

Abstract
Tumor cells utilize glucose as a primary energy source and require ongoing lipid biosynthesis for growth. Expression of DecR1, an auxiliary enzyme in the fatty acid β-oxidation pathway, is significantly diminished in numerous spontaneous mammary tumor models and in primary human breast cancer. Moreover, ectopic expression of DecR1 in ErbB2/Neu-induced mammary tumor cells is sufficient to reduce levels of ErbB2/Neu expression and impair mammary tumor outgrowth. This correlates with a decreased proliferative index and reduced rates of de novo fatty acid synthesis in DecR1-expressing breast cancer cells. Although DecR1 expression does not affect glucose uptake in ErbB2/Neu-transformed cells, sustained expression of DecR1 protects mammary tumor cells from apoptotic cell death following glucose withdrawal. Moreover, expression of catalytically impaired DecR1 mutants in Neu-transformed breast cancer cells restored Neu expression levels and increased mammary tumorigenesis in vivo. These results argue that DecR1 is sufficient to limit breast cancer cell proliferation through its ability to limit the extent of oncogene expression and reduce steady-state levels of de novo fatty acid synthesis. Furthermore, DecR1-mediated suppression of tumorigenesis can be uncoupled from its effects on Neu expression. Thus, while downregulation of Neu expression may contribute to DecR1-mediated tumor suppression in certain cell types, this is not an obligate event in all Neu-transformed breast cancer cells.
Normal cells meet the majority of their energy requirements through oxidative phosphorylation. In contrast, cancer cells produce ATP primarily through aerobic glycolysis, even in the presence of normal oxygen levels (8). Such a metabolic shift to aerobic glycolysis within cancer cells has been termed the “Warburg effect” and represents a fundamental process that supports cancer cell growth (31).
Activation of the Akt pathway in cancer cells is thought to be critical for promoting the Warburg effect (10). Tumor cells that possess constitutively activated Akt display very high glycolytic rates and are strictly dependent on glucose for their survival. The biochemical mechanism by which Akt alters carbohydrate metabolism in cancer cells is beginning to be elucidated with the discovery of several key proteins that are phosphorylated by Akt and function not only to increase glucose uptake but also to promote glycolysis (26).
Tumor cells must also undergo high rates of de novo fatty acid synthesis to maintain a constant supply of membrane phospholipids for continuous cell proliferation (30). Moreover, tumor cells obtain the majority of their fatty acids from de novo synthesis despite the presence of abundant exogenous lipid levels (24, 28). To achieve this, cancer cells convert excess pyruvate generated from glycolysis to citrate. Through the action of ATP citrate lyase (ACL), citrate is then converted to oxaloacetate and cytoplasmic acetyl coenzyme A (acetyl-CoA), the latter representing the major substrate for de novo fatty acid synthesis from carbohydrate sources (30). Consistent with a central role for ACL in this process, stable knockdown of ACL in cancer cells results in decreased acetyl-CoA levels and reduced glucose-dependent lipid synthesis. Tumor cells expressing reduced levels of ACL, in turn, have a reduced proliferative capacity and impaired tumor outgrowth in vivo (14). These observations argue that tumor cells must coordinate energy utilization strategies with lipid biosynthetic processes to support high rates of cell growth and proliferation.
Consistent with this concept, proteomic profiling of breast cancer cells expressing the HER2/Neu receptor tyrosine kinase has revealed elevated expression of proteins that are involved in both glycolysis and de novo lipid synthesis (35). Recent gene expression profiling studies of mammary tumors derived from transgenic mice expressing the HER2/Neu oncogene have revealed that a key enzyme involved in the auxiliary pathway of fatty acid β-oxidation, known as 2,4-dienoyl-CoA reductase (DecR1), is repressed during mammary tumor development (19, 20). DecR1 catalyzes the rate-limiting step in a process that prepares polyunsaturated fatty acids to be utilized as substrates for β-oxidation (12). The observation that DecR1 deficiency in humans is lethal, which results in the buildup of fatty acid intermediates due to incomplete β-oxidation and respiratory acidosis (27), demonstrates that DecR1 catalyzes an essential step for the β-oxidation of polyunsaturated fatty acids. Given the requirement for a continuous supply of fatty acids to support ongoing cell proliferation (14), it is conceivable that downregulation of DecR1 would enable efficient phospholipid biosynthesis, which is required for tumor cell proliferation.
Here we demonstrate that DecR1 expression is dramatically reduced in a number of primary murine and human breast cancers by comparison to normal mammary epithelium. Reexpression of DecR1 in ErbB2-expressing mammary tumor cells results in a dramatic suppression of tumor growth that is associated with the coordinate downregulation of ErbB2, a diminished proliferative index, and lower rates of de novo fatty acid synthesis. These observations suggest that modulation of DecR1 expression may play a critical role in coordinating cell transformation and energy utilization.
MATERIALS AND METHODS
Materials.
The full-length rat DecR1 cDNA was isolated from the UniZapXR rat liver cDNA library (Stratagene) and inserted, via TOPO/TA cloning, into pEF6/V5-His (Invitrogen) to generate pEF6/DecR1-V5. The DecR1(N148A) and DecR1(K214A) mutants were generated using the QuikChange XL site-directed mutagenesis kit (Stratagene) and the following primers: for DecR1(N148A), GATAAACAACGCGGCAGGGGCCTTCATTTCCCCCAGTGAGAG and CTCTCACTGGGGGAAATGAAGGCCCCTGCCGCGTTGTTTATC, and for DecR1(K214A), CCAAGTTCTTCAGCCGCATCAGGCGTGGAAGCC and GGCTTCCACGCCTGATGCGGCTGAAGAACTTGG. Both cDNAs were sequenced to confirm the presence of the point mutation and to ensure that no unwanted mutations were introduced.
NMuMG ells were cultured in Dulbecco's modified Eagle's medium (DMEM), 10% fetal calf serum (FCS), 10 μg/ml insulin, 20 mM HEPES, pH 7.5, at 37°C and 5% CO2. TM15 cells were isolated from the breast tumor of an MMTVCre/floxneoNeuNT mouse (4) and were cultured in DMEM, 10% FCS, 5 μg/ml insulin, 10 ng/ml epidermal growth factor, 1 μg/ml hydrocortisone, and 35 μg/ml bovine pituitary extract. NMuMG cells were obtained from the ATCC and transfected with NeuNT/pMSCVpuro. Puromycin-resistant cells were injected into the mammary fat pads of nude mice, and tumors were explanted back into culture in the presence of 1 μg/ml puromycin (NMuMG-NT2196). NMuMG-NT2196 and TM15 mammary epithelial cells were transfected with pEF6/DecR1-V5 and selected in the presence of 8 μg/ml blasticidin. A peptide corresponding to amino acids 315 to 335 of the rat DecR1 protein was injected into rabbits to generate rabbit polyclonal DecR1 antiserum (Covance).
TMA construction, staining, scoring criteria, and image processing.
Duplicate core (0.6-mm) tissue microarrays (TMAs) were constructed to assess the expression of DecR1 in benign and cancer formalin-fixed and paraffin-embedded human breast tissue specimens. The benign breast array included tissues from 29 cases of radial scar, 34 cases of sclerosing adenosis, 12 cases of duct adenoma, 15 cases of nipple duct adenoma, 27 cases of papilloma, two cases of tubular adenoma, and one case each of lactating adenoma and adenomyoepithelioma. The breast cancer array contained tissues from 438 cases of invasive breast carcinoma that were referred to Vancouver General Hospital between 1974 and 1995 (21).
Sections from TMA were cut at 4 μm and immunostained with our rabbit polyclonal DecR1 antibody. Antigen retrieval and detection were performed using a Ventana Systems Discovery XT automated immunostainer at a 1:50 dilution. Slides were counterstained with hematoxylin and mounted. Slides for both arrays were scored according to the intensity of cytoplasmic staining as follows: negative (0), if there was no staining, and weak (+1), moderate (+2), and strong (+3) positive staining. The stained slides were digitally scanned with a BLISS automated digital imaging microscope (Bacus Laboratories, Lombard, IL) and are accessible through https://www.gpecimage.ubc.ca/tma/web/viewer.php. The slides were scored manually by a pathologist, blinded to the clinical outcome as previously described (7).
Immunofluorescence.
Immunofluorescent staining was performed as previously described (15). Cells were stained with E-cadherin (610181; BD Biosciences; 1:200) and ZO-1 (61-7300; Zymed; 1:200) antibodies followed by Alexa Fluor 555 and Alexa Fluor 488 secondary antibodies (Molecular Probes). Cells were visualized on a Zeiss LSM 510 Meta confocal microscope.
Glucose studies.
Glucose levels in the medium were determined using the Amplex Red glucose assay kit (Molecular Probes). Cells were seeded at a density of 1 × 105 cells/ml in 24-well plates in 1% FCS. Medium was collected from each well, and cells were supplied with fresh medium every 24 h. The assay was performed along with a glucose standard.
Cells were serum starved (0.2% FCS) and cultured in medium containing either a high level of glucose (4.5 g/liter) or no glucose (Invitrogen). Cells lacking glucose were also cultured in the absence or presence of 1 mM AICAR (5-aminoimidazole-4-carboxamide ribonucleoside; Toronto Research Chemicals Inc.). The percentage of early apoptotic cells was determined by staining with annexin V-phycoerythrin (BD Pharmingen), and positively stained cells were detected by flow cytometry using a FACSCalibur (BD Biosciences).
In vivo tumorigenesis assay.
Cells (5 × 104) were injected into the fourth mammary fat pad of 8-week-old athymic Ncr nude female mice (NIH). Tumor growth was monitored by biweekly caliper measurements. Tumor volumes (mm3) were calculated as follows: 4/3π(w2 × l). At necropsy, mammary tumors were fixed in 10% buffered paraformaldehyde or flash frozen in liquid nitrogen. All animal studies were approved by the Animal Resources Centre at McGill University and comply with guidelines set by the Canadian Council of Animal Care.
Immunohistochemical staining was performed on mammary tumor sections as previously described (32). Sections were incubated first with a PCNA-specific antibody (catalog no. N1529; Dakocytomation) and then with the Elite anti-mouse immunoglobulin G Vectastain ABC kit (catalog no. PK-6102; Vector). To quantify the percentage of PCNA-positive cells, five independent fields (×400 magnification) were scored using Image J software.
Real-time PCR, immunoblotting, and immunoprecipitation.
Real-time PCR was performed using the SYBR green I RNA amplification kit (Roche) (see Fig. 1A) or the QuantiTect SYBR green real-time PCR kit (QIAGEN) (see Fig. 2C) according to the manufacturer's instructions. The primers used were decR1 (ACCGTGGTCTTCCACTTGTC and TGCCCCTTTTTGTTTTTCAC), gapdh (CATCAAGAAGGTGGTGAAGC and GGGAGTTGCTGTTGAAGTCG), e-cadherin (ACTGTGAAGGGACGCTCAAC and TGTCCCGGGTATCATCATCT), neuNT (GTGCTAGACAACCGAGATCCTCAGG and CCCTTCAGGATCTCTGTGAGACTTCG), and β2-microglobulin (TGGTGCTTGTCTCACTGACC and AGTTCAGTATGTTCGGCTTCC).
FIG. 1.

DecR1 expression is repressed in transgenic mouse-derived mammary tumors and primary human breast cancer. (A) Quantification of decR1 mRNA levels in mammary glands from wild-type mice (FVB/N) and mammary tumors derived from MMTV/PyVmT, MMTV/NeuNDL2-5, MMTV/NeuYB, and MMTV/NeuYD transgenic mice by real-time PCR. The values were normalized either to gapdh (top) or to e-cadherin (bottom). Two independent mammary tumors from each transgenic line were analyzed. (B) Endogenous DecR1 protein levels in FVB/N mammary gland and PyVmT, NeuNDL2-5, NeuYB, and NeuYD mammary tumors were determined by immunoblot analysis of whole-cell lysates using DecR1- and α-tubulin-specific antibodies. (C) Immunohistochemical staining of primary human benign (n = 109) and breast cancer (n = 306) TMAs using a DecR1-specific antibody. The intensity of DecR1 cytoplasmic staining is represented as negative, weak, moderate, or strong. (D) Representative images of TMA images as described in panel C shown at a ×200 magnification.
FIG. 2.

DecR1 overexpression partially restores cell contacts in a Neu-transformed breast cancer cell line. (A) Immortalized NMuMG mouse mammary epithelial cells were stably transfected with NeuNT, pooled stable transfectants were injected into the mammary fat pads of nude mice, and tumor cells were explanted back into culture (NMuMG-NT2196). Parental NMuMG, NMuMG/NeuNT-expressing pooled stable transfectants, and NMuMG-NT2196 cells (5 × 104) were injected into the mammary fat pads of nude mice. The data are represented as tumor volumes (mm3) ± standard deviations (n = 4 mice). (B) Generation of NT2196 cells stably expressing DecR1-V5. Whole-cell lysates from NMuMG, NT2196, and three independent NT2196/DecR1-V5 stable cell lines (NT2196/DecR1.1, NT2196/DecR1.7, and NT2196/DecR1.8) were subjected to immunoblot analysis using V5-, Neu-, and α-tubulin-specific antibodies. (C) Quantification of neuNT mRNA levels in NMuMG, NT2196, NT2196/DecR1.1, NT2196/DecR1.7, and NT2196/DecR1.8 cells by real-time PCR. The values were normalized to β2-microglobulin. The values are representative of triplicate samples. (D) DecR1 overexpression partially restores cell contacts in NeuNT-transformed cells. Shown are bright-field images of the indicated cells at confluence (left panels). Cells were also stained with E-cadherin (red) and ZO-1 (green) antibodies and analyzed by confocal microscopy (right panels).
In vitro-cultured cells or flash-frozen tumor tissue was lysed in phospholipase Cγ lysis buffer (9). Lysates were analyzed by immunoblotting using the following antibodies: DecR1 (1:2,500), V5 (R960-25; Invitrogen; 1:5,000), Neu (sc-284; Santa Cruz; 1:1,000), GLUT1 (sc-7903; Santa Cruz; 1:1,000), and α-tubulin (T9026; Sigma; 1:5,000). The blots were incubated with horseradish peroxidase-conjugated secondary antibodies (Jackson Labs; 1:10,000 dilution) and visualized by ECL (Amersham).
De novo lipid synthesis assay.
De novo lipid synthesis was assayed as previously described (6). Briefly, cells were plated in 48-well plates, grown to 80 to 90% confluence, and cultured in serum-free DMEM for 2 h. Cells were labeled with [3H]acetate or [3H]oleic acid for 4 h, and then total lipids were extracted and incorporated radioactivity was measured by scintillation counting. Phosphatidylcholine and sphingomyelin were separated by thin-layer chromatography using lipid standards and visualized with iodine vapor. The lipids were scraped from the thin-layer chromatography plate, and samples were counted in a scintillation counter.
RESULTS
Reduced DecR1 expression in breast cancer cells.
To determine whether DecR1 expression was regulated during mammary tumorigenesis, we compared DecR1 mRNA and protein levels in control FVB mammary glands with mammary tumors derived from transgenic mice expressing polyomavirus middle T antigen (MT), activated Neu (NDL2-5), or two oncogenic Neu variants that signal through the Grb2 (YB) or Shc (YD) adaptor protein (9, 13, 29). Indeed, decR1 mRNA levels were significantly reduced in all mammary tumors relative to wild-type mammary glands. decR1 transcript levels were fivefold lower when normalized to gapdh and 12-fold lower following normalization to e-cadherin (Fig. 1A). DecR1 protein levels were also correspondingly decreased in both Neu- and MT-induced tumors relative to normal mammary epithelium (Fig. 1B). To determine whether DecR1 is regulated in primary human breast cancer, immunohistochemical staining of benign (109 cases) and breast cancer (306 cases) TMAs was performed using a DecR1-specific antibody. In the benign breast array, the majority of sections displayed moderate (51.4%) or strong (41.3%) DecR1 cytoplasmic staining, while only a small fraction of the samples (7.3%) were weakly DecR1 positive (Fig. 1C and D). In contrast, the frequency of negative (6.9%) or weak (35.3%) DecR1 staining was significantly increased in the breast cancer array, while moderate (35.6%) and strong (22.2%) DecR1 staining of the tumor cytoplasm was correspondingly decreased (Fig. 1C and D). Decreased DecR1 staining in the breast cancer array versus the benign array is statistically significant to a 99% confidence interval. This demonstrates that repression of DecR1 expression during mammary tumorigenesis is a common event that is observed both in transgenic mouse models and in primary human breast cancer.
DecR1 overexpression impairs Neu-mediated proliferation and tumorigenesis.
To address the impact of DecR1 overexpression on mammary tumorigenesis, we transfected immortalized NMuMG mammary epithelial cells with an oncogenic Neu receptor (NeuNT) and injected the cells into the mammary fat pads of nude mice. One tumor was explanted in culture (NT2196) to generate an established cell line overexpressing Neu that is capable of inducing tumors when injected into the mammary fat pads of recipient animals (Fig. 2A and B). NT2196 cells were observed to lose the organized cobblestone morphology characteristic of NMuMG epithelial cells. This correlates with the mislocalization of proteins found at adherens junctions (E-cadherin) and tight junctions (ZO-1), responsible for the formation of cell contacts (Fig. 2D).
To assess the consequence of ectopic DecR1 expression on Neu-mediated tumorigenesis, we generated NT2196 cells stably expressing V5-tagged DecR1. Three independent stable cell lines (NT2196/DecR1.1, NT2196/DecR1.7, and NT2196/DecR1.8) were selected for further analysis (Fig. 2B). DecR1-expressing cells were morphologically distinct from NT2196 cells and more closely resembled the cobblestone appearance of NMuMG cells, including the partial relocalization of E-cadherin and ZO-1 to the membrane and restoration of cell contacts (Fig. 2D). Coincident with these changes, DecR1-V5 expression diminished Neu expression levels in NT2196 cells (Fig. 2B). Moreover, NT2196/DecR1.8 cells, which express the lowest levels of exogenous DecR1, display a partial reduction of Neu expression compared to the high DecR1 expressors (Fig. 2B). This decrease in Neu expression is not associated with a corresponding reduction in neu transcript levels in NT2196/DecR1-expressing cells (Fig. 2C). This suggests that ectopic expression of DecR1 induces Neu downregulation in NT2196 cells at the posttranscriptional level.
To determine whether DecR1 overexpression affects Neu-mediated tumorigenesis in vivo, NMuMG, NT2196, and DecR1-expressing NT2196 cells were injected into the mammary fat pads of athymic mice and monitored for tumor formation. DecR1-expressing cells showed a two- to fourfold decrease in tumor volume compared to NT2196 cells, demonstrating that DecR1 overexpression is sufficient to significantly limit the extent and severity of tumor outgrowth in all three stable cell lines examined (Fig. 3A; P = 0.007 to 0.05). DecR1-V5 expression is retained in the majority of breast tumor samples, and all DecR1-expressing tumors exhibited decreased Neu levels compared to NT2196-derived tumors, with the most significant effects observed in the high-expressing DecR1 clones (Fig. 3B). These data suggest that enforced DecR1-V5 expression during tumor outgrowth suppresses Neu expression and hence the extent of Neu signaling, which may contribute to the diminished tumorigenic capacity of NT2196/DecR1-expressing breast cancer cells in vivo.
FIG. 3.

DecR1 overexpression impairs proliferation and tumor formation of NT2196 breast cancer cells. (A) The indicated cell populations (5 × 104 cells) were injected into the mammary fat pads of nude mice, and tumor formation was monitored by biweekly palpation and caliper measurements. The data are represented as tumor volumes (mm3) ± standard deviations (n = 4 mice). Statistical significance was determined using Student's t test. (B) DecR1-V5 expression is retained in NT2196/DecR1-V5 tumors. Whole-cell lysates from NT2196, NT2196/DecR1.1, NT2196/DecR1.7, and NT2196/DecR1.8 mammary tumors were analyzed by immunoblotting using V5-, Neu-, and α-tubulin-specific antibodies. Two independent tumors from each cell type were examined. (C) Immunohistochemical staining of paraffin-embedded breast tumors with PCNA-specific antibodies. For each cell line, three independent tumors were analyzed. Photographs were taken at ×400 magnification, and the percentage of PCNA-positive cells was quantified using Image J software. Data are represented as the percentages of PCNA-positive cells ± standard deviations (n = five independent fields for each tumor). Statistical significance was determined using Student's t test. (D) DecR1-expressing NT2196 cells have a lower proliferative rate. Cells were plated in 0.2% FCS, cells were counted for the indicated times, and the data are represented as numbers of viable cells ± standard deviations (n = 4 wells).
A significantly lower percentage of PCNA-positive cells was observed in all DecR1-expressing tumors compared to NT2196 breast tumors (P = 0.01) (Fig. 3C), without any significant changes in the rates of apoptosis (data not shown). Consistent with their lower rates of tumor outgrowth and proliferation in vivo, DecR1-expressing NT2196 cells also grew more slowly than did NT2196 cells in vitro (two- to fivefold) (Fig. 3D). This suggests that DecR1 overexpression lowers the proliferative capacity of breast cancer cells and contributes to the suppressed outgrowth of Neu-induced mammary tumors.
Elevated DecR1 expression rescues breast cancer cells from their glucose dependency.
Cancer cells possess high glycolytic rates and are strictly dependent on a constant source of glucose for their survival (10). Moreover, mobilization of energy sources through fatty acid β-oxidation is sufficient to protect cancer cells from death following glucose withdrawal (5). To investigate the requirement for glucose in DecR1-expressing NT2196 cells, we first measured glucose uptake in these cells relative to NMuMG and NT2196 cells. In comparison to NT2196 cells, which progressively deplete glucose from the medium, glucose uptake by NMuMG cells is not significantly increased over time. DecR1-expressing NT2196 cells also increasingly deplete glucose from the medium, albeit at a lower rate than that of NT2196 cells (Fig. 4A), which reflects the decreased proliferative capacity of NT2196/DecR1-expressing cells (Fig. 3D). We also examined expression levels of the glucose transporter GLUT1 in NMuMG, NT2196, and NT2196/DecR1-expressing cells. GLUT1 is the most widely expressed glucose transporter in human tumors with a high proliferative capacity (33, 34) and is localized to the cell membrane, unlike other glucose transporters, which localize mostly to cytoplasmic compartments within the cell (23). Indeed, GLUT1 levels are comparable between NT2196 and NT2196/DecR1-expressing cells and modestly decreased in NMuMG cells (Fig. 4B).
FIG. 4.

DecR1-V5-expressing breast cancer cells display increased survival following glucose withdrawal. (A) DecR1 overexpression does not significantly alter the rate of glucose uptake in NT2196 cells. Medium was collected at the indicated times, fresh medium was added daily to ensure that glucose was not limiting, and glucose levels in the medium were determined. Data are represented as glucose levels in medium (mM) ± standard deviations (n = 3 wells). (B) Whole-cell lysates of the indicated cells were subjected to immunoblot analysis with GLUT1- and α-tubulin-specific antibodies. (C) NMuMG, NT2196, and DecR1-expressing cells were cultured for 8 h in 0.2% FCS in medium containing high levels of glucose or no glucose. Whole-cell lysates were probed with DecR1-, Neu-, and α-tubulin-specific antibodies. (D) The percentage of annexin V-positive cells was determined following 1 day in culture medium containing high levels of glucose or no glucose or containing no glucose but supplemented with 1 mM AICAR. Data are represented as percent annexin V-positive cells ± standard deviations (n = 3 wells).
We next determined the effect of glucose availability on the expression levels of Neu and endogenous DecR1 in NMuMG, NT2196, and NT2196/DecR1-expressing cells. Regardless of glucose availability, endogenous DecR1 levels are consistently reduced in NT2196 cells, compared to NMuMG cells, and correspondingly increased in the presence of ectopically expressed DecR1 (Fig. 4C). However, Neu expression levels are even further decreased in DecR1-V5-expressing NT2196 cells following glucose withdrawal (Fig. 4C). This suggests that steady-state levels of Neu, but not endogenous DecR1, are directly linked to glucose availability in DecR1-overexpressing breast cancer cells.
To examine the consequence of elevated DecR1 expression during glucose withdrawal, NMuMG, NT2196, and NT2196/DecR1-expressing cells were cultured under serum starvation conditions in the presence or absence of glucose. Immortalized NMuMG cells display high rates of apoptosis under serum starvation conditions, regardless of glucose availability (Fig. 4D). Consistent with observations made with other cancer cells (10), NT2196 breast cancer cell viability is unaffected by serum starvation in the presence of glucose but is dramatically reduced following glucose withdrawal, indicating that these cells are strictly dependent on glucose for their survival (Fig. 4D; see also Fig. S1 in the supplemental material). While serum starvation may modestly increase the apoptotic rate of NT2196/DecR1-expressing cells (Fig. 4D), cell viability is similar in parental NT2196 and DecR1-expressing cells at steady state (see Fig. S1 in the supplemental material). However, ectopic expression of DecR1 is sufficient to restore viability to NT2196 cells in the absence of glucose (Fig. 4D; see also Fig. S1 in the supplemental material). Addition of the AMP analog AICAR has been previously used to hyperactivate the AMP kinase pathway in cancer cells, which mobilizes fatty acid stores via β-oxidation and increases cell survival following glucose withdrawal (5). AICAR addition was able to rescue the viability of NT2196 cells in the absence of glucose but had little effect on the basal rates of apoptosis observed with the NT2196/DecR1-V5 expressors (Fig. 4D). This suggests that DecR1 may mobilize additional energy sources in NT2196 cells, which would otherwise be unavailable, to rescue them from their glucose dependency.
Reduced de novo lipid synthesis in DecR1-expressing breast cancer cells.
Ongoing de novo lipid synthesis is required for cancer cell proliferation, and perturbation of this pathway has deleterious effects on tumorigenesis (14). Therefore, we asked whether the ability of DecR1 to attenuate Neu-mediated tumorigenesis is linked to its ability to regulate de novo lipid synthesis in breast cancer cells. We first measured the incorporation of [3H]acetate or [3H]oleate, an unsaturated fatty acid and substrate for DecR1, into total cellular lipid over a 4-hour time period under serum starvation conditions. Interestingly, both [3H]acetate (2.5-fold) and [3H]oleate (3.5-fold) incorporation into total lipid was significantly higher in NT2196 cells than in parental NMuMG cells (Fig. 5A and B, top). Elevated DecR1 expression was sufficient to reduce the rate of both [3H]acetate and [3H]oleate incorporation into total lipid to levels that were at or below those observed with NMuMG cells (Fig. 5A and B). In cancer cells, the bulk of de novo-synthesized fatty acids are incorporated into cellular membranes (17, 25). Moreover, biological membranes are comprised of phospholipids and sphingolipids, which are required for ongoing cell proliferation. Therefore, we separated total lipid into phosphatidylcholine (a major membrane phospholipid) and sphingomyelin (a major membrane sphingolipid) by thin-layer chromatography. Compared to NMuMG cells, the degree of de novo sphingomyelin synthesis was increased two- to threefold and de novo phosphatidylcholine synthesis was elevated 3.5-fold in NT2196 cells (Fig. 5A and B). In contrast, [3H]acetate or [3H]oleate incorporation into sphingomyelin (two- to sixfold) and phosphatidylcholine (4- to 17-fold) was significantly reduced in NT2196/DecR1.1 and NT2196/DecR1.7 expressors relative to the NT2196 cell line (Fig. 5A and B). While de novo sphingomyelin synthesis was not significantly reduced in the lower DecR1-V5-expressing clone (NT2196/DecR1.8), we did observe a threefold decrease in de novo phosphatidylcholine synthesis in these cells relative to NT2196 cells (Fig. 5A and B). These results demonstrate that DecR1 overexpression significantly impairs de novo lipid synthesis in breast cancer cells. Reduced phospholipid levels in DecR1-expressing cells may negatively impact new membrane synthesis, which would limit their proliferative capacity and impair tumor outgrowth.
FIG. 5.

Decreased de novo phospholipid synthesis in DecR1-expressing breast cancer cells. (A) Cells were incubated with [3H]oleate for 4 h, lipids were extracted, and [3H]oleate incorporation was measured by scintillation counting. Phosphatidylcholine and sphingomyelin were separated from the total lipid by thin-layer chromatography, and [3H]oleate incorporation was measured by scintillation counting. Data are from two independent experiments and are represented as [3H]oleate incorporation levels (n-fold) ± standard errors of the means (n = 6 wells). (B) Cells were incubated with [3H]acetate for 4 h and analyzed as described in panel A. For each, differences were evaluated by Student's t test. Statistical significance was determined using analysis of variance. We consider our data to be statistically significant when the P value is at or below 0.05.
DecR1 catalytic function is required for its ability to downregulate Neu and impair mammary tumor outgrowth.
To elucidate the potential mechanisms through which DecR1 impairs tumor outgrowth and decreases Neu expression, we generated DecR1 mutants with impaired catalytic activities (3). Mutation of asparagine-148 to an alanine results in a molecule that displays tighter binding to substrate, reduced binding to NADPH cofactor, and only 3% catalytic activity relative to the wild-type protein. A lysine-to-alanine alteration at residue 214 of DecR1 reduces the catalytic activity to 0.03% of that of the wild-type protein without affecting its affinity for substrate or cofactor (3). Therefore, both DecR1(N148A) and DecR1(K214A) behave as dominant-negative proteins.
Stable NT2196 cell lines overexpressing V5-tagged DecR1(N148A) and DecR1(K214A) were generated, and two independent cell lines stably expressing each DecR1 mutant [NT2196/DecR1(N148A)-3,5 and NT2196/DecR1(K214A)-22,23] were selected for further analysis (Fig. 6A). While the N148A mutant comigrates with wild-type DecR1, the K214A mutant appears as a faster-migrating band (Fig. 6A). Both the DecR1(N148A) and DecR1(K214A) cDNAs were sequenced in their entirety and did not contain additional mutations, suggesting that the DecR1(K214A) mutant is lacking a posttranslational modification that is present in both the wild-type and DecR1(N148A) mutant proteins.
FIG. 6.

DecR1 catalytic activity is required for the repression of Neu expression and mammary tumor outgrowth. (A) Generation of NT2196 cells stably expressing DecR1(N148A) and DecR1(K214A) mutants. Whole-cell lysates from the indicated cells were subjected to immunoblot analysis using DecR1-, Neu-, and α-tubulin-specific antibodies. (B) The indicated cell populations (5 × 104 cells) were injected into the mammary fat pads of nude mice, and tumor formation was monitored by biweekly caliper measurements. The data are represented as tumor volumes (mm3) ± standard deviations (n = 5). (C) The indicated cells were incubated with [3H]acetate for 4 h, lipids were extracted, and [3H]acetate incorporation was measured by scintillation counting. Phosphatidylcholine and sphingomyelin were separated from the total lipid by thin-layer chromatography, and [3H]acetate incorporation was measured by scintillation counting. Data are from two independent experiments and are represented as [3H]acetate incorporation (n-fold) ± standard errors of the means (n = 6 wells).
We first characterized the NT2196/DecR1 mutant cell lines for their morphological appearance and Neu expression levels. NT2196 cells overexpressing either the DecR1(N148A) or the DecR1(K214A) mutant lacked cell contacts due to the mislocalization of E-cadherin and ZO-1 (data not shown). Moreover, expression of the DecR1(N148A) or DecR1(K214A) mutant restored Neu levels to that observed in parental NT2196 cells (Fig. 6A). These results demonstrate that DecR1 catalytic function is required for its ability to reduce Neu expression levels in breast cancer cells.
To determine whether DecR1 catalytic activity is required for repression of Neu-mediated tumorigenesis in vivo, NT2196, NT2196/DecR1.7, NT2196/DecR1(N148A), and NT2196/DecR1(K214A) cells were injected into the mammary fat pads of athymic mice and monitored for tumor formation. As shown previously, NT2196/DecR1.7 cells exhibited a fourfold decrease in tumor volume compared to NT2196 cells. Overexpression of either the DecR1(N148A) or DecR1(K214A) mutant partially restored its transforming ability to 40 to 75% of that observed with NT2196 cells (Fig. 6B). In order to determine whether the observed effects on tumor outgrowth correlated with increased lipid levels, we also measured de novo lipid synthesis in the NT2196/DecR1(N148A) and NT2196/DecR1(K214A) mutants relative to the NT2196 cell line. Indeed, [3H]acetate incorporation into both total lipid and phosphatidylcholine was completely restored in NT2196/DecR1(N148A) and NT2196/DecR1(K214A) cells relative to the parental NT2196 cell line (Fig. 6C). Expression of the DecR1 catalytic mutants also increased de novo sphingomyelin synthesis, albeit to variable levels, compared to NT2196 cells (Fig. 6C). These data suggest that DecR1 catalytic function contributes to its ability to repress Neu expression, reduce steady-state phospholipid levels, and impair tumor outgrowth of Neu-transformed breast cancer cells.
DecR1-mediated suppression of mammary tumorigenesis in the absence of Neu downregulation.
To verify that the ability of DecR1 to repress mammary tumorigenesis was not restricted to NMuMG cells, we utilized a spontaneously arising mouse mammary tumor cell line (TM15) derived from a transgenic mouse model that drives mammary cell-specific expression of NeuNT under the control of the endogenous mouse erbB2 promoter (4). Ectopic expression of DecR1-V5 in numerous TM15-derived stable cell lines was sufficient to increase endogenous DecR1 expression levels, in the absence of a corresponding decrease in Neu expression levels relative to parental TM15 cells (Fig. 7A). To determine whether DecR1-mediated suppression of mammary tumorigenesis could be uncoupled from its ability to reduce Neu expression levels, parental and DecR1-expressing TM15 cells were injected into the mammary fat pads of nude mice. Indeed, mammary tumor outgrowth was significantly reduced in four independent DecR1-expressing stable cell lines relative to that observed with parental TM15 cells (Fig. 7B). This suggests that much of DecR1's ability to suppress mammary tumorigenesis may be uncoupled from its ability to reduce Neu expression and most likely results from general effects on fatty acid metabolism and tumor cell proliferation. However, this does not preclude the possibility that DecR1-mediated repression of Neu expression may further contribute to impairment of Neu-mediated breast cancer cell transformation.
FIG. 7.
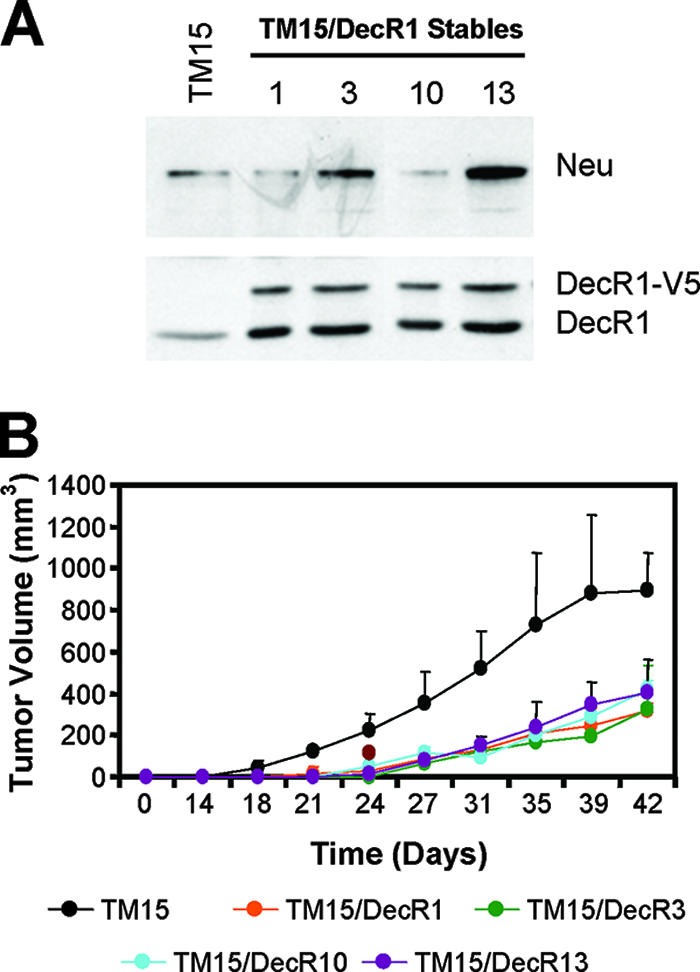
DecR1-mediated suppression of mammary tumorigenesis in the absence of Neu downregulation. (A) Generation of TM15 cells stably expressing DecR1-V5. Whole-cell lysates from parental TM15 and four independent TM15/DecR1-V5 stable cell lines (TM15/DecR1.1, TM15/DecR1.3, TM15/DecR1.10, and TM15/DecR1.13) were subjected to immunoblot analysis using DecR1- and Neu-specific antibodies. (B) The indicated cell populations (2.5 × 105 cells) were injected into the mammary fat pads of nude mice, and tumor formation was monitored by biweekly palpation and caliper measurements. The data are represented as tumor volumes (mm3) ± standard deviations (n = 5 mice).
DISCUSSION
In the present study, we demonstrate that DecR1 overexpression in Neu-transformed breast cancer cells impairs their proliferation and mammary tumor outgrowth. We suggest that these effects may, in certain situations, result from reduced Neu expression and diminished rates of de novo fatty acid synthesis in DecR1-expressing breast cancer cells. However, DecR1 overexpression is sufficient to suppress mammary tumorigenesis in the absence of a corresponding reduction in Neu expression in an independent breast cancer cell line. Thus, DecR1 likely negatively regulates cell proliferation primarily by depriving the cell of necessary phospholipids that are required for cell division. However, this does not preclude additional effects of DecR1 that limit the extent of Neu-mediated signal transduction by downregulating the expression of the transforming oncogene itself (Fig. 8). This is supported by the observation that DecR1 mutants with impaired catalytic function undergo high rates of de novo fatty acid synthesis and are less effective in repressing Neu-mediated tumorigenesis than is wild-type DecR1. The incomplete rescue of mammary tumor outgrowth observed with the catalytically impaired DecR1 mutants suggests either that the reductase activity of these mutants is only partially attenuated in vivo or that DecR1 also functions to impair tumorigenesis through an uncharacterized mechanism.
FIG. 8.

Model depicting role of DecR1 in the suppression of Neu-dependent transformation. DecR1 expression is decreased in breast cancer cells, thus increasing the rates of de novo fatty acid synthesis and stabilizing Neu expression. This not only provides downstream proliferative signals but also increases the availability of phospholipids, which are required for cell proliferation. Ectopic DecR1 expression is sufficient to repress Neu and decrease steady-state rates of fatty acid synthesis, which may contribute, in part, to reduced levels of cancer cell proliferation.
Regardless, it is clear that the ability of DecR1 to increase steady-state levels of β-oxidation is involved in its ability to repress Neu-dependent mammary gland transformation. Given that DecR1 functions in the auxiliary pathway of fatty acid β-oxidation, it is remarkable that elevated DecR1 expression can have such a profound effect on tumor cell growth. The sensitivity of cancer cells to ectopic DecR1 expression may reflect their high proliferative capacity. Given that cancer cells require high levels of phospholipids to support continual membrane synthesis and new cell division, hyperactivation of the fatty acid β-oxidation pathway by ectopic DecR1 expression may be sufficient to limit the availability of phospholipids within the cancer cell and thus impair tumor cell proliferation.
Consistent with the strong antiproliferative effects of ectopic DecR1 expression in breast cancer cells, we have also shown that endogenous DecR1 is consistently and significantly repressed in several primary mouse mammary tumors and in primary human breast cancers. Indeed, previous gene profiling studies have demonstrated that DecR1 levels are decreased during the progression from a normal mammary epithelium to hyperplasia and ultimately to neoplasia in MMTV/Neu mice (19, 20). Examination of a number of published databases reveals that DecR1 expression is consistently repressed in multiple primary epithelial tumors relative to matched normal tissues (http://www.oncomine.org). These include seminoma, hepatocellular carcinoma, clear renal cell carcinoma, pancreatic adenocarcinoma, ovarian adenocarcinoma, and colon carcinoma, all of which exhibit dramatically reduced levels in DecR1 expression relative to their normal tissue counterparts. We also observe that repression of DecR1 expression in primary human breast cancers does not correlate with the HER2 status of the tumor (data not shown). Furthermore, we have also demonstrated that DecR1 overexpression is sufficient to suppress MT-mediated transformation of Rat1 fibroblasts in an in vitro focus-forming assay (data not shown). Taken together, these observations argue that downregulation of DecR1 expression is likely to be a common event in breast cancer cells.
While DecR1 overexpression attenuates fatty acid synthesis in breast cancer cells, we did not observe appreciable differences in the rates of glucose metabolism compared to Neu-transformed cells. Both parental and DecR1-expressing breast cancer cells (i) express similar levels of GLUT1, (ii) deplete glucose from the medium, (iii) hyperactivate the Akt pathway following glucose withdrawal (data not shown), and (iv) proliferate, with little effect on survival, under serum starvation conditions in the presence of glucose. These data suggest that parental and DecR1-expresssing cancer cells utilize glucose as their primary energy source. However, DecR1 overexpression is sufficient to protect Neu-transformed breast cancer cells from apoptosis following glucose withdrawal. This may reflect the ability of DecR1 to increase the availability of fatty acid substrates for β-oxidation, which is consistent with previous observations demonstrating that activation of fatty acid β-oxidation through pharmacological agents increases the survival of cancer cells following glucose withdrawal (5).
The observation that key metabolic pathways play an important role in tumorigenesis is supported by an increasing body of evidence. For example, ACL, which catalyzes the production of cytoplasmic acetyl-CoA for de novo lipid synthesis, has been implicated as a key molecule for tumor growth. Indeed, stable knockdown of ACL in lung adenocarcinoma cells impaired tumor outgrowth in vivo and diminished the proliferative capacity of the tumor cells (14). More recently, stable knockdown of lactate dehydrogenase A (LDH-A) in Neu-initiated mammary tumor cell lines blocked tumor outgrowth and cancer cell proliferation (11). LDH-A catalyzes the conversion of pyruvate to lactate and generates a continuous supply of NAD+, which is a necessary cofactor for glycolysis. In contrast to ACL knockdown, which attenuates tumorigenesis by limiting the availability of fatty acids for new membrane synthesis, LDH-A knockdown does so by decreasing the mitochondrial membrane potential and increasing the cellular respiration rate of cancer cells. In the present study, we demonstrate that overexpression of DecR1 impairs tumor outgrowth, decreases the proliferative rate of breast cancer cells both in vitro and in vivo, and negatively influences steady-state fatty acid levels in breast cancer cells.
Another potential explanation for the potent antiproliferative effects of ectopic DecR1 expression in breast cancer cells may be due to its direct effects on ErbB2 expression. In this regard, Neu levels are consistently reduced, at the posttranscriptional level, in NT2196 breast cancer cells overexpressing wild-type DecR1 but not the catalytically inactive DecR1 mutants. This suggests that increased rates of de novo fatty acid synthesis are associated with elevated Neu expression in breast cancer cells. The observation that TM15 breast cancer cells are repressed by ectopically expressed DecR1, in the absence of a corresponding reduction in Neu expression, suggests that they lack the posttranscriptional machinery to downregulate Neu. Restoration of this machinery in TM15 cells will determine whether downregulation of Neu can further enhance the suppressive effects of DecR1 on mammary tumor outgrowth.
Several observations suggest a close correlation between Neu and fatty acid synthase (FAS) levels in cancer cells. FAS, which catalyzes de novo fatty acid synthesis from carbohydrate sources, is overexpressed in breast cancer cells and correlates not only with a poor clinical prognosis but also with Her-2/Neu amplification in human breast cancer (2, 16, 35). Overexpression of Her-2/Neu in breast cancer cells upregulates FAS gene expression and leads to increased de novo fatty acid synthesis (18). Pharmacological inhibition of FAS activity (1, 22) or downregulation of FAS expression (22) suppressed Neu expression in breast cancer cell lines (22) and in MMTV/Neu transgenic mice (1). Importantly, administration of pharmacological FAS inhibitors to MMTV/Neu mice significantly delayed the onset of mammary tumor formation (1). Together, these data demonstrate that disruption of steady-state levels of de novo fatty acid synthesis in Neu-overexpressing breast cancer cells is sufficient to negatively impact both Neu expression and mammary tumorigenesis.
The observation that DecR1 overexpression in mammary tumor cells is sufficient to impair tumor outgrowth in vivo provides further evidence that modulation of pathways controlling lipid biosynthesis has a major impact on tumor outgrowth. We have demonstrated that DecR1 may exert these suppressive effects through multiple mechanisms including decreased availability of phospholipids for new membrane synthesis and reduced expression of the transforming oncogene. Generation of MMTV/DecR1-transgenic mice will conclusively demonstrate whether this protein exerts a negative regulatory role in mammary tumor development in response to numerous oncogenes. Another interesting aspect of these studies is that ectopic DecR1 expression can rescue tumor cells from death following glucose deprivation. Given the glucose dependency of cancer cells and the nutrient-deprived nature of the tumor microenvironment, activation of pathways that provide additional energy sources may, paradoxically, be important for tumor cell persistence.
Supplementary Material
Acknowledgments
We gratefully acknowledge members of the Muller laboratory for helpful discussions along with Peter Siegel and Richard Marcotte for critical reading of the manuscript. We are also grateful to Bart Maslikowski for isolation of the DecR1 cDNA, Marilene Paquet for histological evaluation, David Dankort and Lina Jung for initial observations pertaining to DecR1 action in fibroblasts, and Samuel Leung for assistance in the TMA analysis. We thank Marcin Bakinowski and Jo-Ann Bader for their histological services.
This work was supported by grants from the CBCRA and CIHR. W.J.M. is supported by a CRC chair in molecular oncology.
Footnotes
Published ahead of print on 16 July 2007.
Supplemental material for this article may be found at http://mcb.asm.org/.
REFERENCES
- 1.Alli, P. M., M. L. Pinn, E. M. Jaffee, J. M. McFadden, and F. P. Kuhajda. 2005. Fatty acid synthase inhibitors are chemopreventive for mammary cancer in neu-N transgenic mice. Oncogene 24:39-46. [DOI] [PubMed] [Google Scholar]
- 2.Alo, P. L., P. Visca, A. Marci, A. Mangoni, C. Botti, and U. Di Tondo. 1996. Expression of fatty acid synthase (FAS) as a predictor of recurrence in stage I breast carcinoma patients. Cancer 77:474-482. [DOI] [PubMed] [Google Scholar]
- 3.Alphey, M. S., W. Yu, E. Byres, D. Li, and W. N. Hunter. 2005. Structure and reactivity of human mitochondrial 2,4-dienoyl-CoA reductase: enzyme-ligand interactions in a distinctive short-chain reductase active site. J. Biol. Chem. 280:3068-3077. [DOI] [PubMed] [Google Scholar]
- 4.Andrechek, E. R., W. R. Hardy, P. M. Siegel, M. A. Rudnicki, R. D. Cardiff, and W. J. Muller. 2000. Amplification of the neu/erbB-2 oncogene in a mouse model of mammary tumorigenesis. Proc. Natl. Acad. Sci. USA 97:3444-3449. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Buzzai, M., D. E. Bauer, R. G. Jones, R. J. Deberardinis, G. Hatzivassiliou, R. L. Elstrom, and C. B. Thompson. 2005. The glucose dependence of Akt-transformed cells can be reversed by pharmacologic activation of fatty acid beta-oxidation. Oncogene 24:4165-4173. [DOI] [PubMed] [Google Scholar]
- 6.Cianflone, K. M., Z. Yasruel, M. A. Rodriguez, D. Vas, and A. D. Sniderman. 1990. Regulation of apoB secretion from HepG2 cells: evidence for a critical role for cholesteryl ester synthesis in the response to a fatty acid challenge. J. Lipid Res. 31:2045-2055. [PubMed] [Google Scholar]
- 7.Dabiri, S., D. Huntsman, N. Makretsov, M. Cheang, B. Gilks, C. Bajdik, K. Gelmon, S. Chia, and M. Hayes. 2004. The presence of stromal mast cells identifies a subset of invasive breast cancers with a favorable prognosis. Mod. Pathol. 17:690-695. [DOI] [PubMed] [Google Scholar]
- 8.Dang, C. V., and G. L. Semenza. 1999. Oncogenic alterations of metabolism. Trends Biochem. Sci. 24:68-72. [DOI] [PubMed] [Google Scholar]
- 9.Dankort, D., B. Maslikowski, N. Warner, N. Kanno, H. Kim, Z. Wang, M. F. Moran, R. G. Oshima, R. D. Cardiff, and W. J. Muller. 2001. Grb2 and Shc adapter proteins play distinct roles in Neu (ErbB-2)-induced mammary tumorigenesis: implications for human breast cancer. Mol. Cell. Biol. 21:1540-1551. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Elstrom, R. L., D. E. Bauer, M. Buzzai, R. Karnauskas, M. H. Harris, D. R. Plas, H. Zhuang, R. M. Cinalli, A. Alavi, C. M. Rudin, and C. B. Thompson. 2004. Akt stimulates aerobic glycolysis in cancer cells. Cancer Res. 64:3892-3899. [DOI] [PubMed] [Google Scholar]
- 11.Fantin, V. R., J. St.-Pierre, and P. Leder. 2006. Attenuation of LDH-A expression uncovers a link between glycolysis, mitochondrial physiology, and tumor maintenance. Cancer Cell 9:425-434. [DOI] [PubMed] [Google Scholar]
- 12.Fillgrove, K. L., and V. E. Anderson. 2001. The mechanism of dienoyl-CoA reduction by 2,4-dienoyl-CoA reductase is stepwise: observation of a dienolate intermediate. Biochemistry 40:12412-12421. [DOI] [PubMed] [Google Scholar]
- 13.Guy, C. T., R. D. Cardiff, and W. J. Muller. 1992. Induction of mammary tumors by expression of polyomavirus middle T oncogene: a transgenic mouse model for metastatic disease. Mol. Cell. Biol. 12:954-961. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Hatzivassiliou, G., F. Zhao, D. E. Bauer, C. Andreadis, A. N. Shaw, D. Dhanak, S. R. Hingorani, D. A. Tuveson, and C. B. Thompson. 2005. ATP citrate lyase inhibition can suppress tumor cell growth. Cancer Cell 8:311-321. [DOI] [PubMed] [Google Scholar]
- 15.Kim, H., R. Chan, D. L. Dankort, D. Zuo, M. Najoukas, M. Park, and W. J. Muller. 2005. The c-Src tyrosine kinase associates with the catalytic domain of ErbB-2: implications for ErbB-2 mediated signaling and transformation. Oncogene 24:7599-7607. [DOI] [PubMed] [Google Scholar]
- 16.Kuhajda, F. P. 2000. Fatty-acid synthase and human cancer: new perspectives on its role in tumor biology. Nutrition 16:202-208. [DOI] [PubMed] [Google Scholar]
- 17.Kuhajda, F. P., K. Jenner, F. D. Wood, R. A. Hennigar, L. B. Jacobs, J. D. Dick, and G. R. Pasternack. 1994. Fatty acid synthesis: a potential selective target for antineoplastic therapy. Proc. Natl. Acad. Sci. USA 91:6379-6383. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Kumar-Sinha, C., K. W. Ignatoski, M. E. Lippman, S. P. Ethier, and A. M. Chinnaiyan. 2003. Transcriptome analysis of HER2 reveals a molecular connection to fatty acid synthesis. Cancer Res. 63:132-139. [PubMed] [Google Scholar]
- 19.Landis, M. D., D. D. Seachrist, F. W. Abdul-Karim, and R. A. Keri. 2006. Sustained trophism of the mammary gland is sufficient to accelerate and synchronize development of ErbB2/Neu-induced tumors. Oncogene 25:3325-3334. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Landis, M. D., D. D. Seachrist, M. E. Montanez-Wiscovich, D. Danielpour, and R. A. Keri. 2005. Gene expression profiling of cancer progression reveals intrinsic regulation of transforming growth factor-beta signaling in ErbB2/Neu-induced tumors from transgenic mice. Oncogene 24:5173-5190. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Makretsov, N. A., D. G. Huntsman, T. O. Nielsen, E. Yorida, M. Peacock, M. C. Cheang, S. E. Dunn, M. Hayes, M. van de Rijn, C. Bajdik, and C. B. Gilks. 2004. Hierarchical clustering analysis of tissue microarray immunostaining data identifies prognostically significant groups of breast carcinoma. Clin. Cancer Res. 10:6143-6151. [DOI] [PubMed] [Google Scholar]
- 22.Menendez, J. A., L. Vellon, I. Mehmi, B. P. Oza, S. Ropero, R. Colomer, and R. Lupu. 2004. Inhibition of fatty acid synthase (FAS) suppresses HER2/neu (erbB-2) oncogene overexpression in cancer cells. Proc. Natl. Acad. Sci. USA 101:10715-10720. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Moadel, R. M., R. H. Weldon, E. B. Katz, P. Lu, J. Mani, M. Stahl, M. D. Blaufox, R. G. Pestell, M. J. Charron, and E. Dadachova. 2005. Positherapy: targeted nuclear therapy of breast cancer with 18F-2-deoxy-2-fluoro-D-glucose. Cancer Res. 65:698-702. [PubMed] [Google Scholar]
- 24.Ookhtens, M., R. Kannan, I. Lyon, and N. Baker. 1984. Liver and adipose tissue contributions to newly formed fatty acids in an ascites tumor. Am. J. Physiol. 247:R146-R153. [DOI] [PubMed] [Google Scholar]
- 25.Pizer, E. S., F. D. Wood, G. R. Pasternack, and F. P. Kuhajda. 1996. Fatty acid synthase (FAS): a target for cytotoxic antimetabolites in HL60 promyelocytic leukemia cells. Cancer Res. 56:745-751. [PubMed] [Google Scholar]
- 26.Plas, D. R., and C. B. Thompson. 2005. Akt-dependent transformation: there is more to growth than just surviving. Oncogene 24:7435-7442. [DOI] [PubMed] [Google Scholar]
- 27.Roe, C. R., D. S. Millington, D. L. Norwood, N. Kodo, H. Sprecher, B. S. Mohammed, M. Nada, H. Schulz, and R. McVie. 1990. 2,4-Dienoyl-coenzyme A reductase deficiency: a possible new disorder of fatty acid oxidation. J. Clin. Investig. 85:1703-1707. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Sabine, J. R., S. Abraham, and I. L. Chaikoff. 1967. Control of lipid metabolism in hepatomas: insensitivity of rate of fatty acid and cholesterol synthesis by mouse hepatoma BW7756 to fasting and to feedback control. Cancer Res. 27:793-799. [PubMed] [Google Scholar]
- 29.Siegel, P. M., E. D. Ryan, R. D. Cardiff, and W. J. Muller. 1999. Elevated expression of activated forms of Neu/ErbB-2 and ErbB-3 are involved in the induction of mammary tumors in transgenic mice: implications for human breast cancer. EMBO J. 18:2149-2164. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Swinnen, J. V., K. Brusselmans, and G. Verhoeven. 2006. Increased lipogenesis in cancer cells: new players, novel targets. Curr. Opin. Clin. Nutr. Metab. Care 9:358-365. [DOI] [PubMed] [Google Scholar]
- 31.Warburg, O. 1956. On the origin of cancer cells. Science 123:309-314. [DOI] [PubMed] [Google Scholar]
- 32.White, D. E., N. A. Kurpios, D. Zuo, J. A. Hassell, S. Blaess, U. Mueller, and W. J. Muller. 2004. Targeted disruption of beta1-integrin in a transgenic mouse model of human breast cancer reveals an essential role in mammary tumor induction. Cancer Cell 6:159-170. [DOI] [PubMed] [Google Scholar]
- 33.Younes, M., R. W. Brown, D. R. Mody, L. Fernandez, and R. Laucirica. 1995. GLUT1 expression in human breast carcinoma: correlation with known prognostic markers. Anticancer Res. 15:2895-2898. [PubMed] [Google Scholar]
- 34.Younes, M., L. V. Lechago, J. R. Somoano, M. Mosharaf, and J. Lechago. 1996. Wide expression of the human erythrocyte glucose transporter Glut1 in human cancers. Cancer Res. 56:1164-1167. [PubMed] [Google Scholar]
- 35.Zhang, D., L. K. Tai, L. L. Wong, L. L. Chiu, S. K. Sethi, and E. S. Koay. 2005. Proteomic study reveals that proteins involved in metabolic and detoxification pathways are highly expressed in HER-2/neu-positive breast cancer. Mol. Cell. Proteomics 4:1686-1696. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.