

Abstract
The V protein of parainfluenza virus 5 (PIV5) plays an important role in the evasion of host immune responses. The V protein blocks interferon (IFN) signaling in human cells by causing degradation of the STAT1 protein, a key component of IFN signaling, and blocks IFN-β production by preventing nuclear translocation of IRF3, a key transcription factor for activating IFN-β promoter. Interleukin-6 (IL-6), along with tumor necrosis factor (TNF)-α and IL-1β, are major pro-inflammatory cytokines that play important roles in clearing virus infection through inflammatory responses. Many viruses have developed strategies to block IL-6 expression. Wild type PIV5 infection induces little, if any, expression of cytokines such as IL-6 or TNF-α, whereas infection by a mutant PIV5 lacking the conserved C-terminal cysteine rich domain (rPIV5VΔC) induced high levels of IL-6 expression. Examination of mRNA levels of IL-6 indicated that the transcription activation of IL-6 played an important role in the increased IL-6 expression. Co-infection with wild type PIV5 prevented the activation of IL-6 transcription by rPIV5VΔC, and a plasmid encoding the full-length PIV5 V protein prevented the activation of IL-6 promoter-driven reporter gene expression by rPIV5VΔC, indicating that the V protein played a role in inhibiting IL-6 transcription. The activation of IL-6 was independent of IFN-β even though rPIV5VΔC-infected cells produced IFN-β. Using reporter gene assays and chromatin immuno-precipitation (ChIP), it was found that NF-κB played an important role in activating expression of IL-6. We have proposed a model of activating and inhibiting IL-6 transcription by PIV5.
Introduction
PIV5, formerly known as simian virus 5 (SV5) (Chatziandreou et al., 2004), is a prototypical member of the Rubulavirus genus of the family Paramyxoviridae, which includes many important human and animal pathogens such as mumps virus, respiratory syncytial virus (RSV), Newcastle Disease virus (NDV) and measles virus as well as emerging viruses such as Nipah and Hendra viruses (Lamb and Kolakofsky, 2001). Although PIV5 was originally isolated from cultured primary monkey cells, its likely natural host is the dog in which it causes kennel cough (McCandlish et al., 1978). PIV5 encodes eight viral proteins (Lamb and Kolakofsky, 2001). Nucleocapsid protein (NP), phosphoprotein (P) and large RNA polymerase (L) protein are important for transcription and replication of the viral RNA genome. The V protein plays important roles in viral pathogenesis (details will be discussed below). The fusion (F) protein, a glycoprotein, mediates both cell-to-cell and virus-to-cell fusion in a pH-independent manner that is essential for virus entry into cells. Hemagglutinin-neuraminidase (HN), another viral glycoprotein, is also involved in virus entry and release from the host cells. The matrix (M) protein plays an important role in virus assembly and budding (Schmitt, He, and Lamb, 1999; Schmitt et al., 2002). The small hydrophobic (SH) protein is a 44-residue hydrophobic integral membrane protein and is oriented in membranes with its N terminus in the cytoplasm (Hiebert, Richardson, and Lamb, 1988). Recombinant PIV5 without SH (rPIV5 SH) induces apoptosis in L929 cells through a tumor necrosis factor (TNF)-α mediated extrinsic apoptotic pathway (He et al., 1998; He et al., 2001; Lin et al., 2003). SH expressed alone can block TNF-α signaling in a reporter gene assay (Wilson et al., 2006).
PIV5 has seven genes but encodes eight viral proteins (Lamb and Kolakofsky, 2001). The V/P gene of PIV5 is transcribed into both the V mRNA and the P mRNA through a process of pseudo-templated addition of nucleotides, commonly called “RNA editing” (Thomas, Lamb, and Paterson, 1988). The V mRNA is made when the viral RNA polymerase faithfully transcribes the V/P gene. However, during transcription the viral RNA polymerase complex recognizes a specific RNA sequence in the V/P gene and occasionally inserts two non-templated G residues at the site to generate the P mRNA. As a result, the V/P gene is transcribed into two mRNAs at about the same level and translated into two proteins, V and P, which share identical N-termini but have different C-termini. The P protein is a co-factor of the viral RNA-dependent RNA polymerase complex which contains P and L. The V protein of PIV5, a component of PIV5 virions (~350 molecules per virion), is a multifunctional protein. The V protein C-terminal domain contains seven cysteine residues, resembling a zinc finger domain and binds atomic zinc (Liston and Briedis, 1994; Paterson et al., 1995; Steward et al., 1995; Thomas, Lamb, and Paterson, 1988). The V protein of PIV5 interacts with soluble NP (Randall and Bermingham, 1996) and the N-terminal domain of V binds RNA through a basic region (Lin, Paterson, and Lamb, 1997). The PIV5 V protein interacts with a cellular protein (DDB1), the 127 kD subunit of the damage-specific DNA-binding protein (DDB) that is involved in damaged DNA repair. This interaction requires the presence of the C-terminal domain of V protein (Lin and Lamb, 2000). Expression of the PIV5 V protein, via its C-terminal domain, slows down the cell cycle (Lin and Lamb, 2000). The V protein of PIV5 can cause degradation of STAT1 protein, an essential regulator of IFN signaling, through a proteasome mediated pathway in human cells but not in mouse cells (Didcock et al., 1999). It has been shown that V, DDB1, Cul4A, STAT1 and STAT2 form a complex which is essential for V-mediated STAT1 degradation, and V has an E3 ubiquitin ligase activity (Andrejeva et al., 2002; Parisien et al., 2002; Precious et al., 2005; Ulane and Horvath, 2002). In addition to preventing IFN signaling, V can inhibit IFN-β production through an IRF-3 dependent pathway (He et al., 2002; Poole et al., 2002). Recently, it has been shown that V interacts with MDA-5 and this interaction plays an important role in blocking activation of IFN-β production (Andrejeva et al., 2004). The V protein is also known to play an essential role in blocking apoptosis in virus-infected cells. While the exact mechanism is not clear, it is thought that V blocks ER-stress-induced apoptosis and this blockage function is independent of its involvement with IFN pathways (Sun et al., 2004). Besides playing roles in regulating host cell functions, V also regulates viral RNA synthesis (Lin et al., 2005).
IL-6, together with proinflammatory cytokines TNF-α and IL-1β, plays an essential role in acute inflammatory responses in infection and in tissue repair (Kishimoto, 2005). While IL-6 itself does not have any known anti-viral activity, IL-6 facilitates trafficking of leukocytes to sites of infection, leading to clearance. IL-6 also plays an essential role in eliciting adaptive immune responses to infection: IL-6 knockout mice are defective in the transition from innate to adaptive immunity (Jones, 2005). Virus infections are known to activate IL-6 expression. HHV-8 activates IL-6 expression via AP1 responsive element in the IL-6 promoter (An et al., 2002); HCV activates IL-6 expression through activation of toll-like receptor 4 (TLR4) expression (Machida et al., 2006). HCMV activates IL-6 at a transcriptional level as well as at a post-transcriptional level (Gealy et al., 2005). Interestingly, PIV5 infection does not induce expression of IL-6, indicating PIV5 may encode a protein preventing IL-6 production (Arimilli, Alexander-Miller, and Parks, 2006). In this work, IL-6 expression was not detected in wild type PIV5 infected cells but was readily detected in mutant virus rPIV5VΔC-infected cells, which lacks the conserved C-terminus of the V protein, suggesting that wild type PIV5 infection prevents production of IL-6, and the V protein, through its conserved C-terminus, plays an essential role in blocking the expression of IL-6. The mechanisms of activation of IL-6 expression by rPIV5VΔC and the inhibition of IL-6 expression by V have been investigated in this work.
RESULTS
IL-6 expression in rPIV5VΔC-infected cells
Previously, a large amount of IFN-β has been detected from cells infected by rPIV5VΔC, indicating the V protein plays an important role in blocking IFN-β production (He et al., 2002; Poole et al., 2002). Subsequent studies have demonstrated that the V protein blocks IFN-β expression through IRF3 and MDA5 (Andrejeva et al., 2004). To examine whether the V protein plays a role in regulating expression of other cytokines, expression levels of IL-6, TNF-α and IL-1β, which are major pro-inflammatory cytokines, were measured in mock, PIV5 or rPIV5VΔC-infected cell media using ELISA. Interestingly, increased production of IL-6 was observed from rPIV5VΔC-infected cells while no detectable amount of IL-6 was seen in the media of mock or PIV5-infected HeLa cells (Fig. 1A). No significant differences in growth were observed between PIV5 and rPIV5VΔC in HeLa cells during the same time period (Fig. 1B). No TNF-α or IL-1β was detected in virus or mock-infected cells (data not shown). Thus, rPIV5VΔC, which lacks the conserved C-terminus of the V protein, induced IL-6 expression in HeLa cells, suggesting the V protein played a role in preventing expression of IL-6.
Figure 1. Expression of IL-6 from virus-infected cells.

(A). Expression levels of IL-6 from virus-infected cells. HeLa cells were infected with mock, PIV5 or rPIV5VΔC at 5 MOI. At different time points after infection, media from the infected cells were collected and assayed for IL-6 using ELISA as described in the Materials and Methods. Error bars are standard deviation of mean (SEM). (B). Growth Rates of PIV5 and rPIV5VΔC in HeLa cells. HeLa cells were infected with PIV5 or rPIV5VΔC and collected at time points indicated. Titers of the viruses were determined using plaque assays.
Activation of IL-6 transcription by rPIV5VΔC
To investigate the mechanism of increased IL-6 production in rPIV5VΔC-infected cells, the levels of IL-6 mRNA were examined using quantitative RT-PCR (q-RT-PCR). Shown in Fig. 2A, an increase in the amount of IL-6 mRNA was detected in rPIV5VΔC-infected cells compared with mock or PIV5-infected cells, suggesting rPIV5VΔC activated IL-6 expression by increasing the amount of IL-6 mRNA. To further investigate the role of transcription in the activation of IL-6 by rPIV5VΔC, actinomycin D (ActD), an inhibitor of host cell transcription, was used. Because of ActD’s cytotoxicity to HeLa cells, A549 cells, a human lung carcinoma cells, were used for this experiment (Fig. 2B). As expected, higher levels of IL-6 mRNA were detected in rPIV5VΔC-infected A549 cells (no treatment or DMSO, a solvent used for ActD, treatment) (Fig. 2B), further confirming the activation of IL-6 by rPIV5VΔC in a different cell line, A549. Addition of ActD reduced the level of IL-6 mRNA in rPIV5VΔC-infected cells to that found in mock or PIV5-infected cells, indicating that the transcription activation of IL-6 by rPIV5VΔC played an important role in the increased expression of the IL-6 gene (Fig. 2B). To further confirm the role of transcription activation in activating expression of the IL-6 gene by rPIV5VΔC, a reporter gene system, which contains a firefly luciferase (F-Luc) gene under control of full-length human IL-6 promoter (p1168hIL6-F-Luc) (Fig. 2 and (Vanden Berghe et al., 1998)), was used. This plasmid was transfected into HeLa cells with a second plasmid, phTK-RL, which contains renilla luciferase (R-Luc) under control of HSV TK promoter (Promega, Inc., Madison, WI) as an indicator of transfection efficiency. The transfected cells were then infected with mock, PIV5 or rPIV5VΔC at one day after transfection. The cells were collected and assayed for dual luciferase activities. Increased reporter gene expression was observed in rPIV5VΔC-infected cells, suggesting that rPIV5VΔC infection activated transcription driven by IL-6 promoter (Fig. 2C). The lower induction of the gene system may be due to the low rate co-transfection/infection rate of PIV5-infected and transfected cells (less than 10%) as reported before (He et al., 2002). In addition, the reported system may be less sensitive. It was reported that TNF-α, a known IL-6 inducer, was able only to have three folds of the reporter gene induction using this system (Vanden Berghe et al., 1998).
Figure 2. Activation of IL-6 promoter by rPIV5VΔC.

(A) Levels of IL-6 mRNA in infected cells. HeLa cells were infected with mock, PIV5 or rPIV5VΔC at 5 MOI. At 36 hpi, the cells were collected and total RNAs were purified. The RNAs were used for quantitative real time RT-PCR (q-RT-PCR) as described in the Materials and Methods. The ratio of IL-6 mRNA to actin mRNA from mock-infected cells was set at 1. (B) Inhibition of IL-6 mRNA levels by Actinomycin D. A549 cells were infected as before and treated with DMSO, ActD (2.5 μg/ml), or nothing. The mRNA levels were examined as in (A) using qRT-PCR. (C) Activation of IL-6 promoter in a reporter gene assay. HeLa cells were transfected with p1168hIL-6-F-Luc, which contains a firefly luciferase (F-Luc) under control of full-length IL-6 promoter, and phTK-RL, which contains a renilla luciferase (R-Luc) under control of HSV TK promoter. The transfected cells were then infected with mock, PIV5 or rPIV5VΔC as before. At 1 to 2 dpi, the cells were collected and assayed for luciferase activities as described in the Materials and Methods. The luciferase activity is shown as a ratio of F-Luc to R-Luc. MK, mock infection. Error bars are standard deviation of mean (SEM).
Inhibition of IL-6 expression by V
To examine whether V can block the production of IL-6 in rPIV5VΔC-infected cells, the cells were co-infected with rPIV5VΔC and wild type PIV5, which expresses the full-length V protein. As shown in Fig. 3A, rPIV5VΔC infection caused an increase in the level of IL-6 mRNA and this increased level of IL-6 mRNA was reduced by wild type PIV5 co-infection, suggesting that the full-length V protein blocked activation of the IL-6 promoter in rPIV5VΔC-infected cells. Inhibition of the activation of IL-6 promoter in rPIV5VΔC-infected cells was also examined using the reporter gene system. An expression plasmid encoding the full-length V protein was transfected into the cells together with p1168hIL6-F-Luc and a transfection control plasmid. The cells were then infected with rPIV5VΔC. Transfection with the plasmid encoding V blocked the activation of IL6-promoter-driven reporter gene expression induced by rPIV5VΔC infection (Fig. 3B), indicating V inhibited the activation of IL-6 promoter by rPIV5VΔC.
Figure 3. Inhibition of activation of IL-6.

(A) Inhibition of IL-6 activation by rPIV5VΔC by co-infection with PIV5. HeLa cells were infected with mock, PIV5, rPIV5VΔC or various ratios of PIV5 to rPIV5VΔC. The levels of IL-6 mRNA were measured as in Fig. 2A. MOIs used in the experiment are indicated below the graph. (B) Inhibition of IL-6 activation by rPIV5VΔC by V. HeLa cells were transfected with p1168hIL6-F-Luc, phTK-RL and a plasmid encoding V or GFP as and then infected with viruses as in Fig. 2C. The luciferase activities were measured as described before (Fig. 2C). MK, mock infection. Error bars are standard deviation of mean (SEM).
Virus replication was required for activation of IL-6 expression
To examine whether virus replication is required for the activation of IL-6 transcription by rPIV5VΔC, the ability of UV-inactivated virus to induce IL-6 production was investigated. As shown in Fig. 4, UV-inactivated rPIV5VΔC did not cause an increase in IL-6 mRNA expression as measured using real time PCR, indicating virus replication was required for the activation of the IL-6 promoter by rPIV5VΔC.
Figure 4. Activation of IL-6 expression required live virus.
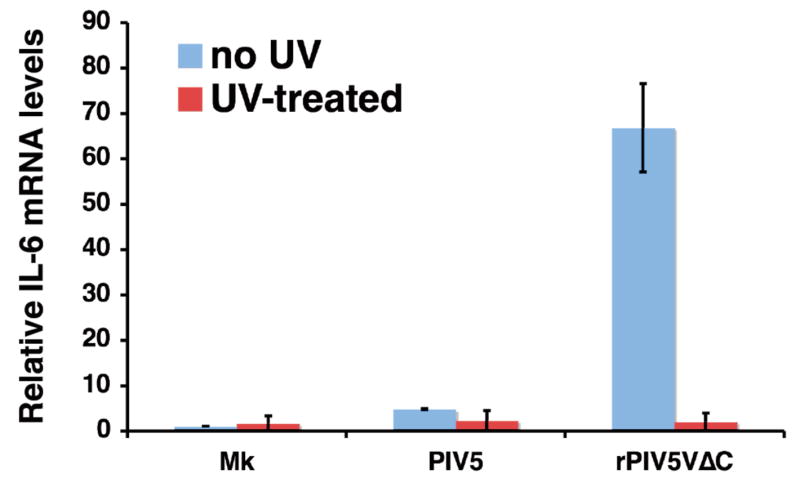
PIV5 or rPIV5VΔC were UV-inactivated as described before in Sun et al (Sun et al., 2004). HeLa cells were incubated with mock, PIV5, rPIV5VΔC or UV-inactivated viruses. The relative levels of IL-6 mRNA were measured using qRT-PCR as in Fig. 2A.
Activation of IL-6 expression was independent of IFN-β
It is known that rPIV5VΔC can activate expression of IFN-β (He et al., 2002; Poole et al., 2002). Thus, it is possible that expression of IL-6 is caused by IFN-β expression. To investigate this possibility, production of IL-6 in U3A cells, which are defective in STAT1 expression (McKendry et al., 1991), a vital transcriptional factor for IFN-β signaling, was examined. The production of IL-6 was enhanced by rPIV5VΔC infection compared with either mock or PIV5 infection in U3A cells (Fig. 5A), indicating that IL-6 expression in rPIV5VΔC-infected cells does not depend on STAT1-dependent IFN signaling, and that expression of IL-6 is likely independent of IFN. The effect of anti-IFN-β antibody on the activation of IL-6 was also examined. Even in the presence of a vast amount of anti-IFN-β antibody, IL-6 mRNA level was increased by rPIV5VΔC infection (Fig. 5B).
Figure 5. Activation of IL-6 was independent of IFN-β.

(A) Activation of IL-6 expression in the U3A cells. The U3A cells were infected with mock, PIV5 or rPIV5VΔC at 5 MOI as before. Media were collected from infected cells at time points indicated. Amount of IL-6 in the media were measured using ELISA. (B) Effect of anti-IFN-β on the activation of IL-6. HeLa cells were infected and treated with 1 μg/ml of anti-IFN-β antibody or equal amount of control antibody. The relative levels of IL-6 mRNA were measured using qRT-PCR. (C) Effect of IRF-3DN on the activation of IL-6. HeLa cells were transfected with plasmids encoding V, VΔC, P or IRF-3DN and p1168hIL-6-F-Luc and phTK-RL. The transfected cells were then infected with, mock, PIV5 or rPIV5VΔC. Dual luciferase assays were carried out as described in Materials and Methods. The relative luciferase activity indicates ratio of F-Luc to R-Luc. Error bars are standard deviation of mean (SEM).
It is known that IRF-3 plays an important role in activation of IFN-β production by rPIV5VΔC. To examine whether IRF-3 plays a role in rPIV5VΔC-activated IL-6 expression, a plasmid encoding a dominant negative mutant of IRF-3 (IRF-3DN) was used in the reporter gene assay. As shown in Fig. 5C, IRF-3DN did not block the activation of IL-6 promoter in rPIV5VΔC-infected cells (Fig. 5C), suggesting that IL-6 expression was independent of IFN-β.
The role of NF-κB in the activation of IL-6 promoter by rPIV5VΔC
The IL-6 promoter contains many binding sites for transcription factors including NF-κB. It is known that NF-κB plays an essential role in pro-inflammatory responses including the activation of IL-6 expression. To examine whether NF-κB played a role in the activation of IL-6 by rPIV5VΔC infection, a plasmid containing mutations in the NF-κB binding site was used in the reporter gene assay system. As shown in Fig. 6A, abolishing the NF-κB binding site within the IL-6 promoter abrogated the activation of IL-6 promoter by rPIV5VΔC, indicating that NF-κB played an essential role in the activation of IL-6 promoter in rPIV5VΔC-infected cells. There are five subunits of NF-κB factors. To investigate which NF-κB factor is responsible for the activation of IL-6 promoter, activation of NF-κB factors were examined using an ELISA-based assay as described in Fuentes et al (Fuentes et al., 2007). It was observed that rPIV5VΔC infection activated p65, p50 and p52, to a lesser extent. To further examine which NF-κB factor is involved in the activation of IL-6 expression in rPIV5VΔC, association of IL-6 promoter with individual NF-κB factors were examined using chromatin immuno-precipitation (ChIP). As shown in Fig. 6C, anti-p50 antibody precipitated the most IL-6 promoter, a finding that is consistent with the possibility that p50 played a role in the activation of IL-6 promoter by rPIV5VΔC. Interestingly, anti-p52 also precipitated a low level IL-6 promoter-containing DNA.
Figure 6. NF-κB played an essential role in the activation of IL-6.

(A) NF-κB was required for the activation of IL-6. A reporter gene assay similar to Fig. 2A was used. Besides using a plasmid containing a full-length human IL-6 promoter driven luciferase gene (p1168hIL-6-F-Luc, IL6P-Wt in figure), an identical plasmid except with two point mutations in the NF-κB binding site of the IL-6 promoter (pNF-κBmut-IL-6-F-Luc, IL6P-NF-κBmut in figure) or a plasmid containing only the TATA box of the IL-6 promoter (p50hIL-6-F-Luc, IL6P-deletion in figure) was used. (B) Activation of NF-κB factors. Nuclear extract from mock, PIV5 or rPIV5VΔC-infected HeLa cells were used in an ELISA-based NF-κB transcription assay from Active Motif (TransAM NF-κB family kit, Active Motif, Carlsbad, California) following manufacture’s instructions as described in the Materials and Methods. (C) ChIP of IL-6 promoter. HeLa cells were infected with rPIV5VΔC as before and the cells were collected for ChIP as described in the Materials and Methods.
Effect of V on TNF-α and LPS-induced NF-κB activation
To examine whether the V protein could block the activation of the IL-6 promoter by other stimuli other than virus infection such as LPS, an LPS-responsive system (HEK 293 cells containing TLR4) was employed. The V protein expression had no effect on LPS-activated IL-6 promoter-driven reporter gene expression, indicating that the V protein does not block LPS-activated IL-6 expression (Fig. 7A). To examine whether the V protein could block the activation of NF-κB factor by stimuli such as TNF-α, a reporter gene assay was used. The V protein had no effect on TNF-α-induced activation of NF-κB (Fig. 7B), in contrast to the SH protein of PIV5, which is known to inhibit TNF-α-mediated NF-κB activation (Wilson et al., 2006).
Figure 7. Effects of the V protein on IL-6 activation by LPS and NF-κB activation by TNF-α.

(A) Effect of the V protein on IL-6 activation by LPS. 293 cells stably expressing TLR4 were transfected with plasmids encoding V, P, SH or GFP and plasmids p1186hIL-6-F-Luc, phTK-RL and a plasmid encoding CD14. The transfected cells were then treated with LPS for 16 hours and assayed for luciferase activities as described in the Materials and Methods. (B) Effect of the V protein on NF-κB activation by TNF-α. L929 cells were transfected with plasmids encoding V, P, SH or GFP and plasmids pNF-κB-TATA-F-Luc and phTK-RL. The transfected cells were then treated with TNF-α for 16 hours and assayed for luciferase activities. The relative luciferase activity indicates ratio of F-Luc to R-Luc. Error bars are standard deviation of mean (SEM).
DISCUSSION
Here, we provide evidence that the PIV5 V protein prevents the induction of IL-6 expression in PIV5-infected cells, as IL-6 expression was induced on infection with a recombinant virus lacking the cysteine-rich C-terminus of V protein, but was not induced on infection with wt virus harboring full-length V protein. Previously, a mutant PIV5, CPI-, was reported to induce expression of IL-6 (Young, Dillon, and Parks, 2006). However, because mutations in the CPI- virus V/P gene are located in the shared region that encodes both V protein and P protein, it was not clear whether increased expression of IL-6 was caused by mutation to V or to P or to both. It was also unclear whether the increased IL-6 expression resulted from a loss of function of the V protein to inhibit IL-6 expression, or a gain of function by the mutant V protein or the mutant P protein to activate IL-6 expression. In this study, we used a mutant PIV5 that lacks the conserved C-terminus of the V protein, but has no changes to the amino acid sequence of P protein. This virus induced IL-6 expression. Thus, the increased expression of IL-6 in this case can be directly attributed to the V protein. To determine whether IL-6 expression is blocked by wt V protein or alternatively is induced by the truncated VΔC protein, we tested the ability of full-length V protein, expressed either through co-infection with wild type PIV5 or via an expression plasmid, to block production of IL-6 resulting from PIV5 VΔC infection. In both cases, the full-length V protein was effective in preventing IL-6 expression in PIV5 VΔC-infected cells, indicating that the V protein functions to block production of IL-6. Strahle et al have reported that different strains of Sendai virus have different abilities to induce IL-6 expression (Strahle et al., 2003). Interestingly, the main differences among these Sendai virus strains include changes in the V protein. Thus, it is possible that the V protein also plays a role in Sendai virus-induced IL-6 expression. It is tempting to speculate that all paramyxoviruses may have developed means to prevent IL-6 expression, and that the V proteins may play an important role in this process.
We hypothesize that one or more PIV5 proteins and/or the viral replication process activates IL-6 expression. While it is not known at present which viral protein activated IL-6 expression, or alternatively whether viral RNA synthesis activated IL-6 expression, it is clear NF-κB factor played an essential role in the activation of IL-6 expression because mutant IL-6 promoter without an NF-κB binding site was not activated by rPIV5VΔC infection. Nuclear factor κB (NF-κB) is a family of transcription factors that binds to a specific DNA sequence. NF-κB plays important roles in inflammatory and innate immune responses (Ghosh, May, and Kopp, 1998; Karin et al., 2002). The family of NF-κB includes NF-κB1 (p105), NF-κB2 (p100), Rel A (p65), Rel B and c-Rel which can be divided into two classes dependent on their transactivation properties and mode of synthesis. p65 has the strongest transactivation domain and p65 is responsible for most of NF-κB transcriptional activities. p65, Rel B and C-Rel are translated as mature forms and contain a Rel-homology domain (RHD) that is essential for dimerization and DNA binding at their N-termini and a transactivation domain at their C-termini. p105 and p100 that contain RHD domain at their N-termini and ankyrin repeats at their C-termini are precursors for p50 and p52 respectively. The precursors undergo ubiquitin-dependent proteolysis to remove the C-terminal domains to generate p50 and p52 which only have the RHD domain that enables them to dimerize and to bind DNA but lack the C-terminal transactivation domain. Interestingly, two structurally similar NF-κB factors, p50 and to a lesser extent, p52 were found to be associated with the IL-6 promoter in rPIV5VΔC-infected cells. Previously, it has been reported that p65 is activated in L929 cells infected with PIV5 lacking the SH gene (rPIV5 SH) (Lin et al., 2003). In this work, p50 of NF-κB was associated with the IL-6 promoter in rPIV5VΔC-infected HeLa cells, indicating that p50 of NF-κB played a role in activating IL-6 expression. It is not clear how p50 or p52 activates IL-6 transcription since neither has an activation domain. While NF-κB factors are required for activation of IL-6 expression, they may not be sufficient. It is known that different stimuli activate IL-6 expression differently through activation of different transcription factors. For instance, TNF-α activates IL-6 expression through activation of NF-κB factors and C/EBP factors (Vanden Berghe et al., 1999). IL-1 activates IL-6 expression through NF-IL6 factor (also known as C/EBP-β) (Akira et al., 1990). While mutations of NF-κB binding site in IL-6 promoter resulted in reduction of IL-6 promoter activity without reducing the basal expression level of the IL-6-driven reporter (Fig. 6A), further deletion of the promoter resulted in a severe reduction in IL-6 driven reporter gene expression, suggesting that promoter elements in addition to the NF-κB site are important for IL-6 expression. Previously, it was shown that rPIV5VΔC activates IFN-β expression as well as NF-κB factors (He et al., 2002; Poole et al., 2002). It is not surprising that activation of IL-6 and IFN-β occur in different ways. IRF3 plays an essential role in activating IFN-β expression but did not affect IL-6 expression. IFN-β promoter contains an IRF3 binding site while IL-6 promoter does not. Activation of IL-6 by rPIV5VΔC relied on p50 of NF-κB, but not p65, the strongest NF-κB factor. It is likely that other transcription factors are activated in rPIV5VΔC-infected cells, leading to an enhanced expression of IL-6. Further studies on the activation of NF-κB factors and their role in the production of IL-6 have the potential to provide new knowledge relevant to paramyxovirus pathogenesis.
Whereas the V protein blocked rPIV5VΔC-activated IL-6 expression, interestingly it had no effect on LPS-activated IL-6 production or TNF-α-activated NF-κB activity, indicating that the V protein blocked a virus-specific pathway leading to NF-κB activation and IL-6 expression. Many NF-κB target genes, including IL-6, are important for host defense against virus infection. Therefore, it is not surprising that many viruses have developed means to block NF-κB activation at various stages of NF-κB activation cascades (Tato and Hunter, 2002). Because the V protein did not affect the activation of IL-6 promoter by LPS, or the activation of NF-κB by TNF-α, we speculate that the V protein blocks a step in the pathway upstream of NF-κB activation. However, it is possible that the activation of NF-κB by LPS or TNF-α is different from that caused by rPIV5VΔC infection and that the V protein blocks a step downstream of NF-κB activation that is ciritical for virus-stimulated induction, but not for LPS or TNF-α-mediated induction.
MATERIALS AND METHODS
Plasmids
pIRF3-DN was generated from a plasmid encoding IRF3-GFP (a gift from Dr. Peter Howley and (Ronco et al., 1998)) by deleting N-terminal residues of IRF3 following description by Lin et al (Lin et al., 1999) using standard molecular cloning techniques. phTK-RL, which contains a modified renilla luciferase gene with potential enhancer elements changed within its coding sequence under control of TK promoter of HSV, was from Promega (Madison, WI). pNF-κB-TATA-F-Luc, which contains NF-κB binding sites and firefly luciferase gene, was described before by Sun et al (Sun et al., 1994). p1168hIL-6-F-Luc, which contains full-length human IL-6 promoter and reporter gene firefly luciferase under its control, p50hIL-6-F-Luc, which contains only the TATA box region of the IL-6 promoter, and phIL-6-NF-κBmut-F-Luc, which contains the full-length IL-6 promoter with two point mutations in the NF-κB binding site, were described by Vanden Berghe et al (Vanden Berghe et al., 1999; Vanden Berghe et al., 1998). pCD14, which expresses CD14, was described in Chow et al (Chow et al., 1999). Expression vector pCAGGS was first described by Niwa et al (Niwa, Yamamura, and Miyazaki, 1991). pCAGGS-V, pCAGGS-P and pCAGGS-GFP were described before (Lin et al., 1998; Lin et al., 2005; Waning et al., 2002). Plasmids were prepared using QiaGen Maxiprep kit according to manufacture’s protocol (Valencia, CA).
Viruses and Cells
The PIV5 lacking the conserved C-terminus of the V protein (rPIV5VΔC) was first described in He et al (He et al., 2002). The virus was amplified in Vero cells as described before (He et al., 2002). To ensure the validity of the virus used, RNA from the virus-infected cells were purified and viral genome sequence of the V/P gene was determined using RT-PCR sequencing. When the batch of virus gave rise to the expected viral genome sequence in infected cells, the virus was then used in the experiments. Wild type PIV5 was grown in MDBK cells as described before (Paterson and Lamb, 1993). Titers of the viruses were determined using plaque assay with BHK cells.
HeLa, A549, MDBK and U3A were maintained in Dulbecco’s modified Eagle’s medium (DMEM) (Invitrogen) with 10% fetal bovine serum (FBS), 100 I.U/ml Penicillin and 100 μg/ml Streptomycin at 37 °C with 5% CO2. BHK cells were maintained in the same media plus 10% tryptose phosphate. 293T-TLR4 cells were maintained in the same media as HeLa cells plus 0.4 μg/ml G418 (Genetecin, Invitrogen Inc., San Diego, CA) to maintain TLR4 expression as described in Chow et al (Chow et al., 1999). After infection, FBS concentration was reduced to 2%.
ELISA
Amount of IL-6 protein in media were measured using ELISA. 96-well plate was coated with reconstituted mouse anti-human IL-6 mAb (#MAB206, R&D Systems, Minneapolis, MN) at 500 μg/ml in buffer containing 0.1 M NaHCO3, pH 8.2 in a humidified container and incubated overnight at 4°C. The plate was washed 4 times with PBST (0.5% TWEEN-20 in phosphate-buffered saline, PBS, pH 7.0). The plate was then blocked with 200 μl of 1% bovine serum albumin (BSA) in PBS for at least 2 hours at room temperature and washed 3 times with PBST, pH 7.0. Samples, as well as standards and blanks in volume of 100 μl were added o the plate. IL-6 standards ranging from 10,000 to 9.8 pg/ml were obtained by 1:1 series dilution using recombinant human IL-6 (rh IL-6, #206-IL, R&D Systems). The plate was incubated inside a humidified container overnight at 4°C. The plate was then washed 4 times with PBST, pH 7.0 and the reconstituted biotinylated affinity purified goat anti-human IL-6 pAb (R&D Systems #BAF206) in Tris-buffered saline at 50 μg/ml in 1% BSA/PBS were added. The plate was then incubated for 2 hours at room temperature and washed 6 times with PBST, pH 7.0. Streptavidin peroxidase at 1 μg/ml in PBS were added to the plate and the plate was incubated for 30 minutes at room temperature. The plate was washed 6 times with PBST, pH 7.0 and 100 μl of the ABTS/peroxide substrate solution (150 mg of 2,2′-Azino-bis(3-ethylbenzthiazoline-6-sulfonic acid, Sigma) to 500 ml of 0.1 M citric acid and adjust pH to 4.35 with NaOH.) was added to each well. The plate was incubated for 60 to 90 minutes at room temperature in the dark and read at a wavelength of 405 nm using a plate reader.
Quantitative RT-PCR
Monolayer of HeLa or A549 cells in 6 cm dishes were washed with PBS and inoculated with mock, rPIV5 and rPIV5VΔC viruses in DMEM with 1% bovine serum albumin (BSA) at a multiplicity of infection (MOI) of 5 for 1 to 2 hours at 37 °C. Cells were then washed and incubated in DMEM with 2% FBS at 37 °C with 5% CO2. At indicated time points post infection, cells were lysed, total RNA extracted with RNeasy Mini Kit (Qiagen) and eluted in RNase-free H2O. RNA concentration was determined by using a spectraphotometer. 500 ng of total RNA for each sample was used for reverse transcription with random primers (Applied Biosystems) and Superscricpt III reverse transcriptase (Invitrogen) according to manufacture’s protocol. 8% of the cDNA from each sample was then used for real time PCR reaction. Real Time PCR was carried out on ABI 7300 Real Time PCR System using Taqman Universal PCR Master Mix (Applied Biosystems) and pre-validated Taqman Gene Expression Assays kit (Applied Biosystems), with human IL6 (Assay ID: Hs00174131_m1) as target gene and human beta actin as endogenous control (Applied Biosystems 4326315E). Each sample was done in triplicates. Results were analyzed with RQ study software (Applied Biosystems). The ΔΔCT method recommended by the manufacturer (Applied Biosystems) was used to compare the relative expression levels of IL-6 mRNA. Expression of IL6 gene was normalized based on the levels of mRNA for human beta actin. The normalized IL6 expression level of mock-infected cells was set as 1, to which the expression levels of samples were compared and then presented as fold changes.
UV-inactivation of the viruses was carried out as before (Lin et al., 2003; Sun et al., 2004). Viruses were placed in 3.5 cm dishes with lids removed inside a Fisher Hamilton Biological Safety cabinet Class II and were UV irradiated for 30 min before inoculation. Cells were then washed and incubated in the UV-inactivated media at 37 °C with 5% CO2. In samples that were further treated with other reagents, the DMEM with 2% FBS was supplemented with DMSO, or 2.5 μg/ml Actinomycin D (A. G. Scientific, San Diego, CA); 1 μg/ml normal goat IgG (Santa Cruz Biotechnology, Santa Cruz, CA), or 1 μg/ml anti-Interferon β antibody (Sigma, St. Louis, MO).
Dual luciferase reporter gene assay
HeLa cells were seeded in 96-well MicroClear plates (Greiner Bio-One) at about 1:3 dilution. Cells at about 80–90% confluence were transfected (about 16 to 21 h after passage). Opti-MEM medium (0.3 ml) were mixed with 12 μl Fugene 6 transfection reagent (Roche Diagnostics, Indianapolis, IN) at room temperature for five minutes. DNA was added to each mixture and incubated at room temperature for 15 minutes. Plasmid amounts were as follows: 0.06 μg phTK-RL with 4.68 μg p1168hIL-6-F-Luc, 4.68 μg p50hIL-6-F-Luc, or 4.68 μg phIL-6-NF-κBmut-F-Luc. To transfect cells, 12 μl mixture was added to each well of 24 wells in 96-well plate. For transfection with pCAGGS-V or pIRF3DN, amount of IL-6-reporter plasmid was reduced to accommodate the addition of pCAGGS-V or other plasmids. The ratio of pCAGGS-V or other plasmids expressing GFP or IRF3DN to p1168hIL6P-Luc was 3 to 1. For virus infection, media was aspirated 24h after transfection and then transfected cells were infected with mock, wild-type, or rPIV5VΔC virus in DMEM plus 1% BSA at 3 MOI for 1hr at 37°C plus 5% CO2. Media was then changed to DMEM plus 2% FBS, and incubation was continued at 37°C plus 5% CO2. Luciferase assays were carried out at 24 to 48 h after infection. Media was aspirated and cells were lysed in 30 μl 1x Passive Lysis Buffer (Promega, Madison, WI). Thirty microliters of Luciferase Assay Buffer II mixed with Luciferase Assay substrate (Promega) were added to each well, activity was measured using Veritas Microplate Luminometer.
NF-κB assays and ChIP
HeLa cells were infected at an MOI of 3. One-day post-infection cells were trypsonized and washed with PBS without calcium or magnesium, counted and 2×106 cells were used for the nuclear extraction. Cells were pelleted at 1,000 rpm for 5 minutes at 4 °C and resuspended in 400 μL of cold buffer A (10 mM HEPES, pH 7.9, 10 mM KCl, 0.1 mM EDTA, 0.1 mM EGTA, 1 mM DTT and 0.5 mM phenylmethylsulfonyl fluoride) and incubated for 15 minutes on ice. 25 μL of 10 % IGEPAL was added to the tubes, the cells were vortexed for 15 seconds and centrifuged at 1,000 rpm for 5 minutes at 4 °C. The pellet was washed with cold buffer A and finally resuspended in 100 μL of cold buffer C (20 mM HEPES, pH 7.9, 400 mM NaCl, 1 mM EDTA, 1 mM EGTA, 1mM DTT, 1mM phenylmethylsulfonyl fluoride). Nuclear extract was incubated at 4 °C over night and the debris was pelleted at 14, 000 rpm for 5 minutes. Supernatants were frozen at −80 °C until use. Amount of protein in the extract was measured using the BCA Protein Assay reagent kit from PIERCE (PIERCE, Rockford, IL) according to the manufacturer’s instructions. To examine activation of the p65, p50 and p52 subunits of NF-κB the Active Motif TransAM NF-κB family kit (Active Motif, California, Cat no. 43296) was used according to the manufacturer’s recommendations. One microgram of protein was added to the wells of a 96 well plate coated with immobilized oligonucleotide containing the NF-κB consensus site (5′-GGGACTTTCC-3′). After a one hour incubation period at room temperature the wells were washed 3 times with the washing buffer included on the kit and 100 μL of the provided anti-p65, anti-p50 or anti-p52 antibodies was added at a 1:1000 dilution. The plate was incubated at room temperature for 1 hour and the wells were washed 3 times. HRP-conjugated anti-rabbi t IgG was added at a 1:1000 dilution and incubated for 1 hour at room temperature. Cells were washed 4 times and developing solution was added, followed by the stop solution. The amount of p65, p50 or p52 activation was measured at 405 nm in a HTS 700 BioAssay reader (Perkin Elmer, Norwalk, CT).
To examine association of transcription factors NF-κB and IL-6 promoter, ChIP was used following standard ChIP procedure. HeLa cells (3×107) (in six T150 flasks) were infected by rPIV5VΔC at M.O.I of 5 for two hours. At 24 hpi, the cells were digested with Trypsin +EDTA for 10 min and then 20ml of 10% formaldehyde were added to each T150 to cross-link the cells at room temperature for 10 min. Glycine was added to make the final concentration at 0.1M and the cells were incubated at room temperature for 5 min. The cells were then collected, washed with PBS and then lysed with cell lysis buffer (5mM Pipes (KOH), pH 8.0/85mM KCl/0.5% NP-40) containing 1mM PMSF on ice for 10 min. To remove the supernatant, the cell lysate were spun at 5000 rpm for 5 min in a table top centrifuge. The pellet was resuspended in nuclear lysis buffer (50mM Tris, pH 8.1/10mM EDTA/1%SDS) and incubated on ice for 10 min. Chromatin was then sonicated to an average length of about 600 bp using a Branson Sonifier 250 at 3–4 output, 90% duty cycle, 5 second for six times while keeping samples on ice. After sonication, the samples were spun using a tabletop centrifuge at 14000rpm for 1 min to remove debris and then diluted in CHIP dilution buffer (0.01%SDS/1.1% Triton-X-100/1.2mM EDTA/16.7mM Tris, pH 8.1/167mM NaCl plus 1mM PMSF). The sample was then divided into seven fractions, one was saved for use as total input control and the others were incubated with 8μg of Rabbit IgG, anti-p65, anti-p50, anti-p52, Rel-B and C-Rel (Santa Cruz Biotechnology). At the same time Sepharose-A beads were added and incubated at 4°C overnight. The Sepharose-A beads were then washed with: Low salt wash buffer (0.1%SDS/1%Triton X-100/2mM EDTA, 20mM Tris, pH 8.1/150mM NaCl) once, High salt wash buffer (0.1%SDS/1% Triton-X-100, 2mM EDTA, 20mM Tris, pH 8.1, 500mM NaCl) once, and LiCl wash buffer (0.25M LiCl/1%NP-40/1% deoxycholate, 1mM EDTA/10mM Tris, pH 8.0) once. After washing, the beads were treated with 250 μl fresh-made elution buffer (1%SDS/0.1M NaHCO3) and incubated at 65°C for 15 min. 1 μl of RNase (10mg/ml) in 0.3M NaCl was added to the sample and the sample was incubated at 65°C for 4 hours to reverse cross-linking. Afterwards, 2 volumes of 100% ethanol were added to precipate the DNA overnight. The DNA was spun down, resuspended and treated with protease K at 45°C for 1 hour. DNA was then cleaned up using Qiagen PCR purification kit. To detect the presence of IL-6 promoter in the processed sample, PCR reactions were carried out using forward primer: 5′ gcgctagcctcaatgacgacctaag 3′; reverse primer: 5′ gtcctatatttattgggggttgag 3′, which are specific for human IL-6 promoter. PCR reactions were performed at 94°C for 45 seconds, 55°C for 45 seconds and 72°C for 45 seconds for 40 cycles. The PCR products were resolved in 1% agarose gel.
Acknowledgments
We thank Dr. Andrew Henderson’s lab for providing help with ChIP assay. We thank Dr. Shao-Cong Sun for providing pNF-κB-TATA-F-Luc. We thank Dr. Douglas T. Golenbock for providing 293T/TLR4 cells and CD14 plasmid and Dr. George Stark for providing U3A cells. We are grateful to Dr. Anthony Schmitt for critically reading the manuscript. We appreciate other members of Biao He’s lab for discussion and technical help. The service provided by the General Clinical Research Center of the Pennsylvania State University are appreciated and the center was partially supported by NIH grant M01 RR 10732. The work was supported by a grant from the National Institute of Allergy and Infectious Disease to B.H. (R01 AI 051372).
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
REFERENCES CITED
- Akira S, Isshiki H, Sugita T, Tanabe O, Kinoshita S, Nishio Y, Nakajima T, Hirano T, Kishimoto T. A nuclear factor for IL-6 expression (NF-IL6) is a member of a C/EBP family. Embo J. 1990;9(6):1897–906. doi: 10.1002/j.1460-2075.1990.tb08316.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- An J, Lichtenstein AK, Brent G, Rettig MB. The Kaposi sarcoma-associated herpesvirus (KSHV) induces cellular interleukin 6 expression: role of the KSHV latency-associated nuclear antigen and the AP1 response element. Blood. 2002;99(2):649–54. doi: 10.1182/blood.v99.2.649. [DOI] [PubMed] [Google Scholar]
- Andrejeva J, Childs KS, Young DF, Carlos TS, Stock N, Goodbourn S, Randall RE. The V proteins of paramyxoviruses bind the IFN-inducible RNA helicase, mda-5, and inhibit its activation of the IFN-{beta} promoter. Proc Natl Acad Sci U S A. 2004;101(49):17264–17269. doi: 10.1073/pnas.0407639101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Andrejeva J, Poole E, Young DF, Goodbourn S, Randall RE. The p127 subunit (DDB1) of the UV-DNA damage repair binding protein is essential for the targeted degradation of STAT1 by the V protein of the paramyxovirus simian virus 5. J Virol. 2002;76(22):11379–86. doi: 10.1128/JVI.76.22.11379-11386.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Arimilli S, Alexander-Miller MA, Parks GD. A simian virus 5 (SV5) P/V mutant is less cytopathic than wild-type SV5 in human dendritic cells and is a more effective activator of dendritic cell maturation and function. J Virol. 2006;80(7):3416–27. doi: 10.1128/JVI.80.7.3416-3427.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chatziandreou N, Stock N, Young D, Andrejeva J, Hagmaier K, McGeoch DJ, Randall RE. Relationships and host range of human, canine, simian and porcine isolates of simian virus 5 (parainfluenza virus 5) J Gen Virol. 2004;85(Pt 10):3007–16. doi: 10.1099/vir.0.80200-0. [DOI] [PubMed] [Google Scholar]
- Chow JC, Young DW, Golenbock DT, Christ WJ, Gusovsky F. Toll-like receptor-4 mediates lipopolysaccharide-induced signal transduction. J Biol Chem. 1999;274(16):10689–92. doi: 10.1074/jbc.274.16.10689. [DOI] [PubMed] [Google Scholar]
- Didcock L, Young DF, Goodbourn S, Randall RE. The V protein of simian virus 5 inhibits interferon signalling by targeting STAT1 for proteasome-mediated degradation. J Virol. 1999;73:9928–9933. doi: 10.1128/jvi.73.12.9928-9933.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fuentes S, Tran KC, Luthra P, Teng MN, He B. Function of the Respiratory Syncytial Virus small hydrophobic protein. J Virol. 2007 doi: 10.1128/JVI.02717-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gealy C, Denson M, Humphreys C, McSharry B, Wilkinson G, Caswell R. Posttranscriptional suppression of interleukin-6 production by human cytomegalovirus. J Virol. 2005;79(1):472–85. doi: 10.1128/JVI.79.1.472-485.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ghosh S, May MJ, Kopp EB. NF-kappa B and Rel proteins: evolutionarily conserved mediators of immune responses. Annu Rev Immunol. 1998;16:225–60. doi: 10.1146/annurev.immunol.16.1.225. [DOI] [PubMed] [Google Scholar]
- He B, Leser GP, Paterson RG, Lamb RA. The paramyxovirus SV5 small hydrophobic (SH) protein is not essential for virus growth in tissue culture cells. Virology. 1998;250:30–40. doi: 10.1006/viro.1998.9354. [DOI] [PubMed] [Google Scholar]
- He B, Lin GY, Durbin JE, Durbin RK, Lamb RA. The sh integral membrane protein of the paramyxovirus simian virus 5 is required to block apoptosis in mdbk cells. J Virol. 2001;75(9):4068–79. doi: 10.1128/JVI.75.9.4068-4079.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- He B, Paterson RG, Stock N, Durbin JE, Durbin RK, Goodbourn S, Randall RE, Lamb RA. Recovery of paramyxovirus simian virus 5 with a V protein lacking the conserved cysteine-rich domain: the multifunctional V protein blocks both interferon-beta induction and interferon signaling. Virology. 2002;303(1):15–32. doi: 10.1006/viro.2002.1738. [DOI] [PubMed] [Google Scholar]
- Hiebert SW, Richardson CD, Lamb RA. Cell surface expression and orientation in membranes of the 44 amino acid SH protein of simian virus 5. J Virol. 1988;62:2347–2357. doi: 10.1128/jvi.62.7.2347-2357.1988. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jones SA. Directing transition from innate to acquired immunity: defining a role for IL-6. J Immunol. 2005;175(6):3463–8. doi: 10.4049/jimmunol.175.6.3463. [DOI] [PubMed] [Google Scholar]
- Karin M, Cao Y, Greten FR, Li ZW. NF-kappaB in cancer: from innocent bystander to major culprit. Nat Rev Cancer. 2002;2(4):301–10. doi: 10.1038/nrc780. [DOI] [PubMed] [Google Scholar]
- Kishimoto T. IL-6: from laboratory to bedside. Clin Rev Allergy Immunol. 2005;28(3):177–86. doi: 10.1385/CRIAI:28:3:177. [DOI] [PubMed] [Google Scholar]
- Lamb RA, Kolakofsky D. Paramyxoviridae: The viruses and their replication. In: Knipe DM, Howley PM, editors. Fields Virology . 4. Lippincott, Williams and Wilkins; Philadelphia: 2001. [Google Scholar]
- Lin GY, Lamb RA. The paramyxovirus simian virus 5 V protein slows progression of the cell cycle. J Virol. 2000;74(19):9152–66. doi: 10.1128/jvi.74.19.9152-9166.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lin GY, Paterson RG, Lamb RA. The RNA binding region of the paramyxovirus SV5 V and P proteins. Virology. 1997;238:460–469. doi: 10.1006/viro.1997.8866. [DOI] [PubMed] [Google Scholar]
- Lin GY, Paterson RG, Richardson CD, Lamb RA. The V protein of the paramyxovirus SV5 interacts with damage-specific DNA binding protein. Virology. 1998;249(1):189–200. doi: 10.1006/viro.1998.9317. [DOI] [PubMed] [Google Scholar]
- Lin R, Heylbroeck C, Genin P, Pitha PM, Hiscott J. Essential role of interferon regulatory factor 3 in direct activation of RANTES chemokine transcription. Mol Cell Biol. 1999;19(2):959–66. doi: 10.1128/mcb.19.2.959. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lin Y, Bright AC, Rothermel TA, He B. Induction of apoptosis by paramyxovirus simian virus 5 lacking a small hydrophobic gene. J Virol. 2003;77(6):3371–83. doi: 10.1128/JVI.77.6.3371-3383.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lin Y, Horvath F, Aligo JA, Wilson R, He B. The role of simian virus 5 V protein on viral RNA synthesis. Virology. 2005;338(2):270–80. doi: 10.1016/j.virol.2005.05.014. [DOI] [PubMed] [Google Scholar]
- Liston P, Briedis DJ. Measles virus V protein binds zinc. Virology. 1994;198:399–404. doi: 10.1006/viro.1994.1050. [DOI] [PubMed] [Google Scholar]
- Machida K, Cheng KT, Sung VM, Levine AM, Foung S, Lai MM. Hepatitis C virus induces toll-like receptor 4 expression, leading to enhanced production of beta interferon and interleukin-6. J Virol. 2006;80(2):866–74. doi: 10.1128/JVI.80.2.866-874.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McCandlish IA, Thompson H, Cornwell HJ, Wright NG. A study of dogs with kennel cough. Vet Rec. 1978;102(14):293–301. doi: 10.1136/vr.102.14.293. [DOI] [PubMed] [Google Scholar]
- McKendry R, John J, Flavell D, Muller M, Kerr IM, Stark GR. High-frequency mutagenesis of human cells and characterization of a mutant unresponsive to both alpha and gamma interferons. Proc Natl Acad Sci U S A. 1991;88(24):11455–9. doi: 10.1073/pnas.88.24.11455. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Niwa H, Yamamura K, Miyazaki J. Efficient selection for high-expression transfectants by a novel eukaryotic vector. Gene. 1991;108:193–200. doi: 10.1016/0378-1119(91)90434-d. [DOI] [PubMed] [Google Scholar]
- Parisien JP, Lau JF, Rodriguez JJ, Ulane CM, Horvath CM. Selective STAT protein degradation induced by paramyxoviruses requires both STAT1 and STAT2 but is independent of alpha/beta interferon signal transduction. J Virol. 2002;76(9):4190–8. doi: 10.1128/JVI.76.9.4190-4198.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Paterson RG, Lamb RA. The molecular biology of influenza viruses and paramyxoviruses. In: Davidson A, Elliott RM, editors. Molecular Virology: A Practical Approach. IRL Oxford University Press; Oxford: 1993. pp. 35–73. [Google Scholar]
- Paterson RG, Leser GP, Shaughnessy MA, Lamb RA. The paramyxovirus SV5 V protein binds two atoms of zinc and is a structural component of virions. Virology. 1995;208:121–131. doi: 10.1006/viro.1995.1135. [DOI] [PubMed] [Google Scholar]
- Poole E, He B, Lamb RA, Randall RE, Goodbourn S. The V proteins of simian virus 5 and other paramyxoviruses inhibit induction of interferon-beta. Virology. 2002;303(1):33–46. doi: 10.1006/viro.2002.1737. [DOI] [PubMed] [Google Scholar]
- Precious B, Childs K, Fitzpatrick-Swallow V, Goodbourn S, Randall RE. Simian virus 5 V protein acts as an adaptor, linking DDB1 to STAT2, to facilitate the ubiquitination of STAT1. J Virol. 2005;79(21):13434–41. doi: 10.1128/JVI.79.21.13434-13441.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Randall RE, Bermingham A. NP:P and NP:V interactions of the paramyxovirus simian virus 5 examined using a novel protein:protein capture assay. Virology. 1996;224:121–129. doi: 10.1006/viro.1996.0513. [DOI] [PubMed] [Google Scholar]
- Ronco LV, Karpova AY, Vidal M, Howley PM. Human papillomavirus 16 E6 oncoprotein binds to interferon regulatory factor-3 and inhibits its transcriptional activity. Genes Dev. 1998;12(13):2061–72. doi: 10.1101/gad.12.13.2061. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schmitt AP, He B, Lamb RA. Involvement of the cytoplasmic domain of the hemagglutinin-neuraminidase protein in assembly of the paramyxovirus simian virus 5. J Virol. 1999;73:8703–8712. doi: 10.1128/jvi.73.10.8703-8712.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schmitt AP, Leser GP, Waning DL, Lamb RA. Requirements for budding of paramyxovirus simian virus 5 virus-like particles. J Virol. 2002;76(8):3952–64. doi: 10.1128/JVI.76.8.3952-3964.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Steward M, Samson ACR, Errington W, Emmerson PT. The Newcastle disease virus V protein binds zinc. Arch Virol. 1995;140:1321–1328. doi: 10.1007/BF01322759. [DOI] [PubMed] [Google Scholar]
- Strahle L, Garcin D, Le Mercier P, Schlaak JF, Kolakofsky D. Sendai virus targets inflammatory responses, as well as the interferon-induced antiviral state, in a multifaceted manner. J Virol. 2003;77(14):7903–13. doi: 10.1128/JVI.77.14.7903-7913.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sun M, Rothermel TA, Shuman L, Aligo JA, Xu S, Lin Y, Lamb RA, He B. Conserved cysteine-rich domain of paramyxovirus simian virus 5 V protein plays an important role in blocking apoptosis. J Virol. 2004;78(10):5068–78. doi: 10.1128/JVI.78.10.5068-5078.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sun SC, Elwood J, Beraud C, Greene WC. Human T-cell leukemia virus type I Tax activation of NF-kappa B/Rel involves phosphorylation and degradation of I kappa B alpha and RelA (p65)-mediated induction of the c-rel gene. Mol Cell Biol. 1994;14(11):7377–84. doi: 10.1128/mcb.14.11.7377. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tato CM, Hunter CA. Host-pathogen interactions: subversion and utilization of the NF-kappa B pathway during infection. Infect Immun. 2002;70(7):3311–7. doi: 10.1128/IAI.70.7.3311-3317.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thomas SM, Lamb RA, Paterson RG. Two mRNAs that differ by two nontemplated nucleotides encode the amino coterminal proteins P and V of the paramyxovirus SV5. Cell. 1988;54:891–902. doi: 10.1016/S0092-8674(88)91285-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ulane CM, Horvath CM. Paramyxoviruses SV5 and HPIV2 assemble STAT protein ubiquitin ligase complexes from cellular components. Virology. 2002;304(2):160–6. doi: 10.1006/viro.2002.1773. [DOI] [PubMed] [Google Scholar]
- Vanden Berghe W, De Bosscher K, Boone E, Plaisance S, Haegeman G. The nuclear factor-kappaB engages CBP/p300 and histone acetyltransferase activity for transcriptional activation of the interleukin-6 gene promoter. J Biol Chem. 1999;274(45):32091–8. doi: 10.1074/jbc.274.45.32091. [DOI] [PubMed] [Google Scholar]
- Vanden Berghe W, Plaisance S, Boone E, De Bosscher K, Schmitz ML, Fiers W, Haegeman G. p38 and extracellular signal-regulated kinase mitogen-activated protein kinase pathways are required for nuclear factor-kappaB p65 transactivation mediated by tumor necrosis factor. J Biol Chem. 1998;273(6):3285–90. doi: 10.1074/jbc.273.6.3285. [DOI] [PubMed] [Google Scholar]
- Waning DL, Schmitt AP, Leser GP, Lamb RA. Roles for the cytoplasmic tails of the fusion and hemagglutinin-neuraminidase proteins in budding of the paramyxovirus simian virus 5. J Virol. 2002;76(18):9284–97. doi: 10.1128/JVI.76.18.9284-9297.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wilson RL, Fuentes SM, Wang P, Taddeo EC, Klatt A, Henderson AJ, He B. Function of Small Hydrophobic Proteins of Paramyxovirus. J Virol. 2006;80(4) doi: 10.1128/JVI.80.4.1700-1709.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Young VA, Dillon PJ, Parks GD. Variants of the paramyxovirus Simian virus 5 with accelerated or delayed viral gene expression activate proinflammatory cytokine synthesis. Virology. 2006;350(1):90–102. doi: 10.1016/j.virol.2006.01.006. [DOI] [PubMed] [Google Scholar]