

Abstract
Cockayne syndrome (CS) is a progressive childhood neurodegenerative disorder associated with a DNA repair defect caused by mutations in either of two genes, CSA and CSB. These genes are involved in nucleotide excision repair (NER) of DNA damage from ultraviolet (UV) light, other bulky chemical adducts and reactive oxygen in transcriptionally active genes (transcription coupled repair, TCR). For a long period it has been assumed that the symptoms of CS patients are all due to reduced TCR of endogenous DNA damage in the brain, together with unexplained unique sensitivity of specific neural cells in the cerebellum. Not all the symptoms of CS patients are however easily related to repair deficiencies, so we hypothesize that there are additional pathways relevant to the disease, particularly those that are downstream consequences of a common defect in the E3 ubiquitin ligase associated with the CSA and CSB gene products. We have found that the CSB defect results in altered expression of anti-angiogenic and cell cycle genes and proteins at the level of both gene expression and protein lifetime. We find an over-abundance of p21 due to reduced protein turnover, possibly due to the loss of activity of the CSA/CSB E3 ubiquitylation pathway. Increased levels of p21 can result in growth inhibition, reduced repair from the p21-PCNA interaction, and increased generation of reactive oxygen. Consistent with increased reactive ozygen levels we find that CS-A and -B cells grown under ambient oxygen show increased DNA breakage, as compared to xeroderma pigmentosum cells. Thus the complex symptoms of CS may be due to multiple, independent downstream targets of the E3 ubiquitylation system that results in increased DNA damage, reduced transcription coupled repair, and inhibition of cell cycle progression and growth.
Keywords: Cockayne syndrome, p21, Reactive oxygen, H2AX, protein expression, gene expression
Introduction
CS is an autosomal recessive disease characterized by cachectic dwarfism, retinopathy, microcephaly, dysmyelination, ganglial calcification, deafness, neural defects, retardation of growth and development after birth (Nance and Berry, 1992). The disease has been classified according to severity into several classes, but these classes have not yet been correlated with molecular defects. CS patients are sun sensitive but do not develop cancers, setting this disease apart from xeroderma pigmentosum (XP) where sun sensitivity is mainly expressed as an increase in skin cancer of all types (Kraemer et al., 1994). CS patients have mutations in one of two genes, CSA and CSB (Bootsma et al., 1998; Lehmann, 1982) and mutations in XPB, XPD and XPG genes can also give rise to combined XP/CS symptoms, (Weeda et al., 1990; Wood et al., 2001) (Fig 1). The CSA & B gene products were originally identified as regulating repair in actively transcribed genes (TCR). They have subsequently been ascribed other functions such as ubiquitylation and transcription regulation that may themselves be proximate causes of the TCR and other cellular defects (Bohr, 1991; Groisman et al., 2003; Licht et al., 2003).
Figure 1.

Induction of p53 in normal and CS cells by 10 or 20 J.m−2 UV light; western blot (antibody SC126) of cells harvested 16 hr after UV irradiation.
CSA (ERCC8) is located on chromosome 5 and encodes a 396 amino acid WD repeat protein (Henning et al., 1995). CSB (ERCC6) is located on chromosome 10q11-21 and encodes a 1493 amino acid protein with a nucleotide-binding site, ATPase activity and helicase motifs, but the protein does not display helicase activity in vitro (Citterio et al., 1998; Selby and Sancar, 1997). The CSB protein can actively wrap the DNA (Beerens et al., 2005) and its association with transcription elongation is stabilized by DNA damage (Boom et al., 2004). CSA is also a regulator of an E3 ubiquitin ligase, which could explain the role of CS genes in ubiquitylation of RNA pol II (Groisman et al., 2003). The GGR and TCR pathways interact through DDB1 that also acts as a cofactor of the E3 ligase complexes associated with CSA and DDB2 (Groisman et al., 2003).
The association of CSA and B with transcription elongation proteins suggests that CS mutations could impair basal transcription (Balajee et al., 1997; Boom et al., 2004). Therefore, if specific genes have sequences or structures that present natural impediments to RNA pol II progression, reductions in expression might be expected in CS cells even in the absence of of DNA damage. Mutations in XPB and XPD affect specific expression of nuclear hormone receptors that are important in development including the retinoic acid receptors RARalpha, ERalpha, and AR (Keriel et al., 2002), which may account for part of the wasting phenotype of XP/CS patients. Microarray analysis of CSB cells has detected multiple changes in gene expression associated with the response to oxidative stress, signal transduction, ribosomal synthesis and uracil-D-glycosylase (Kyng et al., 2003). We have also found several specific gene expression markers of CSB in a microarray analysis that compared parent child pairs from CS-B families (Hefner et al., 2006). These include several that may have clinical relevance such as collagen 15a1 that is a secreted collagen that is cleaved to have anti-angiogenic activity, and latrophilin that is an orphan neural receptor for the black widow spider neurotoxin, whose endogeneous ligand is unknown (Hefner et al., 2006).
When cells are exposed to UV damage the C-terminal domain of a fraction of RNA pol II molecules is phosphorylated and then ubiquitylated. CS cells fail to ubiquitylate RNA pol II and in consequence cannot remove and degrade the transcription complex stalled at a damaged site in DNA (Bregman et al., 1996; McKay et al., 2001; Tu et al., 1998; Woudstra et al., 2002). CSA and CSB are required for ubiquitylation of RNA Pol II. Ubiquitin is a 76-residue polypeptide that is conjugated to target protein substrates via a three-step enzymatic process: a ubiquitin-activating enzyme (E1), a ubiquitin-conjugating enzyme (E2), a ubiquitin protein ligase (E3), followed by a de-ubiquitylation enzyme (E4). Mono-ubiquitylation alters the specificity of protein functions and poly-ubiquitylation marks proteins for destruction. The E3 ligases are primarily responsible for conferring substrate specificity (Hochstrasser, 1996; Peng et al., 2003; Semple et al., 2003; Wilkinson, 2000). CSA is part of an SCF complex that mainly ubiquitylate phosphorylated substrates, but additional criteria confer a high degree of specificity toward the substrates (Ciechanover and Brundin, 2003). The RNA pol II subunit is ubiquitylated specifically on the active subunit Rbp1 on those molecules that have been hyperphosphorylated (Lee et al., 2002). The DNA binding protein DDB2, specific for GGR, is part of closely similar SCF complex. The CSA and DDB2 components appear to be responsible for the specificity of these versions of two similar SCF complexes (Groisman et al., 2003).
We have therefore conducted a search for genes and proteins that are consistently dysregulated (over or under expressed) in CS cells. The detailed results of our gene expression study are being reported elsewhere (Hefner et al., 2006); this report describes proteins that we have found in higher levels in CS cells. We designed the present study on the assumption that proteins that are normally targets for CS-dependent ubiquitylation would be found at above normal levels if the ubiquitylation was part of the cellular degradation pathway and the proteins were consequently not degraded normally. We would not detect proteins that were monoubiquitylated for regulatory purposes rather than degradation. We have, however, identified several proteins in this way, notably p21 among others that may be relevant to the developmental and neurodegenerative symptoms of CS patients.
Materials and methods
Lymphoblastoid cells lines GM10902 (CS-B), GM01712 (CS-B), GM12496 (CS-B), GM02498 (XP-C) and GM02345 (XP-A) were obtained from the Coriel Cell Repository (Camden, NJ). The wild type control lymphoblastoid cell line (PP034) was created at UCSF. Primary fibroblast cell lines GM10903 (CS-B) and GM10901 (unaffected parent of GM10903) were obtained from Coriel Cell Repository (Camden, NJ). Lymphoblastoid cells were cultured in RPMI media with 15% heat inactivated foetal bovine serum, 100μg/mL penicillin, 100 μg/mL streptomycin and 2 mM glutamine. Primary fibroblasts were cultured in MEM media with 15% foetal bovine serum, 100 μg/mL Penicillin, 100 μg/mL Streptomycin and 2 mM Glutamine. Cells were grown at 37° C with 5% CO2. A set of fibroblasts were transfected with either hTERT to immortalize them or SV40 for transformation; the lymphoid cells were transformed with Epstein Barr virus. CS-B lymphoblasts (GM10903) were electroporated with a plasmid expressing cDNA for CSB linked to the neomycin resistance gene and selected for continuous expression, and function was confirmed by measuring restoration of UV resistance.
Whole cell protein lysates were produced from cells 6 hours after UV irradiation or from mock treated controls. The extraction buffer contained 150 mM NaCl (unless other wise stated), 50 mM Tris-HCl, pH:7.5, 0.5 % SDS, 0.5 % Nonident P-40, 0.5 % Sodium Deoxycholate. The buffer was supplemented with PMSF, and Protease Inhibitor #2 (Calbiochem). Cells were suspended in extraction buffer at a concentration of 1×106 cells/100 μL of extraction buffer. The lysate suspention was sonicated 3×30 sec on ice allowed to mix by nutation for 20 min at 4° C. After nutation samples were quantitated using the Lowry assay (Bio-Rad) then stored at −80° C until used.
For each sample 30 μg of lysate was electrophoresed through a 4–15 % polyacrylamide gel then transferred to a 0.20 μM pore size nitrocellulose membrane (Bio-Rad). Membranes were blocked overnight in 0.5 % nonfat milk at 4° C. Blots were probed with 1:250 α p21 antibody (BD Pharmingen, cat # 556431), anti-p53 (1:500, Santa Cruz biotechnology), or anti-βactin (1:10000, Sigma) followed by HRP conjugated goat anti-rabbit (1:2000, Santa Cruz Biotechnology) or HRP conjugated goat anti-mouse secondary antibody diluted 1:2000 (Santa Cruz, cat # sc-2064). Chemiluminescent visualization was performed with Visualizer (Upstate Biotechnology) or ECL Plus (Amersham). The anti-βactin was used as the loading control for total protein in PAGE. For analysis of p21, cell extracts were prepared in a range of salt concentrations from 50 to 500 mM. Gels were photographed and presented, as observed, without preparation of any composites run on different occasions. The gel band intensities should therefore be compared within each gel image, rather than between images prepared on separate occasions. Gels were quantified by densitometry and mean and standard errors calculated for the ratio of p21 levels in CS as compared to normal cell lines.
DNA breakage was determined in SV40 transformed cells grown under ambient oxygen by measuring the formation of foci of γH2AX as a marker characteristic of double strand breaks (Lowndes and Toh, 2005). Cells were grown on dual-chambered slides (Nalge Nunc) and fixed with 2% paraformaldehyde for 15 minutes and permeabilized with 0.2% Triton X-100 for 15 minutes. Slides were air dried for 5 minutes and stored at −80°C. After storage, cells were re-permeabilized with ice-cold 50:50 acetone: methanol for 30 minutes. Slides were blocked for 1 hour in 1X PBS, 10%FBS at 37°C, incubated for 1 hr at 37°C with a rabbit polyclonal γH2AX antibody 1:400 (Novus Biologicals #100-384A) in 1% BSA 0.5% Tween 20 and 1X PBS; followed by incubation for 1 hr at 37°C with 1:100 dilution of the secondary FITC-labeled antibody (ImmunoPure fluorescein conjugated goat antirabbit IgG (H+L) Pierce #31853) in 1X PBS 1X BSA. Three washes in PBS were made between each step in fixation and antibody application described above. Finally slides were mounted overnight with ProLong Gold Antifade reagent containing 4′-6-diamidino-2-phenylindole (DAPI) (Molecular probes). 400–500 cells were scored to determine the fractions that were positive for at least 10 foci per cell. Replicates were pooled to calculate the means and standard errors.
Differences in the level of proteins in CS cells as compared to normal cells were determine using a protein array (BD Clontech #631791) that consisted of approximately 500 monoclonal antibodies on a glass microarray. This was used according to manufacturer’s instructions using the supplied reagents. Proteins were extracted from a pair of CS and normal cell types and labeled with Cy3 and Cy5 fluorescent dyes, respectively. The proteins from the two cell types were applied to the array that produced green or red signals for over or under expression, and yellow for similar expression. The observed color signals were analyzed using the commercial software to quantify the proteins that were over-expressed in CS cells. Those that appeared to be over-expressed were subsequently analyzed by standard western blots using antibodies different from those that were on the array.
Results
Expression of p53 and p21 in CS cells
We first examined the p53, p21 damage response pathway in normal, XP and CS cells because of its importance in the cell cycle and apoptotic response of human cells. Previous studies have shown that after UV irradiation of normal cells, p21 shows a transient p53-dependent increase and is then degraded through ubiquitylation to facilitate NER (Bendjennat et al., 2003).
The human lymphoid cells of all genotypes showed a normal induction of p53 by UV (Fig 1). In the absence of irradiation, control cells of all genotypes had similar low levels of p53 (Fig 1). A significant increase in p53 levels was observed at a dose of 10 J.m−2, which declined at higher dose of 20 J.m−2 in normal and CS-B, but not XP-C (Fig 1). We then analyzed the expression of p21, an important mediator of p53 function (Fotedar et al., 2004); in unirradiated normal and CS cells, p21 aggregated in high salt concentrations (Fig 2) and was about 3 fold over-abundant in both CS-B lymphoblasts and fibroblasts (Fig 3). The aggregation was less for p21 from CSB cells than normal cells (Fig 2), suggesting that the aggregating species of p21 was modified in a manner that reduced protein-protein interactions at high salt. Over-abundance of p21 was not seen in XP-A (Fig 3A). Correction of CS-B with cDNA restored the UV resistance of CS-B cells (Fig 4A) and reduced the abundance of p21 to that of control cells (Fig 4B), indicating that p21 abundance was related to the functional activity of CSB. These results represent protein accumulation, because no corresponding differences were seen in p21 mRNA expression when analyzed by quantitative RT-PCR (data not shown). This observation in CSB cells was specific for p21, and the related family member p27 was not elevated when analyzed under the same conditions.
Figure 2.

Western analysis of p21 (antibody: SX118 mouse IgG, epitope last 20 aa) as a function of salt concentration. Upper band a high molecular weight aggregate that moved slightly below the loading well; lower band normal size p21. Salt concentration left to right: 50, 150, 250, and 500 mM.
Figure 3.

A. Western analysis of p21 (antibody: SX118 mouse IgG, epitope last 20 aa) in lymphoid cells: normal (PP034), CS-B (GM10902, GM1712 respectively) and XP-A (GM2345) grown in 15% serum. Right panel shows fibroblasts from normal and CSB (GM10903). B. Histogram showing the ratios of p21 expression levels in two CSB cell lines GM10902 and GM01712 as compared to the normal PP034 cell line. Error bars represent the standard errors of the means.
Figure 4.
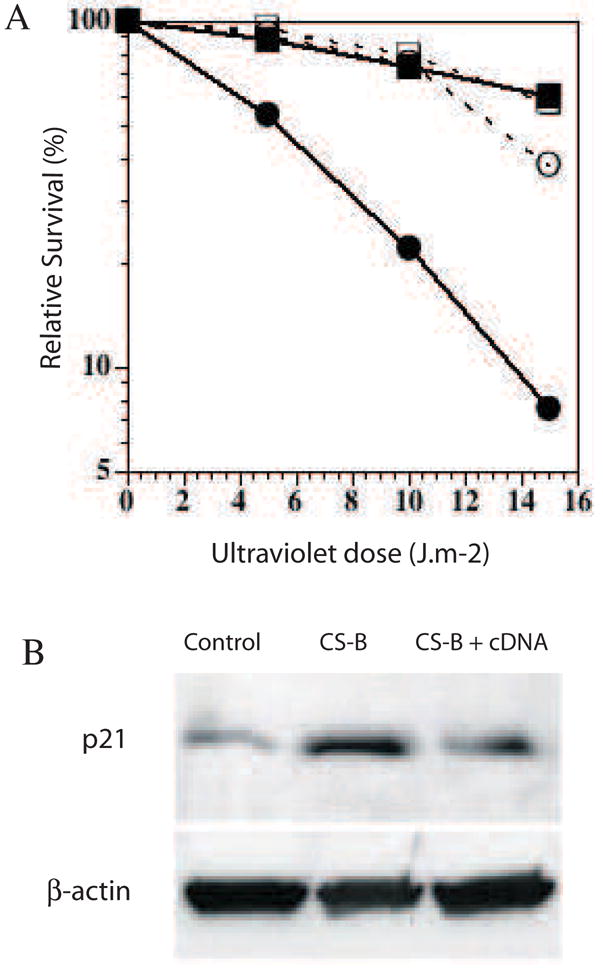
A. Complementation of CS-B lymphoblast with cDNA for CSB corrects the UV sensitivity of CSB cells. control; CS-B; CS-B corrected with cDNA clone 1; CS-B corrected with cDNA clone 2. B. Complementation of CS-B lymphoblast with cDNA for CSB corrects the over-abundance of p21. Upper bands p21, lower bands βactin loading controls. Wt lane PP034 cells, CS-B lane GM10903, cDNA correction of GM10903 plus cDNA, clone 1, transfected and selected for continuous expression.
We determined the half-life of p21 in human lymphoid cells by inhibiting new protein synthesis with cycloheximide (50 μg/ml) and measuring the presence of p21 in a western blot (Fig 5). The westerns were quantified by densitometry and corrected for loading using βactin as a marker. These experiments indicated that the half-life of p21 was 1.5 hr in normal cells and 3.0 hr in CS-B cells, consistent with a slower rate of degradation (Fig 5).
Figure 5.
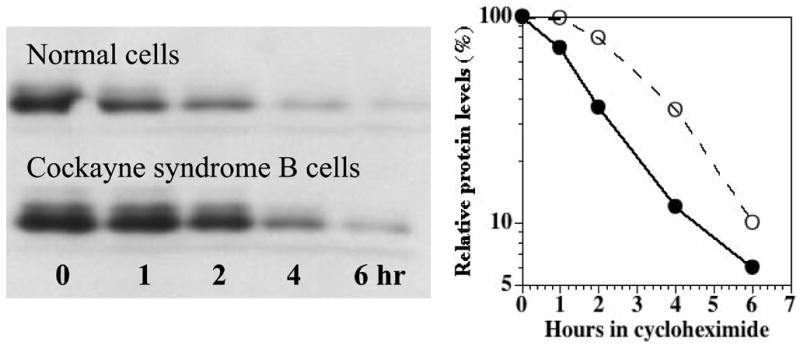
Relative levels of p21 in normal and CSB lymphoblastoid cells (GM10902) as a function of time in cycloheximide (50microgram/ml). Primary mouse monoclonal anti-p21 (1:250 dilution), secondary horse-radish peroxidase anti-mouse (1/2000 dilution). Values normalized to the ambient level in each cell type without cycloheximide.
p21 is a cell cycle regulatory protein and a negative regulator of PCNA and DNA replication. Consistent with this function, the CS cells showed a slower growth rate than normal cells. The doubling times of normal cells ranged from 0.8 to 1.0 days, days, XP-A 1.5 days, and CS-B was 2.6 days. Re-expression of CSB cDNA in CS-B cells increased the growth rate to a near normal doubling time of 1.0 to 1.5 days, and restored UV resistance (Fig 4A).
DNA damage in CS cells under ambient growth conditions
Reactive oxygen species (ROS) can be generated as a consequence of high levels of p21, possibly through interaction with mitochondrial apoptosis-inducing factors (Macip et al., 2002) and can consequently damage DNA causing single and double strand breaks. We therefore scored the presence of γH2AX foci in normal, XP and CS cells, as a general marker of DNA damage from ROS (Lowndes and Toh, 2005). Previous studies have shown that, under ambient conditions, naturally-occurring DNA damage becomes converted into double strand breaks detected as γH2AX foci, when replication forks encounter unrepaired single strand lesions (Bryant et al., 2005; Farmer et al., 2005). Normal and XP-V cells had negligible levels of foci-positive cells, XP-A and XP-C cells had 4 to 5% foci-positive cells, but CS-A had 12% and CS-B 22% foci-positive cells (Fig 5). These results indicate a higher level of spontaneous DNA damage in CS cells, consistent with either increased damage or reduced repair under ambient conditions (Macip et al., 2002).
Protein expression in CS cells
We hypothesized that if proteins were not degraded fully in CS cells due to an E3 ligase deficiency (Groisman et al., 2003) they could accumulate to abnormal levels. We have shown this for the specific case of p21 (Fig 2–5), and we therefore chose to screen using a protein array to identify other possible targets for CS-dependent over-expression. We examined the level of a series of proteins in CS cells as compared to wild type using a mouse monoclonal antibody array (BD Clontech #631791; for complete description see http://bioinfo.clontech.com/abinfo/array-list-action.do) that consists of approximately 500 monoclonal antibodies on a glass microarray. Using the commercial software for analysis we identified five proteins that were expressed at increased levels in CSB cells. These were: DP-1 a transcription factor (7.9 fold); CA-150 a transcription elongation factor involved in RNA pol II (3.2 fold); Hsp90, a heat shock protein (2.2 fold); CTB1, a negative regulator of proliferation (1.7 fold); CAF-1, a chromatin assembly factor (1.55 fold). We did not detect p21 because the extraction method used for the antibody arrays involve a high-speed centrifugation (14,000g) that removes aggregates that would have contained over-expressed p21 under these salt conditions.
Validation of proteins detected in an array is important before we attempt to determine their possible role in the CS phenotype. We have validated over-expression of Hsp90 and Dp-1 by western analysis, and results are shown for Hsp90 (Fig 7). The observation of an increased level of a heat shock protein is consistent with recent studies showing that dysregulation of the ubiquitylation and proteasome systems results in induction of several heat shock proteins and accumulation of tau proteins in oligodendrocytes, precursors for myelin synthesizing cells (Goldbaum and Richter-Landsberg, 2004). The increase in CAF-1 may be related to the slow recovery of DNA synthesis after DNA damage to CS cells because this protein interacts with PCNA (Cleaver, 1982). DP-1 is a G1 cell cycle regulated protein that forms dimers with E2F, a transcription factor that controls many factors involved with the G1 to S transition. In the western analysis we note than there were differences in migration rates between normal, heterozygotes and CS homozygotes that we need to investigate further.
Figure 7.
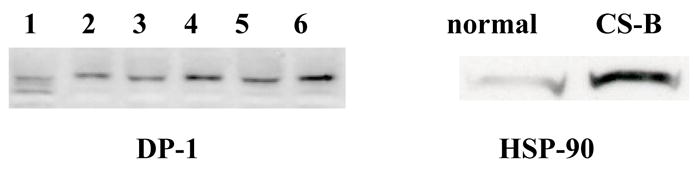
Western blots from normal and CSB fibroblasts showing over-abundance of Hsp90 protein; normal (PPO34) and CSB (GM10903) lymphoblasts.
Discussion
CSA has been show to be part of an E3 ubiquitin ligase (Groisman et al., 2003), and both CSA and CSB are required for ubiquitylation of RNA pol II (Bregman et al., 1996). In addition CSB can wrap DNA and associate with RNA pol II thereby influencing transcription (Beerens et al., 2005). The interaction with RNA pol II may be a mechanism that leads to defective TCR in CS cells, but other effects on protein life-time and gene expression may occur independent of repair. We therefore hypothesize that there are several targets for the CS gene products that will contribute to the clinical symptoms of CS. Our results are consistent with this hypothesis, but we have yet to confirm that the protein targets identified are directly ubiquitylated by the CS E3 system, and the initial results obtained with the protein array need to be extended and further validated.
In screens for gene and protein expression we have identified several biomarkers that are over- or under-expressed. We have identified proteins that are found at high levels (e.g. p21) in CS cells, due to defects at the level of protein processing (Fig 2–5). We have also shown, elsewhere, that other genes are regulated at the transcription level (e.g. col15a1, latrophilin) in CS cells (Hefner et al., 2006). Our results provide an approach to solving a major issue in CS: the absence of correlations between the severity of the clinical disorder and the biochemical and genetic defects. Although patients show a wide range of severity, this does not yet show any correlation with sites of mutation in the gene and cell cultures show a uniform UV sensitivity and TCR deficiency. One reason may be that certain phenotypes are dependent on the downstream targets of CS-dependent regulation, not on the CS proteins directly.
We have shown that total cell contents of p21 are about 3 fold greater in CSB cells (Fig 3B), and we hypothesize that this is because of insufficient ubiquitylation by the CS-dependent E3 ligase activity. The C-terminal 148–157 amino acids of p21 are ubiquitylated during cellular stress responses (Fukuchi et al., 2002) and during S phase by the SCF/Skp2 ligase (Bornstein et al., 2003). Whether this is the region that is targeted by the CS-ubiquitylation system remains to be established. We have identified that p21 aggregates at high salt with other cellular components to create a high molecular weight complex that is more prominent in normal cells than CS cells (Fig 2). At low salt concentrations (below 150mM which is near physiological) no aggregates were formed. The elevated level of p21 was found in an aggregate at high salt with material that would not enter a gel (Fig 2) and would pellet when centrifuged at 14,000g. This phenomenon was observed in other studies of CS-dependent ubiquitylation, and could be eliminated by digestion of the DNA in the pellet with micrococcal nuclease (Groisman et al., 2003).
Previous studies have shown that after UV irradiation in normal cells p21 is transiently increased and then degraded to facilitate NER (Bendjennat et al., 2003). Since CS cells carry out normal levels of GGR we would expect that UV induction and degradation of a subset of p21 is normal in CS cells. Our decay rate may therefore represent an average over several sub-populations of p21 molecules with different functional roles in repair and cell cycle regulation.
The observations of increased levels of p21 in CS cells raise the questions of the functional significance for the CS phenotype. P21 may be either (a) a growth inhibitor, thereby causing the reduced growth rate in CS patients; (b) a repair inhibitor through interaction with PCNA, contributing to the low level of TCR; (c) a tumor suppressor, perhaps explaining the absence of cancer in CS cells as compared to XP patients; and (d) a cause of increased ROS causing more in vivo damage including apoptosis in neural cells (Macip et al., 2002). Recent studies have shown that p21 can play a paradoxical role in both promoting and inhibiting programmed cell death under a variety of conditions (Borgne and Golsteyne, 2003). Increased expression of p21 occurs in cells of the brain under stress but it does not appear to act then as a cell cycle inhibitor (Macleod et al., 1996). Reactive oxygen can be generated by high levels of p21, possibly through interaction with mitochondrial apoptosis-inducing factors (Macip et al., 2002). It would be particularly intriguing if the CS phenotype results not only from a repair deficiency toward oxidative damage, but also, through p21 over-expression, an enhanced intracellular production of ROS. We are in the process or confirming and extending these results by correction of the phenotypical changes associated with p21 using CSB cDNA and siRNA targeting p21. We also need to demonstrate directly that there is an elevated level of intracellular ROS in CSB cells associated with increased p21 levels (Macip et al., 2002).
In parallel experiments we performed an Affymetrix microarray analysis of CS-B and wild type primary fibroblasts, comparing 6 pairs of parents and affected children. The genes that were positive on the arrays were then subjected to direct expression analysis using the Taqman method. We found that the extracellular matrix protein type XV collagen (Col 15a1) was under-expressed in 5/6 CS-B cells. We also found that the receptor for the black widow spider neurotoxin, latrophilin, was over-expressed in 3/6 CS-B cells. The reasons for the negative results in some of the cell lines are being further evaluated. We also found that the difference in expression of Col15a1 between CS-B and normal cells was suppressed if the cell lines were immortalized by hTERT. We also found that after transformation with SV40, Col15a1 was not expressed in either normal or CS-B cells. This represents a cautionary observation about the potential significance of results on gene expression when immortalized or transformed cells are used. The use SV40-transformed cells may consequently hide or reveal (see next section) important differences associated with the disease.
A positive application of transformed cells was in our analysis of spontaneous damage in CS cells with the double strand break marker γH2AX. DNA breakage in the S phase is suppressed by normal levels of p53, and we have reported previously that the ability to detect double strand breaks in the S phase resulting from replication fork arrest can be enhanced through inactivation of p53 by transformation or suppression with siRNA (Limoli et al., 2000). Therefore we used transformed cells from normal, XP and CS cells deliberately to enhance our detection of endogenous DNA damage. Under normal growth conditions that represent a high level of oxygenation, we observed a much greater level of DNA damage in CSA and B cells than either of XP-A or XP-C (Fig 6). Since the XP cells are devoid of either GGR (XP-C) or both GGR and TCR (XP-A) these observations indicate an increased level of spontaneous DNA damage in CS cells resulting from more than simply the repair deficiency. The high level of p21 in CS-B cells may for example generate increased ROS (Macip et al., 2002).
Figure 6.
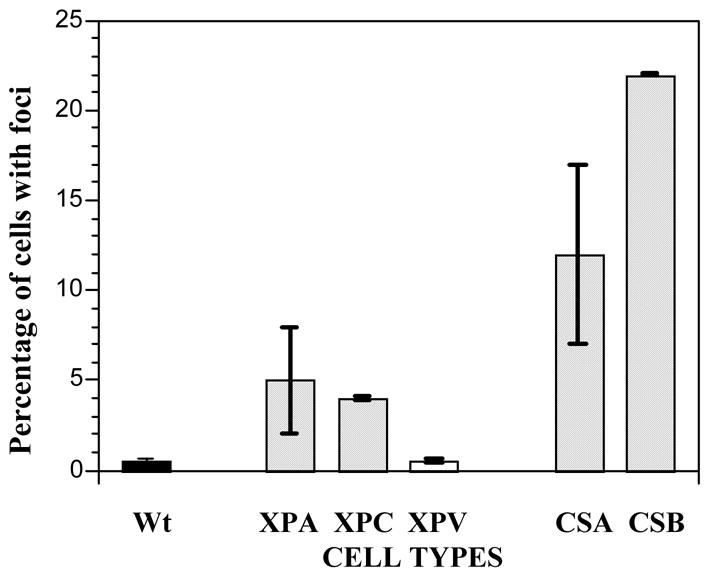
CS-A and CS-B cells show increased DNA breakage, as determined by γH2AX foci formation, when grown under normal ambient oxygen. Values represent the standard errors of the means.
The absence of cancer in human CS patients is difficult to explain because CS cells specifically fail to repair cyclobutane dimers in UV damaged episomal plasmids and show increased mutagenesis, but do repair [6–4] photoproducts (Barrett et al., 1991; Parris and Kraemer, 1993). Since XP cells fail to repair [6–4] photoproducts as well as dimers, they could be the more important carcinogenic damage for XP patients. Alternatively, episomal vectors may not reflect the full spectrum of damage responses that occur when whole cells are subject to UV damage. One possible explanation for the skin and neurological symptoms may lie in the cell cycle arrest and apoptotic response associated with over-expression of p21 as well as reduced TCR. The TCR defect in CS cells may trigger an apoptotic response via the JNK pathway that removes damaged cells from the proliferating pool of cells in the skin (Hamdi et al., 2005; Ljungman and Zhang, 1996), thereby eliminating premalignant cells; but a similar loss would be pathological in the brain. The over-abundance of p21 in CS cells may further act as a tumor suppressor via its inhibition of cell cycle progression.
The photosensitivity of CS cells and patients can easily be attributed to the DNA repair defect, but the neurodegeneration remains difficult to explain in other than the broadest terms. Human CS cells are sensitive to UV (Licht et al., 2003), and the transport of CSA to nuclear locations for TCR is stimulated by UV, H2O2, and cisPt (Kamiuchi et al., 2002). The CSB protein interacts with PARP-1, an important sensor of single strand breaks arising from oxidative damage (Flohr et al., 2003; Thorsland et al., 2005). The neurodegeneration may therefore be supposed as due to a failure to repair endogenous ROS damage to DNA, but this must be tied to the observation that the cellular damage in mouse and human CS brains is predominantly in the cerebellum and Purkinje cell layers (de Boer et al., 2002; Murai et al., 2001; Shiomi et al., 2005). High levels of oxidative metabolism occur in the brain and are increased in many instances of neurodegeneration (Kruman, 2004). Endogenous oxidative damage has been reported in the brains of repair deficient patients, although more prominently in XPA than CS patients (Hayashi et al., 2005; Hayashi et al., 2001). The granule and Purkinje cells of the cerebellum appear to be targets for many cellular toxins (Fonnum and Lock, 2000; Fonnum and Lock, 2004) and the Purkinje cells have unique deficits of several enzymes that present a low threshold for cell death from intrinsic or extrinsic sources (Welsh et al., 2002).
The possibility that ubiquitin ligase and transcription regulation may be important activities of CS-A and B implies that the varied phenotypes observed in CS cells may be due to the targets for ubiquitylation and transcription regulation (Groisman et al., 2003). The lack of correspondence between mutations in CSA and CSB and patient phenotypes is not surprising, if the CS genes are not directly involved in the phenotype, but rather in modifying genes and gene products more directly involved. Defects in protein processing would make CS a disease similar in principle to other neurodegenerative disorders such as Alzheimer’s, Parkinson’s, Lou-Gehrig’s and prion diseases (Ciechanover and Brundin, 2003). CS would also resemble an acceleration of normal aging, in which the brain accumulates oxidative damage (Lu et al., 2004). Recently a complete absence of CSB protein was found in a very mildly photosensitive patient, implying that mutant CSB proteins in other patients could be the cause of more severe disease (Horibata et al., 2004). This is reminiscent of other neurodegenerative diseases in which the mutant proteins are toxic and their loss can relieve some neurodegenerative symptoms (Arrasate et al., 2004; Harper et al., 2005; Santacruz et al., 2005).
The complex phenotype of CS can therefore be ascribed to a DNA repair-dependent defect and associated transcription regulation and protein degradation defects. The challenge is to discriminate between the phenotypes that can be attributed to these differing mechanisms. Resolution of some, at least, would be a major step forward and could suggest ways to develop targeted intervention and treatment methods.
Acknowledgments
The work described here was supported by grants from the National Institutes of Environmental Health Sciences grant 1 RO1 ES 8061 (JEC), the Cancer Center Support grant P30 CA82103 (PI: McCormick). Postdoctoral support was provided by a UCSF Dermatology Training Grant T32 AR007175-27 (E. Hefner) and a National Cancer Institute Ruth Kirstensen Postdoctoral Fellowship 1 F32 CA099499-01A1 (R.R. Laposa). Dr D. Karentz conducted this work while on sabbatical with additional support from the Lily Drake Cancer Research Fund from the University of San Francisco. We are also grateful to the XP Society, Poughkeepsie, NY, and the Luke O’Brien Foundation for their continued support and encouragement for the UCSF research program (JEC), especially in furthering the work on Cockayne Syndrome.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Arrasate M, Mitra S, Schweitzer ES, Segal MR, Finkbeiner S. Inclusion body formation reduces levels of mutant huntingtin and the risk of neuronal death. Nature. 2004;431:805–810. doi: 10.1038/nature02998. [DOI] [PubMed] [Google Scholar]
- Balajee AS, May A, Dianov GL, Friedberg EC, Bohr VA. Reduced RNA polymerase II transcription in intact and permeabilized Cockayne syndrome group B cells. Proc Natl Acad Sci USA. 1997;94:4306–4311. doi: 10.1073/pnas.94.9.4306. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barrett SF, Robbins JH, Tarone RE, Kraemer KH. Evidence for defective repair of cyclobutane dimers with normal repair of other photoproducts in a transcriptionally active gene transfected into Cockayne syndrome cells. Mutation Research. 1991;255:281–291. doi: 10.1016/0921-8777(91)90032-k. [DOI] [PubMed] [Google Scholar]
- Beerens N, Hoeijmakers JH, Kanaar R, Vermeulen W, Wyman C. The CSB Protein Actively Wraps DNA. J Biol Chem. 2005;280:4722–4729. doi: 10.1074/jbc.M409147200. [DOI] [PubMed] [Google Scholar]
- Bendjennat M, Boulaire JRM, Jascur T, Brickner H, Barbier V, Sarasin A, Fotedar A, Fotedar R. UV Irradiation triggers Ubiquitin-Dependent degradation of p21 WAF1 to promote DNA repair. Cell. 2003;114:599–610. doi: 10.1016/j.cell.2003.08.001. [DOI] [PubMed] [Google Scholar]
- Bohr VA. Gene specific DNA repair. Carcinogen. 1991;12:1983–1992. doi: 10.1093/carcin/12.11.1983. [DOI] [PubMed] [Google Scholar]
- Boom Vvd, Citterio E, Hoogstrraten D, Zotter A, Egly JM, Cappellen VAv, Hoeijmakers HJH, Hooutsmuller AB, Vermeulen W. DNA damage stabilizes interaction of CSB wiht the transcription elongation machinery. J Cell Biol. 2004;166:27–36. doi: 10.1083/jcb.200401056. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bootsma D, Kraemer KH, Cleaver JE, Hoeijmakers JHJ. Nucleotide excision repair syndromes: xeroderma pigmentosum, Cockayne syndrome, and trichothiodystrophy. In: Vogelstein B, Kinzler KW, editors. The Genetic Basis of Human Cancer. McGraw-Hill; 1998. pp. 245–274. [Google Scholar]
- Borgne A, Golsteyne RM. The role of cyclin-dependent kinases in apoptosis. Prog cell cycle res. 2003;5:453–459. [PubMed] [Google Scholar]
- Bornstein G, Bloom J, Sitry-Shevah D, Nakayama K, Pagano M, Hershko A. Role of the SCF/Skp2 ubiquitin ligase in the degradation of p21/Cip1 in the S phase. J Biol Chem. 2003;278:25752–25757. doi: 10.1074/jbc.M301774200. [DOI] [PubMed] [Google Scholar]
- Bregman DB, Halaban R, van Gool AJ, Henning KA, Friedberg EC, Warren SL. UV-induced ubiquitination of RNA polymerase II: a novel modification deficient in Cockayne syndrome cells. Proc Natl Acad Sci U S A. 1996;93:11586–11590. doi: 10.1073/pnas.93.21.11586. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bryant HE, Schultz N, Thomas HD, Parker KM, Flower D, Lopez E, Kyle S, Meuth M, Curtin NJ, Helleday T. Specific killing of BRCA2-deficient tumours with inhibitors of poly(ADP-ribose) polymerase. Nature. 2005;434:913–917. doi: 10.1038/nature03443. [DOI] [PubMed] [Google Scholar]
- Ciechanover A, Brundin P. The ubiquitin proteasome system in neurodegenerative diseases: sometimes the chicken, sometimes the egg. Neuron. 2003;40:427–446. doi: 10.1016/s0896-6273(03)00606-8. [DOI] [PubMed] [Google Scholar]
- Citterio E, Rademakers S, van der Horst GT, van Gool AJ, Hoeijmakers JHJ, Vermeulen W. Biochemical and biological characterization of wild-type and ATPase- deficient Cockayne syndrome B repair protein. J Biological Chemistry. 1998;273:11844–11851. doi: 10.1074/jbc.273.19.11844. [DOI] [PubMed] [Google Scholar]
- Cleaver JE. Normal reconstruction of DNA supercoiling and chromatin structure in Cockayne syndrome cells during repair of damage from ultraviolet leight. American Journal of Human Genetics. 1982;34:566–575. [PMC free article] [PubMed] [Google Scholar]
- de Boer J, Andressoo JO, de Wit J, Huijmans J, Beems RB, van Steeg H, Weeda G, van der Horst GTJ, van Leeuwan W, Themmen APN, et al. Premature aging in mice deficient in DNA repair and transcription. Science. 2002;296:1276–1279. doi: 10.1126/science.1070174. [DOI] [PubMed] [Google Scholar]
- Farmer H, McCabe N, Lord CJ, Tutt AN, Johnson DA, Richardson TB, Santarosa M, Dillon KJ, Hickson I, Knights C, et al. Targeting the DNA repair defect in BRCA mutant cells as a therapeutic strategy. Nature. 2005;434:917–921. doi: 10.1038/nature03445. [DOI] [PubMed] [Google Scholar]
- Flohr C, Burkle A, Radicella JP, Epe B. Poly(ADP-ribosyl)ation accelerates DNA repair in a pathway dependent on Cockayne syndrome B protein. Nucleic Acids Res. 2003;31:5332–5337. doi: 10.1093/nar/gkg715. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fonnum F, Lock EA. Cerebellum as a target for toxic substances. Toxicology letters . 2000;112–113:9–16. doi: 10.1016/s0378-4274(99)00246-5. [DOI] [PubMed] [Google Scholar]
- Fonnum F, Lock EA. The contributions of excitotoxicity, glutathione depletion and DNA repair in chemically induced injury to neurones: exemplified with toxic effects on cerebellar granule cells. J Neurosci. 2004;88:513–531. doi: 10.1046/j.1471-4159.2003.02211.x. [DOI] [PubMed] [Google Scholar]
- Fotedar R, Bendjennat M, Fotedar A. Functional anaysis of CDK inhibitor p21WAF1. Methods Mol Biol. 2004;281:55–71. doi: 10.1385/1-59259-811-0:055. [DOI] [PubMed] [Google Scholar]
- Fukuchi K, Hagiwara T, Nakammura K, Ichimura S, Tatsumi K, Guomi K. Identification of the regulatory region required for ubiquitination of the cyclin kinase inhibitor, p21. Biochem Biophys Res Commun. 2002;293:120–125. doi: 10.1016/S0006-291X(02)00198-5. [DOI] [PubMed] [Google Scholar]
- Goldbaum O, Richter-Landsberg C. Proteolytic stress causes heat shock protein induction, tau ubiquitination, and the recruitment of ubiquitin to tau-positive aggregates in oligodendrocytes in culture. J Neurosci. 2004;24:5748–5757. doi: 10.1523/JNEUROSCI.1307-04.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Groisman R, Polanowski J, Kuraoka I, Sawada JI, Saijo M, Drapkin R, Kisselev AF, Tanaka K, Nakatani Y. The ubiquitin ligase activity in the DDB2 and CSA complexes is differentially regulated by the COP9 signalosome in response to DNA damage. Cell. 2003;113:357–367. doi: 10.1016/s0092-8674(03)00316-7. [DOI] [PubMed] [Google Scholar]
- Hamdi M, Kool J, Cornelissen-Steijger P, Carlotti F, Popeijus HE, van der Burgt C, Janssen JM, Yasui A, Hoeben RC, Terleth C, et al. DNA damage in transcribed genes induces apoptosis via the JNK pathway and the JNK-phosphatase MKP-1. Oncogene. 2005 doi: 10.1038/sj.onc.1208875. Epub ahead of print. [DOI] [PubMed] [Google Scholar]
- Harper SQ, Staber PD, He X, Eliason SL, Martins IH, Mao Q, Yang L, Kotin RM, Paulson HL, Davidson BL. RNA interference improves motor and neuropathological abnormalities in a Huntington’s disease mouse model. Proc Natl Acad Sci U S A. 2005;102:5820–5825. doi: 10.1073/pnas.0501507102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hayashi M, Araki S, Kohyama J, Shioda K, Fukatsu R. Oxidative nucleotide damage and superoxide dismutase expression in the brains of xeroderma pigmentosum group A and Cockayne syndrome. Brain Dev. 2005;27:34–38. doi: 10.1016/j.braindev.2004.04.001. [DOI] [PubMed] [Google Scholar]
- Hayashi M, Itoh M, Araki S, Kumada S, Shioda K, Tamagawa K, Mizutani T, Morimatsu Y, Minagawa M, Oda M. Oxidative stress and disturbed glutamate transport in hereditary nucleotide repair disorders. J Neuropathol Exp Neurol. 2001;60:350–356. doi: 10.1093/jnen/60.4.350. [DOI] [PubMed] [Google Scholar]
- Hefner E, Fridlyander J, Cleaver JE, Natale V. Collagen type XV is deficient in primary fibroblast cell lines from patients with Cockayne Syndrome. Genomics. 2006 in press. [Google Scholar]
- Henning KA, Li L, Iyer N, McDaniel D, Reagan MS, Legerski R, Schultz RA, Stefanini M, Lehmann AR, Mayne LV, Friedberg EC. The Cockayne syndrome group A gene encodes a WD repeat protein that interacts with the CSB protein and a subunit of RNA polymerase II, TFIIH. Cell. 1995;82:555–564. doi: 10.1016/0092-8674(95)90028-4. [DOI] [PubMed] [Google Scholar]
- Hochstrasser M. ubiquitin-dependent protein degradation. Ann Rev Genet. 1996;30:403–409. doi: 10.1146/annurev.genet.30.1.405. [DOI] [PubMed] [Google Scholar]
- Horibata K, Iwamoto Y, Kuraoka I, Jaspers NG, Kurimasa A, Oshimura M, Ichihashi M, Tanaka K. Complete absence of Cockayne syndrome group B gene product gives rise to UV-sensitive syndrome but not Cockayne syndrome. Proc Natl Acad Sci USA. 2004;101:15410–15415. doi: 10.1073/pnas.0404587101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kamiuchi S, Saijo M, Citterio E, de Jager M, Hoeijmakers JH, Tanaka K. Translocation of Cockayne syndrome group A protein to the nuclear matrix: possible relevance to transcription-coupled DNA repair. Proc Natl Acad Sci USA. 2002;99:201–206. doi: 10.1073/pnas.012473199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Keriel A, Stary A, Sarasin A, Rochette-Egly C, Egly JM. XPD mutations prevent TFIIH-dependent transactivation by nuclear receptors and phosphorylation of RARα. Cell. 2002;109:125–135. doi: 10.1016/s0092-8674(02)00692-x. [DOI] [PubMed] [Google Scholar]
- Kraemer KH, Lee MM, Andrews AD, Lambert WC. The role of sunlight and DNA repair in melanoma and nonmelanoma skin cancer. The xeroderma pigmentosum paradigm. Archives of Dermatology. 1994;130:1018–1021. [PubMed] [Google Scholar]
- Kruman I. Why do neurons enter the cellcycle? Cell Cycle. 2004;3:769–773. [PubMed] [Google Scholar]
- Kyng KJ, May A, Brosh RMJ, Cheng WH, Chen C, Becker KG, Bohr VA. The transcriptional response after oxidative stress is defective in Cockayne syndrome group B cells. Oncogene. 2003;22:1135–1149. doi: 10.1038/sj.onc.1206187. [DOI] [PubMed] [Google Scholar]
- Lee KB, Wang D, Lippard SJ, Sharp PA. Transcription-coupled and DNA damage-dependent ubiquitination of RNA polymerase II in vitro. ProcNatlAcadSciUSA. 2002;99:4239–4244. doi: 10.1073/pnas.072068399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lehmann AR. Three complementation groups in Cockayne syndrome. Mutation Research. 1982;106:347–356. doi: 10.1016/0027-5107(82)90115-4. [DOI] [PubMed] [Google Scholar]
- Licht CL, Stevnser T, Bohr VA. Cockayne syndrome group B cellular and biochemical functions. Amer J Human Genetics. 2003;73:1217–1239. doi: 10.1086/380399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Limoli CL, Giedzinski E, Morgan WF, Cleaver JE. Polymerase η deficiency in the XP variant uncovers an overlap between the S phase checkpoint and double strand break repair. Proc Nat Acad Sci USA. 2000;97:7939–7946. doi: 10.1073/pnas.130182897. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ljungman M, Zhang F. Blockage of RNA polymerase as a possible trigger for u.v. light-induced apoptosis. Oncogene. 1996;13:823–831. [PubMed] [Google Scholar]
- Lowndes NF, Toh GWL. DNA repair: the importance of phosphorylating histone H2AX. Curr Biol. 2005;15:R99–R102. doi: 10.1016/j.cub.2005.01.029. [DOI] [PubMed] [Google Scholar]
- Lu T, Pam Y, Kao SY, Li C, Kohane I, Chan J, Yankner BA. Gene regulation and DNA damage in the ageing human brain. Nature. 2004;429:883–891. doi: 10.1038/nature02661. [DOI] [PubMed] [Google Scholar]
- Macip S, Igarashi M, Fanf L, Chen A, Pan ZQ, Lee SW, Aaronson SA. Inhibition of p21-mediated ROS accumulation can rescue p21-induced sensescence. EMBO J. 2002;21:2180–2188. doi: 10.1093/emboj/21.9.2180. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Macleod KF, Hu Y, Jacks T. Loss of Rb activates both p53-dependent and independent cell death pathways in the developing mouse nervous system. EMBO J. 1996;15:6178–6188. [PMC free article] [PubMed] [Google Scholar]
- McKay BC, Chen F, Clark ST, Wiggin HE, Harley LM, Lljungman M. UV-light-induced degradation of RNA polymerase II is dependent on the Cockayne’s syndrome A or B proteins but not p53 or MHL1. Mutat Res. 2001;485:93–105. doi: 10.1016/s0921-8777(00)00064-1. [DOI] [PubMed] [Google Scholar]
- Murai M, Enokido Y, Inamura N, Yoshino M, Nakatsu Y, van der Horst GT, Hoeijmakers JH, Tanaka K, Hatanaka H. Early postnatal ataxia and abnormal cerebellar development in mice lacking Xeroderma pigmentosum Group A and Cockayne syndrome Group B DNA repair genes. Proc Nat Acad Sci USA. 2001;98:13379–13384. doi: 10.1073/pnas.231329598. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nance MA, Berry SA. Cockayne syndrome:review of 140 cases. Amer J Med Genet. 1992;42:68–84. doi: 10.1002/ajmg.1320420115. [DOI] [PubMed] [Google Scholar]
- Parris CH, Kraemer KH. Ultraviolet-light induced mutations in Cockayne syndrome cells are primarily caused by cyclobutane dimer photoproducts while repair of other photoproducts is normal. Proceedings of the National Academy of Sciences USA. 1993;90:7260–7264. doi: 10.1073/pnas.90.15.7260. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peng J, Schwartz D, Elias JE, Thoreen CC, Marsischky G, Roelofs J, Finley D, Gygi SP. A proteomics approach to understanding protein ubiquitination. Nature Biotech. 2003;21:921–926. doi: 10.1038/nbt849. [DOI] [PubMed] [Google Scholar]
- Santacruz K, Lewis J, Spires T, Paulson J, Kotilinek L, Ingelsson M, Guimaraes A, DeTure M, Ramsden M, McGowan E, et al. Tau suppression in a neurodegenerative mouse model improves memory function. Science. 2005;309:476–481. doi: 10.1126/science.1113694. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Selby CP, Sancar A. Human transcription-repair coupling factor CSB/ERCC6 is a DNA- stimulated ATPase but is not a helicase and does not disrupt the ternary transcription complex of stalled RNA polymerase II. Journal of Biological Chemistry. 1997;272:1885–1890. doi: 10.1074/jbc.272.3.1885. [DOI] [PubMed] [Google Scholar]
- Semple CA, Group RG, Members G. The comparative proteomics of ubiquitination in mouse. Genome Research. 2003;13:1389–1394. doi: 10.1101/gr.980303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shiomi N, Mori M, Kito S, Harada Y-N, Tanaka K, Shiomi T. Severe growth retardation and short life-span of double mutant mice lacking Xpa and exon 15 of Xpg. DNA repair. 2005 doi: 10.1016/j.dnarep.2004.10.009. in press. [DOI] [PubMed] [Google Scholar]
- Thorsland T, von Kobbe C, Harrigan JA, Indig FE, Christiansen M, Stevsner T, Bohr VA. Cooperation of the Cockayne syndrome group B protein and poly(ADP-ribose) polymerase I in the response to oxidative damage. Mol Cell Biol. 2005;25:7625–7636. doi: 10.1128/MCB.25.17.7625-7636.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tu Y, Bates S, Pfeiffer GP. The transcription-repair coupling factor CSA is required for efficent repair only during the elongation stages of RNA polymerase II transcription. Mutat Res. 1998;400:143–151. doi: 10.1016/s0027-5107(98)00038-4. [DOI] [PubMed] [Google Scholar]
- Weeda G, Ham RCAv, Vermeulen W, Bootsma D, Eb AJvd, Hoeijmakers JHJ. A presumed helicase encoded by ERCC-3 is involved in the human repair disorders xeroderma pigmentosum and Cockayne’s syndrome. Cell. 1990;62:777–791. doi: 10.1016/0092-8674(90)90122-u. [DOI] [PubMed] [Google Scholar]
- Welsh JP, Yuen G, Placantonakis DG, Vu TQ, Haiss F, O’Hearn E, Molliver ME, Aicher SA. Why do Purkinje cells die os easily after global brain ischemia? Aldolase C, EAAT4, and the cerebellar contribution to posthypoxic myoclonus. Adv Neurol. 2002;89:331–359. [PubMed] [Google Scholar]
- Wilkinson KD. Ubiquitination and deubiquitination: targeting of proteins for degradation by the proteasome. Cell and develop biology. 2000;11:141–148. doi: 10.1006/scdb.2000.0164. [DOI] [PubMed] [Google Scholar]
- Wood RD, Mitchell M, Sgouros J, Lindahl T. Human DNA repair genes. Science. 2001;291:1284–1289. doi: 10.1126/science.1056154. [DOI] [PubMed] [Google Scholar]
- Woudstra EC, Gilbert C, Fellows J, Jansen L, Brouwer J, Erdjument-Bromage H, Tempst P, Svejstrup JQ. A Rad26-Def1 complex coordinates repair and RNA pol II proteolysis in response to DNA damage. Nature. 2002;415:929–933. doi: 10.1038/415929a. [DOI] [PubMed] [Google Scholar]