

Abstract
Background & Aims
The endogenous opioid system is involved in modulating the experience of pain, the response to stress and the action of analgesic therapies. Recent human imaging studies have demonstrated a significant tonic modulation of visceral pain, raising the question of whether endogenous opioids tonically modulate the pain of visceral cancer.
Methods
Transgenic mice expressing the first 127 amino acids of simian virus 40 large T antigen, under the control of the rat elastase-1 promoter that spontaneously develop pancreatic cancer were used to investigate the role of endogenous opioids in the modulation of pancreatic cancer pain. Visceral pain behaviors were assessed as degree of hunching and vocalization.
Results
Whereas, mice with late stage pancreatic cancer displayed spontaneous, morphine-reversible, visceral pain-related behaviors such as hunching and vocalization, these behaviors were absent in mice with early stage pancreatic cancer. Following systemic administration of the central nervous system (CNS) penetrant opioid receptor antagonists naloxone or naltrexone, mice with early stage pancreatic cancer, displayed significant visceral pain-related behaviors, while systemic administration of the CNS non-penetrant opioid antagonist naloxone-methiodide did not induce an increase in visceral pain behaviors.
Conclusions
Our findings suggest that a CNS opioid-dependent mechanism tonically modulates early and late stage pancreatic cancer pain. Understanding the mechanisms that mask this pain in early stage disease and drive this pain in late stage disease may allow improved diagnosis, treatment, and care of patients with pancreatic cancer.
Keywords: pain inhibition, visceral pain behavior, opioid antagonists, naloxone, opioid receptors, descending modulation
Introduction
Spinal cord excitability is directly influenced by descending inputs originating in higher brain centers and this descending modulation can be inhibitory or facilitatory in nature1, 2. The ability of higher brain centers to modulate the transmission of nociceptive information in the central nervous system (CNS) was demonstrated by Sherrington (1906) who showed that somatic nociceptive reflexes were enhanced following spinal cord transection. Over the last several decades evidence has accumulated that a variety of brain regions (including thalamus, amygdala, hypothalamus, periaqueductal grey (PAG) and raphe magnus) are involved in this descending modulation1, 3. This descending inhibitory system can be remarkably effective at modulating the perception of nociceptive stimuli; for example, stimulation of the rat PAG allowed abdominal surgery to be conducted without the use of general anesthesia4. In humans, stimulation of the PAG has also been shown to result in significant pain relief, and this stimulation-produced analgesia is antagonized by the opioid receptor antagonist naloxone5.
Although most of early studies on the circuitry and neurochemistry of the descending modulation of nociceptive inputs focused on somatic pain, there is evidence that a similar descending modulation is also present for visceral pain6-8. For example, a recent study using functional magnetic resonance to image the neural correlates of visceral and somatic pain in healthy human subjects concluded that while noxious somatic or visceral stimulations resulted in significant activation in the PAG and nucleus cuneiformis (NCF), significantly greater activation of both areas was observed in the visceral pain group than somatic pain group7. These and other data3, 9-11 suggest that there is significant tonic modulation of visceral pain and that activation of specific cortical and subcortical areas can produce a marked reduction in visceral pain.
A major reason why pancreatic cancer remains such a lethal disease is that visceral pain is not perceived by the patient until the disease is highly advanced and the tumor has metastasized to other organs at which point therapeutic treatment no longer has a significant effect on survival12-14. As with most cancers, when pancreatic tumors are detected early patient survival is significantly enhanced15. Using mice that spontaneously develop pancreatic cancer, we observed an approximately ten-week gap between the appearance of markers of pancreatic cancer (tumor growth, increased microvascular density, infiltration of macrophages that express nerve growth factor, and increased density of sensory and sympathetic fiber innervation) and the onset of visceral pain-related behaviors16. As the normal pancreas receives an extensive sensory and sympathetic innervation17, 18, and acute chemical stimulation19 or pathology of the pancreas can produce significant pain20, it has remained unclear why pain due to pancreatic cancer is perceived only in late stage disease. The present study investigates the possibility that the endogenous opioid system modulates the initial presentation and long term experience of pancreatic cancer pain.
Materials and Methods
Experimental mice
Transgenic mice expressing the N-terminal 127 amino acids of simian virus 40 large T antigen, under the control of the rat elastase-1 promoter (ET) were generated as previously described21, 22. Homozygosity for the mutant allele is embryonic lethal; consequently, heterozygous mice (ET) and their wild type littermates (WT) were used for all experiments and breeding. A total of 221 mice were used in the present experiments.
ET mice with a C57BL/6J background (Mayo Clinic, Scottsdale, AZ) were bred with female C57BL/6J mice (The Jackson Laboratory, Bar Harbor, ME). Pups were weaned at 3-4 weeks, and the DNA extracted from tail sections were screened for the presence of the transgene using RT-PCR analysis. Mice included in this study were genotyped twice to ensure accuracy of the experimental condition.
All mice were maintained in a vivarium at 22°C with a 12-hour alternating light-dark cycle and given food and water ad libitum. The mice were housed in accordance with The National Institutes of Health guidelines and all procedures were approved by the Institutional Animal Care and Use Committee at the University of Minnesota.
Mice with no embryological or developmental abnormalities and male mice not used in colony development were included. Testing and data analysis exclusion criteria included observation of physical abnormalities such as blindness or severe alopecia, or data resulting in greater than two standard deviations from the mean test variable. Mice were not assessed behaviorally prior to 5 weeks of age due to low body weight and small body structure which resulted in difficulty in assessing visceral pain-related behaviors. Euthanasia criteria included weight loss of 20-25%, weakness, inability to obtain food or water, loss of coat luster, and/or excessive bite or scratch wounds. These criteria have been set by and are followed in accordance with Research Animal Resources of the University of Minnesota.
Behavioral analysis
Male ET mice and age-matched WT littermates were behaviorally observed from 5 weeks of age for visceral pain-related symptoms. Visceral pain-related behaviors were assessed as degree of hunching, time spent hunching (over 300 sec) and the number of palpation-induced vocalization events (over 60 sec). The hunching behavior is similar to what has been previously described as hunching or hump-backed behavior in rats with visceral pain23. Displays of vocalization have also been described as measures of pain in other rodent pain models24-26. To monitor the general health of the mice, body weights were recorded weekly prior to behavioral tests.
Hunching behavior was assessed at 5-7, 9-12 and 14-24 weeks or until euthanasia criteria were met. In this study, the same mice were tested at 5-7, 9-12 and 14-24 weeks. In addition, we also used a separate group of mice which were tested only at each time period. We observed the same results in both groups of animals. Mice were placed individually in the center of an open field arena and observed over a 300 second period. The hunching score is based on amount of accumulated time, in seconds, engaged in the hunching behavior (up to 300sec observation period) multiplied by the scoring factor associated with that behavior's observed degree of impairment. The possible range of scores is from 0 (300 sec × 0=0 for normal animals that do not hunch) to 1200 (300 sec × 4 for animals which continuously hunch throughout the entire 300sec test period).
The scoring factors for the observed hunching behaviors were as follows: 0 = normal coat luster, displays exploratory behavior, lack of a rounded-back posture. 1 = mild rounded-back posture, displays exploratory behavior, normal coat luster. 2 = severe rounded-back posture, displays slightly reduced exploratory behavior, slight piloerection, intermittent abdominal contractions. 3 = severe rounded-back posture, displays considerably reduced exploratory behavior, moderate piloerection, intermittent abdominal contractions. 4 = severe rounded-back posture, displays little or no exploratory behavior, whole body piloerection with head immobile, intermittent abdominal contractions. For example, mice that spent the entire 5-min period in a severe rounded-back posture, displayed considerably reduced exploratory behavior, had moderate piloerection with intermittent abdominal contractions would have a score of 900. These methods were adapted from Lantèri-Minet, et al. 199527. The arena was washed between each trial with a 10% bleach solution to eliminate possible bias due to odors left by the previous mouse. In all cases, observations were performed by two independent observers blinded as to the experimental status of the mouse.
Quantification of ultrasonic and audible vocalization was performed prior to and 15 minutes following drug or saline administration. Mice were gently restrained and, using a five millimeter diameter cotton tipped applicator (Solon MFG., Solon, ME), palpated approximately one centimeter below the xiphoid process to the left of the midline with a palpation occurring approximately every three seconds over a one-minute observation period. Data recording was performed in a manner similar to methods described previously24. Briefly, recordings were made with a D 980 bat detector (Pettersson Elektronik AB, Uppsala, Sweden) (detection range; 20 Hz to 180 kHz ± 5 kHz). This range was chosen because others have reported mice to vocalize in both the audible and high ultrasonic ranges28, 29. The recorded signals were filtered and amplified (Ultravox 4-channel filter system, Noldus Information Technology, Wageningen, The Netherlands). Analyses of the number of palpation-evoked vocalizations over the observation period were made using Ultravox 2.0 software (Noldus Information Technology). The detector shifts a user-defined, high-frequency band to the audible range while suppressing non-relevant sounds and ignoring ultrasounds outside the defined frequency range. For as long as the animal is emitting sound, the detector and audio filter records the signal. Only sounds within the frequency range of 10kHz-180kHz and exceeding a -6 dBm amplitude threshold value are recorded by the UltraVox software as an event. Experiments were carried out in a quiet area and appropriate filtering levels were used to avoid the recording of background noise. One mouse was in the testing room at a time during recordings.
Drug administration
Mice were administered opioid antagonists naloxone hydrochloride (3 mg/kg, s.c.; Sigma, St. Louis, MO), naloxone methiodide (3 mg/kg, s.c.; Sigma), naltrexone hydrochloride (10 mg/kg, s.c.; Sigma), opioid agonist morphine sulfate (10 mg/kg, s.c.; MS, Elkins-Sinn, Cherry Hills, NJ), opioid agonist loperamide (10 mg/kg s.c.; Sigma) or vehicle (0.9% saline; Sal) following baseline hunching and vocalization quantification at each time point. Dose-response experiments using 0.1,1.0, 3.0 mg/kg naltrexone; 1.0,1.5, 3.0 mg/kg naloxone; 1.0, 3.0mg/kg naloxone methiodide were performed to assess side effects profiles. These experiments, in addition to doses of naloxone methiodide up to 30.0 mg/kg, did not produce any observable side effects.
After drug administration, mice were again placed in the open field arena and the degree of hunching and the time spent hunching over a 300 second period were quantified. The number of vocalization events was quantified following observation of the hunching behaviors. Behavioral analysis was performed 15-50 minutes post-injection to ensure that mice were tested within the therapeutic time window of the antagonists30, 31, and within 30 minutes for MS32. From observations and previous preliminary experiments, the robust effects of the agonists and antagonists were observed starting at 10 minutes (min) post-injection. Each drug was tested at least 24 hours after the previous drug in order to assure clearance of previous drug from the body. All antagonists and agonists used in the present study were well tolerated and did not induce side effects.
Euthanasia and processing of tissue
Euthanasia and processing of tissue was performed as previously described33, 34. Mice were sacrificed with CO2 at 5-7, 9-12 and 14-24 weeks following the established euthanasia criteria (WT 5-7 weeks n=3; ET 5-7 weeks n=7; WT 9-12 weeks n=8; ET 9-12 weeks n=11; WT 14-20 weeks n=5; ET 14-24 weeks n=26).
Mice were perfused intracardially with 12 mL of 0.1 M phosphate buffered saline (PBS) solution, followed by 25 ml 4% formaldehyde/12.5% picric acid solution in 0.1 M PBS. Following euthanasia, a necroscopy was performed on all animals and the abdominal cavity was examined for visual evidence of gross lesions or metastases as previously described21. At 18 - 24 weeks, which was the age at which ET mice were euthanized due to weight loss or deterioration of the general health, 12% of the ET mice showed visual evidence of metastases to the liver, duodenum or peritoneum. Mice found to have visual evidence of lesions or metastases at the time of euthanasia were analyzed separately. The pancreas was dissected from the peritoneal viscera, leaving the spleen and duodenum associated for anatomical reference.
Immunohistochemistry
The pancreas was embedded in TissueTek (Sakura, Torrance, CA) and cut on a cryostat at a thickness of 40 μm in the frontal plane. Serial tissue sections were collected, thaw mounted, and processed on gelatin-coated slides. These sections were incubated for 30 min at room temperature (RT) in a blocking solution of 3.0% normal donkey serum in PBS with 0.3% Triton X-100, and then incubated overnight at RT in the primary antiserum. Small to medium diameter peptidergic primary afferent sensory nerve fibers were immunostained for the neurotransmitter calcitonin gene-related peptide (polyclonal rabbit anti-rat CGRP, 1:15,000, Sigma, St. Louis, MO). Blood vessels were immunostained for the platelet endothelial cell adhesion molecule CD31 (monoclonal rat anti-mouse CD31, 1:500, BD Pharmingen, San Diego, CA). Macrophages were immunostained for the myeloid specific lysosomal antigen CD68 (monoclonal rat anti-mouse CD68, 1:5000, Serotec, Oxford, UK). Following incubation, tissue sections were washed three times for 10 min each in PBS and incubated in the secondary antibody solution for 3 hours at RT. Secondary antibodies conjugated to fluorophores Cy3 and FITC (Jackson ImmunoResearch, West Grove, PA) were used at 1:600 and 1:150, respectively. To confirm specificity of primary antibodies, controls included pre-absorption with the corresponding antigen or omission of the primary antibody. To control for interactions between antibodies in double immunostaining experiments, all results were confirmed on singly immunostained sections. After secondary incubation, sections were washed three times for 10 min each in PBS and coverslipped with p-phenylenediamine in glycerol. Following immunohistochemical quantification, sections were counterstained with hematoxylin and eosin (H&E) for histological identification of pancreatic structures.
Histological analysis and quantification
Quantification was performed on a total of 21 sections per marker; 3 sections were sampled at 400 μm intervals from the pancreas of 12 male mice (WT 9-12 weeks n = 3; ET 5-7 weeks n = 3; ET 9-12 weeks n = 3; ET 14-24 weeks n = 3). To determine association of nerve fibers with pancreatic structures, sections were viewed using a MRC-1024 confocal imaging system (Bio-Rad, Richmond, CA). Blood vessel, macrophage and nerve fiber density and distribution was determined using an Olympus BH2 microscope equipped for epifluorescence and a SPOT2 digital camera (Diagnostic Instruments, Inc., Sterling Heights, MI).
Quantification of CD31-immunoreactive (CD31-ir) blood vessels was performed using methodology adapted from previous studies35-38. Briefly, sections were initially scanned at low power (100X) to identify four areas (hot spots) with the highest capillary density. These areas of high vascularization were observed throughout the tumor, but were most frequent at the margins of the tumor36. Hot spots were defined as areas with a microvessel density (MVD) greater than 10 intersections/mm. Each hot spot was then viewed at 400X magnification and profile counts were performed using the entire z-plane of the 40 μm thick section. Microvessel profiles were identified using criteria described by Weidner and colleagues36, where the presence of a closed vessel lumen was not required for identification. Only vessel profiles that were 2-10 μm in diameter were counted. A 1 cm2 eyepiece grid containing ten 250-μm long horizontal gridlines separated at 25 μm intervals was used to quantify microvessel density, and the number of intersections between microvessel profiles and horizontal gridlines was determined37-40. Four hot spots on three separate sections were assessed for each mouse and results are expressed as the mean number of gridline intersections/mm.
Nerve fiber density was quantified using methodology adapted from previous studies41-43 as follows: sections were viewed at 200X magnification using a one cm2 eyepiece grid that was divided into 100 1-mm2 units. At 200X magnification this grid defined a 500 μm × 500 μm observation field. For each section, areas representing the head, body and tail of the pancreas were identified using the following criteria: 1) fields proximal to the duodenum (within 3mm) were classified as within the head, 2) fields proximal to the hilus of the spleen (within 3mm) were classified as the tail, and 3) fields lying between the head and tail were classified as body. Five, random non-overlapping observation fields were selected within each region, and fiber density was assessed as the number of immunostained nerve fibers per mm2. Only nerve fibers greater than 30 μm in length were included in the analysis and fibers were counted in all focal planes within an observation field.
Quantification of CD68-immunoreactive (CD68-ir) macrophage density was performed using methodology adapted from previous studies35, 44. Briefly, sections were initially scanned at low power (100X) to identify four areas (hot spots) with the highest macrophage density35. Hot spots were defined as areas with a macrophage density greater than 600 cells/mm2. A 250 μm × 250 μm observation field was viewed for each hot spot at 400X magnification and the number of CD68 cells was counted. Only cells with a clearly visible cell body were counted. The number of hot spots on three separate sections was determined for each mouse, and results are expressed as the mean number of CD68 cells/mm2. Quantification of these markers was performed by an observer blinded to the status of each mouse.
Digital images of pancreas tissue from WT (n=4) and ET (n=4) mice were obtained using a Canon Digital Rebel EOS 300D (Canon U.S.A., Inc., Lake Success, New York). The area and height of each pancreas were measured using Image Pro Plus software version 3.0 (Media Cybernetics, Silver Spring, MD). The volume of each pancreas was then calculated according to volume = area × height.
Statistical analysis
Hierarchical (mixed effects) regression modeling, which can accommodate missing values and subjects measured at differing time intervals, was used to analyze the longitudinal weight data. Hunching and vocalization data with non-normal frequency distributions were statistically analyzed using Mann-Whitney test for between-group and Wilcoxon matched-pairs signed-ranks test for within group comparisons, and data with normal distribution were analyzed using 2-way ANOVA. Results were considered statistically significant at p<0.05. The SPSS version 13 computer statistics package (SPSS, Chicago, IL) was used to perform statistical analyses.
Results
Gross and histopathological changes associated with the development of pancreatic cancer
To examine the progression of pancreatic cancer in ET mice, a variety of morphological, histochemical and immunohistochemical techniques were used to compare the pancreas tissue of 9-12 and 14-24 week WT and ET mice (Fig. 1). The overall histomorphology of the pancreas of 9-12 and 14-24 week WT animals remains remarkably similar as assessed by gross morphology, palpation, hematoxylin and eosin staining16 or assessment of the density of CD31-immunoreactive (ir) blood vessels, thus 9-12 week data is presented. The gross morphology of the pancreas of 9-12 week ET animals is similar to that observed in age-matched WT mice. However, palpation of the dissected pancreas of 9-12 week ET mice revealed the presence of 5-10 small nodules scattered throughout the head, body and tail of the pancreas (Fig. 1B). In ET mice, the pancreatic tumor is grossly observable in situ at 14-24 weeks (Fig. 1C), and by this time, metastases were present in the adjacent liver and duodenum in 12% of the animals, which is similar to the number of metastases previously reported using this same model of pancreatic cancer21. Volumetric analysis of the pancreas reveals a significant increase in the size of the pancreas in the late stage as compared to early stages of disease (Fig. 2A) (WT=760±156 mm3; ET 5-7 wks, 551±104mm3; ET 9-12 wks, 521±136 mm3; ET 14-24 wks, 2854±962* mm3; *p<0.01).
Figure 1.
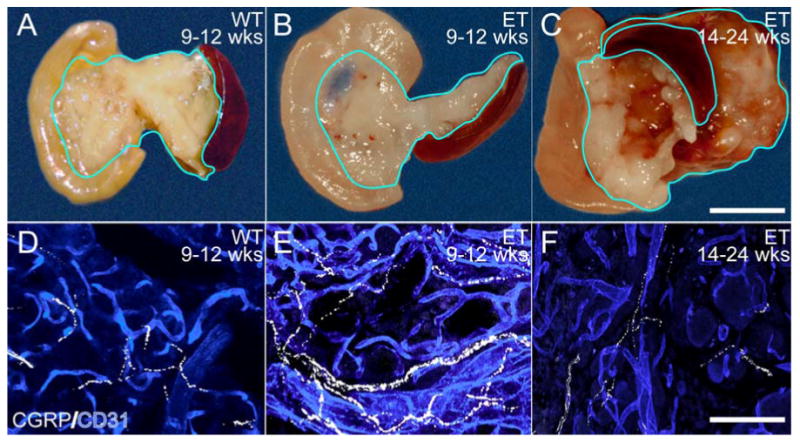
The gross pathology and histopathological changes associated with the development of pancreatic cancer. Low power photographs of the pancreas (A, B, C) and confocal photomicrographs of CGRP-immunoreactive (CGRP-ir) small diameter peptidergic sensory nerve fibers (white) and CD31-immunoreactive (CD31-ir) blood vessels (blue) (D, E, F) in sections of WT and ET pancreas. Upon dissection, major pathological changes were not observed until 14-24 weeks (wks) (C), although palpation of the pancreas at 9-12 wks revealed small nodules corresponding to small well-circumscribed tumors or adenomas (B). In the pancreas of WT mice at 9-12 wks there is a smooth and organized appearance of capillaries and CGRP fibers (D). At early stage in ET mice (9-12 wks), there is a significant increase in capillary and CGRP fiber density (E) within the parenchyma of the pancreas as compared to the age-matched WT controls. At late timepoints (F; 14-24 wks), capillary and CGRP fiber density in central necrotic areas of the tumor declined; however, areas of higher capillary and CGRP fiber density were still observed near the margins of the tumor (shown) where active growth was still occurring. Scale bar: 5 mm, A, B, C; 100 μm, D, E, F.
Figure 2.
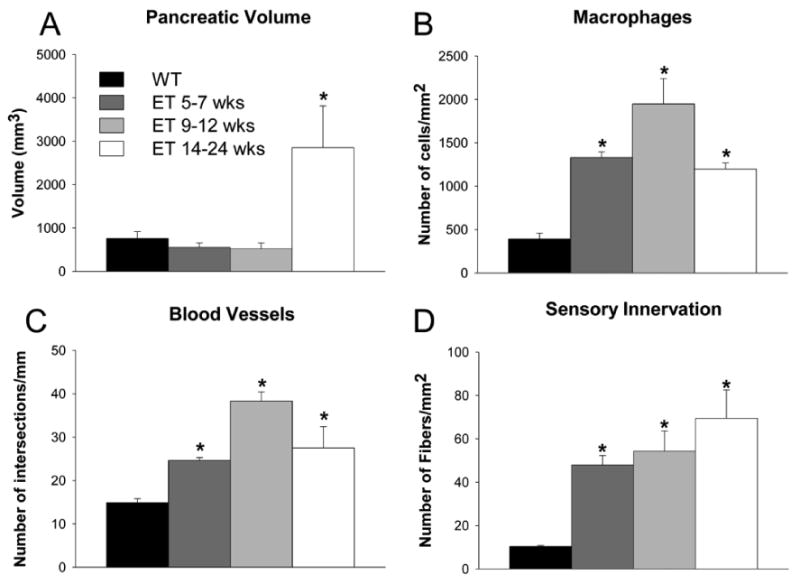
Quantification of the gross and histopathological changes associated with the development of pancreatic cancer. (A) Pancreatic volume remains constant until 14-24 weeks (wks) where a significant increase is observed. (B) The number of CD68-immunoreactive (CD68-ir) macrophages in the pancreas increases significantly at 5-7 wks, 9-12 wks, and 14-24 wks disease (p<0.05 vs. WT). (C) In the pancreas of ET mice at 5–7 wks, 9-12 wks and 14-24 wks there is an increased density of CD31-immunoreactive (CD31-ir) vascular endothelial cells. These increases in microvascular density were significant as compared to WT at all timepoints. (D) The density of CGRP-immunoreactive (CGRP-ir) sensory nerve fibers increases with disease progression and first becomes significantly different from WT at 5-7 wks and continues to be significantly different until 14-24 wks or euthanasia criteria are met. Note, however, that while changes in all other pathological endpoints (B-D) are rapidly increasing as early as 5-7 wks, changes in pancreatic volume do not become significant until 14-24 wks (A). *p<0.05 vs. WT.
Immunohistochemical quantification of the density of CD68-ir macrophages and CD31-ir endothelial cells also indicated that significant cellular changes occur as early as 5-7 and 9-12 weeks in the pancreas of ET mice (Fig. 2B-C). The number of CD68-ir macrophages/mm2 increased dramatically (WT: 266.8±8.0; ET 5-7 wks, 1277.2±155. 0*; ET 9-12 wks, 1410.7±358.0*; ET 14-24 wks, 983.3±99.0*; *p<0.01 in comparing ET to WT controls) (Fig. 2B) and the number of CD31-ir blood vessels/mm2 significantly increased as compared to WT controls (WT, 13.1±0.2; ET 9-12 wks 41.5±3.4*, ET 14-24 wks, 32.8±5.6*; *p<0.01) (Fig. 2C). To quantify potential changes in sensory innervation with disease progression, sections of WT and ET pancreas were immunostained for calcitonin gene-related peptide (CGRP), a neuropeptide that is expressed by the majority of sensory fibers that innervate the normal pancreas. While the density of CGRP-expressing primary afferent fibers was not significantly different in WT mice of different ages (9-12 and 14-24 weeks), there was a clear increase in the density of CGRP-expressing nerve fibers in ET mice with disease progression (WT 16.3±2.8; ET 9-12 wks 64.9±9.0*; ET 14-24 wks 67.4±16.8 fibers/mm2*; *p<0.01 WT vs. ET) (Fig. 2D).
Endogenous CNS opioid-dependent mechanisms inhibits hunching and vocalization behaviors in mice with pancreatic cancer
Spontaneous visceral pain-related behaviors such as hunching (Fig, 3A-J) became evident only in late stage disease (14-24 weeks) when pancreatic cancer is highly advanced (Fig. 4A). This behavior appeared to mirror the “pancreatic position” commonly assumed by humans with pancreatic pain45. In this position, patients bend forward or lie in the knee-chest position, presumably to minimize movement of the pancreas and thus minimize pancreatic cancer pain. Whereas significant hunching behavior was not observed in WT control mice at any timepoint examined, beginning at approximately 14 weeks, all ET mice spontaneously hunched (Fig 4A). To investigate whether this mechanism might also be active in early stage pancreatic cancer and contribute to the disconnect between pancreatic pathology and pancreatic pain, opioid antagonists were administered to 5-7, 9-12, and 14-24 week WT and ET mice. In the WT mice, administration of naloxone did not induce significant hunching behaviors at any timepoint examined (Fig. 4B). In contrast, administration of naloxone to ET mice with early stage pancreatic cancer (5-7, 9-12 weeks) induced significant hunching behavior that was not observed in age-matched WT littermate controls (Fig. 4B).
Figure 3.
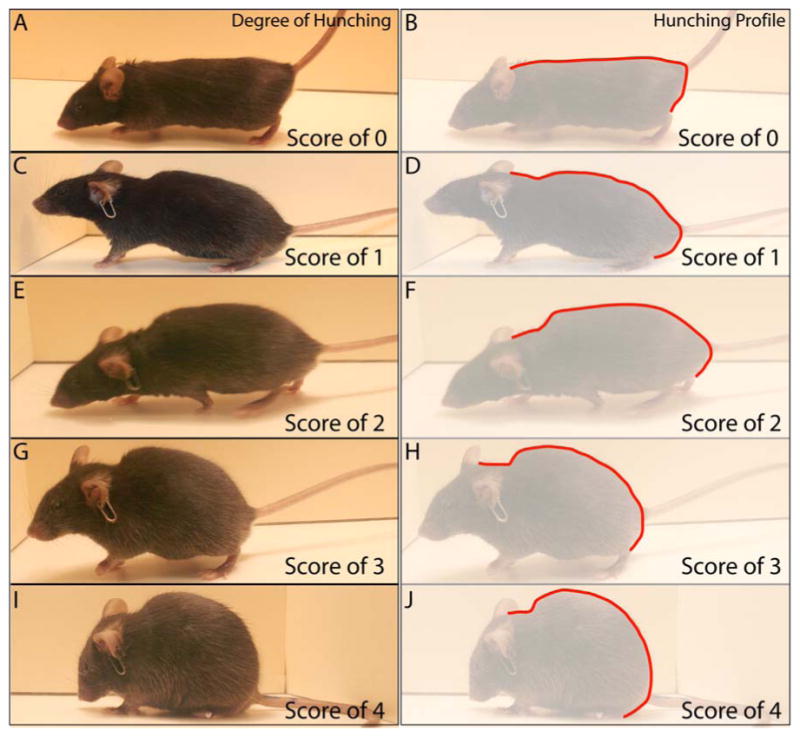
Representative images and silhouette outlines of the hunching behavioral characteristics of mice with varying stages of pancreatic cancer. The scores for the observed behaviors were as follows: 0 = lack of a rounded-back posture, displays exploratory behavior and normal coat luster (A, B displays hunching silhouette for a score of 0). 1 = mild rounded-back posture, displays exploratory behavior, normal coat luster (C, D displays hunching silhouette for a score of 1). 2 = severe rounded-back posture, displays slightly reduced exploratory behavior, slight piloerection and intermittent abdominal contractions (E, F displays hunching silhouette for a score of 2). 3 = severe rounded-back posture, displays considerably reduced exploratory behavior, moderate piloerection and intermittent abdominal contractions (G, H displays hunching silhouette for a score of 3). 4 = severe rounded-back posture, displays little or no exploratory behavior, whole body piloerection with head immobile and intermittent abdominal contractions (I, J displays hunching silhouette for a score of 4). Note the silhouette outlines (B, D, F, H, J; red line) comparing the progressive rounded-back posture that occurs with disease progression in this mouse model of pancreatic cancer pain.
Figure 4.
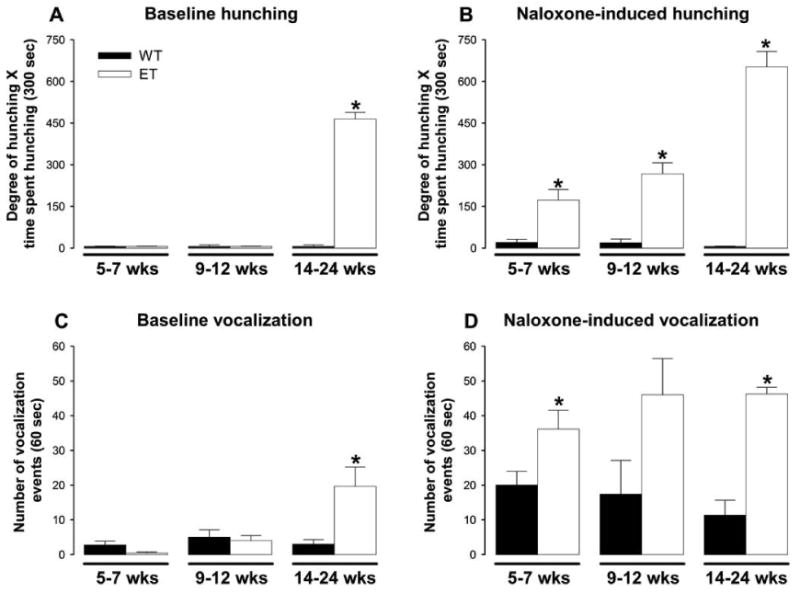
The effect of the CNS-penetrant opioid antagonist naloxone on pancreatic cancer pain-related behaviors. (A) Spontaneous hunching behavior first became evident in ET mice at approximately 14 weeks (wks), increased in severity until euthanasia and was rarely observed in WT mice at any timepoint examined (n≥8 WT, n≥8 ET). (B) When ET mice were administered the opioid antagonist, naloxone (3 mg/kg, s.c), the hunching behavior became evident at 9-12 wks, increased dramatically at 14-24 wks and was significantly different from WT controls that also received naloxone (n≥8 WT, n≥8 ET). (C) Spontaneous vocalization behavior first became slightly evident in ET mice at approximately 12 wks, increased at 14-24 wks and was observed minimally in WT mice (n≥5 WT, n≥5 ET). (D) The number of vocalization events over a one minute observation period increased at 9-12 wks and 14-24 wks in ET mice following injection of naloxone when compared to age-matched WT mice that also received naloxone (n≥5 WT, n≥5 ET). All error bars shown in A-D represent S.E.M. *p<0.05 vs. WT.
To determine whether the gross lesions or metastases to other organs were correlated with pain-related behaviors in the ET animals at the time of euthanasia (18-24 weeks), we performed a post-hoc analysis on spontaneous hunching behaviors exhibited by ET animals with (n=7) and age matched ET animals without visually obvious extra-pancreatic lesions or metastases (n=9). In all cases, these extra-pancreatic lesions or metastases were found in the liver, duodenum and adjacent peritoneum. In comparing spontaneous hunching behaviors exhibited by ET animals with visually observable extra-pancreatic lesions or metastases (707±43; n=7) vs. ET age matched animals lacking such extra-pancreatic lesions or metastases (705±50; n=9), there was no significant difference between the two groups (p=0.98). It should be emphasized that there is currently no unequivocal method to identify and quantify all micro-metastases present in animals or humans with pancreatic cancer. Thus, it is possible in any ET animal that micro-metastases within and outside the pancreas could be contributing to the pain behaviors observed in these animals.
It has been shown that animals frequently vocalize when experiencing pain24. In the ET animals, spontaneous vocalization was similar to spontaneous hunching behavior in that ET mice exhibited significantly enhanced spontaneous vocalization only in late stage disease (14-24 weeks), whereas no significant increase in spontaneous vocalization was observed with age in WT animals (vocalization events in WT vs. ET animals: 9-12 wks, 4.6±3.2 vs. 8.6±1.6; 14-24 wks, 3.0±1.3 vs. 19.7±5.5*, *p<0.01 in comparing ET to WT controls) (Fig. 4D). Administration of naloxone to ET mice with early stage pancreatic cancer (5-7, 9-12 weeks) induced significant vocalization behaviors that were not observed in age-matched WT control (Fig. 4D) mice that also received naloxone.
Pharmacology of opioid-dependent mechanisms that inhibits pancreatic cancer pain
A major question raised by these studies is whether endogenous opioid masking of early stage pancreatic pain occurs in peripheral tissues or in the CNS. Whereas subcutaneous injection of 3 mg/kg of the CNS-impermeant opioid antagonist naloxone methiodide did not induce any significant hunching or vocalization behaviors in WT or ET animals, subcutaneous injection of 3 mg/kg of the CNS-penetrant analog naloxone rapidly induced significant hunching and vocalization behaviors in 9-12 week ET mice with early stage pancreatic cancer (hunching behaviors/vocalization events in 9-12 week ET mice: saline alone, 0.0±0.0/0.5±0.2; naloxone methiodide, 25.0±25.0/0.2±0.2; naloxone, 300±0.0*/22.3±4.0*; *p<0.01 in comparing saline to drug treatment at 30 min post injection) (Fig. 5A-B). In addition, 10 and 30mg/kg of naloxone methiodide was administered and similar effects were observed as that at 3 mg/kg (data not shown).
Figure 5.
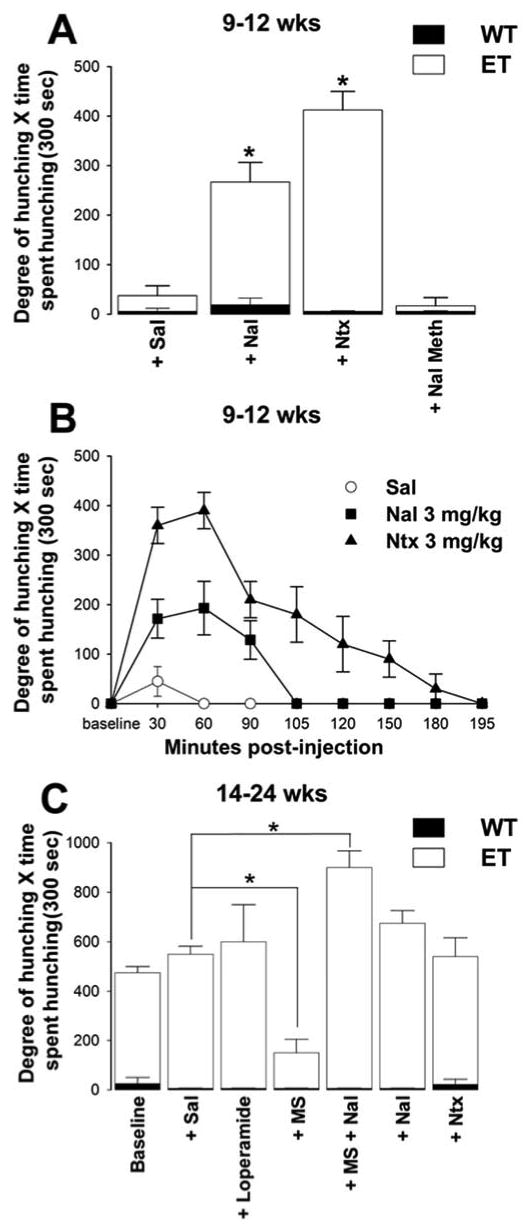
The effects of CNS-penetrant and non-penetrant opioid receptor agonists and antagonists on visceral pain-related behaviors in mice with early and late stage pancreatic cancer. (A) In WT mice, spontaneous, saline (Sal), naloxone (Nal) or naltrexone (Ntx)-induced hunching behavior was rarely observed at any timepoint, and there was no significant difference between WT + Sal vs. WT+ Nal or WT + Ntx (n≥5). Spontaneous hunching of ET mice at 9-12 wks was not significantly increased by s.c. injection of Sal alone; however, following injection of Nal or Ntx (3 mg/kg, s.c.), ET mice had a higher degree of hunching. Following injection of the peripherally acting naloxone methiodide in early stage disease, hunching behavior was not significantly elevated from WT + Sal or from ET + Sal. (B) Following administration of Nal (3 mg/kg, s.c.) or Ntx (3mg/kg, s.c.), ET mice at 9-12 wks exhibited hunching behavior that continued for 105 or 195 minutes, respectively, and the area under each response-time curve was significantly larger than for Sal controls (n≥5). In preliminary experiments, the naloxone and naltrexone-induced visceral pain behaviors in mice with early stage pancreatic cancer were robust at 10 minutes post-injection. (C) In late stage disease (14-24wks), baseline hunching in ET mice was drastically elevated from WT. Loperamide, a peripherally acting opioid agonist, did not reduce hunching behaviors in late stage ET mice. Note that morphine sulfate (MS) administration (10 mg/kg, s.c.) attenuates hunching compared to Sal (p=0.03); however, after Nal (3 mg/kg, s.c.) reversal of MS, this hunching behavior is significantly increased when compared to Sal. Naltrexone administration did not significantly increase hunching behaviors at late stage disease compared to saline. All error bars shown in A-C represent S.E.M. *p<0.05 vs. Sal.
As in humans with pancreatic cancer46, administration of morphine to mice with late stage pancreatic cancer attenuated this hunching behavior which was blocked by the opioid antagonist naloxone (Fig. 5C). Naloxone administration not only reversed the morphine attenuation of hunching in ET mice, but resulted in an even greater hunching behavior than that observed in the same animals before any drug treatment (Fig. 5C). Loperamide, a peripherally acting opioid agonist, did not reduce hunching behaviors in late stage ET mice (ET 14-24 wks spontaneous, 450±73 degree of hunching × time spent hunching; ET 14-24 wks + 10 mg/kg s.c. loperamide, 557±63 degree of hunching × time spent hunching). Furthermore, the naloxone-induced hunching behavior in ET mice with early stage cancer reflected the known pharmacokinetics of these drugs in mice (Fig. 5B)30. Naloxone and naltrexone have similar potencies, but the latter is more bioavailable and has a longer half-life.
Discussion
The endogenous modulation of visceral pain
Previous studies have suggested that descending modulation of nociceptive input can be effectively engaged with a slowly evolving disease or tissue injury10, 47, 48. The present study suggests that in mice with either early or late stage pancreatic cancer there is an endogenous CNS opioid analgesic system that masks the pain in early stage disease and attenuates the pain in late stage disease. Although it has been shown that naloxone administration induces a behavioral response in animals49, behavioral responses in age-matched wild type control mice that received naloxone were not statistically significant. The opioid receptors that mediate this inhibition of pancreatic cancer pain appear to reside in the CNS, as CNS-penetrant opioid antagonists increase visceral pain-related behaviors in both early and late stage pancreatic cancer while a non-CNS penetrant opioid receptor antagonist does not. Similarly, the exogenous opioid agonist morphine is effective at attenuating late stage pancreatic cancer pain-related behaviors whereas the non-CNS penetrant opioid agonist loperamide was ineffective. These data suggest that the opioid receptors mediating the opioid modulation of pancreatic cancer pain are located primarily in the CNS.
While the present study has demonstrated that there is a tonic endogenous opioid-dependent inhibition of pancreatic pain in both early and late stage disease, there are several outstanding questions as to the mechanism(s) by which endogenous opioids exert this action. These include the source(s) of the endogenous opioids that drive the tonic inhibition, the specific subtypes and locations of the opioid receptors that mediate the CNS inhibition of pancreatic cancer pain, and whether the inhibition of pancreatic cancer pain is in part due to descending inhibition and/or blockade of descending facilitation.
Previous studies have shown that multiple endogenous CNS opioids (including beta-endorphin, dynorphin, and met- and leu-enkephalin) are involved in modulating visceral pain11. Many of these peptides have been shown to be present and released in brain areas such as the PAG, rostral ventral medulla (RVM) and spinal cord following nociceptive stimuli and to be involved in mediating endogenous opioid analgesia50, 51. Interestingly, it has been shown that met-enkephalin is also expressed in pancreatic tumor cells and that humans with pancreatic cancer have significantly higher plasma levels of met-enkephalin than normal controls52, 53. While it is not clear whether met-enkephalin released from the pancreas could cross the blood-brain barrier and have a pharmacological effect on opioid receptors in the CNS, these observations raise the possibility that endogenous opioids released from a peripheral tissue could play a role in the inhibition of pancreatic cancer pain.
The past two decades have seen a major effort to define the specific subtypes and locations of the opioid receptors involved in the inhibition of pain. While substantial information exists on opioid modulation of somatic sensory transmission, much less is known about opioid modulation of visceral sensory transmission. Opioid receptors of the mu, kappa and delta classes are extensively expressed in the mouse brain54 as well as in the areas of the dorsal horn of the spinal cord where primary visceral afferent neurons terminate and exogenously administered opioids from all three major opioid classes have been shown to modulate visceral noxious information55, 56. Indeed, while naloxone is considered a mu-preferring opioid receptor antagonist, it has been shown to have lesser but significant affinity for the both the delta and kappa opioid receptors57; thus, none of the known opioid receptors could be definitively excluded from a role in the endogenous tonic inhibition of pancreatic cancer pain.
While significant attention has focused on the ability of cortical and subcortical brain areas to inhibit the transmission of pain, there is significant evidence that many of these same sites can also facilitate the transmission of pain3, 10, 50. Thus, behavioral and autonomic reactions to visceral stimulation are blocked by spinal cord transection while general, reactions to cutaneous stimuli typically persist or are enhanced58. For example, it has been shown that colorectal distension evokes a pressor response, tachycardia and hunching behaviors and that cold block at the high cervical spinal cord level largely abolishes all these responses58. In light of these and other reports, the present study is fully consistent with the interpretation that administration of naloxone in early or late stage pancreatic cancer increases pain by reducing descending inhibition and/or by promoting facilitation of the transmission of visceral noxious information arising from the pancreas.
In the case of pancreatic cancer, the slow but persistent progress of the disease, either from the pancreas itself or micrometastases to other organs, could provide a persistent noxious stimulation that would gradually induce the activation of modulatory circuits in the CNS. These neural circuits would suppress the transmission of the nociceptive information from the pancreas to the cerebral cortex2 until late stage disease when the tumor-induced activation of nociceptors would “break through” this modulation, resulting in the perception of cancer-induced pain. Whether a similar descending inhibition masks the detection of pancreatic cancer in humans or in other visceral cancers (such as lung or ovarian), which are also characterized by late stage detection59, 60, is currently unknown.
Mechanisms that drive pancreatic cancer pain
Although multiple factors have been hypothesized to induce pain in patients with pancreatic cancer, remarkably little is known about the specific mechanism(s) and cascade(s) of events that drive this visceral pain. Factors hypothesized to drive pancreatic pain include acute inflammation61, elevated pancreatic pressure62, 63, ongoing ischemia, local tissue hypoxia and acidosis64, and excess release and activation of endogenous proteases65.
In human pancreatic cancer, it has been reported that 1) there is a noticeable increase in macrophages throughout the parenchyma of the pancreas44, 2) many of the macrophages and pancreatic ductal adenocarcinoma cells express nerve growth factor (NGF), and 3) there is significant perineural invasion by tumor cells66-69. In human and mouse somatic tissues such as skin, many of the above changes would be accompanied by significant pain70. In contrast, although many of the above changes are observed relatively early in both humans and mice with pancreatic cancer only when the disease is highly advanced and has metastasized to the liver, peritoneum and GI tract, is the associated pain apparent. These findings are consistent with the hypothesis that while both the nociceptors and the pathology to drive a persistent pain state are present in the cancerous pancreas and adjacent effected tissues, there is a modulation of visceral cancer pain that tonically inhibits the transmission of this visceral pain.
The presentation and clinical outcome of patients with pancreatic cancer
In most patients with pancreatic cancer, the initial presenting symptoms are vague, and include weight loss, fatigue and non-specific gastrointestinal symptoms, all of which go unnoticed until there is an obvious need to seek medical care71. Pain referred to the upper abdomen and/or mid-back is a common symptom that brings the patient to the physician that ultimately results in the diagnosis of pancreatic cancer72. A major problem with pancreatic cancer is that by the time pain is perceived by the patient, the disease is well advanced, has metastasized to other organs and the long-term benefits of resection, chemotherapy and radiation therapies are limited71. Although pancreatic cancer represents only 2% of diagnosed cancers, it is the fourth leading cause of cancer-related death in the United States15. In the United States alone, 32,000 new patients will be diagnosed with pancreatic cancer in 2005 with an average one year survival of 24% and five year survival of 4%15, 73.
The present study suggests that in pancreatic cancer an endogenous opioid dependent pain modulation is active in both early and late stage pancreatic cancer. As humans with acinar or ductal pancreatic cancer exhibit the same stereotypic set of symptoms and disease progression as do the mice in the present study, defining the mechanisms that both inhibit and drive this pain may aid in earlier diagnosis, survival and increased quality of life of patients with pancreatic cancer.
Acknowledgments
We thank Prisca Honore, Ph.D., and Gilbert Y. Wong, M.D., for their helpful comments on the manuscript. We also thank Therese Schachtele for excellent secretarial assistance and Judy Bradley and Nathan Koewler for exceptional technical assistance. Supported by the National Institutes of Health grants NS23970, NS048021 and a VA Merit Review.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Vanegas H, Schaible HG. Descending control of persistent pain: inhibitory or facilitatory? Brain Res Brain Res Rev. 2004;46:295–309. doi: 10.1016/j.brainresrev.2004.07.004. [DOI] [PubMed] [Google Scholar]
- 2.Apkarian AV, Bushnell MC, Treede RD, Zubieta JK. Human brain mechanisms of pain perception and regulation in health and disease. Eur J Pain. 2005;9:463–84. doi: 10.1016/j.ejpain.2004.11.001. [DOI] [PubMed] [Google Scholar]
- 3.Gebhart GF. Descending modulation of pain. Neurosci Biobehav Rev. 2004;27:729–37. doi: 10.1016/j.neubiorev.2003.11.008. [DOI] [PubMed] [Google Scholar]
- 4.Reynolds DV. Surgery in the rat during electrical analgesia induced by focal brain stimulation. Science. 1969;164:444–5. doi: 10.1126/science.164.3878.444. [DOI] [PubMed] [Google Scholar]
- 5.Hosobuchi Y, Adams JE, Linchitz R. Pain relief by electrical stimulation of the central gray matter in humans and its reversal by naloxone. Science. 1977;197:183–6. doi: 10.1126/science.301658. [DOI] [PubMed] [Google Scholar]
- 6.Young RF, Brechner T. Electrical stimulation of the brain for relief of intractable pain due to cancer. Cancer. 1986;57:1266–72. doi: 10.1002/1097-0142(19860315)57:6<1266::aid-cncr2820570634>3.0.co;2-q. [DOI] [PubMed] [Google Scholar]
- 7.Dunckley P, Wise RG, Fairhurst M, Hobden P, Aziz Q, Chang L, Tracey I. A comparison of visceral and somatic pain processing in the human brainstem using functional magnetic resonance imaging. J Neurosci. 2005;25:7333–41. doi: 10.1523/JNEUROSCI.1100-05.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Friedrich AE, Gebhart GF. Modulation of visceral hyperalgesia by morphine and cholecystokinin from the rat rostroventral medial medulla. Pain. 2003;104:93–101. doi: 10.1016/s0304-3959(02)00469-4. [DOI] [PubMed] [Google Scholar]
- 9.Brink TS, Mason P. Role for raphe magnus neuronal responses in the behavioral reactions to colorectal distension. J Neurophysiol. 2004;92:2302–11. doi: 10.1152/jn.00374.2004. [DOI] [PubMed] [Google Scholar]
- 10.Porreca F, Ossipov MH, Gebhart GF. Chronic pain and medullary descending facilitation. Trends Neurosci. 2002;25:319–25. doi: 10.1016/s0166-2236(02)02157-4. [DOI] [PubMed] [Google Scholar]
- 11.Borgbjerg FM, Frigast C, Madsen JB. Tonic endogenous opioid inhibition of visceral noxious information in rabbits. Gastroenterology. 1996;111:78–84. doi: 10.1053/gast.1996.v111.pm8698228. [DOI] [PubMed] [Google Scholar]
- 12.Pour PM. The silent killer. Int J Pancreatol. 1991;10:103–4. [PubMed] [Google Scholar]
- 13.Kelsen DP, Portenoy R, Thaler H, Tao Y, Brennan M. Pain as a predictor of outcome in patients with operable pancreatic carcinoma. Surgery. 1997;122:53–9. doi: 10.1016/s0039-6060(97)90264-6. [DOI] [PubMed] [Google Scholar]
- 14.White RR, Hurwitz HI, Morse MA, Lee C, Anscher MS, Paulson EK, Gottfried MR, Baillie J, Branch MS, Jowell PS, McGrath KM, Clary BM, Pappas TN, Tyler DS. Neoadjuvant chemoradiation for localized adenocarcinoma of the pancreas. Ann Surg Oncol. 2001;8:758–65. doi: 10.1007/s10434-001-0758-1. [DOI] [PubMed] [Google Scholar]
- 15.Jemal A, Murray T, Ward E, Samuels A, Tiwari RC, Ghafoor A, Feuer EJ, Thun MJ. Cancer statistics, 2005. CA Cancer J Clin. 2005;55:10–30. doi: 10.3322/canjclin.55.1.10. [DOI] [PubMed] [Google Scholar]
- 16.Lindsay TH, Jonas BM, Sevcik MA, Kubota K, Halvorson KG, Ghilardi JR, Kuskowski MA, Stelow EB, Mukherjee P, Gendler S, Wong GY, Mantyh PW. Pancreatic cancer pain and its correlation with changes in tumor vasculature, macrophage infiltration, neuronal innervation, body weight and disease progression. PAIN. 2005;119:233–246. doi: 10.1016/j.pain.2005.10.019. [DOI] [PubMed] [Google Scholar]
- 17.Sternini C, De Giorgio R, Anderson K, Watt PC, Brunicardi FC, Widdison AL, Wong H, Reber HA, Walsh JH, Go VL. Species differences in the immunoreactive patterns of calcitonin gene-related peptide in the pancreas. Cell Tissue Res. 1992;269:447–58. doi: 10.1007/BF00353900. [DOI] [PubMed] [Google Scholar]
- 18.Oomori Y, Iuchi H, Ishikawa K, Satoh Y, Ono K. Immunocytochemical study of tyrosine hydroxylase and dopamine beta-hydroxylase immunoreactivities in the rat pancreas. Histochemistry. 1994;101:313–23. doi: 10.1007/BF00268992. [DOI] [PubMed] [Google Scholar]
- 19.Wang CC, Westlund KN. Responses of rat dorsal column neurons to pancreatic nociceptive stimulation. Neuroreport. 2001;12:2527–30. doi: 10.1097/00001756-200108080-00047. [DOI] [PubMed] [Google Scholar]
- 20.Tenenbein MS, Tenenbein M. Acute pancreatitis due to erythromycin overdose. Pediatr Emerg Care. 2005;21:675–6. doi: 10.1097/01.pec.0000181419.49106.ec. [DOI] [PubMed] [Google Scholar]
- 21.Tevethia MJ, Bonneau RH, Griffith JW, Mylin L. A simian virus 40 large T-antigen segment containing amino acids 1 to 127 and expressed under the control of the rat elastase-1 promoter produces pancreatic acinar carcinomas in transgenic mice. J Virol. 1997;71:8157–66. doi: 10.1128/jvi.71.11.8157-8166.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Ornitz DM, Hammer RE, Messing A, Palmiter RD, Brinster RL. Pancreatic neoplasia induced by SV40 T-antigen expression in acinar cells of transgenic mice. Science. 1987;238:188–93. doi: 10.1126/science.2821617. [DOI] [PubMed] [Google Scholar]
- 23.Wesselmann U, Czakanski PP, Affaitati G, Giamberardino MA. Uterine inflammation as a noxious visceral stimulus: behavioral characterization in the rat. Neurosci Lett. 1998;246:73–6. doi: 10.1016/s0304-3940(98)00234-1. [DOI] [PubMed] [Google Scholar]
- 24.Han JS, Bird GC, Li W, Jones J, Neugebauer V. Computerized analysis of audible and ultrasonic vocalizations of rats as a standardized measure of pain-related behavior. J Neurosci Methods. 2005;141:261–9. doi: 10.1016/j.jneumeth.2004.07.005. [DOI] [PubMed] [Google Scholar]
- 25.Swedberg MD. The mouse grid-shock analgesia test: pharmacological characterization of latency to vocalization threshold as an index of antinociception. J Pharmacol Exp Ther. 1994;269:1021–8. [PubMed] [Google Scholar]
- 26.Giamberardino MA, Valente R, de Bigontina P, Vecchiet L. Artificial ureteral calculosis in rats: behavioural characterization of visceral pain episodes and their relationship with referred lumbar muscle hyperalgesia. Pain. 1995;61:459–69. doi: 10.1016/0304-3959(94)00208-V. [DOI] [PubMed] [Google Scholar]
- 27.Lanteri-Minet M, Bon K, de Pommery J, Michiels JF, Menetrey D. Cyclophosphamide cystitis as a model of visceral pain in rats: model elaboration and spinal structures involved as revealed by the expression of c-Fos and Krox-24 proteins. Exp Brain Res. 1995;105:220–32. doi: 10.1007/BF00240958. [DOI] [PubMed] [Google Scholar]
- 28.Gourbal BE, Barthelemy M, Petit G, Gabrion C. Spectrographic analysis of the ultrasonic vocalisations of adult male and female BALB/c mice. Naturwissenschaften. 2004;91:381–5. doi: 10.1007/s00114-004-0543-7. [DOI] [PubMed] [Google Scholar]
- 29.Ko SW, Chatila T, Zhuo M. Contribution of CaMKIV to injury and fear- induced ultrasonic vocalizations in adult mice. Mol Pain. 2005;1:10. doi: 10.1186/1744-8069-1-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Raehal KM, Lowery JJ, Bhamidipati CM, Paolino RM, Blair JR, Wang D, Sadee W, Bilsky EJ. In Vivo Characterization of 6{beta}-Naltrexol, an Opioid Ligand with Less Inverse Agonist Activity Compared with Naltrexone and Naloxone in Opioid-Dependent Mice. J Pharmacol Exp Ther. 2005;313:1150–62. doi: 10.1124/jpet.104.082966. [DOI] [PubMed] [Google Scholar]
- 31.Shimizu N, Kishioka S, Maeda T, Fukazawa Y, Dake Y, Yamamoto C, Ozaki M, Yamamoto H. Involvement of peripheral mechanism in the verapamil-induced potentiation of morphine analgesia in mice. J Pharmacol Sci. 2004;95:452–7. doi: 10.1254/jphs.fp0040252. [DOI] [PubMed] [Google Scholar]
- 32.Hasselstrom J, Svensson JO, Sawe J, Wiesenfeld-Hallin Z, Yue QY, Xu XJ. Disposition and analgesic effects of systemic morphine, morphine-6-glucuronide and normorphine in rat. Pharmacol Toxicol. 1996;79:40–6. doi: 10.1111/j.1600-0773.1996.tb00239.x. [DOI] [PubMed] [Google Scholar]
- 33.Schwei MJ, Honore P, Rogers SD, Salak-Johnson JL, Finke MP, Ramnaraine ML, Clohisy DR, Mantyh PW. Neurochemical and cellular reorganization of the spinal cord in a murine model of bone cancer pain. J Neurosci. 1999;19:10886–97. doi: 10.1523/JNEUROSCI.19-24-10886.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Honore P, Rogers SD, Schwei MJ, Salak-Johnson JL, Luger NM, Sabino MC, Clohisy DR, Mantyh PW. Murine models of inflammatory, neuropathic and cancer pain each generates a unique set of neurochemical changes in the spinal cord and sensory neurons. Neuroscience. 2000;98:585–98. doi: 10.1016/s0306-4522(00)00110-x. [DOI] [PubMed] [Google Scholar]
- 35.Esposito I, Menicagli M, Funel N, Bergmann F, Boggi U, Mosca F, Bevilacqua G, Campani D. Inflammatory cells contribute to the generation of an angiogenic phenotype in pancreatic ductal adenocarcinoma. J Clin Pathol. 2004;57:630–6. doi: 10.1136/jcp.2003.014498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Weidner N, Semple JP, Welch WR, Folkman J. Tumor angiogenesis and metastasis--correlation in invasive breast carcinoma. N Engl J Med. 1991;324:1–8. doi: 10.1056/NEJM199101033240101. [DOI] [PubMed] [Google Scholar]
- 37.Nagy K, Toth S, Palfia Z, Rez G. Angiogenesis is continuous with two peaks during azaserine-induced rat pancreatic adenocarcinoma progression: an electron microscopic morphometrical study. Oncol Rep. 2003;10:1999–2004. [PubMed] [Google Scholar]
- 38.Weibel ER. Stereological principles for morphometry in electron microscopic cytology. Int Rev Cytol. 1969;26:235–302. doi: 10.1016/s0074-7696(08)61637-x. [DOI] [PubMed] [Google Scholar]
- 39.Parsons-Wingerter P, Lwai B, Yang MC, Elliott KE, Milaninia A, Redlitz A, Clark JI, Sage EH. A novel assay of angiogenesis in the quail chorioallantoic membrane: stimulation by bFGF and inhibition by angiostatin according to fractal dimension and grid intersection. Microvasc Res. 1998;55:201–14. doi: 10.1006/mvre.1998.2073. [DOI] [PubMed] [Google Scholar]
- 40.Rieder MJ, O'Drobinak DM, Greene AS. A computerized method for determination of microvascular density. Microvasc Res. 1995;49:180–9. doi: 10.1006/mvre.1995.1014. [DOI] [PubMed] [Google Scholar]
- 41.McCarthy BG, Hsieh ST, Stocks A, Hauer P, Macko C, Cornblath DR, Griffin JW, McArthur JC. Cutaneous innervation in sensory neuropathies: evaluation by skin biopsy. Neurology. 1995;45:1848–55. doi: 10.1212/wnl.45.10.1848. [DOI] [PubMed] [Google Scholar]
- 42.Kennedy WR, Nolano M, Wendelschafer-Crabb G, Johnson TL, Tamura E. A skin blister method to study epidermal nerves in peripheral nerve disease. Muscle Nerve. 1999;22:360–71. doi: 10.1002/(sici)1097-4598(199903)22:3<360::aid-mus9>3.0.co;2-j. [DOI] [PubMed] [Google Scholar]
- 43.Mach DB, Rogers SD, Sabino MC, Luger NM, Schwei MJ, Pomonis JD, Keyser CP, Clohisy DR, Adams DJ, O'Leary P, Mantyh PW. Origins of skeletal pain: sensory and sympathetic innervation of the mouse femur. Neuroscience. 2002;113:155–66. doi: 10.1016/s0306-4522(02)00165-3. [DOI] [PubMed] [Google Scholar]
- 44.Emmrich J, Weber I, Nausch M, Sparmann G, Koch K, Seyfarth M, Lohr M, Liebe S. Immunohistochemical characterization of the pancreatic cellular infiltrate in normal pancreas, chronic pancreatitis and pancreatic carcinoma. Digestion. 1998;59:192–8. doi: 10.1159/000007488. [DOI] [PubMed] [Google Scholar]
- 45.Pitchumoni CS. Pathogenesis and managenent of pain in chronic pancreatitis. World J Gastroenterol. 2000;6:490–496. doi: 10.3748/wjg.v6.i4.490. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Foley KM. Treatment of cancer-related pain. J Natl Cancer Inst Monogr. 2004:103–4. doi: 10.1093/jncimonographs/lgh034. [DOI] [PubMed] [Google Scholar]
- 47.Ren K, Dubner R. Descending modulation in persistent pain: an update. Pain. 2002;100:1–6. doi: 10.1016/s0304-3959(02)00368-8. [DOI] [PubMed] [Google Scholar]
- 48.Hunt SP. Pain control: breaking the circuit. Trends Pharmacol Sci. 2000;21:284–7. doi: 10.1016/s0165-6147(00)01496-6. [DOI] [PubMed] [Google Scholar]
- 49.Harris AC, Gewirtz JC. Elevated startle during withdrawal from acute morphine: a model of opiate withdrawal and anxiety. Psychopharmacology (Berl) 2004;171:140–7. doi: 10.1007/s00213-003-1573-0. [DOI] [PubMed] [Google Scholar]
- 50.Fields HL. Pain modulation: expectation, opioid analgesia and virtual pain. Prog Brain Res. 2000;122:245–53. doi: 10.1016/s0079-6123(08)62143-3. [DOI] [PubMed] [Google Scholar]
- 51.Basbaum AI, Fields HL. Endogenous pain control systems: brainstem spinal pathways and endorphin circuitry. Annu Rev Neurosci. 1984;7:309–38. doi: 10.1146/annurev.ne.07.030184.001521. [DOI] [PubMed] [Google Scholar]
- 52.Zagon IS, Verderame MF, McLaughlin PJ. The biology of the opioid growth factor receptor (OGFr) Brain Res Brain Res Rev. 2002;38:351–76. doi: 10.1016/s0165-0173(01)00160-6. [DOI] [PubMed] [Google Scholar]
- 53.Smith JP, Conter RL, Demers TM, McLaughlin PJ, Zagon IS. Elevated levels of opioid growth factor in the plasma of patients with pancreatic cancer. Pancreas. 2000;21:158–64. doi: 10.1097/00006676-200008000-00009. [DOI] [PubMed] [Google Scholar]
- 54.Gaveriaux C, Peluso J, Simonin F, Laforet J, Kieffer B. Identification of kappa- and delta-opioid receptor transcripts in immune cells. FEBS Lett. 1995;369:272–6. doi: 10.1016/0014-5793(95)00766-3. [DOI] [PubMed] [Google Scholar]
- 55.Ness TJ, Gebhart GF. Visceral pain: a review of experimental studies. Pain. 1990;41:167–234. doi: 10.1016/0304-3959(90)90021-5. [DOI] [PubMed] [Google Scholar]
- 56.Borgbjerg FM, Frigast C, Madsen JB, Mikkelsen LF. The effect of intrathecal opioid-receptor agonists on visceral noxious stimulation in rabbits. Gastroenterology. 1996;110:139–46. doi: 10.1053/gast.1996.v110.pm8536850. [DOI] [PubMed] [Google Scholar]
- 57.Lewanowitsch T, Irvine RJ. Naloxone and its quaternary derivative, naloxone methiodide, have differing affinities for mu, delta, and kappa opioid receptors in mouse brain homogenates. Brain Res. 2003;964:302–5. doi: 10.1016/s0006-8993(02)04117-3. [DOI] [PubMed] [Google Scholar]
- 58.Ness TJ, Gebhart GF. Colorectal distension as a noxious visceral stimulus: physiologic and pharmacologic characterization of pseudaffective reflexes in the rat. Brain Res. 1988;450:153–69. doi: 10.1016/0006-8993(88)91555-7. [DOI] [PubMed] [Google Scholar]
- 59.Granville CA, Dennis PA. An overview of lung cancer genomics and proteomics. Am J Respir Cell Mol Biol. 2005;32:169–76. doi: 10.1165/rcmb.F290. [DOI] [PubMed] [Google Scholar]
- 60.Stoff-Khalili MA, Dall P, Curiel DT. From gene therapy to virotherapy for ovarian cancer. Minerva Ginecol. 2004;56:503–14. [PubMed] [Google Scholar]
- 61.Di Sebastiano P, di Mola FF, Bockman DE, Friess H, Buchler MW. Chronic pancreatitis: the perspective of pain generation by neuroimmune interaction. Gut. 2003;52:907–11. doi: 10.1136/gut.52.6.907. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Okazaki K, Yamamoto Y, Kagiyama S, Tamura S, Sakamoto Y, Nakazawa Y, Morita M, Yamamoto Y. Pressure of papillary sphincter zone and pancreatic main duct in patients with chronic pancreatitis in the early stage. Scand J Gastroenterol. 1988;23:501–7. doi: 10.3109/00365528809093901. [DOI] [PubMed] [Google Scholar]
- 63.Ebbehoj N. Pancreatic tissue fluid pressure and pain in chronic pancreatitis. Dan Med Bull. 1992;39:128–33. [PubMed] [Google Scholar]
- 64.Braganza JM. The pathogenesis of chronic pancreatitis. Qjm. 1996;89:243–50. doi: 10.1093/qjmed/89.4.243. [DOI] [PubMed] [Google Scholar]
- 65.Hoogerwerf WA, Zou L, Shenoy M, Sun D, Micci MA, Lee-Hellmich H, Xiao SY, Winston JH, Pasricha PJ. The proteinase-activated receptor 2 is involved in nociception. J Neurosci. 2001;21:9036–42. doi: 10.1523/JNEUROSCI.21-22-09036.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Ohta T, Numata M, Tsukioka Y, Futagami F, Kayahara M, Kitagawa H, Nagakawa T, Yamamoto M, Wakayama T, Kitamura Y, Terada T, Nakanuma Y. Neurotrophin-3 expression in human pancreatic cancers. J Pathol. 1997;181:405–12. doi: 10.1002/(SICI)1096-9896(199704)181:4<405::AID-PATH786>3.0.CO;2-3. [DOI] [PubMed] [Google Scholar]
- 67.Zhu Z, Friess H, diMola FF, Zimmermann A, Graber HU, Korc M, Buchler MW. Nerve growth factor expression correlates with perineural invasion and pain in human pancreatic cancer. J Clin Oncol. 1999;17:2419–28. doi: 10.1200/JCO.1999.17.8.2419. [DOI] [PubMed] [Google Scholar]
- 68.Zhu Z, Kleeff J, Kayed H, Wang L, Korc M, Buchler MW, Friess H. Nerve growth factor and enhancement of proliferation, invasion, and tumorigenicity of pancreatic cancer cells. Mol Carcinog. 2002;35:138–47. doi: 10.1002/mc.10083. [DOI] [PubMed] [Google Scholar]
- 69.Pour PM, Bell RH, Batra SK. Neural invasion in the staging of pancreatic cancer. Pancreas. 2003;26:322–5. doi: 10.1097/00006676-200305000-00002. [DOI] [PubMed] [Google Scholar]
- 70.Koltzenburg M, Bennett DL, Shelton DL, McMahon SB. Neutralization of endogenous NGF prevents the sensitization of nociceptors supplying inflamed skin. Eur J Neurosci. 1999;11:1698–704. doi: 10.1046/j.1460-9568.1999.00590.x. [DOI] [PubMed] [Google Scholar]
- 71.Hawes RH, Xiong Q, Waxman I, Chang KJ, Evans DB, Abbruzzese JL. A multispecialty approach to the diagnosis and management of pancreatic cancer. Am J Gastroenterol. 2000;95:17–31. doi: 10.1111/j.1572-0241.2000.01699.x. [DOI] [PubMed] [Google Scholar]
- 72.Matsuyama T, Ogata S, Sugiura Y, Yoshizumi Y, Aiko S, Aida S, Maehara T. Acinar cell carcinoma of the pancreas eroding the pylorus and duodenal bulb. J Hepatobiliary Pancreat Surg. 2004;11:276–9. doi: 10.1007/s00534-003-0875-2. [DOI] [PubMed] [Google Scholar]
- 73.Wray CJ, Ahmad SA, Matthews JB, Lowy AM. Surgery for pancreatic cancer: recent controversies and current practice. Gastroenterology. 2005;128:1626–41. doi: 10.1053/j.gastro.2005.03.035. [DOI] [PubMed] [Google Scholar]