

Abstract
This article, dedicated to Edward Stiefel, reviews three nickel enzymes that play important roles in the carbon cycle: CO dehydrogenase, acetyl-CoA synthase, and methyl-coenzyme M reductase. After a short discussion of the carbon cycle, the structures of the active centers of the proteins and their proposed mechanisms are discussed. A brief description of future research areas is presented for each enzyme system. A short perspective on future research on nickel enzymes ends this contribution.
Keywords: Acetogenesis, iron-sulfur, methanogenesis, CO2 fixation
The Carbon Cycle
Carbon is the key marker for life on Earth. The carbon cycle (Figure 1) involves the oxidation of energy-rich organic molecules to CO2 and the reduction of CO2 back to organic carbon. Like the burning of fuel in an internal combustion engine to provide the power for movement, the oxidation of organic carbon by cells provides the energy required to sustain life. Common oxidative processes like glycolysis directly synthesize ATP via substrate-level phosphorylation or generate reducing equivalents that indirectly couple to ATP synthesis by oxidative phosphorylation. On the other hand, CO2 reduction uses energy from light for phototrophs or inorganic sources for chemolithotrophs to fix carbon into the biomolecules of life.
Figure 1.

The carbon cycle.
Carbon is cycled in huge amounts among its various reservoirs on Earth. For example, plants and microbes fix about 1017 g of CO2 every year, while respiration releases a nearly equal amount. The major reservoirs of carbon are the organic matter found in the cells of living organisms and degradation products, inorganic carbon in rocks like limestone (CaCO3), and gases in the atmosphere. Among the major carbon-containing atmospheric gases, CO2 is present in the largest amount (3 × 1012 metric tonnes or 3 × 1018 g), reaching a total atmospheric concentration of 383 ppm (Jan. 2007) [1]. Of the other two major atmospheric gases, methane levels have reached approximately 1.75 ppm [2] and CO levels vary widely from 0.1 ppb in rural areas up to dangerous 200 ppb in some cities [3]. CO2, CO, and CH4 are produced and utilized by microbes and nickel enzymes play a key role in the cycling of these gases.
The major pathway for CO2 incorporation into organic compounds is the Calvin Cycle. The CO2 fixation step is catalyzed by ribulose biophosphate carboxylase/oxygenase (Rubisco), which may be the most abundant protein on earth. In C-4 plants, PEP carboxylase is a key enzyme that is involved in delivering CO2 to Rubisco. The reverse TCA cycle is a second CO2 fixation cycle. In a third mechanism of CO2 fixation, the 3-hydroxypropionate cycle, the CO2 incorporation step is catalyzed by acetyl-CoA carboxylase [4,5]. The fourth CO2 fixation pathway is the Wood-Ljungdahl or reductive acetyl-CoA pathway. Two nickel proteins, CO dehydrogenase (CODH) and acetyl-CoA synthase (ACS), which are the key enzymes in this pathway, are a major focus of this review.
Besides the CO2 fixation reactions that lead to organic carbon, CO2 also undergoes reduction to one- (CO, CH3NH2, CH4, etc.) and two- (CH3-COOH, CH3CH2OH, etc.) cparbon compounds in processes that generate metabolic energy. The reduction of CO2 to methane by methanogenic archaea (Equation 1) and the reduction of CO2 to acetic acid by acetogenic bacteria (Equation 2) are exergonic reactions that drive ATP synthesis. Nickel enzymes catalyze the key reactions in these processes. The acetate and methane are then oxidatively recycled to CO2 by other microbes; for example, methanotrophs convert CH4 to CO2 in a highly exergonic process that uses O2 as an electron acceptor (Equation 3). Furthermore, although the ΔG0′ values indicate that the reactions shown in Equations 1 and 2 are irreversible, organisms have found ways to perform anaerobic methane oxidation and anaerobic oxidation of acetate. Thus, by enlisting the different electron donors and acceptors available in diverse ecosystems, microbes can make a living on the cycling of oxidized and reduced carbon, an illustration of how the carbon and oxygen cycles are tightly integrated in nature. Although the importance of nickel in one-carbon metabolism is the central topic of this review article, cobalt in vitamin B12, iron in iron-sulfur and heme, and even selenium and tungsten play important roles in the carbon cycle.
| (1) |
| (2) |
| (3) |
Three nickel-containing enzymes that play a key role in the carbon cycle, CODH, ACS, and methyl-CoM reductase (MCR), are the focus of this review. The Ni-metallocenters that are the catalytic centers of these proteins are illustrated in Figure 2, and will be discussed in this order.
Figure 2.

Metalloclusters at the active site of CODH, ACS, and MCR. (A). NiFe4S4 C-Cluster in CODH, (B) Ni2Fe4S4 A-Cluster in ACS, (C). Ni-tetrahydrocorphin (F430) in MCR, from Figure 1 of [95].
CO Dehydrogenase (CODH)
CODH allows microbes to use carbon monoxide as their sole source of carbon and energy. Microbial CO metabolism is globally important since 108 tons of CO are removed from the lower atmosphere of the earth by bacterial oxidation every year [6], helping to maintain ambient CO below toxic levels. It has been proposed that CO can be produced and utilized as part of a bioenergetic cycle [7], similar to the proposed H2 cycling mechanism [8]. In some organisms, CO oxidation is also coupled to H2 production [9]. CO also is proposed to have been present in the early earth and current CO metabolism may be a highly successful descendent of a bioenergetic mechanism that fueled the evolution of life [10].
In catalyzing CO oxidation to CO2, CODH generates high-energy electrons with a redox potential for the CO2/CO couple that is much more negative than those of the 2H+/H2 or NAD/NADH couples (Equation 4). Thus, CO is approximately 1000-times more potent as an electron donor than NADH. The CO2 that is generated can be fixed into cellular carbon by one of the reductive CO2 fixation pathways described above. The rate of CO oxidation to CO2 varies widely; the turnover numbers for the Ni-containing Carboxydothermus hydrogenoformans CODH-I and CODH-II reach 40,000 s−1 with diffusion-controlled values of kcat/Km of 2 × 109 M−1s−1 [11], while the turnover number of the Mo-containing Oligotropha carboxidovorans enzyme is ~50 s−1 [12] at the relevant growth temperatures for these organisms. One of the most remarkable aspects of CODH is that it can perform CO oxidation or CO2 reduction at the thermodynamic potential [13,14]. This contrasts with other nonbiological CO2 reduction catalysts, which require a significant over-potential to drive this reaction.
| (4) |
There are two major classes of CODHs (Table 1). One is the aerobic CODH from carboxydobacteria, which is a monofunctional molybdopterin copper iron-sulfur enzyme; the Ni-CODHs constitute the other major class. Ni-CODHs are broken down into two classes: the monofunctional nickel CODH containing 10 Fe and 1 Ni per monomer; and the bifunctional CODH/ACS, containing 14 Fe and 3 Ni per monomeric unit.
Table 1.
CODHs in nature
| Type of CODH | Reactions catalyzed | Metal Centers |
|---|---|---|
| Aerobic or Mo-CODH | ||
| Oligotropha carboxidovorans | CO oxidation | Mo-Pterin Cu
2 [2Fe-2S] FAD |
| Anaerobic or Ni-CODH | ||
| Monofunctional CODH | C-Cluster, [NiFe4-S5] | |
| Rhodospirillum rubrum | CO oxidation | B-Cluster, [4Fe-4S] |
| C. hydrogenoformans | D-Cluster, bridged [4Fe-4S] | |
| Bifunctional CODH/ACS | ||
| Acetogenic (M. thermoacetica) | CO2 reduction & Acetyl-CoA synthesis | B, C, D (like R. rubrum) |
| Methanogenic (M. thermophila) | Acetyl-CoA → CO2+CH4 | NiNi-[4Fe-4S]A |
The molybdopterin Cu CODH is a 250 kDa (αβγ)2 protein [16–18], with an active site containing molybdopterin cytosine dinucleotide linked to Cu, which is proposed to bind CO [17]. These Mo-CODHs also contain (per monomeric unit) 1 mol of FAD and 2 [2Fe-2S] centers [19] that transfer electrons from the catalytic Cu-Mo-pterin center to external electron acceptors [20].
The monofunctional Ni-CODH is a mushroom-shaped homodimer containing five metal clusters (two B-, two C-, and one D-Cluster per dimer) [21–24]. The catalytic site for CO oxidation or CO2 reduction is the C-Cluster (Figure 2A), which can be described as a [3Fe-4S] cluster bridged to a binuclear NiFe cluster and is buried 18 Å below the protein surface. Figure 2A shows only the Rhodospirillum rubrum CODH structure with a bridging Cys between Ni and Fe, but there is controversy about the existence and importance of a bridging sulfide residue [25,26] and evidence for a catalytically important persulfide at the C-Cluster [27]. Issues related to the different structures are discussed in a recent review [28]. Rees noted that CODH shares a common structural motif for binding the FeS metalloclusters with nitrogenase and the Fe-only hydrogenase consisting of a four-stranded, parallel β-sheet surrounded by α-helices [29]. The Band D-Clusters are [4Fe-4S]2+/1+ clusters that transfer electrons between the C-Cluster and external redox proteins. The CODH reaction is a ping-pong reaction: CODH is reduced by CO in the “Ping” step (Steps 1–3, Figure 3) and the reduced enzyme transfers electrons to an external redox mediator like ferredoxin in the “Pong” step (Step 4). The reduced electron acceptors then couple to other energy requiring cellular processes.
Figure 3.

Proposed mechanism of CODH.
Step 1 of the CODH reaction involves binding of CO and water to the C-Cluster (Figure 3). The proposal that CO binds to Ni [21,23] is supported by Fourier transfer infrared (FTIR) studies with the M. thermoacetica CODH in which several IR bands were attributed to a Ni-CO complex [30] and by EXAFS studies of the C. hydrogenoformans CODH-II in which a linear Ni-CN bond was identified [31]. Based on evidence that CN is a competitive inhibitor with respect to CO [31], the results suggest a Ni-CO intermediate; though with some CODHs, CN binding can be rather compex [32,33]. ENDOR studies indicate that cyanide and hydroxide (or water) binds to the Fe site (called Ferrous component II, FCII in Figure 4) that is bridged to Ni [34]. Binding of water to iron would be expected to lower its pKa, which would facilitate formation of an active hydroxide, as in carbonic anhydrase [35]. Furthermore, there are several basic residues (Lys563, His113, not shown) near the C-Cluster that have been proposed to participate in the proton transfer steps in the reaction [21,23]. Accordingly, mutagenesis of Lys587 and His113 (M. thermoacetica numbering) abolishes catalysis [27].
Figure 4.
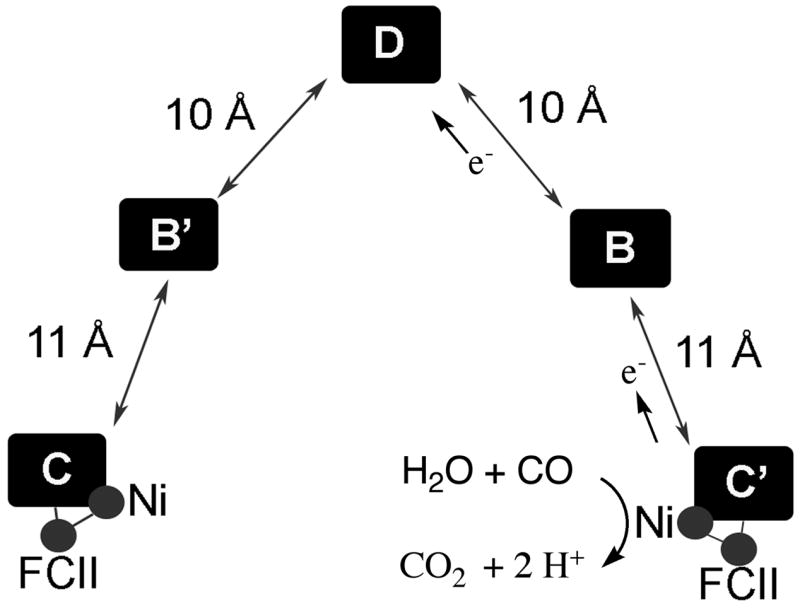
Metalloclusters in CODH based on the 1OAO structure from [23].
In Step 2, the active metal-hydroxide is proposed to attack the M-CO complex and a proton is eliminated to form a Ni-carboxylate complex that Ha et al. propose to bridge the Ni and Fe atoms [31]. In Step 3, CO2 is generated and released and the C-Cluster undergoes two-electron reduction. The two-electron reduction could generate a transient Ni0 state of the cluster as proposed by Lindahl [36] (depicted as “0+” in the diagram); however, with so many redox active metal centers near the Ni, it seems more likely that the electrons are distributed among the Ni, Fe or S components of the C-Cluster. Freeze quench EPR studies show that when CODH in its resting state (called Cred1) is reacted with CO, it converts rapidly (2 × 108 M−1s−1) to another distinct paramagnetic state called Cred2, before electrons are transferred to the chain of redox centers (the B- and D-Clusters) [37,38] in Step 4. Transfer of electrons from the C-Cluster to the B- and D-Clusters in the redox chain (Figure 4) would return the C-Cluster to its resting form and leave the B- and D-Clusters in a reduced state. The three clusters are approximately 10 Å apart, which is an ideal distance for rapid electron transfer [39]. Rapid kinetic studies have followed this internal electron transfer reaction and shown that at high CO concentrations, electron transfer can become rate limiting [37,38]. Finally, in the “Pong” step, electrons would be transferred from the reduced [4Fe-4S] clusters on CODH to the external mediators, e.g., ferredoxin or other carriers.
The reaction sequence shown in Figure 3 is similar to a well-known reaction known as the water-gas shift reaction, which also is proposed to occur through M-CO, M-OH, and M-COOH intermediates; however in the water gas reaction, CO2 and H2, instead of CO2, 2 electrons, and 2 protons are formed. Unlike the water gas shift reaction, in CODH, electrons are transferred very rapidly out of the active site. It is likely that having the catalytic C-Cluster so near the electron transfer chain (the B- and D-Clusters) is what prevents H2 formation. Generation of protons and electrons allows flexibility for CODH to transfer electrons to various cellular electron acceptors; then, when the cell requires H2, CODH can couple through a ferredoxin to a dedicated hydrogenase.
Thus, CODH is an important enzyme in the carbon cycle, allowing organisms to utilize CO as well as CO2 as a source of carbon and energy. It contains an unusual nickel-iron-sulfur cluster that is located near a redox wire, an architecture that allows the transfer of low-potential electrons from CO oxidation to external redox mediators at diffusion-controlled rates. The identity of the acid base catalysts in the mechanism are not yet known, the redox state of the various components of the C-Cluster at the various stages of catalysis have not been unambiguously determined, and the mechanism by which electron transfer from CODH is coupled to ATP synthesis by oxidative phosphorylation is not yet known. Given the high activity of enzymatic CO oxidation and CO2 reduction, perhaps the efficiency of purely inorganic CO2 reduction catalysts could be enhanced by incorporating aspects of the biochemical design, such as placing one-electron redox centers between the catalyst and the electrode.
Acetyl-CoA Synthase
Some anaerobic microbes contain the acsB gene that encodes ACS. In nature, ACS appears to be found in a complex with CODH. The acsB gene also appears to be always within a gene cluster that includes the other key functions of the Wood Ljungdahl pathway for anaerobic CO2 fixation: acsA (CODH), acsCD (the corrinoid iron-sulfur protein, CFeSP), and methyltransferase (acsE). ACS catalyzes the synthesis of acetyl-CoA from CO, a methyl group donated by the methylated CFeSP, and Coenzyme A (CoA) (Equation 5). Furthermore, ACS associates tightly with CODH to form a heterotetrameric (α2β2) machine that can convert CO2 to the carbonyl group of acetyl-CoA (Reaction 6). In this reaction, CO is formed as an intermediate [40] that is channeled between the CODH and ACS active sites. Encompassing hydrophobic residues within CODH and ACS, this channel is 70 Å in length and functions to sequester CO and facilitate its movement to the ACS active site [41]. The channel has been identified biochemically [42,43] and by X-ray crystallography [22,24]. Several reviews that focus on the structure, function, and mechanism of ACS are available [28,44–46].
| (5) |
| (6) |
The only metallocenter in ACS is the A-Cluster (Figure 2B); thus, CODH/ACS contains all the centers (B, C, and D) found in the monofunctional CODH plus the A-Cluster. Each monomeric unit of CODH/ACS, therefore, contains 3 discrete Ni sites. The A-Cluster was the first published example of a NiFeS cluster [47] and consists of a [4Fe-4S] cluster bridged to a Ni site (Nip) that is thiolate bridged to another Ni ion in a thiolato- and carboxamido-type N2S2 coordination environment (Figure 2B) [22,24,48]. Thus, one can describe the A-Cluster as a binuclear NiNi center bridged by a cysteine residue (Cys509) to a [4Fe-4S] cluster. This arrangement is similar to the Fe-only hydrogenases in which a [4Fe-4S] cluster and a binuclear Fe site are bridged by a Cys residue [49,50].
Two proposed mechanisms for acetyl-CoA synthesis are shown in Figure 5. The major difference between the two mechanisms is in the assignment of the electronic structure of the intermediates; Figure 5A proposes a paramagnetic Ni(I)-CO species as a central intermediate and Figure 5B proposes a Ni(0) intermediate. Either mechanism could be alternatively written with the methyl group binding before CO; for example, Lindahl supports a strictly ordered mechanism with methylation first, then carbonylation followed by carbonylation, as described in a recent review [44]. One issue that is not explicitly addressed in either of these cycles is the apparent requirement for a conformational change during the catalytic cycle. CODH/ACS was crystallized in two states: (1) a closed state [22] in which CO has access through the channel to the A cluster, however, there is insufficient space for binding the methylated corrinoid protein, and (2) an open state [24] in one of the domains of ACS is rotated into a position that partially exposes the A-cluster but, which pinches the CO channel shut.
Figure 5.

Proposed ACS mechanisms. (A) the paramagnetic mechanism and (B) the diamagnetic mechanism. The redox states of the metals has not been unambiguously determined in the catalytic mechanism
Whether the carbonyl group binds first or second, it is clear that CO is derived from the CO2 reduction reaction that occurs at the CODH active site. CO then migrates through the intersubunit channel from the C-Cluster to the A-Cluster where it binds to Ni. Movement of CO to the A-cluster requires the “closed” conformation of ACS. As proposed in Figure 5A, the carbonylation step involves reaction of Cluster A with CO to form an organometallic complex, called the NiFeC species, which has been characterized by a number of spectroscopic approaches [51]. The electronic structure of the paramagnetic A-Cluster-CO adduct (the NiFeC species) is described as a [4Fe-4S]2+ cluster linked to a Ni1+ center at the proximal metal site, which is called the proximal Nip, and is labeled in blue [45,52]. The other Ni is considered to remain in the Ni2+ state throughout the catalytic cycle. The unpaired spin density in this complex resides predominantly on Nip, with some delocalization into the terminal carbonyl group, based on the observation of 13CO hyperfine splittings in the EPR signal [47,53]. Based on detection of a diamagnetic product of the reaction of methylated ACS with CO and experiments that indicated electrons were not transferred to ACS during this reaction, it has been argued that the NiFeC species is not a true catalytic intermediate in acetyl-CoA synthesis, that it may be an inhibitory state, and that the Ni(0) state is the catalytically relevant state [44,54]. However, the NiFeC species does meet the criteria of catalytic competence in that its characteristic EPR signal forms (when treated with CO) and decays (upon reaction with the methyl group donor) at a rate consistent with its involvement as a catalytic intermediate in acetyl-CoA synthesis [55,56]. Furthermore, it forms at the same rate as the sole metal-carbonyl species formed upon reaction of ACS with CO, based on stopped-flow infrared experiments [56]. Thus, if a metal carbonyl intermediate is part of the catalytic cycle, the paramagnetic NiFeC species is the only feasible intermediate. On the other hand, if the reaction is strictly ordered with the methyl group binding before CO, the NiFeC species could be of less relevance since CO would bind to a methyl-Ni intermediate. Thus, it is important to unambiguously determine the order of substrate binding, which is rather complicated for this three-substrate enzyme that can exist in various redox states.
The transmethylation step in the ACS mechanism involves transfer of the methyl group from a methylated CFeSP to the A-Cluster. As mentioned above, this methyl transfer reaction requires ACS to assume the “open” conformation. The methyl group binds to Nip site of the A-Cluster [57–59]. The transmethylation reaction could occur by transfer of a methyl radical or an SN2-type nucleophilic mechanism involving a methyl cation. Rapid kinetic studies and stereochemical studies using a chiral methyl donor indicate that the transmethylation reaction involves the nucleophilic attack of the reduced form of ACS on CH3-Co3+ to form a diamagnetic methylated state of ACS and generate Co1+ [55,58,60–62]. The generation of a diamagnetic (likely methyl-Ni(II)) product on ACS is difficult to explain if the active form of ACS contains a Ni(I) nucleophile, since one would expect a methyl-Ni(III) product. Therefore, an internal electron transfer cycle is proposed in Figure 5A that would reduce the methyl-Ni(III) to methyl-Ni(II). Linking the SN2 reaction to an electron-transfer step would be equivalent to a radical methyl transfer, which is supported by model studies of the reaction between methyl-Co3+ (CH3-Co3+ dimethylglyoximate) and a Ni1+ macrocycle [63,64]. Since it is clear that the ACS reaction does not require net input of electrons [65], Figure 5A proposes that the electron that is donated during the methylation step is returned at a latter step in the reaction (after CoA binds). There is no need for an internal electron shuttle in the mechanism described in Figure 5B, since the reaction involves attack of Ni(0) to form a methyl-Ni(II).
The next step in the ACS mechanism involves carbon-carbon bond formation, which occurs by condensation of the methyl and carbonyl groups to form an acetyl-metal species. Then, CoA binds, triggering thiolysis of the acetyl-metal bond. There is evidence that the sulfur of CoA binds to Nip in the A-Cluster, based on EXAFS studies of the binding of seleno-CoA to CODH/ACS [66].
In summary, the ACS catalytic cycle involves metal centered organometallic catalysis involving intermediates (methyl-metal, metal-CO, and acetyl-metal) that resemble those proposed for the Monsanto process for industrial acetate production. The use of a low-valent metal center as a nucleophile is rare and is similar to the reactions of cobalamin-dependent methyltransferases [67,68]. In fact, kinetic studies indicate that the Ni1+ site on ACS appears to be about as effective as Co1+-CFeSP as a methyl group acceptor [69]. Coordination of the activities of the ACS and CODH active sites is essential to ensure that each molecule of CO that is generated is incorporated into acetyl-CoA. As proposed on the basis of the CODH/ACS structure, conformational changes appear to be required during the catalytic cycle to prevent escape of CO from the channel and to allow access of the methylated CFeSP to the A-Cluster [22]. Understanding these features of CODH/ACS will require unambiguous establishment of the order of substrate binding and studies of the conformational changes that occur during catalysis. Generation of crystal structures of intermediate states is also an important goal for the understanding of this important set of reactions in the carbon cycle.
Methyl-CoM Reductase
All biologically produced methane is formed by methanogenic organisms through the catalytic action of methyl-coenzyme M reductase (MCR). Anaerobic methane oxidation also appears to be catalyzed by the same enzyme, or an isozyme of MCR. MCR catalyzes the conversion of methyl-coenzyme M (methyl-SCoM) and N-7-mercaptoheptanoylthreonine phosphate (CoBSH) to methane and the CoB-SS-CoM heterodisulfide (Eq. 7) [70]. In this reaction, CoBSH serves as the electron donor [71] and has also been proposed to donate the proton to a methyl-enzyme intermediate to form methane [72]. Catalysis requires a nickel hydrocorphin called coenzyme F430 (Figure 2C) [73–75], which is located at the base of a narrow hydrophobic well that accommodates the two substrates and shields the reaction from solvent [76]. Containing only five double bonds, F430 is the most reduced tetrapyrrole in nature.
| (7) |
MCR contains three nonidentical subunits in an (αβγ)2 structure that is predominantly composed of helices. F430 is found in the α subunit. The active state of MCR, called MCRred1, contains the Ni(I) state of F430 [72,77,78], which is green (λmax ~ 390 nm) and paramagnetic, exhibiting the fairly typical EPR spectra of a d9 system with g|| (2.2 – 2.3) > g⊥ (2.05), and both are g|| and g⊥ are greater than ge (2.0) [79], where the “parallel” and “perpendicular” subscripts designate the symmetry of the EPR signals (the subscript “e” is the free electron g-value). Most of the unpaired spin density is on nickel in the MCRred1 state [80]. Because of the low redox potential of the Ni(II)/(I) couple, great care must be taken to isolate and maintain the enzyme in the Ni(I) oxidation state; otherwise, it undergoes oxidative inactivation to Ni(II), turning bright yellow (λmax ~ 420 nm). High resolution X-ray crystal structures have been obtained of the inactive nickel (II) enzyme complexed with CoM and CoB (MCRox1-silent) and in complex with the heterodisulfide CoM-S-S-CoB product (MCRsilent) [81].
Methane formation by MCR occurs with a turnover number of approximately 100 s−1 and a kcat/Km (methyl-SCoM) of ~1 × 105 M−1s−1 [82,83]. Two mechanisms have been considered for this reaction (Figure 6): Mechanism I involving a organometallic methyl-Ni intermediate and Mechanism II involving a methyl radical and hydrogen atom abstraction from CoBSH. To summarize the key tenets of Mechanism I, the Ni(I) center on MCR performs an SN2-type displacement of the methyl group of methyl-CoM to form a methyl-Ni(III) intermediate [72,82,84–86]. A proton transfer from CoBSH to CoM would then place a proton near the site of methane formation so that CoM-dependent protonation of the methyl group would generate Ni(III) and methane. Then, electron transfer from CoB or CoM followed by linkage of the two thiol-containing cofactors would generate Ni(II) and a disulfide radical anion. Electron transfer from the radical anion to Ni(II) would then yield the CoM-SS-CoB heterodisulfide and regenerate the active Ni(I) form of MCR.
Figure 6.

Proposed MCR mechanisms. Mechanism I involves a methyl-Ni intermediate and Mechanism II involves a methyl radical.
Experimental support for Mechanism I includes model studies of the reaction between the Ni(I) pentamethyl ester of F430 and activated methyl donors (e.g., methyl sulfonium ions and methyl iodide) to yield methane through protonation of a methylnickel intermediate [87–89]. Furthermore, alkyl-Ni intermediates, formed by reaction of MCRred1 with bromopropanesulfonate, have been characterized as a high-spin Ni(II)/alkyl radical species [90,91]. This intermediate undergoes protonation to form the corresponding alkane or to react with various thiol groups (including CoM) to form the methylthioether (mimicking the reverse of the first step in methane formation or the final step in methane oxidation) [92].
Mechanism II, based on computational studies, avoids the methyl-Ni species and proposes that a methyl radical is formed during methanogenesis [93]. According to this mechanism, which is summarized in Figure 6, reaction of MCRred1 with methyl-SCoM leads to cleavage of the C-S bond to generate a methyl radical and a Ni(II)-thiol. The methyl radical is proposed to abstract a H atom from CoBSH to form methane and a thiyl radical on CoB. The next step involves formation of a disulfide anion radical involving CoBS• and CoMS−. Finally, reduction of Ni(II) by the anion radical regenerates the active Ni(I) state and the heterodisulfide. In this mechanism, the major role of the Ni is to facilitate C-S bond cleavage by a redox process and to stabilize the product of C-S homolytic bond cleavage by forming a coordination complex with the sulfur of CoM.
Distinction between the two mechanisms will require characterization of the intermediates in the MCR reaction, which have only recently begun to yield to rapid kinetic and spectroscopic identification [92]. Future studies may need to use substrate analogs and/or variant forms of MCR in order to trap and accumulate postulated intermediates in the reaction cycle. Another important area of future study is determination of the crystal structure of the highly labile active Ni(I) or perhaps an alkyl-Ni intermediate.
The reaction of MCR appears to run in reverse in a process called anaerobic methane oxidation. Methane can be oxidized anaerobically where it is found in high concentrations, like in the deep ocean and near pools of methane hydrates [94]. Anaerobic methane oxidation occurs in microbial consortia where it can be coupled to processes like sulfate oxidation to sulfide. Anaerobic methane oxidation is also called reverse methanogenesis, because the key enzymes involved in methanogenesis appear to be responsible for this process. Characterization of the MCR (and other enzymes) involved in methane oxidation may reveal what controls the catalytic bias of the enzymatic system(s) toward methane oxidation versus methane production.
Future Directions
Besides the biological questions that remain, there are a number of interesting unsolved problems in the bioinorganic chemistry of CODH, ACS, and MCR. It is interesting to reflect on the wide range of coordination environments found in nickel enzymes and on why Ni was selected for these key one-carbon transformations. ACS and MCR involve reactions with methyl groups, while CODH and ACS involve transformations involving CO. One might consider that acetyl-CoA synthesis and methane formation could occur on a B12 enzyme, since Co(I) (like Ni(I)) is a strong nucleophile that forms important biological organometallic complexes with methyl groups. It will be interesting to determine if MCR reconstituted with a Co-analog of F430 might have any activity in methane synthesis or if ACS might have acetyl-CoA synthesis activity if the proximal Ni site in the A-Cluster can be substituted with Co (or other metals). Such studies may reveal special properties of Ni that have been enlisted for these important reactions. Why does Ni(I) in F430 catalyze methane formation while Ni(I) in ACS reacts with CO and methyl-cobalamin? Why does methyl-Ni in MCR undergo conversion to methane, while methyl-Ni in ACS cannot be converted to methane, but can rapidly be converted to acetyl-CoA? Issues of metal specificity and reactivity at the active sites of CODH, ACS, and MCR can be resolved by a combination of biochemical and inorganic model chemical studies. Computational work will also provide important information related to the reactivity of Ni (and other transition metals) in various coordination environments and redox states.
It is important to identify all intermediates in the CODH, ACS, and MCR reactions. This should be achieved by a combination of site-directed mutagenesis and rapid kinetics using natural substrates and substrate analogs. Though mutagenesis of MCR will be difficult, studies using substrate analogs of MCR are beginning to yield fruit. Studying variants of CODH and ACS are providing important information about the identity of the acid-base catalysts and allow accumulation of intermediates in the CODH and ACS reaction mechanisms. It is important to unambiguously establish the order of substrate binding in the ACS mechanism and the role of the various metal components in the reaction. It is difficult to believe that the only important component among the 6 metal sites in the A-Cluster is the proximal nickel; thus, the roles of the other metals should be established. Conformational changes have been proposed for the ACS and MCR reactions, yet these structural changes have not been studied with the vast array of techniques available.
I would encourage any student to take up bioinorganic chemistry because of the many methods that can be used to address our problems. There is still much to learn about the important roles that metals and metalloenzymes play in the carbon cycle and in other key life processes. Furthermore, the number of novel metalloenzymes is expanding rapidly as metagenomics studies sample increasingly diverse environmental samples.
Acknowledgments
I am grateful to NIH (Grant # GM39451) for supporting our work on CODH and ACS and to DOE (Grant # DE-FG02-04ER15532) for supporting our work on MCR. I am grateful to have had the opportunity to work with wonderful collaborators, students, postdoctors, and competitors on these projects. I have learned a tremendous amount from these enthusiastic colleagues.
Abbreviations
- CODH
CO dehydrogenase
- ACS
acetyl-CoA synthase
- MCR
methyl-coenzyme M reductase
- methyl-coenzyme M
methyl-SCoM
- N-7-mercaptoheptanoylthreonine phosphate
CoBSH
- CFeSP
corrinoid iron-sulfur protein
- MeTr
methyltransferase
- EXAFS
Extended X-ray absorption fine structure
- ENDOR
electron nuclear double resonance
Footnotes
This article is dedicated to Ed Stiefel, who had a strong interest in the roles of trace metals and metalloenzymes in the major biogeochemical cycles. Though his major research efforts were in the nitrogen cycle, Ed had a keen understanding of the role of metalloenzymes in microbial transformations and in the various linkages among the carbon, nitrogen, and oxygen cycles. I wish that I could rely on his painstaking and insightful suggestions on this present article; unfortunately, however, this is written as a memorial.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Tans P NOAA/ESRL. Trends in Atmospheric Carbon Dioxide - Mauna Loa. www.cmdl.noaa.gov/gmd/ccgg/trends.
- 2.Dlugokencky EJ, Masarie KA, Lang PM, Tans PP. Nature. 1998;393:447–450. [Google Scholar]
- 3.Meyer O. In: Microbial gas metabolism, mechanistic, metabolic, and biotechnological aspects. Poole RK, Dow CS, editors. Academic press; London: 1985. pp. 131–151. [Google Scholar]
- 4.Holo H, Sirevag R. Arch Microbiol. 1986;145:173–180. [Google Scholar]
- 5.Strauss G, Fuchs G. Eur J Biochem. 1993;215:633–643. doi: 10.1111/j.1432-1033.1993.tb18074.x. [DOI] [PubMed] [Google Scholar]
- 6.Bartholomew GW, Alexander M. Appl Environ Microbiol. 1979;37:932–937. doi: 10.1128/aem.37.5.932-937.1979. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Voordouw G. J Bacteriol. 2002;184:5903–5911. doi: 10.1128/JB.184.21.5903-5911.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Odom JM, Peck HD., Jr Annu Rev Microbiol. 1984;38:551–592. doi: 10.1146/annurev.mi.38.100184.003003. [DOI] [PubMed] [Google Scholar]
- 9.Soboh B, Linder D, Hedderich R. Eur J Biochem. 2002;269:5712–5721. doi: 10.1046/j.1432-1033.2002.03282.x. [DOI] [PubMed] [Google Scholar]
- 10.Wächtershäuser G, Huber C. Science. 1997;276:245–247. doi: 10.1126/science.276.5310.245. [DOI] [PubMed] [Google Scholar]
- 11.Svetlitchnyi V, Peschel C, Acker G, Meyer O. J Bacteriol. 2001;183:5134–5144. doi: 10.1128/JB.183.17.5134-5144.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Meyer O, Gremer L, Ferner R, Ferner M, Dobbek H, Gnida M, MeyerKlaucke W, Huber R. Biol Chem. 2000;381:865–876. doi: 10.1515/BC.2000.108. [DOI] [PubMed] [Google Scholar]
- 13.Kumar M, Lu WP, Ragsdale SW. Biochemistry. 1994;33:9769–9777. doi: 10.1021/bi00198a048. [DOI] [PubMed] [Google Scholar]
- 14.Lindahl PA, Münck E, Ragsdale SW. J Biol Chem. 1990;265:3873–3879. [PubMed] [Google Scholar]
- 15.Grahame DA, Demoll E. Biochemistry. 1995;34:4617–4624. doi: 10.1021/bi00014a015. [DOI] [PubMed] [Google Scholar]
- 16.Meyer O, Jacobitz S, Kruger B. FEMS Microbiol Rev. 1986;39:161–179. [Google Scholar]
- 17.Gnida M, Ferner R, Gremer L, Meyer O, Meyer-Klaucke W. Biochemistry. 2003;42:222–230. doi: 10.1021/bi026514n. [DOI] [PubMed] [Google Scholar]
- 18.Dobbek H, Gremer L, Kiefersauer R, Huber R, Meyer O. Proc Natl Acad Sci USA. 2002;99:15971–15976. doi: 10.1073/pnas.212640899. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Bray RC, George GN, Lange R, Meyer O. Biochem J. 1983;211:687–694. doi: 10.1042/bj2110687. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Meyer O, Frunzke K, Mörsdorf G. In: Microbial growth on C1 compounds. Murrell JC, Kelly DP, editors. Intercept, Ltd; Andover, Mass: 1993. pp. 433–459. [Google Scholar]
- 21.Drennan CL, Heo J, Sintchak MD, Schreiter E, Ludden PW. Proc Natl Acad Sci USA. 2001;98:11973–11978. doi: 10.1073/pnas.211429998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Doukov TI, Iverson T, Seravalli J, Ragsdale SW, Drennan CL. Science. 2002;298:567–572. doi: 10.1126/science.1075843. [DOI] [PubMed] [Google Scholar]
- 23.Dobbek H, Svetlitchnyi V, Gremer L, Huber R, Meyer O. Science. 2001;293:1281–1285. doi: 10.1126/science.1061500. [DOI] [PubMed] [Google Scholar]
- 24.Darnault C, Volbeda A, Kim EJ, Legrand P, Vernede X, Lindahl PA, Fontecilla-Camps JC. Nat Struct Biol. 2003;10:271–279. doi: 10.1038/nsb912. [DOI] [PubMed] [Google Scholar]
- 25.Dobbek H, Svetlitchnyi V, Liss J, Meyer O. J Am Chem Soc. 2004;126:5382–5387. doi: 10.1021/ja037776v. [DOI] [PubMed] [Google Scholar]
- 26.Sun J, Tessier C, Holm RH. Inorg Chem. 2007;46:2691–2699. doi: 10.1021/ic062362z. [DOI] [PubMed] [Google Scholar]
- 27.Kim EJ, Feng J, Bramlett MR, Lindahl PA. Biochemistry. 2004;43:5728–5734. doi: 10.1021/bi036062u. [DOI] [PubMed] [Google Scholar]
- 28.Drennan CL, Doukov TI, Ragsdale SW. J Biol Inorg Chem. 2004;9:511–515. doi: 10.1007/s00775-004-0563-y. [DOI] [PubMed] [Google Scholar]
- 29.Rees DC. Annu Rev Biochem. 2002;71:221–246. doi: 10.1146/annurev.biochem.71.110601.135406. [DOI] [PubMed] [Google Scholar]
- 30.Chen J, Huang S, Seravalli HG J, Jr, Swartz DJ, Ragsdale SW, Bagley KA. Biochemistry. 2003;42:14822–14830. doi: 10.1021/bi0349470. [DOI] [PubMed] [Google Scholar]
- 31.Ha SW, Korbas M, Klepsch M, Meyer-Klaucke W, Meyer O, Svetlitchnyi V. J Biol Chem. 2007;282:10639–10646. doi: 10.1074/jbc.M610641200. [DOI] [PubMed] [Google Scholar]
- 32.Ensign SA, Hyman MR, Ludden PW. Biochemistry. 1989;28:4973–4979. doi: 10.1021/bi00438a011. [DOI] [PubMed] [Google Scholar]
- 33.Anderson ME, Lindahl PA. Biochemistry. 1994;33:8702–8711. doi: 10.1021/bi00195a011. [DOI] [PubMed] [Google Scholar]
- 34.DeRose VJ, Telser J, Anderson ME, Lindahl PA, Hoffman BM. J Am Chem Soc. 1998;120:8767–8776. [Google Scholar]
- 35.Bertini I, Luchinat C. In: Bioinorganic Chemistry. Bertini I, Gray HB, Lippard SJ, Valentine JS, editors. University Science Books; Mill Valley, CA: 1994. pp. 37–106. [Google Scholar]
- 36.Lindahl PA. Biochemistry. 2002;41:2097–2105. doi: 10.1021/bi015932+. [DOI] [PubMed] [Google Scholar]
- 37.Kumar M, Lu WP, Liu L, Ragsdale SW. J Am Chem Soc. 1993;115:11646–11647. [Google Scholar]
- 38.Seravalli J, Kumar M, Lu WP, Ragsdale SW. Biochem. 1997;36:11241–11251. doi: 10.1021/bi970590m. [DOI] [PubMed] [Google Scholar]
- 39.Page CC, Moser CC, Chen XX, Dutton PL. Nature. 1999;402:47–52. doi: 10.1038/46972. [DOI] [PubMed] [Google Scholar]
- 40.Menon S, Ragsdale SW. Biochemistry. 1996;35:12119–12125. doi: 10.1021/bi961014d. [DOI] [PubMed] [Google Scholar]
- 41.Tan X, Loke HK, Fitch S, Lindahl PA. J Am Chem Soc. 2005;127:5833–5839. doi: 10.1021/ja043701v. [DOI] [PubMed] [Google Scholar]
- 42.Maynard EL, Lindahl PA. J Am Chem Soc. 1999;121:9221–9222. [Google Scholar]
- 43.Seravalli J, Ragsdale SW. Biochemistry. 2000;39:1274–1277. doi: 10.1021/bi991812e. [DOI] [PubMed] [Google Scholar]
- 44.Lindahl PA. J Biol Inorg Chem. 2004;9:516–524. doi: 10.1007/s00775-004-0564-x. [DOI] [PubMed] [Google Scholar]
- 45.Brunold TC. J Biol Inorg Chem. 2004;9:533–541. doi: 10.1007/s00775-004-0566-8. [DOI] [PubMed] [Google Scholar]
- 46.Riordan C. J Biol Inorg Chem. 2004;9:542–549. doi: 10.1007/s00775-004-0567-7. [DOI] [PubMed] [Google Scholar]
- 47.Ragsdale SW, Wood HG, Antholine WE. Proc Natl Acad Sci USA. 1985;82:6811–6814. doi: 10.1073/pnas.82.20.6811. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Svetlitchnyi V, Dobbek H, Meyer-Klaucke W, Meins T, Thiele B, Romer P, Huber R, Meyer O. Proc Natl Acad Sci USA. 2004;101:446–451. doi: 10.1073/pnas.0304262101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Peters JW, Lanzilotta WN, Lemon BJ, Seefeldt LC. Science. 1998;282:1853–1858. doi: 10.1126/science.282.5395.1853. [DOI] [PubMed] [Google Scholar]
- 50.Nicolet Y, Lemon BJ, Fontecilla-Camps JC, Peters JW. Trends Biochem Sci. 2000;25:138–143. doi: 10.1016/s0968-0004(99)01536-4. [DOI] [PubMed] [Google Scholar]
- 51.Ragsdale SW. CRC Crit Rev Biochem Mol Biol. 2004;39:165–195. doi: 10.1080/10409230490496577. [DOI] [PubMed] [Google Scholar]
- 52.Schenker RP, Brunold TC. J Am Chem Soc. 2003;125:13962–13963. doi: 10.1021/ja037893q. [DOI] [PubMed] [Google Scholar]
- 53.Ragsdale SW, Ljungdahl LG, DerVartanian DV. Biochem Biophys Res Commun. 1983;115:658–665. doi: 10.1016/s0006-291x(83)80195-8. [DOI] [PubMed] [Google Scholar]
- 54.Bramlett MR, Stubna A, Tan X, Surovtsev IV, Munck E, Lindahl PA. Biochemistry. 2006;45:8674–8685. doi: 10.1021/bi060003+. [DOI] [PubMed] [Google Scholar]
- 55.Seravalli J, Kumar M, Ragsdale SW. Biochemistry. 2002;41:1807–1819. doi: 10.1021/bi011687i. [DOI] [PubMed] [Google Scholar]
- 56.George SJ, Seravalli J, Ragsdale SW. J Am Chem Soc. 2005;127:13500–13501. doi: 10.1021/ja0528329. [DOI] [PubMed] [Google Scholar]
- 57.Shin W, Anderson ME, Lindahl PA. J Am Chem Soc. 1993;115:5522–5526. [Google Scholar]
- 58.Barondeau DP, Lindahl PA. J Am Chem Soc. 1997;119:3959–3970. [Google Scholar]
- 59.Seravalli J, Xiao Y, Gu W, Cramer SP, Antholine WE, Krymov V, Gerfen GJ, Ragsdale SW. Biochemistry. 2004;43:3944–3955. doi: 10.1021/bi036194n. [DOI] [PubMed] [Google Scholar]
- 60.Menon S, Ragsdale SW. Biochemistry. 1998;37:5689–5698. doi: 10.1021/bi9727996. [DOI] [PubMed] [Google Scholar]
- 61.Menon S, Ragsdale SW. J Biol Chem. 1999;274:11513–11518. doi: 10.1074/jbc.274.17.11513. [DOI] [PubMed] [Google Scholar]
- 62.Lebertz H, Simon H, Courtney LF, Benkovic SJ, Zydowsky LD, Lee K, Floss HG. J Am Chem Soc. 1987;109:3173–3174. [Google Scholar]
- 63.Ram MS, Riordan CG. J Am Chem Soc. 1995;117:2365–2366. [Google Scholar]
- 64.Ram MS, Riordan CG, Yap GPA, Liable-Sands L, Rheingold AL, Marchaj A, Norton JR. J Am Chem Soc. 1997;119:1648–1655. [Google Scholar]
- 65.Ragsdale SW, Wood HG. J Biol Chem. 1985;260:3970–3977. [PubMed] [Google Scholar]
- 66.Seravalli J, Gu W, Tam A, Strauss E, Begley TP, Cramer SP, Ragsdale SW. Proc Natl Acad Sci USA. 2003;100:3689–3694. doi: 10.1073/pnas.0436720100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Matthews RG. Acc Chem Res. 2001;34:681–689. doi: 10.1021/ar0000051. [DOI] [PubMed] [Google Scholar]
- 68.Banerjee R, Ragsdale SW. Ann Rev Biochem. 2003;72:209–247. doi: 10.1146/annurev.biochem.72.121801.161828. [DOI] [PubMed] [Google Scholar]
- 69.Tan X, Sewell C, Yang Q, Lindahl PA. J Am Chem Soc. 2003;125:318–319. doi: 10.1021/ja028442t. [DOI] [PubMed] [Google Scholar]
- 70.DiMarco AA, Bobik TA, Wolfe RS. Ann Rev Biochem. 1990;59:355–394. doi: 10.1146/annurev.bi.59.070190.002035. [DOI] [PubMed] [Google Scholar]
- 71.Ellermann J, Kobelt A, Pfaltz A, Thauer RK. FEBS Lett. 1987;220:358–362. doi: 10.1016/0014-5793(87)80846-3. [DOI] [PubMed] [Google Scholar]
- 72.Thauer RK. Microbiol. 1998;144:2377–2406. doi: 10.1099/00221287-144-9-2377. [DOI] [PubMed] [Google Scholar]
- 73.Diekert G, Klee B, Thauer RK. Arch Microbiol. 1980;124:103–106. doi: 10.1007/BF00407036. [DOI] [PubMed] [Google Scholar]
- 74.Diekert G, Jaenchen R, Thauer RK. FEBS Lett. 1980;119:118–120. doi: 10.1016/0014-5793(80)81011-8. [DOI] [PubMed] [Google Scholar]
- 75.Whitman WB, Wolfe RS. Biochem Biophys Res Commun. 1980;92:1196–1201. doi: 10.1016/0006-291x(80)90413-1. [DOI] [PubMed] [Google Scholar]
- 76.Ermler U, Grabarse W, Shima S, Goubeaud M, Thauer RK. Science. 1997;278:1457–1462. doi: 10.1126/science.278.5342.1457. [DOI] [PubMed] [Google Scholar]
- 77.Holliger C, Pierik AJ, Reijerse EJ, Hagen WR. J Am Chem Soc. 1993;115:5651–5656. [Google Scholar]
- 78.Telser J. In: Structure and Bonding. Williams RJP, editor. Springer Verlag; Heidelberg: 1998. pp. 32–63. [Google Scholar]
- 79.Maki AH, Edelstein N, Davison A, Holm RH. J Am Chem Soc. 1964;86:4580–4587. [Google Scholar]
- 80.Harmer J, Finazzo C, Piskorski R, Bauer C, Jaun B, Duin EC, Goenrich M, Thauer RK, Van Doorslaer S, Schweiger A. J Am Chem Soc. 2005;127:17744–17755. doi: 10.1021/ja053794w. [DOI] [PubMed] [Google Scholar]
- 81.Grabarse WG, Mahlert F, Duin EC, Goubeaud M, Shima S, Thauer RK, Lamzin V, Ermler U. J Mol Biol. 2001;309:315–330. doi: 10.1006/jmbi.2001.4647. [DOI] [PubMed] [Google Scholar]
- 82.Horng YC, Becker DF, Ragsdale SW. Biochemistry. 2001;40:12875–12885. doi: 10.1021/bi011196y. [DOI] [PubMed] [Google Scholar]
- 83.Goubeaud M, Schreiner G, Thauer RK. Eur J Biochem. 1997;243:110–114. doi: 10.1111/j.1432-1033.1997.00110.x. [DOI] [PubMed] [Google Scholar]
- 84.Berkessel A. Biorganic Chem. 1991;19:101–115. [Google Scholar]
- 85.Jaun B. Helv Chem Acta. 1990;73:2209–2216. [Google Scholar]
- 86.Signor L, Knuppe C, Hug R, Schweizer B, Pfaltz A, Jaun B. Chemistry. 2000;6:3508–3516. doi: 10.1002/1521-3765(20001002)6:19<3508::aid-chem3508>3.3.co;2-n. [DOI] [PubMed] [Google Scholar]
- 87.Lin SK, Jaun B. Helv Chem Acta. 1991;74:1725–1738. [Google Scholar]
- 88.Lin SK, Jaun B. Helv Chem Acta. 1992;75:1478–1490. [Google Scholar]
- 89.Jaun B, Pfaltz A. J Chem Soc Chem Commun. 1988;1327:293–294. [Google Scholar]
- 90.Rospert S, Voges M, Berkessel A, Albracht SPJ, Thauer RK. Eur J Biochem. 1992;210:101–107. doi: 10.1111/j.1432-1033.1992.tb17396.x. [DOI] [PubMed] [Google Scholar]
- 91.Goenrich M, Mahlert F, Duin EC, Bauer C, Jaun B, Thauer RK. J Biol Inorg Chem. 2004;9:691–705. doi: 10.1007/s00775-004-0552-1. [DOI] [PubMed] [Google Scholar]
- 92.Kunz RC, Horng YC, Ragsdale SW. J Biol Chem. 2006;281:34663–34676. doi: 10.1074/jbc.M606715200. [DOI] [PubMed] [Google Scholar]
- 93.Pelmenschikov V, Blomberg MRA, Siegbahn PEM, Crabtree RH. J Am Chem Soc. 2002;124:4039–4049. doi: 10.1021/ja011664r. [DOI] [PubMed] [Google Scholar]
- 94.Strous M, Jetten MS. Annu Rev Microbiol. 2004;58:99–117. doi: 10.1146/annurev.micro.58.030603.123605. [DOI] [PubMed] [Google Scholar]
- 95.Dey M, Kunz R, Heuvelen KMV, Craft JL, Horng YC, Tang Q, Bocian DF, George SJ, Brunold TC, Ragsdale SW. Biochemistry. 2006;45:11915–11933. doi: 10.1021/bi0613269. [DOI] [PMC free article] [PubMed] [Google Scholar]