

Case Presentation
An 11-month-old boy was admitted to the hospital for a history of chronic diarrhea and severe failure to thrive. He was born full-term with a birth weight of 2.8 kg, but did not regain his birth weight until 2 months of age. He was noted by his parents to have foul-smelling diarrhea after every feeding for most of his life. He had been fed standard cow's milk protein-based formula and was reported to have a hearty appetite, typically ingesting at least 32 oz per day of formula without any vomiting or arching. He was seen by an endocrinologist at 4 months of age for poor weight gain, but no diagnosis was obtained. It was noted at the time, however, that he had a mildly enlarged liver.
His weight and height paralleled but remained below the third percentile curves. He began eating solids at 7 months of age and was started on a cow's milk protein-based nutritional supplement at 9 months of age without improvement in his weight gain. No hematochezia, fevers, jaundice, or rashes were reported.
His past medical history was remarkable for occipital nodal abscesses at 5 months of age and right lower lobe pneumonia at age 10 months. Both infections responded rapidly to antibiotics. He had no history of prior surgeries and his only medication was ranitidine for presumed gastroesophageal reflux disease. He had no known drug allergies. Both the social and family history were noncontributory. There was no known consanguinity in the family. The patient did have a history of developmental delay, as he rolled over at 7 months of age and was not yet able to sit or stand without support.
On physical examination, the patient weighed 5.16 kg (50th percentile for a 2-month-old) and had a length of 62.5 cm (50th percentile for a 3-month-old). His vital signs were within normal limits. He appeared to be a small, wasted infant with a high-pitched cry. There was no scleral icterus. He had a narrow thorax, and his lungs were clear to auscultation. His cardiac exam was normal. His abdomen was protuberant, nontender, and had normal bowel sounds. His liver edge was palpable 3 cm below the right costal margin, and his spleen was not palpable. There was no jaundice, and his neurologic exam demonstrated mild generalized hypotonia.
Electrolytes and coagulation studies were normal. He had elevated aminotransferases (alanine aminotransferase [ALT] 365 U/L, aspartate aminotransferase [AST] 325 U/L) and a mildly elevated serum gamma-glutamyl transpeptidase (GGT) of 63 U/L. His total bilirubin, alkaline phosphatase, total protein, and albumin levels were normal. His serum amylase and lipase levels were normal. Complete blood count was notable for an elevated white blood cell count of 20,900 cells/mcL (3% neutrophils, 83% lymphocytes, 9% monocytes), hemoglobin of 10.7 g/dL, hematocrit of 32.9%, and platelet count of 433,000 cells/mcL. A urine culture was positive for Escherichia coli.
-
1.Which of the following conditions is the most likely explanation for the patient's failure to thrive, diarrhea, and liver abnormalities?
- Inadequate caloric intake
- Urinary tract infection
- Cow's milk protein allergy
-
Celiac diseaseAlthough inadequate caloric intake and chronic urinary tract infections are causes of failure to thrive, they do not explain the patient's diarrhea and hepatomegaly. Cow's milk protein allergy is a common cause of chronic diarrhea in infancy, although liver enzyme abnormalities are not common. On the basis of the information available at this point in work-up, celiac disease is the most likely diagnosis. Celiac disease may present with severe failure to thrive, diarrhea, and abnormal liver enzymes, although it should be noted that this condition typically manifests at an age when gluten is introduced into the infant's diet. Click on Next Page for a more comprehensive discussion of the initial differential diagnosis.
Initial Differential Diagnosis
In addition to the conditions mentioned in the previous question, there are other processes that should be considered in this patient's differential diagnosis. Conditions causing pancreatic insufficiency, such as cystic fibrosis, may cause chronic diarrhea as well as liver abnormalities. Metabolic disorders such as galactosemia may present with diarrhea, hepatomegaly, and sepsis. The patient's diarrhea could also be explained by an infection or a postinfectious enteropathy that led to intestinal villous damage. Neonatal enteropathies, such as microvillus inclusion disease, tufting enteropathy, autoimmune enteropathy, and IPEX (immunodysregulation, polyendocrinopathy, enteropathy, X-linked) syndrome should also be considered. Conditions causing carbohydrate malabsorption, such as sucrase-isomaltase deficiency, could also be a cause of chronic diarrhea. Patients with an immunodeficiency such as chronic granulomatous disease could present with a history of abscesses and pneumonia, diarrhea, and if the infection involved the liver, increased aminotransferase levels. Endocrine abnormalities and nonorganic causes should always be considered in patients with failure to thrive; however, this patient's clinical history and laboratory abnormalities do not seem to be compatible with these causes.
Further Work-up
Stool studies were negative for infectious etiologies, and testing for stool-reducing substances was negative. A celiac panel and a viral hepatitis panel were normal. Abdominal ultrasound (Figure) showed an echogenic pancreas, possibly representing fatty infiltration. The liver and biliary tree were normal-appearing, and there was nephrocalcinosis with multiple nonobstructing kidney stones. A 72-hour fecal fat collection was consistent with fat malabsorption, and the stool pancreatic elastase level was < 15 mcg/g (normal, 200–500 mcg/g). The patient was also noted to be neutropenic (absolute neutrophil count of 627 cells/mcL).
-
2.Which of the following condition(s) is associated with pancreatic insufficiency and neutropenia?
- Cystic fibrosis
- Shwachman-Diamond syndrome
- Pearson's marrow-pancreas syndrome
- Johansson-Blizzard syndrome
-
Jeune syndromeAlthough all of these conditions are associated with pancreatic insufficiency, only Shwachman-Diamond syndrome and Pearson's marrow-pancreas syndrome are associated with bone marrow failure. Neutropenia is typically associated with Shwachman-Diamond syndrome, although many of these patients may also be anemic and thrombocytopenic.<br/> Click on Next Page for further discussion of this patient's differential diagnosis.
Figure.
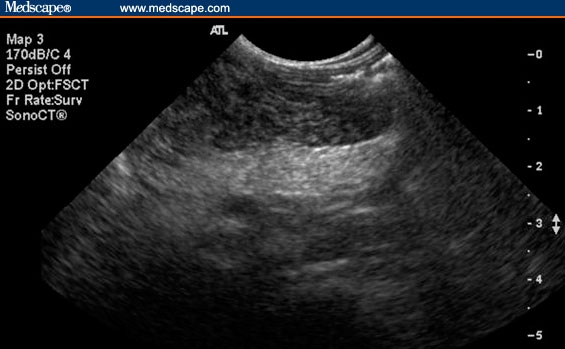
Increased pancreas echogenicity secondary to fatty infiltration.
Differential Diagnosis
The low stool pancreatic elastase level is consistent with severe exocrine pancreatic insufficiency. Cystic fibrosis, the most common cause of pancreatic insufficiency in children, should be strongly considered, especially in light of a past medical history of pneumonia. As noted earlier, the patient was also found to be neutropenic (absolute neutrophil count of 627 cells/mcL), which could be associated with Shwachman-Diamond syndrome. Patients with Pearson's marrow-pancreas syndrome can also have bone marrow failure, hepatomegaly, and exocrine pancreatic insufficiency. Other less common causes of pancreatic insufficiency include Johansson-Blizzard syndrome and Jeune syndrome. The patient's kidney stones are likely related to his fat malabsorption, because the fat binds to calcium in the gastrointestinal tract, increasing the free oxalate to be absorbed. The increased oxalate can be deposited in the kidney to form stones.
Establishing the Diagnosis
The patient underwent a sweat chloride test to rule out cystic fibrosis; results revealed a normal sweat chloride concentration. A bone marrow aspirate showed myeloid asynchrony not diagnostic of a specific pathologic entity, but possibly consistent with a bone marrow failure syndrome such as cyclic neutropenia or Shwachman-Diamond syndrome. Genetic testing demonstrated 2 mutations in the SBDS (Shwachman-Bodian-Diamond syndrome) gene consistent with the diagnosis of Shwachman-Diamond syndrome.
Clinical Course and Outcome
The patient was started on pancreatic enzyme supplementation and was changed to a formula with a higher caloric concentration. He demonstrated good weight gain before being discharged.
Discussion
Shwachman-Diamond syndrome is a rare autosomal recessive disorder characterized by exocrine pancreatic dysfunction, bone marrow failure, and skeletal abnormalities. It has also been known as “Shwachman syndrome,” “Shwachman-Bodian syndrome,” and “congenital lipomatosis of the pancreas.” This disorder was described by Shwachman, Diamond, Oski, and Knaw in 1964 in 5 children showing evidence of exocrine pancreatic insufficiency and leukopenia, and was described a few months afterwards by Bodian and colleagues. Associated skeletal abnormalities of the metaphyseal dysostosis type were observed a few years later, and became the third fundamental feature of the syndrome.
Shwachman-Diamond syndrome is the most common inherited cause of exocrine pancreatic insufficiency after cystic fibrosis. It is also likely the third most common inherited bone marrow failure syndrome after Fanconi's anemia and Diamond-Blackfan anemia. However, Shwachman-Diamond syndrome has a low prevalence, estimated at 1 in 50,000. It has a slight male predominance (ratio 1.7:1) and is reported among all racial and ethnic groups.
All patients with Shwachman-Diamond syndrome have varying degrees of pancreatic insufficiency, which is related to poor acinar cell development in utero. The acinar cells are replaced with fat, although the ductular architecture remains intact, in contrast to cystic fibrosis. Patients often present with steatorrhea and failure to thrive, requiring pancreatic enzyme supplementation. However, the exocrine pancreatic function has been reported to improve with age, resulting in decreased fat malabsorption. Approximately 50% of affected patients will show enough improvement with increasing age such that pancreatic enzyme supplementation becomes unnecessary. Although fat malabsorption certainly contributes to failure to thrive in these patients, other factors that may contribute to poor growth include recurrent infections, skeletal abnormalities, and decreased growth hormone levels.
Neutropenia is the most common manifestation of bone marrow failure in patients with Shwachman-Diamond syndrome, and is found in 88% to 100% of patients. The neutropenia is intermittent in two thirds of patients and is chronic in the remaining one third. Because most Shwachman-Diamond syndrome patients suffer from neutropenia, they are susceptible to recurrent bacterial infections, particularly of the upper respiratory tract, oropharynx, and skin. Not only do Shwachman-Diamond syndrome patients have decreased neutrophil counts, but they also seem to have defective neutrophil chemotaxis, further predisposing these patients to infection. Patients can also have variable degrees of anemia or thrombocytopenia, and almost 50% of patients with Shwachman-Diamond syndrome have pancytopenia. Myelodysplastic syndromes and acute leukemias develop in up to one third of patients.
More than 75% of patients with Shwachman-Diamond syndrome have skeletal abnormalities. These commonly include metaphyseal dysostosis, which usually involves the femoral head. Abnormally shortened ribs with flared anterior ends, costochondral thickening, and a narrow rib cage have also been described. Other skeletal anomalies reported include delayed secondary ossification, generalized osteopenia, clinodactyly, syndactyly, supernumerary digits, pes cavus, kyphosis, and scoliosis. The patient featured in this case did not have a skeletal survey performed to look for metaphyseal dysostosis, although on physical exam he was noted to have a narrow thorax.
Hepatic involvement in children with Shwachman-Diamond syndrome is common. Hepatomegaly and increased aminotransferase levels are commonly seen, with liver biopsies demonstrating microvesicular and macrovesicular steatosis and mild portal fibrosis. Liver biopsy findings in our patient were not consistent with steatosis and were nonspecific. Progressive liver dysfunction is uncommon, and most patients have normalization of liver enzymes as they become older.
Other organ systems involved in Shwachman-Diamond syndrome include the endocrine system, with abnormalities such as insulin-dependent diabetes and growth hormone deficiencies. Dermatologic abnormalities may include eczema, ichthyosis, and petechiae. Delayed dentition of permanent teeth, dental dysplasia, and increased risk for dental caries may occur. Mild-to-moderate psychomotor or developmental delay can be seen in up to 15% of patients with Shwachman-Diamond syndrome.
The diagnostic work-up for a patient suspected of having Shwachman-Diamond syndrome should include complete blood cell counts that may suggest bone marrow dysfunction. A 72-hour fecal fat measurement with concurrent measurement of fat intake to calculate fat clearance can show an increase in fecal fat losses, and a decreased fecal pancreatic elastase level can suggest exocrine pancreatic insufficiency. Serum trypsinogen and isoamylase testing have also proven to be useful in identifying pancreatic insufficiency in Shwachman-Diamond syndrome. A sweat chloride test should be performed, which is normal in Shwachman-Diamond syndrome. Ultrasound examination of the pancreas may demonstrate increased echogenicity, and a computed tomography scan can show lipomatosis of the pancreas. A skeletal survey may uncover skeletal abnormalities, such as thoracic dysostosis, delayed bone age, tubulation of the long bones, and valgus deformities of the elbows and knees. A bone marrow aspiration and biopsy can be performed, and although there are no pathognomonic bone marrow features in Shwachman-Diamond syndrome, the examination may exclude alternative diagnoses, as well as screen for leukemia or myelodysplastic syndromes. Neutrophil chemotaxis assays can be considered for detection of defective chemotaxis. Finally, testing is available for mutations in the SBDS gene, located on chromosome 7q11. Approximately 90% of patients meeting clinical criteria for Shwachman-Diamond syndrome have mutations in the SBDS gene.
Because Shwachman-Diamond syndrome affects multiple organ systems, patients should be referred to the appropriate medical subspecialties for management, including gastroenterology, hematology, endocrinology, and genetics. Nutritional therapy for Shwachman-Diamond syndrome typically involves pancreatic enzyme supplementation. Dietary administration of fat-soluble vitamins, medium-chain triglycerides, and other high-calorie supplements may also be needed. Because most patients with Shwachman-Diamond syndrome are neutropenic, clinicians should have a low threshold for the evaluation and treatment of potential infections. Granulocyte colony-stimulating factor administration can be considered in Shwachman-Diamond syndrome patients with repeated or serious life-threatening infections. Bone marrow transplant is the only curative therapy for severe hematologic manifestations of Shwachman-Diamond syndrome. For those with short stature, growth hormone can be used for Shwachman-Diamond syndrome patients who have growth hormone deficiency.
Footnotes
Reader Comments on: An 11-Month-Old Boy With Chronic Diarrhea, Failure to Thrive, and Hepatomegaly See reader comments on this article and provide your own.
Readers are encouraged to respond to the author at jmarkowitz@ghs.org or to Paul Blumenthal, MD, Deputy Editor of MedGenMed, for the editor's eyes only or for possible publication as an actual Letter in MedGenMed via email: pblumen@stanford.edu
Contributor Information
Steven Liu, The Children's Hospital of Philadelphia, University of Pennsylvania School of Medicine, Philadelphia, Pennsylvania.
Jonathan E. Markowitz, Children's Center for Digestive Health, University of South Carolina School of Medicine, Greenville Hospital System University Medical System, Greenville, South Carolina Author's email: jmarkowitz@ghs.org.
Petar Mamula, Endoscopy Suite; Division of GI & Nutrition, The Children's Hospital of Philadelphia, Philadelphia, Pennsylvania; University of Pennsylvania School of Medicine, Philadelphia, Pennsylvania.
David A. Piccoli, Division of Gastroenterology, Children's Hospital of Philadelphia, University of Pennsylvania School of Medicine, Philadelphia, Pennsylvania.
Suggested Reading
- Boocock GRB, Morrison JA, Popovic M, et al. Mutations in SBDS are associated with Shwachman-Diamond syndrome. Nat Genet. 2003;33:97–101. doi: 10.1038/ng1062. [DOI] [PubMed] [Google Scholar]
- Cipolli M, D'Orazio C, Antonella D, et al. Shwachman's syndrome: pathomorphosis and long-term outcome. J Pediatr Gastroenterol Nutr. 1999;29:265–272. doi: 10.1097/00005176-199909000-00006. [DOI] [PubMed] [Google Scholar]
- Dror Y, Freedman MH. Shwachman-Diamond syndrome. Br J Haematol. 2002;118:701–713. doi: 10.1046/j.1365-2141.2002.03585.x. [DOI] [PubMed] [Google Scholar]
- Sherman PM, Mitchell DJ, Cutz E. Neonatal enteropathies: defining the causes of protracted diarrhea of infancy. J Pediatr Gastroenterol Nutr. 2004;38:16–26. doi: 10.1097/00005176-200401000-00007. [DOI] [PubMed] [Google Scholar]
- Shimamura A. Shwachman-Diamond syndrome. Semin Hematol. 2006;43:178–188. doi: 10.1053/j.seminhematol.2006.04.006. [DOI] [PubMed] [Google Scholar]
- Stormon MO, Durie PR. Pathophysiologic basis of exocrine pancreatic dysfunction in childhood. J Pediatr Gastroenterol Nutr. 2002;35:8–21. doi: 10.1097/00005176-200207000-00004. [DOI] [PubMed] [Google Scholar]