

Abstract
Vaccinia virus (VV) infection is known to inhibit dendritic cells (DC) functions in vitro. Paradoxically, VV is also highly immunogenic and thus has been used as a vaccine. In the present study, we investigated the effects of an in vivo VV infection on DC function by focusing on early innate immunity. Our data indicated that DC are activated upon in vivo VV infection of mice. Splenic DC from VV-infected mice expressed elevated levels of MHC class I and co-stimulatory molecules on their cell surface and exhibited the enhanced potential to produce cytokines upon LPS stimulation. DC from VV-infected mice also expressed a high level of interferon-β. However, a VV infection resulted in the down-regulation of MHC class II expression and the impairment of antigen presentation to CD4 T cells by DC. Thus, during the early stage of a VV infection, although DC are impaired in some of the critical antigen presentation functions, they can promote innate immune defenses against viral infection.
Keywords: vaccinia virus, dendritic cell, MHC class II, gene regulation, cytokines, type I interferon, innate immunity
INTRODUCTION
Vaccini a virus (VV) belongs to the poxviridae family of DNA virus and has been used as a vaccine against smallpox as well as a vector to deliver novel vaccines [1; 2]. Unfortunately, however, immunization with the vaccinia has been associated with significant complications, particularly in immunocompromised individuals [3]. Thus, an increased understanding of the host’s immune responses in the context of viral pathogenesis during a VV infection is of critical importance. Although VV is known to be equipped with multiple strategies that can impair host immune responses [4; 5], the effect of a VV infection on antigen presentation is not well understood.
A typical immune response against a viral infection is mediated by CD8 cytotoxic T cells that require major histocompatibility complex (MHC) class I. Because of this, many viruses down-regulate the level of surface MHC class I antigens following infection [6; 7; 8]. Defects in MHC class I-mediated antigen presentation have been observed with recombinant VV that was engineered to express a heterologous antigen [9; 10]. In this case, however, the virus appears to have altered antigen processing in an epitope specific manner. Less is known regarding MHC class II mediated antigen presentation during a VV infection, although CD4 T cell responses are also thought to be critical in promoting viral clearance and immunity [11]. Mice deficient in MHC class II protein expression displayed slightly decreased cytotoxic T lymphocyte (CTL) responses against VV [12]. In addition, we have recently shown that a VV infection in vitro disrupts antigen presentation by MHC class II [13]. Thus, it is clear that VV can modulate antigen presentation by both MHC class I and class II molecules.
Dendritic cells (DC) are specialized antigen-presenting cells (APC) derived from bone marrow (BM) precursors, and carry out important functions during both innate and adaptive immune responses [14; 15]. Upon encountering infectious agents, immature DC resident in peripheral tissues capture antigens and migrate to secondary lymphoid organs while undergoing maturation [16]. Matured and thus activated DC secrete both inflammatory and anti-inflammatory cytokines that regulate the immune response. Mature DC also typically express high levels of MHC class I, class II, and co-stimulatory molecules. Therefore, mature DC are potent APC set to prime naive CD4 T cells and elicit CD8 T cell responses, bridging the innate and adaptive immune responses [17; 18].
There are multiple DC subsets in both humans and mice, as characterized by surface marker expression and phenotypic functions [19]. In general, DC can be divided into two major populations: plasmacytoid DC (CD11c+B220+; pDC) and conventional DC (CD11c+B220−; cDC) that are preferentially associated with innate and adaptive immune responses, respectively [19; 20]. cDC can be further divided into lymphoid DC (CD11c+CD8+) and myeloid DC (CD11c+CD11b+CD4+/−; mDC), within which CD8+ DC are generally involved in priming CTL immunity to viruses [19; 21]. On the other hand, pDC specialize in the secretion of type I (α/β) interferons (IFN), which play a pivotal role in viral clearance [22; 23]. Therefore, it would not be surprising that one means by which viruses can evade the host’s antiviral immune response is by altering the balance between these DC subsets. Indeed, an lymphocytic choriomeningitis virus infection has been shown to change the predominant DC subset from pDC to mDC to divert host immune response away from anti-viral immunity [24]. On the other hand, it was reported that pDC accumulate in the spleen after infection with murine cytomegalovirus [25], and that mDC present exogenous virus-like particles to CD8 T cells and subsequently become CD8+ [26].
VV infection is also known to inhibit DC functions [4]. VV blocks cytokine signals by expressing decoy receptors for interleukin (IL)-1β, tissue necrosis factor (TNF)-α, and IFN [27; 28]. The VV protein A52R disrupts the formation of Toll-like receptor (TLR) complexes, which are important in triggering some pathways of DC maturation [29]. A VV mutant lacking A52R was shown to be less virulent than the wild type (WT) VV in vivo in a murine intranasal infection model [29]. In addition, a VV infection induces apoptotic cell death and blocks maturation of human DC [30; 31]. However, a majority of the studies demonstrating the impairment of DC function by a VV infection have been carried out in vitro. VV is known to be highly immunogenic, and thus has been successfully used as an attenuated vaccine to eradicate human smallpox [2]. Nonetheless, it has remained unclear whether DC functions were compromised by a VV infection in vivo.
In the present study, we investigated the effects of a VV infection on DC function in vivo by focusing on aspects of innate immunity. Our data indicated that DC are activated upon an in vivo VV infection of mice. Splenic DC from VV-infected mice expressed elevated levels of MHC class I and co-stimulatory molecules on their cell surface and exhibited an enhanced potential to produce cytokines. However, a VV infection resulted in the down-regulation of MHC class II expression and an impairment of antigen presentation to CD4 T cells. Together, our results demonstrate that during early stages of a VV infection, the host’s immune responses are geared toward anti-viral immunity but the host experiences a transient immunosuppression presumably due to deficits in MHC class II presentation by DC.
MATERIALS AND METHODS
Mice, viruses and infection
C57BL/6 mice were purchased from the Jackson Laboratories (Bar Harbor, ME), and maintained under specific pathogen-free conditions at the Indiana University School of Medicine. BM cells from mice deficient in type I IFN receptor (IFNAR−/−) were kindly provided by Dr. Mary O’Riordan (U. Michigan, Ann Arbor). All procedures were approved by the Indiana University School of Medicine Institutional Animal Care and Use Committee.
Stocks of WT VV (Western Reserve strain) were generated in the human osteosarcoma 143B cell line, followed by sucrose purification and titer determination as described [13]. The recombinant mKO mutant VV expressing orange fluorescent protein (OFP) has been previously described [32].
Female C57BL/6 mice were injected i.p. with 1×106 pfu (plaque-forming unit) of VV in HBSS with 0.1% BSA (HBSS/BSA) and sacrificed up to two days after injection. Mice receiving HBSS/BSA alone were used as a mock-infected control.
To prepare splenic DC, CD11c positive cells were isolated from splenocytes using anti-CD11c magnetic beads (Miltenyi Biotech, Auburn, CA). In some experiments, the CD11c negative flow through was used to isolate B cells and macrophages with anti-B220 and anti-CD11b magnetic beads, respectively. To activate DC, CD11c+ enriched DC were cultured for 24 hr at 1×106 cells/ml in RPMI 1640 supplemented with 5% fetal bovine serum (FBS) in the presence of 1 μg/ml lipopolysaccharide (LPS, E. coli O55:B5 serotype, Sigma, St. Louis, MO). BMDC were generated as previously described [33].
In vitro infection
Immature BMDC or total splenocytes were infected with VV at different multiplicities of infection (MOI, 10−4 – 10−1) for 90 min, followed by washing and culturing in fresh RPMI 1640 supplemented with 5% FBS in the presence or absence of 10 ng/ml murine recombinant IFN-β (Endogen) for 24 or 48 hr. To activate BMDC, mock- or VV-infected BMDC were cultured in the presence of 1 μg/ml LPS for 24 hr.
To investigate the bystander effect of VV infection, conditioned medium (CM) were collected from mock- or VV-infected BMDC 24 hr post-infection, either filtered or unfiltered with a 0.2 μm syringe driven filter unit (Millipore, Bedford, MA), and added to uninfected BMDC followed by culturing them for 24 hr.
Fluorescence-activated cell sorter (FACS) analysis
Cells were preincubated for 15 min with the anti-FcγR monoclonal antibody (Ab) 2.4G2 to block non-specific binding before staining with the following fluorescein isothiocyanate (FITC)-, phycoerythrin (PE)-, peridinin chlorophyll a protein (PerCP)-, or allophycocyanin (APC)-conjugated Abs for 30 min at 4 °C: CD11c (clone HL3), CD11b (clone M1/70), CD49b (pan-NK, clone DX5), CD45R (B220, clone RA3-6B2), CD4 (L3T4, clone RM4-5), CD8α (Ly-2, clone 53-6.7), CD40 (clone 3/23), CD80 (B7-1, clone 16-10A1), CD86 (B7-2, clone GL-1), MHC class I (H2-Kb, clone AF6-88.5), MHC class II (I-Ab, clone AF6-120.1), and mouse IgG2a, κ (isotype control), all of which were obtained from BD Biosciences (San Diego, CA). Cells were then washed and fixed with 2% paraformaldehyde. To identify VV-infected cells, the cells were fixed, permeabilized with 0.2% saponin (Sigma), and stained with a biotinylated mouse IgG2a, κ isotype control (eBioscience, San Diego, CA) or a biotinylated TW2.3 monoclonal Ab specific for the E3L viral protein [34], followed by the addition of PerCP- or APC-conjugated streptavidin (BD Biosiences). Flow cytometric analyses were performed using a FACSCalibur and analyzed using CellQuest software (BD Biosciences).
Enzyme-linked immunosorbent assay (ELISA)
Cytokine concentrations in the culture supernatants were detected by ELISA as previously described [35]. Purified anti-mouse capture and biotinylated detection Abs, respectively, were: IL-6 (clone P5-20F3 and MP5-32C11), IL-10 (clone JES5-2A5 and SXC-1), IL-12p70 (clone 9A5 and C17.8), and TNF-α (clone G281-2626 and MP6-XT3). All Abs were purchased from BD Biosciences.
RT-PCR and quantitative real-time PCR (qRT-PCR)
Total RNA preparation, cDNA synthesis, and PCR were conducted as described [36]. The primers used for E3L and hypoxanthine guanine phosphoribosyl transferase (HPRT) were described previously [13; 37].
qRT-PCR was performed by the comparative threshold cycle (ΔCT) method and normalized to glyceraldehyde-3-phosphate dehydrogenase (GAPDH). The primers used for IFN-β, IFN regulatory factor (IRF)-7, MHC class II (I-Aαb) and GAPDH were as described [37; 38].
Antigen presentation assays
CD11c-enriched splenic DC (1×105) from VV- or mock-infected mice were incubated with increasing concentrations of the HEL74–88 peptide for 16 hr, followed by washing and fixation (0.5% paraformaldehyde, 10 min, 4°C). Peptide-loaded DC were cultured with the HEL-peptide-specific T cell hybridoma, B04 (5×104) for 24 h. IL-2 production was measured using HT-2, an IL-2-dependent T cell line, as previously described [13]. HT-2 cell proliferation was quantitated via [3H]thymidine incorporation into DNA and the data are expressed as the net cpm (counts per minute).
Statistical analysis
Differences between VV- and mock-infected groups were examined by a two-way ANOVA followed by Bonferroni-Dunn post hoc test. Statistical significance was defined as a p value < 0.05.
RESULTS
Splenic DC are infected by VV at a low efficiency in vivo
To study the effects of a VV infection on DC function in vivo, we infected C57BL/6 mice with 1×106 pfu VV i.p. Mice were then sacrificed one or two days post-infection, and splenic DC (SPDC) were prepared as described in the Materials and Methods. To determine whether DC were infected with the virus in vivo, we examined total splenocytes from mock- and VV-infected mice using an Ab recognizing E3L, a VV-encoded early gene product. As expected, E3L-positive SPDC were not detectable from mock-infected mice (Figure 1A, top panel). However, SPDC from VV-infected mice had a very small population expressing E3L (Figure 1A, top panel). The low levels of E3L detected after an in vivo infection suggest either a low level of SPDC viral infection or reduced virus synthesis of this antigen in DC. To assess the infection efficiency, we performed in vitro VV infection using both BMDC and SPDC with a range of MOI. A distinct E3L positive BMDC population was present after an in vitro infection with an MOI as low as 10−3 and E3L expression correlated with the dose of VV used for infection (Figure 1A, middle panel). SPDC were also infected by VV in vitro, although the efficiency was less than that of BMDC (Figure 1A, bottom panel). The E3L signal appears to be specific since a control Ab with the same IgG2a, κ isotype did not show a positive cell population from mock- or VV-infected BMDC (Figure 1B).
Figure 1. Detection of VV infection.
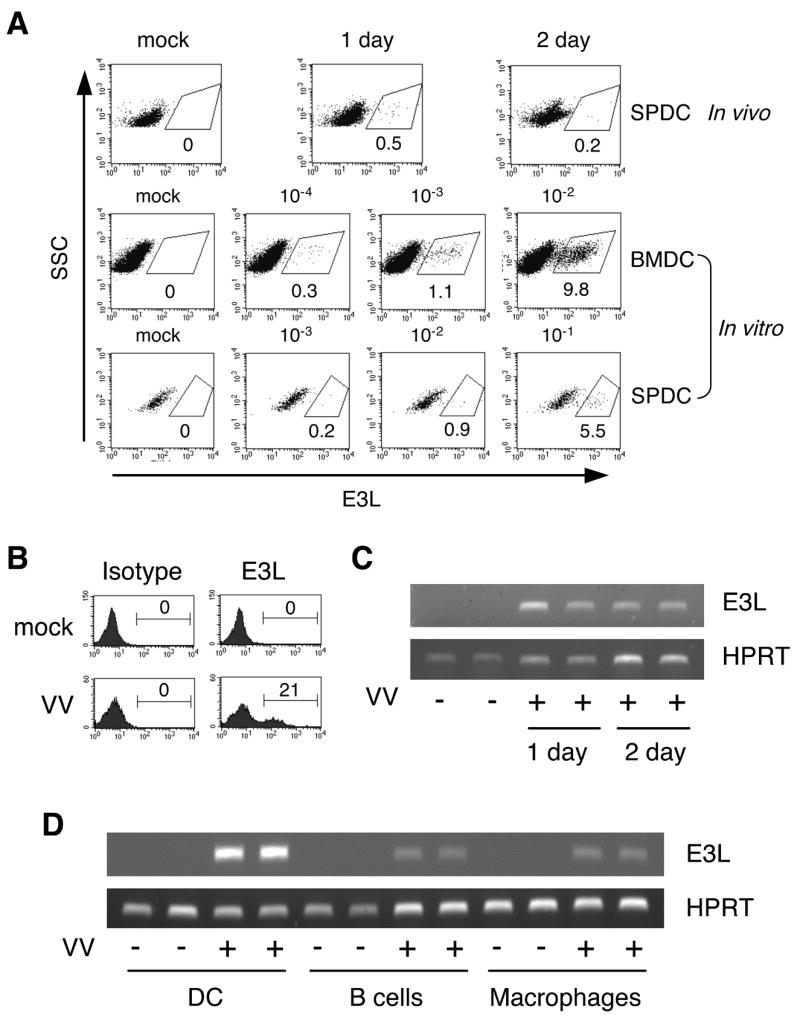
A, In vivo infection was carried out using C57BL/6 mice (four mice/group) that were injected with HBSS/BSA (mock) or 1×106 pfu VV via i.p as described in the Materials and Methods. Mice were sacrificed at the indicated time. Total splenocytes were stained with TW2.3 Ab recognizing E3L. For in vitro infection, BMDC and total splenocytes were infected and cultured for 24 hrs with MOI as indicated. CD11c+ cells were gated and shown. The percentage indicates % positive cells among CD11c+ cells. B, Specificity of TW2.3 staining. BMDC were infected 24 hrs with VV at a MOI of 0.1 and stained with TW2.3 or its isotype-matched control Ab. The numbers indicate % positive cells. C, RNA was prepared from CD11c+ enriched splenic DC from mock- or VV-infected mice as in (A) and analyzed for E3L expression. D, CD11c+ DC cells were prepared as in (C), and B220+ B cells and CD11b+ macrophages were enriched from CD11c− splenocytes of one day infected mice. RT-PCR was performed to detect E3L gene expression. Data are representative of at least 2 independent experiments.
To further confirm that SPDC were infected by VV in vivo, we analyzed mRNA levels of E3L. SPDC from VV- but not mock-infected mice expressed E3L mRNA (Figure 1C). To test whether other APC are also infected, we prepared B cells and macrophages by enriching CD11c−B220+ and CD11c−CD11b+ cells, respectively, and compared E3L mRNA levels among all three APC types. As shown in Figure 1D, both B cells and macrophages expressed E3L mRNA, albeit at lower levels than DC, suggesting that splenic APC including DC are infected by VV in vivo. While E3L mRNA was detected in splenic B cells and macrophages, little, if any, E3L protein was observed in these APC using E3L staining and flow cytometry (data not shown).
Splenic DC from VV-infected mice exhibit an activated phenotype yet display reduced MHC class II expression
It is well established that activated and thus matured DC are potent APC expressing elevated cell surface molecules, such as MHC class I and II and co-stimulatory molecules, as well as producing cytokines. Previously, it has been shown that DC infected with VV in vitro are compromised in their ability to undergo maturation in response to stimulation with TLR ligands [31; 39]. Therefore, we tested whether SPDC were capable of becoming activated after an in vivo VV infection of mice and in vitro exposure to LPS. We assessed the amount of cytokines produced by SPDC that were prepared from mock- and VV-infected mice followed by stimulation with LPS overnight. Unexpectedly, SPDC from VV-infected mice produced a greater amount of IL-10, IL-12, IL-6 and TNF-α than those from mock-infected mice (Figure 2A). Cytokine production peaked on day 1 post-infection, which correlated with the virus load in the DC populations. Thus, SPDC appear to be activated by a VV infection in vivo.
Figure 2. Activated phenotype of SPDC except MHC class II expression.
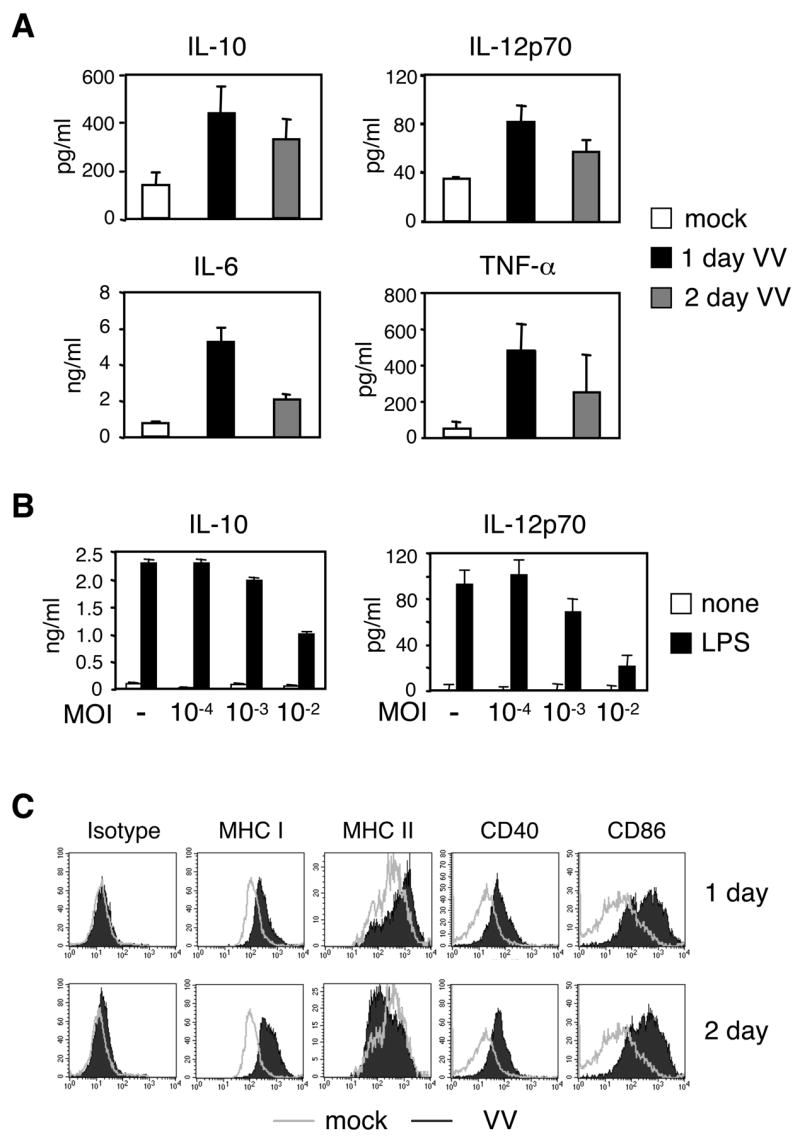
A, SPDC were prepared as in Figure 1 and stimulated with LPS (1 μg/ml) overnight. ELISA was performed to measure the levels of IL-10, IL-12p70, IL-6, and TNF-α. B, BMDC were infected in vitro with VV at the indicated MOI for 24 h followed by LPS stimulation overnight. IL-10 and IL-12p70 production was measured by ELISA. C, SPDC from mock- or VV-infected mice were used to determine cell surface expression levels of MHC and co-stimulatory molecules. DX5−CD11c+ or CD11chigh cells were gated as in Figure 3B and shown. Open and filled histograms are DC from mock- and VV-infected mice, respectively. Data are the mean ± SD (A and B) or representative (C) of at least 2 independent experiments.
The fact that SPDC from VV-infected mice produced more cytokines prompted us to examine the response of in vitro-infected DC. Since the infection efficiency in vivo appears to be equivalent to that of an in vitro infection at an MOI of 10−3–10−4 (Figure 1A), we infected BMDC with VV at various MOI and stimulated them with LPS. We observed that an in vitro infection with a low dose of VV did not inhibit the BMDC’s potential to produce IL-10 or IL-12 upon LPS stimulation (Figure 2B). However, unlike the SPDC infected in vivo that produced more cytokines in response to LPS, BMDC infected in vitro with a low dose of VV had a similar level of cytokine production as compared to that of mock-infected DC upon LPS exposure (Figure 2B).
Next, we examined the levels of MHC and co-stimulatory molecules. SPDC from VV-infected mice as early as one day following infection up-regulated MHC class I, MHC class II, CD40, and CD86 on their cell surface (Figure 2C, top panel). However, two days post-infection, the amount of MHC class II on the cell surface was decreased although the levels of MHC class I and co-stimulatory molecules remained elevated (Figure 2C, bottom panel).
A change in the splenic DC populations in VV-infected mice
Several DC subsets are present in the spleen and each subset is known to perform different functions [19]. Therefore, it is possible that the relative levels of the DC subpopulations could have been altered upon VV infection, which may have been responsible for their apparently activated phenotype. To assess the DC subset composition post-infection, total and CD11c+ enriched splenocytes were prepared from mock- or VV-infected mice and analyzed for the presence of different cell types.
The total number of splenocytes in mice was comparable with or without VV infection (data not shown). However, the proportion of CD4+ T cells was decreased gradually after VV infection, while CD8+ T cell levels remained relatively unchanged at these early stages of infection (Figure 3A). In addition, there was a small but significant increase in the percentage of B cells (B220+CD11c−) and macrophages (CD11b+CD11c−) two days after a VV infection (Figure 3A). The DX5+ natural killer (NK) cell and CD11c+ SPDC population were not altered (Figure 3A).
Figure 3. Changes in cell composition and DC subset in the spleen from VV-infected mice.
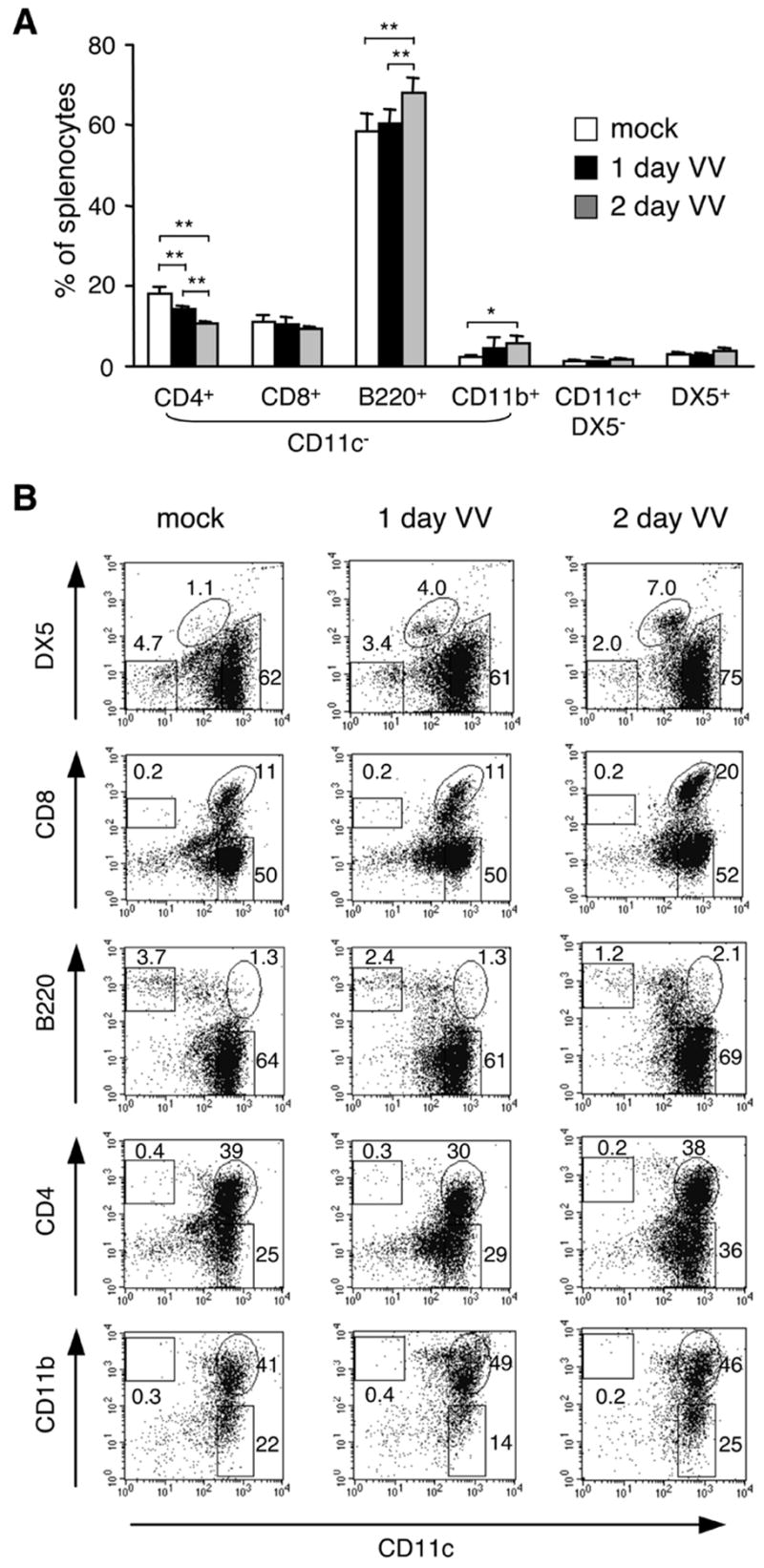
A, Total splenocytes from mock- or VV-infected mice were stained for cell surface markers and analyzed for different cell types. *p < 0.01, **p < 0.001. B, CD11c+ enriched splenocytes were analyzed for purity and DC subpopulations. Numbers indicate each subpopulation as a percentage of live cells. Note that DX5+ cells are CD11clow. Data are the mean ± SD (A) or representative (B) of at least 2 independent experiments.
We next analyzed CD11c+ cells in detail. It is known that NK cells express CD11c upon virus infection or activation [40; 41]. In agreement with these reports, CD11c+ NK cells increased in VV infected mice although the CD11c level of NK cells was lower than that of DC (Figure 3B). A similar observation was made in total splenocytes (data not shown), suggesting that these NK cells might have been activated upon VV infection. We also found that DC subsets expressing CD8 or B220 that are known to increase upon virus infection [25; 26] were indeed enhanced two days post-infection, while CD4+ or CD11b+ DC remained at a comparable level (Figure 3B).
Impaired APC function of DC after a VV infection
Reduced MHC class II on SPDC from VV-infected mice led us to test their potential to present antigens to CD4 T cells. To do this, CD11c+ SPDC were prepared from mock- or VV-infected mice, loaded with a different amount of HEL74–88 peptide, fixed, and used to stimulate HEL peptide-specific CD4 T cells. As expected, SPDC from mock-infected mice stimulated T cells in the presence of the peptide. However, DC from VV-infected mice showed a substantially impaired ability to present antigens to CD4 T cells as early as day 1 post-infection (Figure 4).
Figure 4. Impaired APC function of SPDC from VV-infected mice.
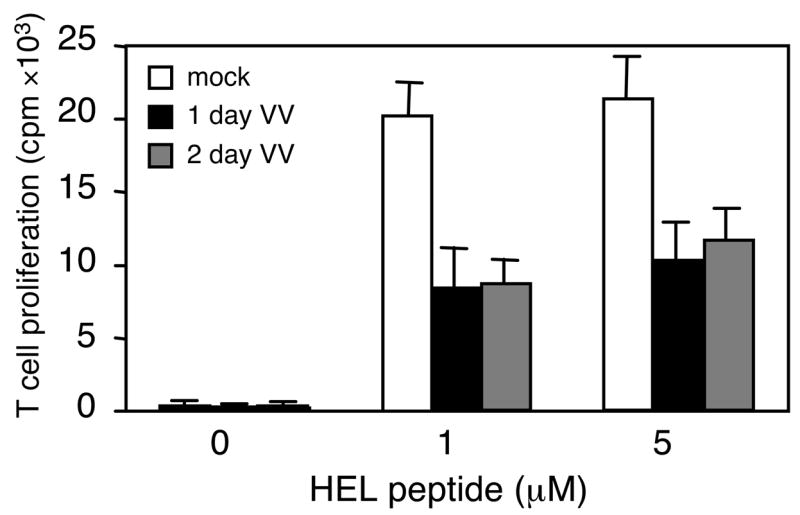
CD11c+ enriched splenic DC from mock- or VV-infected mice were incubated with increasing amounts of the HEL74–88 peptide for 16 h, followed by washing, fixation and cultivation with Ag-specific T cells for 24 h. T cell activation was assessed by measuring IL-2 production using HT-2 cell proliferation. Data are expressed as cpm and are representative of 3 independent experiments with the SD for triplicated samples indicated.
Role of type I IFN in MHC class II expression and viral replication in DC
So far, we have demonstrated that a VV infection appears to down-regulate the amount of MHC class II on the surface of SPDC and disrupts antigen presentation. To understand the mechanism responsible for these effects, we assessed the level of MHC class II mRNA in DC from VV-infected animals. As shown in Figure 5A, SPDC from VV-infected mice expressed less MHC class II mRNA compared to mock-infected mice, implying that the observed decrease in surface MHC class II after two days of infection could have resulted from reduced mRNA. In fact, MHC class II gene expression was gradually declined as the infection progressed during VV infection (Figure 5B). However, the level of MHC class II on the cell surface was increased despite the reduction of mRNA after one day of infection (Figure 2C). This is most likely due to the transport of pre-assembled MHC class II molecules from the endosomal compartments to the cell surface in activated DC [42]. Previous studies have shown that type I IFN can inhibit MHC class II expression in multiple cell types including macrophages [43; 44; 45; 46]. Virus infections are known to induce type I IFN gene expression, raising the possibility that the decline in MHC class II mRNA was linked to the action of type I IFN. Notably, IFN-β expression by SPDC was greatly elevated in SPDC from VV-infected mice (Figure 5A). The induction of IFN-β mRNA was accompanied by the expression of IRF-7, a critical transcription factor for type I IFN [47] (Figure 5A).
Figure 5. Role of type I IFN in MHC class II expression and viral replication in DC.

A, SPDC were enriched from mock- or VV-infected mice to prepare RNA. qRT-PCR was performed to assess MHC class II, IFN-β, and IRF-7 mRNA. The relative mRNA levels were normalized to GAPDH gene expression. B, MHC class II mRNA levels in SPDC enriched from mock- or VV-infected mice at indicated time points were measured by qRT-PCR as in (A). C. BMDC from WT or IFNAR−/− mice were infected with the mKO mutant VV (MOI of 0.01) for 90 min and cultured with or without IFN-β for 48 h. MHC class II mRNA levels were measured by qRT-PCR. D, BMDC were prepared, infected (MOI of 0.1 for 24 hrs and 0.01 for 48 hrs), and cultured as in (C) for 24 or 48 hrs. E3L and OFP expression were analyzed by flow cytometry. Numbers indicate each subpopulation as a percentage of live cells. E and F, BMDC from WT mice were infected with the mKO mutant VV (MOI of 0.1 and 0.01) for 24 hrs (indicated as 1° infection). Conditioned medium (CM) from infected BMDC were collected, filtered or unfiltered, and then added to uninfected BMDC. Cells were analyzed an additional 24 h post-infection. Efficiency of virus depletion was monitored by flow cytometry (E). MHC class II mRNA levels were measured by qRT-PCR (F). Data are the mean ± SD (A, B, C and F) or representative (D and E) of at least 2 independent experiments.
Having observed an inverse relationship between IFN-β and MHC class II gene expression in SPDC after VV infection, we next investigated a possible role for type I IFN in VV-mediated MHC class II mRNA down-regulation. BMDC from wild type and IFNAR−/− mice were mock- or VV-infected, or treated with IFN-β. MHC class II mRNA levels were then assessed. Consistent with the SPDC data from VV-infected mice, the level of MHC class II mRNA was reduced in BMDC by a VV infection in vitro (Figure 5C, left side). In addition, and consistent with a report showing that a viral infection switches non-pDC to IFN producers [48], VV-infected BMDC in vitro also expressed IFN-β, albeit at lower levels compared to SPDC (data not shown). However, IFN-β treatment alone enhanced MHC class II gene expression in BMDC (Figure 5C, left side). When BMDC were infected with VV and then cultured in the presence of IFN-β, they were still able to maintain MHC class II expression. Thus, IFN-β induces an increase in the MHC class II mRNA level in DC, and VV-mediated down-regulation of MHC class II expression is unlikely caused by IFN-β production by infected DC. In fact, the MHC class II mRNA level in IFNAR−/− DC was greatly reduced after VV infection even after receiving IFN-β (Figure 5C, right side).
It has been shown that IFNAR−/− mice are more susceptible to a VV infection than are wild type mice, although DC in these mice were not examined [32; 49]. Therefore, we investigated whether IFNAR−/− DC can control VV infection. To do this, we utilized a recombinant VV strain (mKO) that expresses OFP under the p7.5 early/late promoter of VV [32]. Infection with the mKO VV is comparable to that of wild type VV [32]. BMDC from WT and IFNAR−/− mice were infected with mKO and then cultured with or without IFN-β. As shown in Figure 5C, both WT and IFNAR−/− DC had an equivalent population expressing E3L regardless of the dose of VV or the duration of infection. However, compared to WT DC, IFNAR−/− DC displayed higher OFP positive cells, suggesting that type I IFN is necessary to limit viral replication in this cell population (Figure 5D). In fact, we observed that IFNAR−/− DC were more susceptible to cell death upon VV infection even at a very low dose of VV (data not shown). Although WT DC also underwent cell death after 2 days of infection in vitro, adding IFN-β to the culture prevented cell death and completely abolished virus replication in WT DC (Figure 5D, bottom panel). This effect was not observed in IFNAR−/− DC (Figure 5D, bottom panel).
We next sought whether the down-regulation of MHC class II by VV is a result of VV replication or an indirect effect exerted by host cell factors secreted to culture media. To test this, CM was collected from DC that were mock- or VV-infected at MOI of 0.1 or 0.01 for 24 hrs, filtered to remove virus particles, and then added to uninfected DC. As expected, unfiltered CM from VV-infected DC caused a secondary infection at a dose-dependent manner (Figure 5E). However, filtered and thus virus-free CM was not able to infect DC. Moreover, virus-free CM did not inhibit MHC class II expression, suggesting that the down-regulation of MHC class II expression is likely, at least in in vitro infected BMDC, caused by a VV gene product(s) not secreted host factors (Figure 5F).
DISCUSSION
In the present study, we have demonstrated that SPDC are activated following a VV infection in vivo, as evidenced by elevated levels of cell surface MHC class I and co-stimulatory molecules, and by the greater potential of these cells to produce cytokines. However, despite their activated phenotype, SPDC exposed to VV in vivo are compromised in MHC class II gene expression and antigen presentation by MHC class II molecules.
To our knowledge, this is the first study showing an effect of VV infection on MHC class II gene expression. We propose that there are at least two possible mechanisms responsible for impaired MHC class II APC function in DC following VV infection. First, VV may directly interfere with ligand binding to MHC class II molecules. As the experiments in the current study used antigenic peptides rather than native antigens, the disruption in MHC class II function is unlikely caused by a defect in antigen processing. This hypothesis would be consistent with our previous report that in vitro VV infection disrupts MHC class II-restricted Ag presentation [13]. Secondly, similar to other viruses, VV appears to down-regulate MHC class II gene expression by DC at a low MOI and 1–2 days exposure to virus [6; 50]. Although MHC class II levels were maintained on the cell surface, gene expression is reduced in DC after one day of VV infection (Figure 2C and 5A). The sustained level of surface MHC class II is likely attributed to the longer half-life of proteins than mRNA of MHC class II. Indeed, reduced amounts of MHC class II on the cell surface of DC were only observed after two days of infection.
Currently, it is not clear how MHC class II gene expression is down-regulated by VV. Studies here suggest a possibility that the down-regulation is not mediated by type I IFN, although it has been shown to inhibit IFN-γ-inducible MHC class II gene expression in macrophages, fibrosarcoma, and astrocytoma cell lines [43; 44; 45; 46]. Instead, MHC class II mRNA levels were induced by IFN-β in vitro (Figure 5B). This difference in IFN-β sensitivity could result from the different modes of MHC class II gene expression in DC as compared to these other cell types. DC express MHC class II constitutively, whereas macrophages, fibrosarcoma, and astrocytoma cell lines require an external stimulus, such as IFN-γ, to activate the MHC class II promoter. Regardless, our data using IFNAR−/− DC support the idea that VV-induced type I IFN production is not responsible for the MHC class II down-regulation.
On the other hand, the addition of exogenous IFN-β completely abolished virus replication and restored MHC class II gene expression. Moreover, unfiltered but not filtered CM from VV-infected BMDC caused secondary infection and MHC class II down-regulation. These data suggest that VV-encoded proteins either directly or indirectly, inhibit MHC class II gene expression in BMDC infected in vitro. In SPDC from in vivo infected mice, the down-regulation of MHC class II mRNA appears to be much greater than expected when compared with the relative level of virus detected in cells (Figure 1 and 5). Yet, viral infection in this case, was monitored by expression of the viral early gene product E3L, whose synthesis drops during the progression of infection. Thus, E3L protein levels may offer an underestimate of the amount of virus actually in cells. Some cells might have been infected with very low levels of VV, sufficient to decrease MHC class II gene expression but not detectable by E3L staining. Or, cells were infected but E3L expression was down-regulated during the infection. Lastly, during an in vivo infection, it is very likely that environmental host factors augment or promote down-regulation of MHC class II expression. Our in vitro infection data suggest that the DC-virus interactions lead to inhibition of MHC class II expression. Yet, we cannot rule out any bystander effect from secreted host cell factors in response to infection or stress, as factors driving the down-modulation of class II expression in in vivo infected DC. Future studies are warranted to dissect the specific mechanisms by which VV disrupts MHC class II gene expression. What critically important is our observation that while the virus can promote innate pathways for DC maturation, the loss of class II antigen presentation by these cells compromises or weakens, host protective immunity against VV. We also suspect that the enhanced susceptibility of IFNAR−/− mice to a VV infection is at least partly due to the limited capability of their DC to control virus replication. As the virus infection progresses in vivo, the viral load would increase likely causing more DC death. Consequently, the number of DC declines and T cell priming could thus be compromised.
Our data demonstrated that DC are activated during an in vivo infection, differing from studies suggesting in vitro the virus does not activate DC [30; 31; 39]. In concert with those reports, it has been reported that DC infected with a recombinant modified vaccinia Ankara (MVA) retain their immunogenicity in vivo despite in vitro dysfunction [51]. This is in contrast to another report suggesting that MVA can at least moderately activate human DC [52]. This inconsistency between in vitro and in vivo infection studies may be attributed to two factors: virus load and the DC subset infected. It is likely that low doses of VV activate DC, whereas a high virus load is detrimental. With i.p delivery of virus, the amount of VV entering to the spleen directly, or indirectly carried by infected cells, appears to be low based upon the detection of virus in splenocytes. Delivery of 106 pfu via the i.p route was equivalent to infecting cells with an MOI of 10−3–10−4 in vitro (Figure 1A). At this low dose of VV, DC functions were not compromised and activation was observed (Figure 2B). However, an infection with an MOI greater than 10−3 in vitro inhibited DC maturation (Figure 2B), consistent with published studies [13; 31]. Therefore, it seems that DC with the activated phenotype observed from in vivo infected VV mice are likely bystanders that were not directly infected by VV.
The other factor possibly influencing maturation is the subtype of the infected DC. DC resident in the spleen are comprised of at least two subsets: cDC and pDC. This is in contrast to DC prepared from BM or blood that are primarily mDC (19). pDC are known to produce type I IFN that can control subsequent virus replication and induce DC maturation [53; 54]. As we have shown (Figure 1A), SPDC were more resistant to VV infection in vitro than BMDC, likely due to the ability of DC to produce type I IFN. This idea is further supported by our data demonstrating that adding IFN-β to VV-infected BMDC in vitro completely abolished virus replication (Figure 5C). However, other factor(s), in addition to type I IFN, seem to be required for proper DC activation because IFN-β had no effect on the cytokine production potential of BMDC (data not shown). Moreover, virus-free CM from VV-infected BMDC did not alter the cytokine production by BMDC (data not shown). Together, the response of DC differs depending on the dose of virus, the subset of DC used, and other environmental factors. In this regard, it should be noted that lung DC from mice infected with VV two days previously by the intratracheal route also showed decreased antigen presentation potential (unpublished data). Although the type of DC in the lung and the levels of MHC class II mRNA still need to be analyzed in that model, it is clear that an in vivo VV infection appears to modulate antigen presentation function of DC regardless of the route of infection.
What would be the immunological significance of down-regulating antigen presentation by MHC class II during VV infection? Clinically, a transient decrease in human T cell proliferative responses has also been reported following immunization with the vaccinia vaccine [55]. Such suppressed immune responses could result in part from the decrease in DC’s potential to present antigens by MHC class II. With this, it would be important to determine when DC re-express MHC class II genes and recover antigen presentation function to initiate CD4 T cell activation.
In sum, we showed that select DC functions can be activated by VV infection yet antigen presentation by MHC class II is compromised by this virus. Further investigations are needed to provide insights into the molecular mechanisms of host pathogenesis by VV, and improved vaccine design for poxviruses.
Acknowledgments
We are grateful to Mr. Travis Dickendesher and Dr. Mary O’Riordan (U. Michigan, Ann Arbor, MI) for providing BM cells from IFNAR−/− mice. We also thank Brain McCarthy, Kristin Gillett, Beau Champ and Jeremy Eltz for technical assistance and Dr. Constantin T. Yiannoutsos for the statistical analysis.
This work was supported in part by National Institutes of Health grants AI056097 (to C-H.C, J.S.B, S-C.H, and R.R.B), and an American Heart Association postdoctoral fellowship award 0520111z (to Y.Y.).
Footnotes
The authors have no conflicting financial interests.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Moss B. Genetically engineered poxviruses for recombinant gene expression, vaccination, and safety. Proc Natl Acad Sci U S A. 1996;93:11341–8. doi: 10.1073/pnas.93.21.11341. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Wollenberg A, Engler R. Smallpox, vaccination and adverse reactions to smallpox vaccine. Curr Opin Allergy Clin Immunol. 2004;4:271–5. doi: 10.1097/01.all.0000136758.66442.28. [DOI] [PubMed] [Google Scholar]
- 3.Rosenthal SR, Merchlinsky M, Kleppinger C, Goldenthal KL. Developing new smallpox vaccines. Emerg Infect Dis. 2001;7:920–6. doi: 10.3201/eid0706.010602. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Smith GL, Symons JA, Khanna A, Vanderplasschen A, Alcami A. Vaccinia virus immune evasion. Immunol Rev. 1997;159:137–54. doi: 10.1111/j.1600-065x.1997.tb01012.x. [DOI] [PubMed] [Google Scholar]
- 5.McFadden G. Smallpox: an ancient disease enters the modern era of virogenomics. Proc Natl Acad Sci U S A. 2004;101:14994–5. doi: 10.1073/pnas.0406207101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Yewdell JW, Hill AB. Viral interference with antigen presentation. Nat Immunol. 2002;3:1019–25. doi: 10.1038/ni1102-1019. [DOI] [PubMed] [Google Scholar]
- 7.Brutkiewicz RR, Klaus SJ, Welsh RM. Window of vulnerability of vaccinia virus-infected cells to natural killer (NK) cell-mediated cytolysis correlates with enhanced NK cell triggering and is concomitant with a decrease in H-2 class I antigen expression. Nat Immun. 1992;11:203–14. [PubMed] [Google Scholar]
- 8.Boshkov LK, Macen JL, McFadden G. Virus-induced loss of class I MHC antigens from the surface of cells infected with myxoma virus and malignant rabbit fibroma virus. J Immunol. 1992;148:881–7. [PubMed] [Google Scholar]
- 9.Bennink JR, Yewdell JW. Recombinant vaccinia viruses as vectors for studying T lymphocyte specificity and function. Curr Top Microbiol Immunol. 1990;163:153–84. doi: 10.1007/978-3-642-75605-4_6. [DOI] [PubMed] [Google Scholar]
- 10.Townsend A, Bastin J, Gould K, Brownlee G, Andrew M, Coupar B, Boyle D, Chan S, Smith G. Defective presentation to class I-restricted cytotoxic T lymphocytes in vaccinia-infected cells is overcome by enhanced degradation of antigen. J Exp Med. 1988;168:1211–24. doi: 10.1084/jem.168.4.1211. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Xu R, Johnson AJ, Liggitt D, Bevan MJ. Cellular and humoral immunity against vaccinia virus infection of mice. J Immunol. 2004;172:6265–71. doi: 10.4049/jimmunol.172.10.6265. [DOI] [PubMed] [Google Scholar]
- 12.Battegay M, Bachmann MF, Burhkart C, Viville S, Benoist C, Mathis D, Hengartner H, Zinkernagel RM. Antiviral immune responses of mice lacking MHC class II or its associated invariant chain. Cell Immunol. 1996;167:115–21. doi: 10.1006/cimm.1996.0014. [DOI] [PubMed] [Google Scholar]
- 13.Li P, Wang N, Zhou D, Yee CS, Chang CH, Brutkiewicz RR, Blum JS. Disruption of MHC class II-restricted antigen presentation by vaccinia virus. J Immunol. 2005;175:6481–8. doi: 10.4049/jimmunol.175.10.6481. [DOI] [PubMed] [Google Scholar]
- 14.Lanzavecchia A, Sallusto F. Regulation of T cell immunity by dendritic cells. Cell. 2001;106:263–6. doi: 10.1016/s0092-8674(01)00455-x. [DOI] [PubMed] [Google Scholar]
- 15.Guermonprez P, Valladeau J, Zitvogel L, Thery C, Amigorena S. Antigen presentation and T cell stimulation by dendritic cells. Annu Rev Immunol. 2002;20:621–67. doi: 10.1146/annurev.immunol.20.100301.064828. [DOI] [PubMed] [Google Scholar]
- 16.Austyn JM. New insights into the mobilization and phagocytic activity of dendritic cells. J Exp Med. 1996;183:1287–92. doi: 10.1084/jem.183.4.1287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Norbury CC, Malide D, Gibbs JS, Bennink JR, Yewdell JW. Visualizing priming of virus-specific CD8+ T cells by infected dendritic cells in vivo. Nat Immunol. 2002;3:265–71. doi: 10.1038/ni762. [DOI] [PubMed] [Google Scholar]
- 18.Inaba K, Turley S, Iyoda T, Yamaide F, Shimoyama S, Reis e Sousa C, Germain RN, Mellman I, Steinman RM. The formation of immunogenic major histocompatibility complex class II-peptide ligands in lysosomal compartments of dendritic cells is regulated by inflammatory stimuli. J Exp Med. 2000;191:927–36. doi: 10.1084/jem.191.6.927. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Shortman K, Liu YJ. Mouse and human dendritic cell subtypes. Nat Rev Immunol. 2002;2:151–61. doi: 10.1038/nri746. [DOI] [PubMed] [Google Scholar]
- 20.Nakano H, Yanagita M, Gunn MD. CD11c(+)B220(+)Gr-1(+) cells in mouse lymph nodes and spleen display characteristics of plasmacytoid dendritic cells. J Exp Med. 2001;194:1171–8. doi: 10.1084/jem.194.8.1171. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Belz GT, Smith CM, Eichner D, Shortman K, Karupiah G, Carbone FR, Heath WR. Cutting edge: conventional CD8 alpha+ dendritic cells are generally involved in priming CTL immunity to viruses. J Immunol. 2004;172:1996–2000. doi: 10.4049/jimmunol.172.4.1996. [DOI] [PubMed] [Google Scholar]
- 22.Haeryfar SM. The importance of being a pDC in antiviral immunity: the IFN mission versus Ag presentation? Trends Immunol. 2005;26:311–7. doi: 10.1016/j.it.2005.04.002. [DOI] [PubMed] [Google Scholar]
- 23.Beignon AS, Skoberne M, Bhardwaj N. Type I interferons promote cross-priming: more functions for old cytokines. Nat Immunol. 2003;4:939–41. doi: 10.1038/ni1003-939. [DOI] [PubMed] [Google Scholar]
- 24.Zuniga EI, McGavern DB, Pruneda-Paz JL, Teng C, Oldstone MB. Bone marrow plasmacytoid dendritic cells can differentiate into myeloid dendritic cells upon virus infection. Nat Immunol. 2004;5:1227–34. doi: 10.1038/ni1136. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Dalod M, Hamilton T, Salomon R, Salazar-Mather TP, Henry SC, Hamilton JD, Biron CA. Dendritic cell responses to early murine cytomegalovirus infection: subset functional specialization and differential regulation by interferon alpha/beta. J Exp Med. 2003;197:885–98. doi: 10.1084/jem.20021522. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Moron G, Rueda P, Casal I, Leclerc C. CD8alpha- CD11b+ dendritic cells present exogenous virus-like particles to CD8+ T cells and subsequently express CD8alpha and CD205 molecules. J Exp Med. 2002;195:1233–45. doi: 10.1084/jem.20011930. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Alcami A, Smith GL. A soluble receptor for interleukin-1 beta encoded by vaccinia virus: a novel mechanism of virus modulation of the host response to infection. Cell. 1992;71:153–67. doi: 10.1016/0092-8674(92)90274-g. [DOI] [PubMed] [Google Scholar]
- 28.Smith GL. Vaccinia virus glycoproteins and immune evasion. The sixteenth Fleming Lecture. J Gen Virol. 1993;74(Pt 9):1725–40. doi: 10.1099/0022-1317-74-9-1725. [DOI] [PubMed] [Google Scholar]
- 29.Harte MT, Haga IR, Maloney G, Gray P, Reading PC, Bartlett NW, Smith GL, Bowie A, O’Neill LA. The poxvirus protein A52R targets Toll-like receptor signaling complexes to suppress host defense. J Exp Med. 2003;197:343–51. doi: 10.1084/jem.20021652. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Drillien R, Spehner D, Bohbot A, Hanau D. Vaccinia virus-related events and phenotypic changes after infection of dendritic cells derived from human monocytes. Virology. 2000;268:471–81. doi: 10.1006/viro.2000.0203. [DOI] [PubMed] [Google Scholar]
- 31.Engelmayer J, Larsson M, Subklewe M, Chahroudi A, Cox WI, Steinman RM, Bhardwaj N. Vaccinia virus inhibits the maturation of human dendritic cells: a novel mechanism of immune evasion. J Immunol. 1999;163:6762–8. [PubMed] [Google Scholar]
- 32.Luker KE, Hutchens M, Schultz T, Pekosz A, Luker GD. Bioluminescence imaging of vaccinia virus: effects of interferon on viral replication and spread. Virology. 2005;341:284–300. doi: 10.1016/j.virol.2005.06.049. [DOI] [PubMed] [Google Scholar]
- 33.Yao Y, Li W, Kaplan MH, Chang CH. Interleukin (IL)-4 inhibits IL-10 to promote IL-12 production by dendritic cells. J Exp Med. 2005;201:1899–903. doi: 10.1084/jem.20050324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Yuwen H, Cox JH, Yewdell JW, Bennink JR, Moss B. Nuclear localization of a double-stranded RNA-binding protein encoded by the vaccinia virus E3L gene. Virology. 1993;195:732–44. doi: 10.1006/viro.1993.1424. [DOI] [PubMed] [Google Scholar]
- 35.Gourley T, Roys S, Lukacs NW, Kunkel SL, Flavell RA, Chang CH. A novel role for the major histocompatibility complex class II transactivator CIITA in the repression of IL-4 production. Immunity. 1999;10:377–86. doi: 10.1016/s1074-7613(00)80037-0. [DOI] [PubMed] [Google Scholar]
- 36.Chang CH, Fontes JD, Peterlin M, Flavell RA. Class II transactivator (CIITA) is sufficient for the inducible expression of major histocompatibility complex class II genes. J Exp Med. 1994;180:1367–74. doi: 10.1084/jem.180.4.1367. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Yao Y, Xu Q, Kwon MJ, Matta R, Liu Y, Hong SC, Chang CH. ERK and p38 MAPK Signaling Pathways Negatively Regulate CIITA Gene Expression in Dendritic Cells and Macrophages. J Immunol. 2006;177:70–6. doi: 10.4049/jimmunol.177.1.70. [DOI] [PubMed] [Google Scholar]
- 38.Toshchakov V, Jones BW, Perera PY, Thomas K, Cody MJ, Zhang S, Williams BR, Major J, Hamilton TA, Fenton MJ, Vogel SN. TLR4, but not TLR2, mediates IFN-beta-induced STAT1alpha/beta-dependent gene expression in macrophages. Nat Immunol. 2002;3:392–8. doi: 10.1038/ni774. [DOI] [PubMed] [Google Scholar]
- 39.Jenne L, Hauser C, Arrighi JF, Saurat JH, Hugin AW. Poxvirus as a vector to transduce human dendritic cells for immunotherapy: abortive infection but reduced APC function. Gene Ther. 2000;7:1575–83. doi: 10.1038/sj.gt.3301287. [DOI] [PubMed] [Google Scholar]
- 40.Werfel T, Witter W, Gotze O. CD11b and CD11c antigens are rapidly increased on human natural killer cells upon activation. J Immunol. 1991;147:2423–7. [PubMed] [Google Scholar]
- 41.Lima M, Almeida J, dos Anjos Teixeira M, Queiros ML, Justica B, Orfao A. The “ex vivo” patterns of CD2/CD7, CD57/CD11c, CD38/CD11b, CD45RA/CD45RO, and CD11a/HLA-DR expression identify acute/early and chronic/late NK-cell activation states. Blood Cells Mol Dis. 2002;28:181–90. doi: 10.1006/bcmd.2002.0506. [DOI] [PubMed] [Google Scholar]
- 42.Turley SJ, Inaba K, Garrett WS, Ebersold M, Unternaehrer J, Steinman RM, Mellman I. Transport of peptide-MHC class II complexes in developing dendritic cells. Science. 2000;288:522–7. doi: 10.1126/science.288.5465.522. [DOI] [PubMed] [Google Scholar]
- 43.Ling PD, Warren MK, Vogel SN. Antagonistic effect of interferon-beta on the interferon-gamma-induced expression of Ia antigen in murine macrophages. J Immunol. 1985;135:1857–63. [PubMed] [Google Scholar]
- 44.Ransohoff RM, Devajyothi C, Estes ML, Babcock G, Rudick RA, Frohman EM, Barna BP. Interferon-beta specifically inhibits interferon-gamma-induced class II major histocompatibility complex gene transcription in a human astrocytoma cell line. J Neuroimmunol. 1991;33:103–12. doi: 10.1016/0165-5728(91)90054-b. [DOI] [PubMed] [Google Scholar]
- 45.Kato T, Kitaura M, Inaba K, Watanabe Y, Kawade Y, Muramatsu S. Suppression of macrophage Ia antigen expression by endogenous interferon-alpha/beta. J Interferon Res Spec No. 1992:29–41. doi: 10.1089/jir.1992.1992.29. [DOI] [PubMed] [Google Scholar]
- 46.Lu HT, Riley JL, Babcock GT, Huston M, Stark GR, Boss JM, Ransohoff RM. Interferon (IFN) beta acts downstream of IFN-gamma-induced class II transactivator messenger RNA accumulation to block major histocompatibility complex class II gene expression and requires the 48-kD DNA-binding protein, ISGF3-gamma. J Exp Med. 1995;182:1517–25. doi: 10.1084/jem.182.5.1517. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Honda K, Yanai H, Negishi H, Asagiri M, Sato M, Mizutani T, Shimada N, Ohba Y, Takaoka A, Yoshida N, Taniguchi T. IRF-7 is the master regulator of type-I interferon-dependent immune responses. Nature. 2005;434:772–7. doi: 10.1038/nature03464. [DOI] [PubMed] [Google Scholar]
- 48.Diebold SS, Montoya M, Unger H, Alexopoulou L, Roy P, Haswell LE, Al-Shamkhani A, Flavell R, Borrow P, Reis e Sousa C. Viral infection switches non-plasmacytoid dendritic cells into high interferon producers. Nature. 2003;424:324–8. doi: 10.1038/nature01783. [DOI] [PubMed] [Google Scholar]
- 49.van den Broek MF, Muller U, Huang S, Aguet M, Zinkernagel RM. Antiviral defense in mice lacking both alpha/beta and gamma interferon receptors. J Virol. 1995;69:4792–6. doi: 10.1128/jvi.69.8.4792-4796.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Cebulla CM, Miller DM, Zhang Y, Rahill BM, Zimmerman P, Robinson JM, Sedmak DD. Human cytomegalovirus disrupts constitutive MHC class II expression. J Immunol. 2002;169:167–76. doi: 10.4049/jimmunol.169.1.167. [DOI] [PubMed] [Google Scholar]
- 51.Behboudi S, Moore A, Gilbert SC, Nicoll CL, Hill AV. Dendritic cells infected by recombinant modified vaccinia virus Ankara retain immunogenicity in vivo despite in vitro dysfunction. Vaccine. 2004;22:4326–31. doi: 10.1016/j.vaccine.2004.04.029. [DOI] [PubMed] [Google Scholar]
- 52.Drillien R, Spehner D, Hanau D. Modified vaccinia virus Ankara induces moderate activation of human dendritic cells. J Gen Virol. 2004;85:2167–75. doi: 10.1099/vir.0.79998-0. [DOI] [PubMed] [Google Scholar]
- 53.Honda K, Sakaguchi S, Nakajima C, Watanabe A, Yanai H, Matsumoto M, Ohteki T, Kaisho T, Takaoka A, Akira S, Seya T, Taniguchi T. Selective contribution of IFN-alpha/beta signaling to the maturation of dendritic cells induced by double-stranded RNA or viral infection. Proc Natl Acad Sci U S A. 2003;100:10872–7. doi: 10.1073/pnas.1934678100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Le Bon A, Tough DF. Links between innate and adaptive immunity via type I interferon. Curr Opin Immunol. 2002;14:432–6. doi: 10.1016/s0952-7915(02)00354-0. [DOI] [PubMed] [Google Scholar]
- 55.Mathew A, Ennis FA, Rothman AL. Transient decreases in human T cell proliferative responses following vaccinia immunization. Clin Immunol. 2000;96:100–7. doi: 10.1006/clim.2000.4887. [DOI] [PubMed] [Google Scholar]