

Abstract
Medical countermeasures to prevent or treat smallpox are needed due to the potential use of poxviruses as biological weapons. Safety concerns with the currently available smallpox vaccine indicate a need for research on alternative poxvirus vaccine strategies. Molecular vaccines involving the use of proteins and/or genes and recombinant antibodies are among the strategies under current investigation. The poxvirus L1 protein, encoded by the L1R open reading frame, is the target of neutralizing antibodies and has been successfully used as a component of both protein subunit and DNA vaccines. L1-specific monoclonal antibodies (e.g., mouse monoclonal antibody mAb-7D11, mAb-10F5) with potent neutralizing activity bind L1 in a conformation-specific manner. This suggests that proper folding of the L1 protein used in molecular vaccines will affect the production of neutralizing antibodies and protection. Here, we co-crystallized the Fab fragment of mAb-7D11 with the L1 protein. The crystal structure of the complex between Fab-7D11 and L1 reveals the basis for the conformation-specific binding as recognition of a discontinuous epitope containing two loops that are held together by a disulfide bond. The structure of this important conformational epitope of L1 will contribute to the development of molecular poxvirus vaccines and also provides a novel target for anti-poxvirus drugs. In addition, the sequence and structure of Fab-7D11 will contribute to the development of L1-targeted immunotherapeutics.
Keywords: Conformational epitope, vaccinia, poxvirus L1R, neutralizing antibody, Fab structure
Introduction
As a result of potential bioterrorism threats from smallpox (caused by Variola virus), there has been renewed interest in poxvirus vaccines and protective immunity (Harrison et al., 2004). Current vaccines against smallpox require inoculation with the related live vaccinia virus (VACV). The infection associated with the live virus vaccine can lead to complications in healthy individuals, but immune-deficient individuals and pregnant women are at special risk (Henderson et al., 1999; Belongia and Naleway, 2003). One current approach to improve the vaccination strategy is to use viral strains that are more attenuated (Coulibaly et al., 2005; Kidokoro et al., 2005). An alternative is to use viral protein subunits and/or genes in a molecular vaccine approach.
Development of molecular vaccines requires the identification of poxvirus immunogens that elicit immune responses contributing to protection (Galmiche et al., 1999; Hooper et al., 2000; Fogg et al., 2004; Davies et al., 2005; Sakhatskyy et al., 2006). Several protective immunogens have been identified, including the poxvirus L1 protein encoded by the L1R open reading frame. L1-based DNA vaccines and protein subunit vaccines can elicit neutralizing antibodies in mice and nonhuman primates and contribute to protection in lethal disease models (Fogg et al., 2004; Hooper et al., 2004; Heraud et al., 2006; Xiao et al., 2007).
L1 is a myristoylated, transmembrane protein found on the surface of the mature virion (MV) form of poxviruses (Ravanello et al., 1993). It is conserved in all orthopoxviruses and its sequence is almost identical among VACV, Variola virus, and monkeypox virus. Deleting the L1R gene blocks morphogenesis and prevents the formation of infectious virus (Ravanello and Hruby, 1994). However, L1 may also play a role (direct or indirect) in viral entry into cells because L1-specific monoclonal antibodies (e.g., mouse monoclonal antibodies: 7D11, 10F5, and 2D5) bind the surface of the virions and efficiently neutralize infectivity (Wolffe et al., 1995; Ichihashi and Oie, 1996; Hooper et al., 2000). These antibodies neutralize infectivity after virion attachment (Ichihashi et al., 1994; Wolffe et al., 1995); however, recent reports indicate that the L1 protein is not a component of the viral fusion complex (Senkevich et al., 2005; Townsley et al., 2005; Moss, 2006). L1 contains three disulfide bonds, which are formed by the virus-encoded redox pathway (Senkevich et al., 2002). Two of the three intramolecular disulphide bonds are essential for production of infectious viral particles (Blouch et al., 2005). When the bonds in L1 are reduced, the neutralizing antibodies, mAb-7D11, mAb-10F5 and 2D5, cannot recognize L1 (Wolffe et al., 1995; Ichihashi and Oie, 1996), (Hooper, J.W., Schmaljohn, A.L., unpublished data). Because L1 is an attractive candidate for potential subunit vaccines and/or immunotherapeutics it is important to understand the nature of the epitopes of L1 that play a crucial role in antibody-mediated protective immunity. In this report, we investigated the nature of the interaction between mAb-7D11 and L1 and the mechanism by which these potent neutralizing antibodies prevent infection. The co-crystal structure of the Fab fragment of 7D11 bound to the L1 protein reveals the basis for the conformation-specific binding as recognition of a discontinuous epitope containing two loops held together by a disulfide bond.
Results
MAb-7D11, 7D11-F(ab′)2, and 7D11-Fab fragments bind purified VACV and recombinant L1 expressed in Escherichia coli
Before performing co-crystallization studies involving recombinant L1 and 7D11-Fab, we tested the differential capacity of mAb-7D11, 7D11-F(ab′)2, and 7D11-Fab to bind VACV and recombinant L1 purified from E. coli. Purified mAb-7D11, 7D11-F(ab′)2, and 7D11-Fab were evaluated for binding on ELISA plates coated with purified VACV (Fig. 1A). MAb-7D11 and 7D11-F(ab′)2 bound the purified viral antigen at all concentrations tested. In contrast, 7D11-Fab only bound to purified virions when the Fab concentration was high. We observed similar ELISA results using recombinant L1 purified from E. coli as antigen (Fig. 1B). Interestingly, when the purified VACV or recombinant L1 was treated with ultraviolet (UV) light, reactivity with mAb-7D11, 7D11-F(ab′)2, and 7D11-Fab was lost (Fig. 1A and 1B). Thus, 7D11-Fab bound both intact virions and recombinant L1 protein albeit with lower reactivity than bivalent forms of the antibody, and the epitope recognized by this antibody could be disrupted when exposed to UV light.
Fig. 1.

Interaction of mAb-7D11, F(ab′)2, and Fab fragments with purified VACV and recombinant L1.
(A) Binding to purified VACV. Serial twofold dilutions of mAb-7D11, 7D11-F(ab′)2, and 7D11-Fab or negative control antibody mAb-3D7 starting at 80 μg/ml were tested for the capacity to bind purified virus by VACV-infected-cell lysate ELISA. Some antigen-coated plates were treated with UV light (grey dashed lines) before incubation with mAb-7D11 (MAb-7D11 UV). Each symbol represents the mean ± standard deviation (SD) of duplicate samples.
(B) Binding to recombinant L1. Serial twofold dilutions of mAb-7D11, 7D11-F(ab′)2, and 7D11-Fab or negative control antibody mAb-10F10 (anti-A33) starting at 80 μg/ml were tested for the capacity to bind recombinant L1 by ELISA. Purified L1 at 100 ng/well was untreated or treated with UV light (grey dashed lines). Each symbol represents the mean ± SD of duplicate samples. The x-axes in A and B are plotted on log(2) scales. The smallest error bars in A and B are hidden beneath the symbols and are not seen.
Neutralization capacity of mAb-7D11, 7D11-F(ab′)2, and 7D11-Fab fragments
We investigated the ability of mAb-7D11, 7D11-F(ab′)2, and 7D11-Fab fragments to neutralize VACV infection by a plaque reduction-neutralization test (PRNT) (Fig. 2A). As expected, mAb-7D11 efficiently neutralized VACV plaque formation as previously described (Wolffe et al., 1995). 7D11-F(ab′)2 fragments were also able to neutralize VACV plaque formation to levels similar to those of the intact immunoglobulin. Even at 0.04 μg/ml, the lowest concentration tested, 7D11-F(ab′)2 yielded ~75% neutralization. In contrast, 7D11-Fab required more than 100x greater concentrations of protein to give a similar level of neutralization. At the highest concentration used, 80 μg/ml, Fab fragments elicited ~75% reduction in plaque number. These findings indicated that bivalency plays a role in the capacity of mAb-7D11 to efficiently neutralize VACV.
Fig. 2.

Plaque reduction-neutralization test using mAb-7D11, 7D11-F(ab′)2, and 7D11-Fab.
(A) VACV was incubated with purified mAb-7D11, 7D11-F(ab′)2, or 7D11-Fab at 37°C for 1 h. Virus-antibody mixtures or untreated virus were adsorbed on to confluent monolayers of BSC-1 cells, followed the addition of a semi-solid overlay of 1.5% methylcellulose as described in Methods. After 4 d, plaques were counted after staining with 3% crystal violet. The percent neutralization of plaque formation by antibody addition relative to untreated virus is plotted versus concentration on a log(2) scale.
(B) VACV was incubated with mAb-7D11 or 7D11-Fab at the concentrations indicated, for 1 h at 37°C. One sample, incubated with Fab, was then incubated with anti-Fab antibodies (1:100) for 1 h. The other samples were incubated for an additional 1 h in the absence of anti-Fab antibodies. After incubation, the virus was added to BSC-1 cells and plaque formation analyzed as described in A.
(C) Confluent monolayers of BSC-1 cells were chilled on ice and incubated with ~50 PFU of VACV strain IHD-J per well for 1 h at 4°C. After adsorption, virus was removed and mAb-7D11 or 7D11-Fab was added to the wells at the indicated concentrations for 1 h at 4°C. After incubation, anti-Fab antibodies (1:100) were added to the wells of some samples for another 1 h at 4°C. Plates were then incubated at 37°C and plaques were visualized as described in A.
To determine if bivalent binding of Fab could increase the neutralizing activity, we incubated 7D11-Fab fragments with VACV, added anti-Fab antibody, and assayed neutralization by the PRNT. As shown in Fig. 2B, incubating 7D11-Fab with anti-Fab antibodies restored neutralizing activity. This finding confirmed that bivalency does indeed play a key role in the ability of mAb-7D11 to neutralize.
The role of bivalency in the enhancement of 7D11-mediated neutralization could be due to intra- and/or inter-particle (aggregation) cross-linking. To investigate this, 50 PFU of VACV were adsorbed onto BSC-1 cell monolayers at 4°C. Since only 50 PFU of VACV were used, we reasoned that the viral particles were not close enough to permit inter-particle interactions and therefore allowed us to determine how much neutralization was due to intra-particle L1 binding. Following adsorption, unbound virus was removed and bound particles were incubated at 4°C with varying concentrations of either mAb-7D11 or 7D11-Fab. Subsequently, anti-Fab antibodies were added to the monolayers. Plaque formation was analyzed four days post-infection. As predicted, mAb-7D11 efficiently neutralized VACV plaque formation when added post-adsorption (Fig. 2C). 7D11-Fab alone neutralized virus poorly, and even at the highest concentration tested, only ~50% of virus was inhibited. Cross-linking of the 7D11-Fab fragments bound to adsorbed virus led to a marked increase in virus neutralization, albeit less efficiently than that of mAb-7D11. The neutralizing effect of 7D11-Fab plus anti-Fab antibodies was rapidly lost as the concentration of Fab fragments was reduced, possibly due to the inefficient binding of 7D11-Fab to L1 at 4°C. That cross-linked 7D11-Fab was capable of neutralizing VACV suggested that a significant portion of the bivalent neutralizing capability of mAb-7D11 is intra-particle L1 binding.
The structure of the complex between L1 and 7D11-Fab
To understand the basis of the conformational specificity of mAb-7D11 recognition, the structure of the complex between 7D11-Fab and L1 was determined by X-ray crystallography (Table 1). Four copies of the Fab/L1 complex were present in the crystal asymmetric unit. The 7D11-Fab bound to a surface on L1 formed by four long loops on the side of the molecule opposite the amino and carboxyl termini of L1 and presumably away from the viral membrane. These four loops connect the helices of the helical bundle to the strands of the beta sheets of L1 (Fig. 3A). The heavy chain of 7D11-Fab comprised most of the recognition site for L1 (Fig. 3B). A total surface area of 1340 Å2 was buried in the interface between the heavy chain and L1, in contrast to the 225 Å2 of buried surface area in the light chain:L1 interaction. The typical range for total surface area buried at the interface between an antigen and both chains of the antibody is 1400–1700 Å2 (Davies et al., 1990). Another measure that qualitatively describes the strength of the interaction is the analysis of the interaction surface as given by the shape complementarity statistic (Sc), where a perfect fit yields a score of 1 (Lawrence and Colman, 1993). The heavy chain:L1 interface gave a Sc of 0.65, which is within the range (0.64–0.68) for antibody:antigen complexes. The light chain:L1 interface only yielded a score of 0.26, indicating poor complementarity with L1.
Table 1.
Crystallographic statistics.
| Resolution (Å) | 50–3.10 (3.21–3.10) |
| X-ray source | 19-ID SBC-CAT |
| Energy (eV) | 12662 |
| Wavelength (Å) | 0.97915 |
| Completeness (%) | 98.6 (99.6) |
| Average redundancy | 3.2 (3.2) |
| Rsym (%) | 12.4 (46.4) |
| <I>/<σI> | 9.3 (2.3) |
| Dimensions of C2 cell | |
| a (Å) | 218.8 |
| b (Å) | 85.6 |
| c (Å) | 211.8 |
| α (°) | 90 |
| β (°) | 119.60 |
| γ (°) | 90 |
| Ramachandran: | |
| Favored | 83.4 % |
| Allowed | 15.6 % |
| Generously Allowed | 1.0 % |
| Disallowed | 0.0 % |
| Rmsd bond lengths | 0.01 Å |
| Rmsd bond angles | 1.4° |
| No. residues | 2466 |
| No. of protein atoms | 18612 |
| No. waters | 14 |
| No. glycerols | 8 |
| No. refl. refined/no. free | 58119/3109 |
| Rwork/Rfree (%) | 23.8(32.3)/25.4(35.7) |
Values in parenthesis are for the highest resolution bin.
Fig. 3.
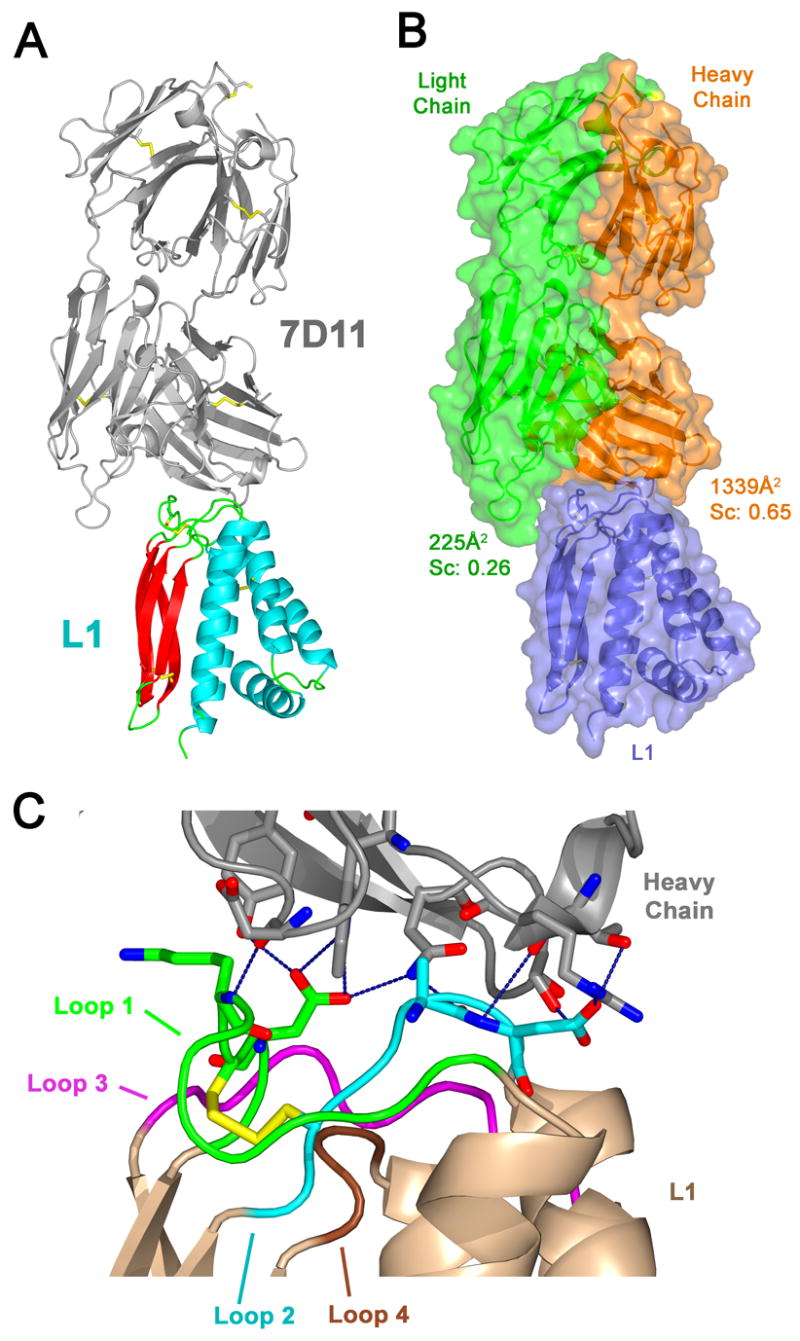
Structure of 7D11-Fab bound to L1.
(A) 7D11-Fab recognizes the loops connecting the helices to the β strands of L1. The ribbon diagram shows L1 bound by the 7D11-Fab (gray). L1 is colored by secondary structure with β sheets in red, helices in blue, and loops in green. Disulfide bonds are shown in yellow.
(B) The heavy chain of 7D11-Fab provides most of the interactions with L1. In the same orientation as in A, the surface diagram is colored by polypeptide chains with the 7D11 heavy chain in orange, the light chain in green, and L1 in blue. The calculated total buried surface area and the shape complementarity statistic between L1 and each of the antibody chains are indicated.
(C) 7D11-Fab makes numerous hydrogen bonds and van der Waals interactions with loops 1 and 2 of L1. Oriented as in parts A and B, and numbered from N-terminus to C-terminus, the four loops of L1 are colored as follows: loop 1 (green), loop 2 (blue), loop 3 (purple), and loop 4 (brown). These loops are held together by a disulfide bond (yellow) between Cys 34 in loop 1 and Cys 57 in loop 2. The carbon atoms are colored based on the loop that the residue is in, nitrogen atoms are blue, and oxygen atoms are red. Hydrogen bonds displayed as dashed lines in blue show extensive bonding with loop 1 (green) and loop 2 (blue) of L1.
Analysis of the molecular interactions between 7D11-Fab and L1 also confirmed that the light chain does not significantly contribute to binding. In the structure, there were no interactions between the light chain and L1 within a distance of 4 Å. In contrast, there were hydrogen bonds (Table 2) formed by the antibody to residues of loops 1 and 2 of L1. These loops are held together by a disulfide bond between Cys34 of loop 1 and Cys57 of loop 2 (Fig. 3C). There were also additional van der Waals interactions between the heavy chain and loops 1–3 of L1 (Table 3). The loops and residues of L1 which mediate interactions with 7D11 are mapped on the L1 sequence in Fig. 4C.
Table 2.
Hydrogen bonds between L1 and 7D11-Fab.
| From L1 | To HC | Distance | ||||||
|---|---|---|---|---|---|---|---|---|
| Residue name | Atom | Loop | Residue name | Atom | CDR | |||
| Lys | 33 | N | 1 | Asp | 102 | O | 3 | 2.7Å |
| Asp | 35 | Oδ1 | 1 | Tyr | 50 | OH | 2 | 2.9Å |
| Asp | 35 | Oδ1 | 1 | Trp | 33 | Nε1 | 1 | 3.0Å |
| Asp | 35 | Oδ2 | 1 | Trp | 33 | Nε1 | 1 | 3.3Å |
| Asp | 35 | Oδ2 | 1 | Asn | 52 | N& δ2 | 2 | 3.1Å |
| Ala | 59 | O | 2 | Asn | 52 | N δ2 | 2 | 2.9Å |
| Asp | 60 | N | 2 | Thr | 30 | O | 1 | 3.6Å |
| Asp | 60 | Oδ1 | 2 | Ser | 54 | Oγ | 2 | 3.4Å |
| Asp | 60 | Oδ2 | 2 | Arg | 31 | Nε | 1 | 3.4Å |
| Asp | 60 | Oδ2 | 2 | Thr | 30 | Oγ1 | 1 | 2.9Å |
Listed are the atoms from L1 and their corresponding loops, the hydrogen bond partner in the heavy chain (HC) and their corresponding complementarity-determining region (CDR) loop, and the distances between them.
Table 3.
Van der Waals interactions between L1 and the heavy chain of 7D11-Fab.
| L1 | Loop | HC | CDR |
|---|---|---|---|
| Asn 27 | 1 | Arg 31 | 1 |
| Gln 31 | 1 | Asp 102 | 3 |
| Gln 31 | 1 | Tyr 104 | 3 |
| Thr 32 | 1 | Asp 102 | 3 |
| Thr 32 | 1 | Trp 33 | 1 |
| Thr 32 | 1 | Tyr 104 | 3 |
| Lys 33 | 1 | Asp 102 | 3 |
| Lys 33 | 1 | Trp 33 | 1 |
| Lys 33 | 1 | Tyr 104 | 3 |
| Asp 35 | 1 | Asn 52 | 2 |
| Asp 35 | 1 | Trp 33 | 1 |
| Asp 35 | 1 | Tyr 50 | 2 |
| Asp 35 | 1 | Tyr 57 | 2 |
| Ser 58 | 2 | Trp 33 | 1 |
| Ala 59 | 2 | Asn 52 | 2 |
| Ala 59 | 2 | Ser 54 | 2 |
| Ala 59 | 2 | Thr 30 | 1 |
| Ala 59 | 2 | Trp 33 | 1 |
| Asp 60 | 2 | Arg 31 | 1 |
| Asp 60 | 2 | Ser 54 | 2 |
| Asp 60 | 2 | Thr 30 | 1 |
| Ala 61 | 2 | Ser 54 | 2 |
| Ala 61 | 2 | Thr 55 | 2 |
| Asp 62 | 2 | Ser 54 | 2 |
| Lys 125 | 3 | Gly 56 | 2 |
| Lys 125 | 3 | Thr 58 | 2 |
| Lys 125 | 3 | Tyr 57 | 2 |
| Lys 127 | 3 | Tyr 57 | 2 |
Residues that participate in van der Waals interactions (4 Å distance cutoff) between L1 and 7D11-Fab are shown. Listed on the left are the atoms from L1 and their corresponding loops. On the right are the interacting atoms in the heavy chain (HC) and their corresponding complementarity-determining region (CDR) loop.
Fig. 4.

Interaction of anti-L1 monoclonal antibodies with mutated L1.
(A) COS-7 cells were transfected with pWRG/TPA-L1R (black line), pWRG/TPA-L1R D35N (grey line), or empty pWRG vector (solid grey). Cells were incubated with 1:100 dilutions of mAb-7D11 (anti-L1), mAb-10F5 (anti-L1), or a control antibody mAb-10F10 (anti-A33). A anti-mouse secondary antibody (1:500) conjugated to Alexa 488 fluorochrome was added and cells were analyzed by flow cytometry using a FACSCalibur flow cytometer. For each sample, 10,000 cells were counted.
(B) Hydrogen bonds involved in the binding by the 7D11-Fab to Asp35 of L1 as determined in the crystal structure are shown in blue. The atoms of critical residues are shown as sticks. The carbon atoms retain the color of the loops to which they belong: loop 1 (green), loop 2 (blue), loop 3 (purple), and loop 4 (brown). In the structure (upper panel), Asp35 (green) makes hydrogen bonds with the side chains of Trp33 and Tyr50 on the heavy chain (gray). Also shown is the hydrogen bond between the carbonyl oxygen of Ala59 on L1 (blue) and Nδ2 of Asn52 of the heavy chain (gray). In the lower panel, the predicted binding of mAb-7D11 to the D35N mutant of L1 (Model) is shown. The hydrogen bond between Asn35 of L1 (green) and Tyr50 of the heavy chain would be analogous to the bond made with Asp35. A new hydrogen bond (red) can be formed between Asn35 of L1 and an alternate rotamer of Asn52 of the Fab heavy chain, while maintaining the hydrogen bond (red) between Ala59 of L1 and Asn52 of the heavy chain. The L1:7D11-Fab structure has been rotated 180° around the vertical axis relative to its orientation in Fig. 3C. (C) Alignment of the L1 ectodomain sequences from VACV, Variola virus, and monkeypox virus in which residues are shown that differ from the vaccinia virus sequence. Residues of the four loops are shaded as in Fig. 3C: loop 1 (green), loop 2 (blue), loop 3 (purple), and loop 4 (brown). Residues involved in van der Waals interactions are marked with one dot and residues that also mediate hydrogen bonds are marked with two dots above the sequence. Cysteines are highlighted in yellow.
mAb-7D11, but not mAb-10F5, binds the 2D5 escape mutant
Previous studies reported the isolation of a mutant VACV that escaped neutralization by the L1-specific mouse mAb, 2D5 (Ichihashi and Oie, 1996). The escape mutation was in the L1R gene and resulted in a change in residue 35 from an Asp to an Asn (D35N). Because mAb-7D11 binds to a similar epitope as 2D5, we wanted to determine if a change from an Asp to Asn would disrupt mAb-7D11 binding. COS-7 cells were transfected with either pWRG/TPA-L1R or a plasmid containing the D35N mutation. pWRG/TPA-L1R is a plasmid in which the tissue plasminogen activator (TPA) secretion signal sequence is inserted upstream and in-frame with the L1R open reading frame. The TPA permits L1 to be processed through the endoplasmic reticulum where it can take advantage of the endogenous cellular disulfide bond formation machinery and fold properly. Because the transmembrane region was not removed, the molecule was targeted to the plasma membrane where it could be detected by anti-L1 antibodies (Hooper et al., 2007). At 48 h post-transfection, the ability of antibodies to bind to surface-expressed L1 or L1 D35N was examined by flow cytometry (Fig. 4A). As indicated by the shift in fluorescence intensity, mAb-7D11 interacted with both L1 and the D35N mutant. In contrast, a second L1-specific mAb with potent neutralizing activity, mAb-10F5, did not efficiently interact with the L1 D35N molecule. Neither mAb-7D11 nor mAb-10F5 interacted with cells transfected with the empty pWRG vector. MAb-7D11 and mAb-10F5 competed for binding to VACV as measured by competition ELISA (Hooper, J.W., Schmaljohn, A.L., unpublished observations). As a control, we used an antibody against the poxvirus A33 protein, mAb-10F10, that did not interact with L1-expressing cells (Fig. 4A). These observations indicated that mutation of Asp35 to Asn did not disrupt the interaction of all potent neutralizing L1-specific monoclonal antibodies even if they bound the same region of L1 as measured by competition ELISA.
Structural basis for the binding of mAb-7D11 to a neutralization escape mutation in L1
In the 7D11-Fab structure, Asp35 is involved in hydrogen bonds with the mAb-7D11 heavy chain. Based on the distances between atoms capable of forming hydrogen bonds (Table 2), the Oδ1 atom of Asp35 can form a hydrogen bond with Tyr50 on the heavy chain, while Oδ2 of Asp35 can form a hydrogen bond with the Nε1 atom of Trp33 on the heavy chain (Fig. 4B, top). If Asp35 was changed to Asn, the hydrogen bond with Tyr50 could still occur (Fig. 4B, bottom). The hydrogen bond formed by Oδ2 of Asp35 with Trp33 could be replaced by a similar hydrogen bond between Nδ2 of Asn35 and Oδ2 of an alternative Asn52 rotamer conformation on the heavy chain. The hydrogen bond made by Asn52 of the heavy chain to the carbonyl oxygen of Ala59 in L1 can still be formed by the alternate rotamer (Fig. 4B, bottom). Thus, based on the structure of the complex, we can provide an explanation for the binding of mAb-7D11 to the D35N mutant, while other L1-specific antibodies (2D5 and mAb-10F5) may not accommodate the hydrogen bond with Nδ2 of Asn35 of the D35N escape mutant.
Discussion
In this study, we characterized the binding of mAb-7D11, a potent neutralizing antibody that recognizes L1 on the surface of poxvirus virions. The 7D11-Fab:L1 co-crystal structure reveals interactions to loops 1 and 2 of L1 that are held together by a disulfide bond between Cys34 and Cys57. The conformations of the loops are stabilized by the disulfide bond, which provides the explanation at the molecular level for previous reports that the 7D11 epitope is disrupted by disulfide bond reduction.
A panel of six anti-L1 antibodies was recently characterized (Aldaz-Carroll et al., 2005). Peptide mapping and blocking assays demonstrated that the mAbs bound two different antigenic sites. Two of the antibodies (VMC-4 and VMC-35) bound a conformational epitope and were non-neutralizing. Four of the antibodies (VMC-2, VMC-3, VMC-5, VMC-6) recognized a linear, or pseudolinear, epitope, which includes residues 118–128 of L1 and were neutralizing (Aldaz-Carroll et al., 2005). In blocking experiments, none of the six mAbs bound to the same conformational epitope recognized by the neutralizing antibodies 7D11, 2D5, and 10F5. MAb-7D11 and mAb-2D5 compete for binding to L1 as measured by BIAcore (Aldaz-Carroll et al., 2005) and mAb-7D11 and mAb-10F5 bind to the same site by competition ELISA. The 7D11:L1 co-crystal structure reveals that there is some overlap between the linear neutralizing epitope that includes residues 118–128 of L1 and the conformational epitope recognized by mAb-7D11. Two residues of loop 3 of L1 (loop 3 is colored purple in Figs. 3C and 4B), Lys125 and Lys127, make van der Waals contacts with mAb-7D11 (Table 3) and are also contained within the linear epitope mapped to antibodies generated by Aldaz-Carroll et al.; however, it is possible that Lys125 and Lys127 do not contribute significantly to mAb-7D11 binding which would allow mAb-7D11 and other antibodies to bind simultaneously. This structure and the previously identified linear epitope suggest that the surface made by the loops at the end of L1 is accessible to antibody and that binding of antibody to this region of L1 interferes with events involved in entry.
None of the six L1-specific mAb characterized by Aldaz-Carroll et al. bind the same epitope recognized by the highly potent neutralizing mAb-7D11. The six antibodies were generated by immunizing mice with the ectodomain of L1 produced in the baculovirus expression system, while mAb-7D11 was generated by vaccinating with infectious virus. The baculovirus-expressed L1 protein was targeted through the endoplasmic reticulum for secretion by the addition of a melittin signal peptide to L1 and appears to be folded correctly as measured by the binding of mAb-7D11 and the presence of three disulfide bonds. Glycosylation, however, is present in the baculovirus-produced protein, unlike the L1 produced in a VACV-infected cell, which does not translocate to the cellular compartments that possess glycosylation machinery. Asn residues 27, 50, and 117 are possible N-linked sites and Asn50 and Asn117 were shown to be glycosylated (Aldaz-Carroll et al., 2005). A glycan on Asn27 could alter the immunogenicity of this epitope as Asn27 makes van der Waals contacts with the 7D11-Fab (Table 2). We found that the correct conformation of the 7D11 epitope can be disrupted by UV light. Exposure of VACV-infected cell lysate or purified L1 protein to UV light ablated binding of mAb-7D11 (Fig. 1) or mAb-10F5 (data not shown). UV radiation likely disrupted the critical disulfide bonds required for generating high-affinity neutralizing epitopes as it is well-known that UV light can break disulfide bonds, especially through the photo-excitation of aromatic residues (Neves-Petersen et al., 2006; Vanhooren et al., 2006). In the past, UV-killed poxvirus vaccines were tested for the capacity to protect (Turner et al., 1970). In retrospect, our data indicate that UV-inactivated poxvirus antigen lacks an important L1-neutralizing epitope. These data emphasize the importance of vaccination with an intact, correctly folded, and processed protein, to provide the discontinuous epitope formed by the loops of L1.
MAb-7D11, 7D11-F(ab′)2, and cross-linked 7D11-Fab exhibit exceedingly potent neutralizing activity, even when added after virus adsorption to cell monolayers. In addition, passive transfer experiments in mice indicate this mAb can elicit protective immunity (Hooper et al., 2002; Lustig et al., 2005). The mechanism by which mAb-7D11 neutralizes virus remains unknown. Here, we identified the precise region of the poxvirus that mAb-7D11 binds and we demonstrated that bivalent binding enhanced the neutralizing activity of this antibody. The ability of high Fab concentration to neutralize virus suggests that the effect does not absolutely require cross-linking of L1 on the surface of virions. The reduced interaction of 7D11-Fabs with intact VACV (Fig. 1A) and purified L1 (Fig. 1B) suggest that the poor neutralizing capacity of 7D11-Fabs was largely due to avidity. This reduced capacity of Fab to neutralize VACV was reported for L1-specific 10F5-Fab (Schmaljohn et al., 1999). Our findings are similar to what has been reported with 7–10A, a mAb that interacts with a discontinuous epitope present in the spike protein of murine hepatitis virus (MHV) (Lamarre and Talbot, 1995). In that study, bivalency significantly enhanced neutralization of MHV both in vitro and in vivo; however, Fab fragments could also neutralize virus but to a much lesser extent. The authors concluded that bivalency might enhance neutralization by steric hindrance or by causing a conformational change in the viral protein due to the increased avidity. It is possible that mAb-7D11 functions by sterically hindering the interaction of L1 with viral or cellular molecules or by blocking a conformational change in the virion required for entry. Further studies will be required to identify the critical step in the infection process blocked by mAb-7D11 and other mAbs binding the same epitope on L1. Post-attachment steps that could be targeted by these mAbs include: the triggering of host-cell signaling pathways involved in particle uptake, the activation or function of the viral fusion complex, or the uncoating events required to deliver active viral replication machinery to the cytoplasm.
The L1:7D11-Fab complex structure provides details of the molecular interactions made by mAb-7D11. With its heavy and light chains sequenced and the specific interacting residues thus mapped, mAb-7D11 can be humanized for development as a therapeutic. The appropriate human antibody scaffold may be found by comparing root mean square deviations (rmsd) of known human antibody structures with mAb-7D11. This could minimize the positional changes of the interacting loops after engineering 7D11 to be like a human immunoglobulin. With the mapped interactions, the specificity-determining residues can also be used to minimize human anti-murine antibody responses (Padlan et al., 1995; Kashmiri et al., 2005).
With knowledge of the molecular interactions, mutations can be made to enhance mAb-7D11 binding. Molecular modeling may determine mutations in mAb-7D11 that would strengthen its interactions with L1. The affinity of mAb-7D11 can also be improved by experimental, molecular techniques such as phage display. Because the wild-type mAb-7D11 light chain does not contribute significantly to binding L1, it may be possible to identify light chain mutations that increase the affinity of mAb-7D11. Although the epitopes of 2D5, mAb-10F5, and mAb-7D11 overlap, there are differences in their binding to the D35N mutant of L1. Thus, combinations of L1-specific mAb would likely neutralize antibody escape mutants should they arise.
In the crystal that was used to determine the structure of L1 alone, two L1 molecules packed together in the asymmetric unit (Su et al., 2005). While others and we observed only monomeric L1 in solution (Aldaz-Carroll et al., 2005), the same packing was observed in the independently derived 7D11-Fab and L1 complex crystal of this study. Two sets of L1 pairs packed in the same manner as in the crystal of L1 alone. One pair of L1 molecules had an rmsd of 1.2 Å and the other pair exhibited an rmsd of 1.3 Å, calculated using 339 Cα–Cα distances (out of 346). If this dimer is physiologically relevant, the complex structure indicates that the same antibody molecule could not bind the two L1 molecules, as the two Fabs bound to each pair of L1 molecules are oriented in opposite directions. More work will be required to determine the exact orientation of L1 on the surface of virions. There are neutralizing anti-L1 antibodies that do not block binding by mAb-7D11 (Aldaz-Carroll et al., 2005). This suggests the interesting possibility that a bi-specific antibody consisting of mAb-7D11 and another specificity that does not block mAb-7D11, may together bind L1 and potentially exhibit a greater neutralizing activity.
Future work will seek to humanize the mAb-7D11 antibody either in the context of a mono-specific or bi-specific construct. Such a molecule used in conjunction with other humanized antibodies that target enveloped particles, such as the humanized chimpanzee B5 antibodies (Chen et al., 2006), may serve as a replacement for vaccinia immune globulin and can be used to treat sequelae arising in individuals vaccinated with live-virus. Alternatively, such a cocktail might function as a standalone anti-poxvirus therapeutic.
Lastly, based on the crystal structure of the mAb-7D11 complex, we identified a critical region on an essential, highly conserved poxvirus protein. Our data indicate that binding of Fab to this region (albeit at high concentrations) can neutralize virus infectivity. It will be possible to model the binding of other compounds to this structure and, in doing so, develop candidate antiviral drugs that may efficiently neutralize virus in serum. If such a compound could cross the cell membrane, it might be possible to neutralize infectivity during morphogenesis and prevent infectious virus from being released and disseminated.
Materials and Methods
Cells and virus
BSC-1, VERO and COS-7 cells were maintained as monolayers in Eagle’s minimal essential medium (EMEM) supplemented to contain 5% (v/v) fetal calf serum (Hyclone; Logan, UT), 10 mM HEPES, 100 U/ml penicillin, and 100 μg/ml of streptomycin. VACV strains IHD-J and Western Reserve (WR) were propagated on VERO monolayers and clarified crude lysates were stored until use at −70°C. WR was purified on a 20–60% sucrose gradient by centrifugation at 25,000 g for 2.5 h in an SW28 rotor (Beckman, Palo Alto, CA). The WR band was harvested from the gradient by using a glass Pasteur pipette.
Generation of mAb-7D11, 7D11-F(ab′)2, and 7D11-Fab
MAb-7D11 was obtained from culture supernatants of mAb-7D11 hybridoma cells and purified on a mAb-Trap antibody-binding column (Amersham Biosciences) according to the manufacturer’s protocol. 7D11-F(ab′)2 was generated using the ImmunoPure F(ab′)2 preparation kit (Pierce, Rockford, IL) following the manufacturer’s recommended protocol. Briefly, mAb-7D11 was concentrated to 10 mg/ml in F(ab′)2 digestion buffer consisting of 20 mM sodium acetate pH 4.5. Concentrated mAb were then incubated overnight with immobilized pepsin at 37°C with constant agitation. Separation of undigested mAb from F(ab′)2 was performed on an affinity Pak protein A binding column. To ensure undigested mAb were removed from the F(ab′)2 preparation, the digested mAb were passed through the column a second time. Recovered F(ab′)2 was concentrated in a 50 kDa molecular weight cut-off (MWCO) concentrator (Millipore, Bedford, MA) in 10 mM Tris-Cl buffer. 7D11-Fab fragments were generated using the Immunopure Fab preparation kit (Pierce) according to the recommended protocol. Purified mAb-7D11 was incubated overnight at 37°C in PBS [pH 10?] containing immobilized papain. Undigested mAb were separated from Fab fragments using a protein A binding column. Fab fragments were run through the column a second time to ensure that intact mAb-7D11 was adsorbed out of the Fab preparation. Fab fragments were concentrated using a 10 MWCO concentrator in 10 mM Tris-Cl buffer.
ELISA
VACV-specific ELISAs were performed essentially as described (Hooper et al., 2000). Purified VACV strain WR was diluted 1:50 in PBS and 50 μL/well was added to the wells of a 96-well, high-binding ELISA plate (Corning, Corning, NY) and dried overnight. “Mock” antigen was also plated. Mock antigen was prepared by loading uninfected cell lysate onto the sucrose gradient and harvesting the same portion of the sucrose gradient from which the purified virus was removed. Some samples were treated with UV light overnight at a distance of 550 cm from a standard safety light used in laminar flow hoods. Plates were blocked for 1 h at 37°C with PBS containing 0.1% Tween-20 (v/v) (PBS-T) and 5% (w/v) dry milk. Serial dilutions of mAb-7D11, 7D11-F(ab′)2, and 7D11-Fab fragments were made in blocking buffer. Dilutions were incubated with bound VACV for 1 h at 37°C. After incubation, plates were washed four times with PBS-T and incubated with a peroxidase-conjugated anti-mouse Fab-specific antibody (Sigma, St. Louis, MO) for 30 min at 37°C. Plates were subsequently washed four times and 100 μL of 2,2′-azinobis-3-ethylbenzthiazoline-6-sulfonic acid (ABTS) substrate (KPL, Gaithersburg, MD) was added to each well. Reactions were stopped by adding 100 μL of ABTS stop solution of 5% (w/v) sodium dodecylsulfate. The optical density (O.D.) at 405 nm was read on a Spectramax ELISA plate reader (Molecular Devices, Sunnyvale, CA). For each dilution series, the O.D. values from the mock antigen wells were subtracted from the experimental values. Each sample was run in duplicate.
L1-specific ELISA was performed essentially as previously described (Hooper et al., 2004). Purified L1 (Su et al., 2005) diluted in 0.1 M carbonate buffer, pH 9.6, was plated at 100 ng/well in 96-well plates. Plates were incubated at room temperature overnight in the presence or absence of UV light as described above. The remainder of the assay was performed as described above.
Plaque reduction-neutralization test
Plaque reduction-neutralization tests (PRNT) were performed as described previously (Hooper et al., 2000). VACV strain IHD-J was incubated with either mAb-7D11, 7D11-F(ab′)2, or 7D11-Fab at the indicated concentrations for 1 h at 37°C in a total volume of 200 μL. Virus mixture (180 μL) was then added to confluent monolayers of BSC-1 cells plated in 6-well plates. Virus was adsorbed to cells for 1 h at 37°C with rocking every 15 min. After adsorption, a 1:1 mixture (2 ml) of 3.0% methylcellulose (w/v) in water and 2x Eagles’ basal medium with Earle’s salts (EBME) was added to each well. Plates were incubated for 4 d, then stained with 3% crystal violet for 15 min and washed with water. In the indicated conditions where anti-mouse Fab antibodies (Sigma) were incubated with 7D11-Fab fragments, the 7D11-Fab fragments were pre-bound to VACV for 1 h, followed by incubation with anti-Fab antibodies (1:100) for 1 h. All experiments were performed in duplicate.
Antibody treatment of pre-bound VACV
To determine if 7D11 molecules inhibited post-attachment events in VACV entry, 50 plaque-forming units (PFU) of VACV were adsorbed to BSC-1 monolayers plated in 6-well plates at 4°C. After 1 h, unbound virus was removed by aspiration and cells were washed once with PBS. Adsorbed virus was next incubated with the indicated concentrations of mAb-7D11 or 7D11-Fab fragments for 1 h at 4°C. After incubation, one group of 7D11-Fab was incubated with anti-Fab antibodies (1:100) for 1 h at 4°C. MAb-7D11 and 7D11-Fab wells were incubated with an equivalent volume of PBS. After 1 h, 1.5% methylcellulose was added to each well and the plates were incubated at 37°C for 4 days. Plates were stained as described for PRNT. Each sample was tested in duplicate.
Site-directed mutagenesis of L1R
The pWRG/TPA-L1R plasmid has been described elsewhere (Hooper et al., 2003). The missense mutation changing the Asp at position 35 to an Asn was introduced into the L1R by performing two separate PCR steps. The first reaction used the primers 5′-GGGGGGCTAGCATGGGTGCCGCAGCAAGC-3′ and 5′-CAAACAAAATGTAACATAGAAATCGGC-3′ and the second reaction used the primers 5′-GGGAGATCTTCAGTTTTGCATATCCG-3′ and 5′-GCCGATTTCTATGTTACATTTTGTTTG-3′. The underlined codon encodes the D35N mutation. Each PCR reaction amplified part of the L1R gene. The two PCR reactions, consisting of parts of the L1R gene containing the mutation, were gel purified and a final PCR reaction was performed by combining both PCR fragments and the primers 5′-GGGGGGCTAGCATGGGTGCCGCAGCAAGC-3′ and 5′-GGGAGATCTTCAGTTTTGCATATCCG-3′. These primers amplified the entire L1R coding region and also introduced NheI and BglII sites into the product. The resultant PCR product was digested with NheI and BglII (New England Biolabs, Ipswich, MA) and then cloned into the pWRG/TPA vector. The mutation to form pWRG/TPA-L1R D35N was verified by DNA sequencing.
Flow cytometry
COS-7 cell monolayers (70–80% confluent) were transiently transfected with pWRG/TPA-L1R, pWRG/TPA-L1R D35N, or empty vector using Fugene6 (Roche, Indianapolis, IN) according to the manufacturer’s recommendations. Transfected cells were incubated at 37°C for 48 h, trypsinized, washed once with EMEM, and 5 × 105 cells were transferred to 1.5-ml tubes. Cells were incubated with the appropriate antibodies (1:100) for 1 h at room temperature with continuous agitation. After incubation with the primary antibody, cells were pelleted by centrifugation at 750 g for 3 min and washed twice with PBS. Cells were next incubated with anti-mouse Alexa 488 (Invitrogen, Carlsbad, CA) (1:500) for 30 min at room temperature. After incubation with the secondary antibody, cells were pelleted by centrifugation at 750 g for 3 min. Washed cells were resuspended in 1 ml of FACS buffer. Flow cytometry was performed on a FACSCalibur flow cytometer (Becton Dickinson, San Jose, CA). Data were collected and analyzed using Cell Quest software.
DNA cloning of the variable domains of 7D11
To obtain the sequence of the mAb-7D11 variable region, RNA was isolated from the hybridoma using Trizol according to the manufacturer’s instructions (Invitrogen, Carlsbad, CA). First-strand DNA was synthesized from the RNA using the Superscript III first-strand kit (Invitrogen, Carlsbad, CA). The product was used as the template for 30 rounds of PCR. Both primers used to PCR the light chain and the 3′ primer used for the heavy chain were from the Novagen mouse Ig-Primer set (EMD Biosciences, San Diego, CA). The 5′ heavy chain primers from the Ig-Primer set did not produce a PCR product so the following four 5′ primers were designed based on the results of N-terminal protein sequencing of 7D11: 5′-CAGGTCCAGCTGCAGCAGTCTGGGGCTG-3′ (Primer HC-1), 5′-CAGCTGCAGCAGTCTGGGGCTGAACTGGC-3′ (Primer HC-2), 5′-GCAGTCTGGGGCTGAACTGGCAAAACCTGG-3′ (Primer HC-3), and 5′-GGGGCTGAACTGGCAAAACCTGGGGC-3′ (Primer HC-4). All four PCR reactions yielded products and the final sequence was confirmed by at least two clones from each of the four PCR products. The two plasmids containing the mAb-7D11 variable region heavy and light chains were designated pCR-7D11-vHC and pCR-7D11-vLC, respectively.
Crystallization of the complex between L1 and 7D11-Fab
VACV L1 protein was expressed and purified as previously described (Su et al., 2005). The purified L1 was mixed with 7D11-Fab in a 1:1 molar ratio with a final total protein concentration of 8 mg/ml (~120 μM). Crystals of the complex were obtained by the hanging drop vapor diffusion method with 1 μl of protein solution mixed with 1 μl of reservoir solution. The best crystals were obtained by streak seeding with small crystals using a reservoir solution of 0.1M Tris-Cl pH 8.0 and 10–12% PEG-10,000. The crystals were flash-frozen for data collection in a solution containing 0.1 M Tris-Cl pH 8.0, 20% PEG-10,000, and 20% glycerol.
Structure determination and analysis
X-ray data were collected, integrated, and scaled using the HKL2000 software (Otwinowski and Minor, 1997). Models for the variable and constant domains of the Fab (PDB code: 1YMH) (Granata et al., 2005) and a model of L1 (PDB code: 1YPY) (Su et al., 2005) were used as search models to determine the structure by molecular replacement. The program PHASER (McCoy et al., 2005) was used in a single search to position four copies of the constant domains, then four copies of the variable domains, and finally four copies of L1.
Iterative model building in O (Jones et al., 1991) based on electron density maps, followed by refinement in Refmac (Murshudov et al., 1997) and CNS (Adams et al., 1997) improved the electron density and assignment of side-chain rotamers. The variable regions of the Fab were rebuilt based on the sequence obtained for 7D11. Non-crystallographic symmetry restraints among the four copies of the L1:7D11-Fab complex in the asymmetric unit were used during refinement. Residues 2–3 and 177–185 at the termini of L1 were disordered in the structure and not built into the model. Buried surface area calculations were performed using CNS (Lee and Richards, 1971). Shape correlation statistics were calculated using SC (Lawrence and Colman, 1993). Figures were generated using Pymol (DeLano, 2002). The coordinates of the L1:7D11-Fab complex have been deposited in the Research Collaboratory for Structural Bioinformatics protein data bank (RCSB pdb code 2I9L).
Acknowledgments
We thank Dr. Alan Schmaljohn (USAMRIID) and Dr. Bernie Moss (NIAID) for supplying the mAb-7D11 hybridoma and purified antibody, and for helpful discussions. We thank Mark Garfield for mass spectrometry and the members of the Structural Biology Section for helpful discussions. We thank Mary Ann Gawinowicz of the Columbia University Protein Core Facility for amino terminal deblocking and sequencing. We thank the staff of SBC-CAT and SER-CAT for assistance during data collection. Data were collected at the Structural Biology Center (SBC-CAT) and Southeast Regional Collaborative Access Team (SER-CAT) beam lines at the Advanced Photon Source, Argonne National Laboratory. Supporting institutions may be found at www.ser-cat.org/members.html. Use of the Advanced Photon Source beam lines was supported by the U. S. Department of Energy, Office of Science, Office of Basic Energy Sciences, under Contract No. W-31-109-Eng-38. Conclusions and recommendations are those of the authors and are not necessarily endorsed by the U.S. Army. This research was supported by the Division of Intramural Research of the NIAID, NIH.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Adams PD, Pannu NS, Read R, Brunger AT. Cross-validated maximum likelihood enhances crystallographic simulated annealing refinement. Proc Natl Acad Sci USA. 1997;94(10):5018–5023. doi: 10.1073/pnas.94.10.5018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aldaz-Carroll L, Whitbeck J, Ponce De Leon M, Lou H, Pannell L, Lebowitz J, Fogg C, White CL, Moss B, Cohen GH, Eisenberg R. Physical and immunological characterization of a recombinant secreted form of the membrane protein encoded by the vaccinia virus L1R gene. Virology. 2005;341(1):59–71. doi: 10.1016/j.virol.2005.07.006. [DOI] [PubMed] [Google Scholar]
- Belongia E, Naleway A. Smallpox vaccine: the good, the bad, and the ugly. Clin Med Res. 2003;1(2):87–92. doi: 10.3121/cmr.1.2.87. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blouch R, Byrd C, Hruby D. Importance of disulphide bonds for vaccinia virus L1R protein function. Virol J. 2005;2:91. doi: 10.1186/1743-422X-2-91. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen Z, Earl P, Americo J, Damon I, Smith SK, Zhou Y, Yu F, Sebrell A, Emerson S, Cohen G, Eisenberg R, Svitel J, Schuck P, Satterfield W, Moss B, Purcell R. Chimpanzee/human mAbs to vaccinia virus B5 protein neutralize vaccinia and smallpox viruses and protect mice against vaccinia virus. Proc Natl Acad Sci USA. 2006;103(6):1882–1887. doi: 10.1073/pnas.0510598103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Coulibaly S, Bruhl P, Mayrhofer J, Schmid K, Gerencer M, Falkner F. The nonreplicating smallpox candidate vaccines defective vaccinia Lister (dVV-L) and modified vaccinia Ankara (MVA) elicit robust long-term protection. Virology. 2005;341(1):91–101. doi: 10.1016/j.virol.2005.06.043. [DOI] [PubMed] [Google Scholar]
- Davies D, Mccausland M, Valdez C, Huynh D, Hernandez J, Mu Y, Hirst S, Villarreal L, Felgner P, Crotty S. Vaccinia virus H3L envelope protein is a major target of neutralizing antibodies in humans and elicits protection against lethal challenge in mice. J Virol. 2005;79(18):11724–11733. doi: 10.1128/JVI.79.18.11724-11733.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Davies DR, Padlan E, Sheriff S. Antibody-antigen complexes. Annu Rev Biochem. 1990;59:439–473. doi: 10.1146/annurev.bi.59.070190.002255. [DOI] [PubMed] [Google Scholar]
- Delano W. The PyMOL molecular graphics system. 2002 ( http://www.pymol.org)
- Fogg C, Lustig S, Whitbeck J, Eisenberg R, Cohen G, Moss B. Protective immunity to vaccinia virus induced by vaccination with multiple recombinant outer membrane proteins of intracellular and extracellular virions. J Virol. 2004;78(19):10230–10237. doi: 10.1128/JVI.78.19.10230-10237.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Galmiche M, Goenaga J, Wittek R, Rindisbacher L. Neutralizing and protective antibodies directed against vaccinia virus envelope antigens. Virology. 1999;254(1):71–80. doi: 10.1006/viro.1998.9516. [DOI] [PubMed] [Google Scholar]
- Granata V, Housden N, Harrison S, Jolivet-Reynaud C, Gore M, Stura EA. Comparison of the crystallization and crystal packing of two Fab single-site mutant protein L complexes. Acta Crystallogr D Biol Crystallogr. 2005;61(Pt 6):750–754. doi: 10.1107/S0907444905007110. [DOI] [PubMed] [Google Scholar]
- Harrison S, Alberts B, Ehrenfeld E, Enquist L, Fineberg H, Mcknight S, Moss B, O’donnell M, Ploegh H, Schmid S, Walter K, Theriot J. Discovery of antivirals against smallpox. Proc Natl Acad Sci USA. 2004;101(31):11178–11192. doi: 10.1073/pnas.0403600101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Henderson D, Inglesby T, Bartlett J, Ascher MS, Eitzen E, Jahrling P, Hauer J, Layton M, Mcdade J, Osterholm M, O’toole T, Parker G, Perl T, Russell P, Tonat K. Smallpox as a biological weapon: medical and public health management. Working Group on Civilian Biodefense. JAMA. 1999;281(22):2127–2137. doi: 10.1001/jama.281.22.2127. [DOI] [PubMed] [Google Scholar]
- Heraud J, Edghill-Smith Y, Ayala V, Kalisz I, Parrino J, Kalyanaraman VS, Manischewitz J, King L, Hryniewicz A, Trindade C, Hassett M, Tsai W, Venzon D, Nalca A, Vaccari M, Silvera P, Bray M, Graham B, Golding H, Hooper J, Franchini G. Subunit recombinant vaccine protects against monkeypox. J Immunol. 2006;177(4):2552–2564. doi: 10.4049/jimmunol.177.4.2552. [DOI] [PubMed] [Google Scholar]
- Hooper J, Custer DM, Schmaljohn C, Schmaljohn A. DNA vaccination with vaccinia virus L1R and A33R genes protects mice against a lethal poxvirus challenge. Virology. 2000;266(2):329–339. doi: 10.1006/viro.1999.0096. [DOI] [PubMed] [Google Scholar]
- Hooper J, Custer D, Thompson E. Four-gene-combination DNA vaccine protects mice against a lethal vaccinia virus challenge and elicits appropriate antibody responses in nonhuman primates. Virology. 2003;306(1):181–195. doi: 10.1016/S0042-6822(02)00038-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hooper J, Golden J, Ferro AM, King A. Smallpox DNA vaccine delivered by novel skin electroporation device protects mice against intranasal poxvirus challenge. Vaccine. 2007;25(10):1814–1823. doi: 10.1016/j.vaccine.2006.11.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hooper J, Schmaljohn A, Schmaljohn C. Prophylactic therapeutic monoclonal antibodies. United States of America; 2002. [Google Scholar]
- Hooper JW, Thompson E, Wilhelmsen C, Zimmerman M, Ichou M, Steffen SE, Schmaljohn C, Schmaljohn A, Jahrling P. Smallpox DNA vaccine protects nonhuman primates against lethal monkeypox. J Virol. 2004;78(9):4433–4443. doi: 10.1128/JVI.78.9.4433-4443.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ichihashi Y, Oie M. Neutralizing epitope on penetration protein of vaccinia virus. Virology. 1996;220(2):491–494. doi: 10.1006/viro.1996.0337. [DOI] [PubMed] [Google Scholar]
- Ichihashi Y, Takahashi T, Oie M. Identification of a vaccinia virus penetration protein. Virology. 1994;202(2):834–843. doi: 10.1006/viro.1994.1405. [DOI] [PubMed] [Google Scholar]
- Jones T, Zou J, Cowan S, Kjeldgaard M. Improved methods for building protein models in electron density maps and the location of errors in these models. Acta Crystallogr A. 1991;47 (Pt 2):110–119. doi: 10.1107/s0108767390010224. [DOI] [PubMed] [Google Scholar]
- Kashmiri S, De Pascalis R, Gonzales N, Schlom J. SDR grafting--a new approach to antibody humanization. Methods. 2005;36(1):25–34. doi: 10.1016/j.ymeth.2005.01.003. [DOI] [PubMed] [Google Scholar]
- Kidokoro M, Tashiro M, Shida H. Genetically stable and fully effective smallpox vaccine strain constructed from highly attenuated vaccinia LC16m8. Proc Natl Acad Sci USA. 2005;102(11):4152–4157. doi: 10.1073/pnas.0406671102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lamarre A, Talbot P. Protection from lethal coronavirus infection by immunoglobulin fragments. J Immunol. 1995;154(8):3975–3984. [PubMed] [Google Scholar]
- Lawrence M, Colman P. Shape complementarity at protein/protein interfaces. J Mol Biol. 1993;234(4):946–950. doi: 10.1006/jmbi.1993.1648. [DOI] [PubMed] [Google Scholar]
- Lee B, Richards F. The interpretation of protein structures: estimation of static accessibility. J Mol Biol. 1971;55(3):379–400. doi: 10.1016/0022-2836(71)90324-x. [DOI] [PubMed] [Google Scholar]
- Lustig S, Fogg C, Whitbeck J, Eisenberg R, Cohen GH, Moss B. Combinations of polyclonal or monoclonal antibodies to proteins of the outer membranes of the two infectious forms of vaccinia virus protect mice against a lethal respiratory challenge. J Virol. 2005;79(21):13454–13462. doi: 10.1128/JVI.79.21.13454-13462.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mccoy AJ, Grosse-Kunstleve RW, Storoni L, Read R. Likelihood-enhanced fast translation functions. Acta Crystallogr D Biol Crystallogr. 2005;61(Pt 4):458–464. doi: 10.1107/S0907444905001617. [DOI] [PubMed] [Google Scholar]
- Moss B. Poxvirus entry and membrane fusion. Virology. 2006;344(1):48–54. doi: 10.1016/j.virol.2005.09.037. [DOI] [PubMed] [Google Scholar]
- Murshudov G, Vagin A, Dodson E. Refinement of macromolecular structures by the maximum-likelihood method. Acta Crystallogr D Biol Crystallogr. 1997;53(Pt 3):240–255. doi: 10.1107/S0907444996012255. [DOI] [PubMed] [Google Scholar]
- Neves-Petersen MT, Snabe T, Klitgaard S, Duroux M, Petersen S. Photonic activation of disulfide bridges achieves oriented protein immobilization on biosensor surfaces. Protein Sci. 2006;15(2):343–351. doi: 10.1110/ps.051885306. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Otwinowski Z, Minor W. Processing of X-ray diffraction data collected in oscillation mode. In: Carter CW, Sweet RM, editors. Methods in Enzymology. Vol. 276. Academic Press; 1997. pp. 307–326. [DOI] [PubMed] [Google Scholar]
- Padlan E, Abergel C, Tipper J. Identification of specificity-determining residues in antibodies. FASEB J. 1995;9(1):133–139. doi: 10.1096/fasebj.9.1.7821752. [DOI] [PubMed] [Google Scholar]
- Ravanello M, Franke CA, Hruby DE. An NH2-terminal peptide from the vaccinia virus L1R protein directs the myristylation and virion envelope localization of a heterologous fusion protein. J Biol Chem. 1993;268(10):7585–7593. [PubMed] [Google Scholar]
- Ravanello M, Hruby D. Conditional lethal expression of the vaccinia virus L1R myristylated protein reveals a role in virion assembly. J Virol. 1994;68(10):6401–6410. doi: 10.1128/jvi.68.10.6401-6410.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sakhatskyy P, Wang S, Chou T, Lu S. Immunogenicity and protection efficacy of monovalent and polyvalent poxvirus vaccines that include the D8 antigen. Virology. 2006;355(2):164–174. doi: 10.1016/j.virol.2006.07.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schmaljohn C, Cui Y, Kerby S, Pennock D, Spik K. Production and characterization of human monoclonal antibody Fab fragments to vaccinia virus from a phage-display combinatorial library. Virology. 1999;258(1):189–200. doi: 10.1006/viro.1999.9701. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Senkevich T, Ojeda S, Townsley A, Nelson GE, Moss B. Poxvirus multiprotein entry-fusion complex. Proc Natl Acad Sci USA. 2005;102(51):18572–18577. doi: 10.1073/pnas.0509239102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Senkevich T, White C, Koonin E, Moss B. Complete pathway for protein disulfide bond formation encoded by poxviruses. Proc Natl Acad Sci USA. 2002;99(10):6667–6672. doi: 10.1073/pnas.062163799. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Su H, Garman S, Allison TJ, Fogg C, Moss B, Garboczi D. The 1.51-Angstrom structure of the poxvirus L1 protein, a target of potent neutralizing antibodies. Proc Natl Acad Sci USA. 2005;102(12):4240–4245. doi: 10.1073/pnas.0501103102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Townsley A, Senkevich T, Moss B. Vaccinia virus A21 virion membrane protein is required for cell entry and fusion. J Virol. 2005;79(15):9458–9469. doi: 10.1128/JVI.79.15.9458-9469.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Turner G, Squires EJ, Murray H. Inactivated smallpox vaccine. A comparison of inactivation methods. J Hyg (Lond) 1970;68(2):197–210. doi: 10.1017/s0022172400028679. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vanhooren A, De Vriendt K, Devreese B, Chedad A, Sterling A, Van Dael H, Van Beeumen J, Hanssens I. Selectivity of tryptophan residues in mediating photolysis of disulfide bridges in goat alpha-lactalbumin. Biochemistry. 2006;45(7):2085–2093. doi: 10.1021/bi0517638. [DOI] [PubMed] [Google Scholar]
- Wolffe EJ, Vijaya S, Moss B. A myristylated membrane protein encoded by the vaccinia virus L1R open reading frame is the target of potent neutralizing monoclonal antibodies. Virology. 1995;211(1):53–63. doi: 10.1006/viro.1995.1378. [DOI] [PubMed] [Google Scholar]
- Xiao Y, Aldaz-Carroll L, Ortiz AM, Whitbeck J, Alexander E, Lou H, Davis HL, Braciale T, Eisenberg R, Cohen GH, Isaacs SN. A protein-based smallpox vaccine protects mice from vaccinia and ectromelia virus challenges when given as a prime and single boost. Vaccine. 2007;25(7):1214–1224. doi: 10.1016/j.vaccine.2006.10.009. [DOI] [PMC free article] [PubMed] [Google Scholar]