris Rhodes for adenoviruses expressing IRS2 and KD-PKB and M. B. Wheeler, H. Y. Gaisano, J. Diao, and Patrick P. L. Lam for providing β-cell lines, MIP-GFP mice, and reagents.
Footnotes
Published ahead of print on 16 July 2007.
REFERENCES
- 1.Allison, J., H. E. Thomas, T. Catterall, T. W. Kay, and A. Strasser. 2005. Transgenic expression of dominant-negative Fas-associated death domain protein in beta cells protects against Fas ligand-induced apoptosis and reduces spontaneous diabetes in nonobese diabetic mice. J. Immunol. 175:293-301. [DOI] [PubMed] [Google Scholar]
- 2.Biden, T. J., D. Robinson, D. Cordery, W. E. Hughes, and A. K. Busch. 2004. Chronic effects of fatty acids on pancreatic beta-cell function: new insights from functional genomics. Diabetes 53(Suppl. 1):S159-S165. [DOI] [PubMed] [Google Scholar]
- 3.Cohen, P. 2006. The twentieth century struggle to decipher insulin signalling. Nat. Rev. Mol. Cell Biol. 7:867-873. [DOI] [PubMed] [Google Scholar]
- 4.Cokol, M., R. Nair, and B. Rost. 2000. Finding nuclear localization signals. EMBO Rep. 1:411-415. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Dickson, L. M., M. K. Lingohr, J. McCuaig, S. R. Hugl, L. Snow, B. B. Kahn, M. G. Myers, Jr., and C. J. Rhodes. 2001. Differential activation of protein kinase B and p70(S6)K by glucose and insulin-like growth factor 1 in pancreatic beta-cells (INS-1). J. Biol. Chem. 276:21110-21120. [DOI] [PubMed] [Google Scholar]
- 6.Dickson, L. M., and C. J. Rhodes. 2004. Pancreatic beta-cell growth and survival in the onset of type 2 diabetes: a role for protein kinase B in the Akt? Am. J. Physiol. Endocrinol. Metab. 287:E192-E198. [DOI] [PubMed] [Google Scholar]
- 7.Diehl, J. A., M. Cheng, M. F. Roussel, and C. J. Sherr. 1998. Glycogen synthase kinase-3beta regulates cyclin D1 proteolysis and subcellular localization. Genes Dev. 12:3499-3511. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Dyson, M. H., S. Rose, and L. C. Mahadevan. 2001. Acetyllysine-binding and function of bromodomain-containing proteins in chromatin. Front. Biosci. 6:D853-D865. [DOI] [PubMed] [Google Scholar]
- 9.Farhang-Fallah, J., V. K. Randhawa, A. Nimnual, A. Klip, D. Bar-Sagi, and M. Rozakis-Adcock. 2002. The pleckstrin homology (PH) domain-interacting protein couples the insulin receptor substrate 1 PH domain to insulin signaling pathways leading to mitogenesis and GLUT4 translocation. Mol. Cell. Biol. 22:7325-7336. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Farhang-Fallah, J., X. Yin, G. Trentin, A. M. Cheng, and M. Rozakis-Adcock. 2000. Cloning and characterization of PHIP, a novel insulin receptor substrate-1 pleckstrin homology domain interacting protein. J. Biol. Chem. 275:40492-40497. [DOI] [PubMed] [Google Scholar]
- 11.Filetici, P., P. Ornaghi, and P. Ballario. 2001. The bromodomain: a chromatin browser? Front. Biosci. 6:D866-D876. [DOI] [PubMed] [Google Scholar]
- 12.Friedrichsen, B. N., H. E. Richter, J. A. Hansen, C. J. Rhodes, J. H. Nielsen, N. Billestrup, and A. Moldrup. 2003. Signal transducer and activator of transcription 5 activation is sufficient to drive transcriptional induction of cyclin D2 gene and proliferation of rat pancreatic beta-cells. Mol. Endocrinol. 17:945-958. [DOI] [PubMed] [Google Scholar]
- 13.Gillespie, K. M. 2006. Type 1 diabetes: pathogenesis and prevention. Can. Med. Assoc. J. 175:165-170. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Hara, M., X. Wang, T. Kawamura, V. P. Bindokas, R. F. Dizon, S. Y. Alcoser, M. A. Magnuson, and G. I. Bell. 2003. Transgenic mice with green fluorescent protein-labeled pancreatic beta-cells. Am. J. Physiol. Endocrinol. Metab. 284:E177-E183. [DOI] [PubMed] [Google Scholar]
- 15.Hennige, A. M., D. J. Burks, U. Ozcan, R. N. Kulkarni, J. Ye, S. Park, M. Schubert, T. L. Fisher, M. A. Dow, R. Leshan, M. Zakaria, M. Mossa-Basha, and M. F. White. 2003. Upregulation of insulin receptor substrate-2 in pancreatic beta cells prevents diabetes. J. Clin. Investig. 112:1521-1532. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Itoh, T., and T. Takenawa. 2002. Phosphoinositide-binding domains: functional units for temporal and spatial regulation of intracellular signalling. Cell. Signal. 14:733-743. [DOI] [PubMed] [Google Scholar]
- 17.Jhala, U. S., G. Canettieri, R. A. Screaton, R. N. Kulkarni, S. Krajewski, J. Reed, J. Walker, X. Lin, M. White, and M. Montminy. 2003. cAMP promotes pancreatic beta-cell survival via CREB-mediated induction of IRS2. Genes Dev. 17:1575-1580. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Kelpe, C. L., P. C. Moore, S. D. Parazzoli, B. Wicksteed, C. J. Rhodes, and V. Poitout. 2003. Palmitate inhibition of insulin gene expression is mediated at the transcriptional level via ceramide synthesis. J. Biol. Chem. 278:30015-30021. [DOI] [PubMed] [Google Scholar]
- 19.Kim, S. J., K. Winter, C. Nian, M. Tsuneoka, Y. Koda, and C. H. McIntosh. 2005. Glucose-dependent insulinotropic polypeptide (GIP) stimulation of pancreatic beta-cell survival is dependent upon phosphatidylinositol 3-kinase (PI3K)/protein kinase B (PKB) signaling, inactivation of the forkhead transcription factor Foxo1, and down-regulation of bax expression. J. Biol. Chem. 280:22297-22307. [DOI] [PubMed] [Google Scholar]
- 20.Koshkin, V., X. Wang, P. E. Scherer, C. B. Chan, and M. B. Wheeler. 2003. Mitochondrial functional state in clonal pancreatic beta-cells exposed to free fatty acids. J. Biol. Chem. 278:19709-19715. [DOI] [PubMed] [Google Scholar]
- 21.Kulkarni, R. N. 2005. New insights into the roles of insulin/IGF-I in the development and maintenance of beta-cell mass. Rev. Endocr. Metab. Disord. 6:199-210. [DOI] [PubMed] [Google Scholar]
- 22.Kulkarni, R. N., M. Holzenberger, D. Q. Shih, U. Ozcan, M. Stoffel, M. A. Magnuson, and C. R. Kahn. 2002. Beta-cell-specific deletion of the Igf1 receptor leads to hyperinsulinemia and glucose intolerance but does not alter beta-cell mass. Nat. Genet. 31:111-115. [DOI] [PubMed] [Google Scholar]
- 23.Kushner, J. A., M. A. Ciemerych, E. Sicinska, L. M. Wartschow, M. Teta, S. Y. Long, P. Sicinski, and M. F. White. 2005. Cyclins D2 and D1 are essential for postnatal pancreatic β-cell growth. Mol. Cell. Biol. 25:3752-3762. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Lazar, D. F., and A. R. Saltiel. 2006. Lipid phosphatases as drug discovery targets for type 2 diabetes. Nat. Rev. Drug Discov. 5:333-342. [DOI] [PubMed] [Google Scholar]
- 25.Mohanty, S., G. A. Spinas, K. Maedler, R. A. Zuellig, R. Lehmann, M. Y. Donath, T. Trub, and M. Niessen. 2005. Overexpression of IRS2 in isolated pancreatic islets causes proliferation and protects human beta-cells from hyperglycemia-induced apoptosis. Exp. Cell Res. 303:68-78. [DOI] [PubMed] [Google Scholar]
- 26.Muise-Helmericks, R. C., H. L. Grimes, A. Bellacosa, S. E. Malstrom, P. N. Tsichlis, and N. Rosen. 1998. Cyclin D expression is controlled post-transcriptionally via a phosphatidylinositol 3-kinase/Akt-dependent pathway. J. Biol. Chem. 273:29864-29872. [DOI] [PubMed] [Google Scholar]
- 27.Neer, E. J., C. J. Schmidt, R. Nambudripad, and T. F. Smith. 1994. The ancient regulatory-protein family of WD-repeat proteins. Nature 371:297-300. [DOI] [PubMed] [Google Scholar]
- 28.Prentki, M., and C. J. Nolan. 2006. Islet beta cell failure in type 2 diabetes. J. Clin. Investig. 116:1802-1812. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Salsali, A., and M. Nathan. 2006. A review of types 1 and 2 diabetes mellitus and their treatment with insulin. Am. J. Ther. 13:349-361. [DOI] [PubMed] [Google Scholar]
- 30.Smith, T. F., C. Gaitatzes, K. Saxena, and E. J. Neer. 1999. The WD repeat: a common architecture for diverse functions. Trends Biochem. Sci. 24:181-185. [DOI] [PubMed] [Google Scholar]
- 31.Stumvoll, M., B. J. Goldstein, and T. W. van Haeften. 2005. Type 2 diabetes: principles of pathogenesis and therapy. Lancet 365:1333-1346. [DOI] [PubMed] [Google Scholar]
- 32.Wadman, I. A., H. Osada, G. G. Grutz, A. D. Agulnick, H. Westphal, A. Forster, and T. H. Rabbitts. 1997. The LIM-only protein Lmo2 is a bridging molecule assembling an erythroid, DNA-binding complex which includes the TAL1, E47, GATA-1 and Ldb1/NLI proteins. EMBO J. 16:3145-3157. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.White, M. F. 2006. Regulating insulin signaling and beta-cell function through IRS proteins. Can. J. Physiol. Pharmacol. 84:725-737. [DOI] [PubMed] [Google Scholar]
- 34.Wrede, C. E., L. M. Dickson, M. K. Lingohr, I. Briaud, and C. J. Rhodes. 2002. Protein kinase B/Akt prevents fatty acid-induced apoptosis in pancreatic beta-cells (INS-1). J. Biol. Chem. 277:49676-49684. [DOI] [PubMed] [Google Scholar]
- 35.Zeng, L., and M. M. Zhou. 2002. Bromodomain: an acetyl-lysine binding domain. FEBS Lett. 513:124-128. [DOI] [PubMed] [Google Scholar]