

Summary
Bacterial tRNA-guanine transglycosylase (Tgt) catalyses the exchange of guanine in the wobble position of particular tRNAs by the modified base preQ1. In vitro, however, the enzyme is also able to insert the immediate biosynthetic precursor, preQ0, into those tRNAs. This substrate promiscuity is based on a peptide switch in the active site, gated by the general acid/base Glu235. The switch alters the properties of the binding pocket to allow either the accommodation of guanine or preQ1. The peptide conformer recognising guanine, however, is also able to bind preQ0. To investigate selectivity regulation, kinetic data for Z. mobilis Tgt were recorded. They show that selectivity in favour of the actual substrate preQ1 over preQ0 is not achieved by a difference in affinity but via a higher turn-over rate. Moreover, a Tgt(Glu235Gln) variant was constructed. The mutation was intended to stabilise the peptide switch in the conformation favouring guanine and preQ0 binding. Kinetic characterisation of the mutated enzyme revealed that the Glu235Gln exchange has, with respect to all substrate bases, no significant influence on kcat. In contrast, KM(preQ1) is drastically increased while KM(preQ0) seems to be decreased. Hence, regarding kcat/KM as an indicator for catalytic efficiency, selectivity of Tgt in favour of preQ1 is abolished or even inverted in favour of preQ0 for Tgt(Glu235Gln). Crystal structures of the mutated enzyme confirm that the mutation strongly favours the binding pocket conformation required for the accommodation of guanine and preQ0. The way this is achieved, however, significantly differs from what was predicted based on crystal structures of wild type Tgt.
Keywords: peptide flip, induced fit, queuine, queuosine, tRNA modification
Introduction
Transfer RNA-guanine transglycosylases (Tgt, E.C. 2.1.2.29) constitute a family of tRNA-modifying enzymes present in all three domains of life. They catalyse the base exchange of specific guanine residues in tRNAs by 7-substituted-7-deazaguanines in a domain specific manner.1,2 Archaeal Tgt inserts preQ0 (for chemical formula see Table 1) at position 15 of the majority of known archaeal tRNAs where it is further converted to archaeosine.3 Bacterial Tgt, in contrast, is involved in a pathway introducing the hypermodified base queuine (Q) into the anticodon position 34 (the “wobble position”) of just four different tRNAs4 (Figure 1). These are specific for His, Tyr, Asp and Asn, respectively, and share a U33G34U35 sequence in common.5,6 The enzyme performs the first tRNA dependent step in queuine biosynthesis replacing guanine34 (G34) by preQ14 (for chemical formula see Table 1). This premodified base is produced from GTP7 by means of the queC, queD, queE and queF gene products.8,9 While the exact functions of QueC, QueD and QueE still have to be figured out, QueF has been identified as an NADPH-dependent oxidoreductase catalysing the reduction of preQ0 to preQ1.10 After incorporation into tRNA, preQ1 is converted to the functional Q base in two subsequent steps performed by S-adenosylmethionine:tRNA ribosyltransferase-isomerase (QueA)11–13 and an unknown coenzyme B12-dependent enzyme14.
Table 1.
Structures of natural substrates of bacterial Tgt and the inhibitor 2-butyl-1H-imidazole-4,5-dicarboxylic acid hydrazide (BIH). In the line “conformation” the functional group of the Leu231/Ala232 peptide bond exposed to the binding pocket is given.
| conformation | carbonyl Leu231H-bonded Glu235 (deprotonated) | amide Ala232H-bonded Glu235 (protonated) | amide Ala232 stacking Glu235 (protonated) |
|---|---|---|---|
| natural substrates |

KM: 0.7 μM |

KM: 1.2 μM |

KM: 0.9 M |
| inhibitor |

Kic: 62 μM |
||
Figure 1. Queuine modification pathway.

AdoMet: S-adenosylmethionine; B12: coenzyme B12; oQ: epoxyqueuine.
The base exchange catalysed by bacterial Tgt follows a ping-pong reaction mechanism15 which is well documented by crystal structure analyses of Tgt from Zymomonas mobilis16–18 (Figure 2). In a first step, tRNA binds to Tgt and the glycosidic bond of G34 is cleaved via a nucleophilic attack of Asp280 producing a covalent Tgt·tRNA complex intermediate. Subsequently, preQ1 replaces guanine within the binding pocket and is incorporated into tRNA in a reverse reaction step. During the base exchange step, replacement of guanine by preQ1 requires an adjustment of the binding pocket geometry (Figures 2(b),(c)). Both guanine and preQ1 are recognised by functional groups of Asp102, Asp156, Gln203, and Gly230. However, to distinguish between the two bases, a peptide bond formed by Leu231 and Ala232 has to flip. G34-tRNA binding results in a water mediated contact of the G34-N7 to the main chain amide of Ala232. Although the moderate resolution of the G34-tRNA bound Tgt crystal structure does not allow to unambiguously extract this structural detail,17 the structure of Tgt bound to the guanine analogous inhibitor 2-butyl-1H-imidazole-4,5-dicarboxylic acid hydrazide (BIH) (Table 1), clearly suggests this binding mode.19 In order to permit efficient preQ1 binding the composition of the binding pocket has to be altered. Proper recognition of the preQ1 amino methyl group requires the CO acceptor functionality of the Leu231/Ala232 peptide bond to be exposed instead of the NH donor (Figure 2(c)). Both peptide bond geometries are stabilised from the protein interior using the side chain carboxyl of Glu235, a residue strictly conserved in all bacterial Tgts. Obviously depending on its protonation state, Glu235 either donates an H-bond to the backwards oriented carbonyl oxygen or accepts an H-bond from the amide of the flipped peptide bond (Figures 2(b),(c)). Consistently, the peptide switch is also observed in crystal structures of the ligand free enzyme in a pH dependent manner. Tgt crystals obtained at pH 5.5 show the amide of the Leu231/Ala232 peptide bond exposed to the binding pocket,18 while the opposite orientation is observed in crystals obtained at pH 8.5 (Figure 3(d)).
Figure 2.

Base exchange mechanism in bacterial Tgt.
Figure 3. Crystal structures of bacterial and archaeal Tgt.
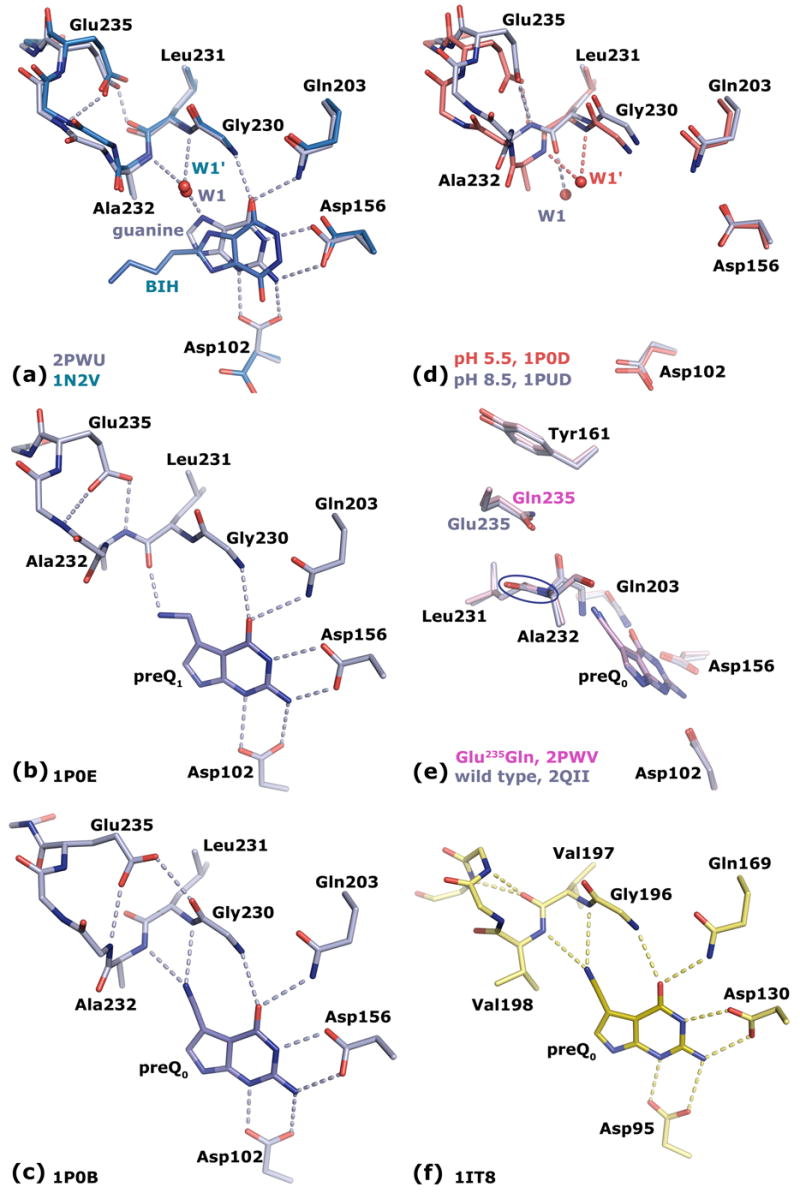
(a) The superposition of bacterial Tgt in complex with the Tgt inhibitor BIH (C atoms in cyan) and guanine (C atoms in grey) shows in both cases an identical conformation for the Leu231/Ala232 peptide bond stabilised by an H-bond of the Leu231 carbonyl to the protonated Glu235 side chain carboxyl. (b) Bacterial Tgt in complex with preQ1. The Leu231/Ala232 peptide bond is switched compared to the guanine/BIH bound situation stabilised by an H-bond of the Ala232 amide to the deprotonated Glu235 carboxyl. (c) Bacterial Tgt in complex with preQ0. The Leu231/Ala232 peptide bond conformation corresponds to that observed for guanine/BIH bound Tgt, but stabilisation surprisingly occurs through a stacking interaction with the protonated Glu235 carboxyl concomitant with a Glu235/Gly236 peptide bond flip. (d) Superposition of ligand-free bacterial Tgt crystallised at pH 5.5 (C atoms in red) and pH 8.5 (C atoms in grey). Protonation of Glu235 at pH 5.5 leads to a binding pocket geometry observed upon binding of guanine/BIH. Deprotonation of Glu235 at pH 8.5 results in a geometry as induced upon preQ1 binding. (e) Superposition of bacterial Tgt and Tgt(Glu235Gln) both in complex with preQ0 reveals identical binding pocket geometries. The Glu235 side chain is stacking between the aromatic ring system of Tyr161 and the Leu231/Ala232 peptide bond (encircled). (f) Archaeal Tgt with bound preQ0. The amide group of the Val197/Val198 peptide bond (corresponding to Leu231/Ala232 in bacterial Tgt) is unable to flip. It permanently exposes the Val198 NH, since the opposing Val197 CO is involved in an invariant backbone H-bond network.
In addition to preQ1, bacterial Tgt in vitro also accepts the biosynthetic precursor of preQ1, namely preQ0, as a substrate.18,20 This substrate promiscuity is a consequence of the above-mentioned peptide switch as binding of preQ0 is facilitated by the Leu231/Ala232 peptide bond conformation required for the accommodation of guanine. Since in bacterial cells both preQ0 and preQ1 are present (even at unknown concentrations), bacterial Tgt must be able to discriminate between these substrates and preferentially incorporate preQ1 into tRNA. For Escherichia coli Tgt there exists evidence that this is achieved through a lower affinity of the enzyme to preQ0, since KM determination revealed a six-fold higher value for preQ0 (2.4 μM) compared to preQ1 (0.4 μM)20 (for Z. mobilis Tgt no kinetic data regarding preQ0 and preQ1 have been reported yet). Although the available complex structures of preQ0 and preQ1 bound to Z. mobilis Tgt do not provide an ostensible explanation for this selectivity, they reveal some significant differences between the binding modes of the two substrates.18
In the Tgt·preQ1 complex structure the Glu235 carboxyl function is clearly present in a deprotonated state, promoting the formation of an H-bond (2.8 Å) to the main chain amide of Ala232. This results in the exposure of the Leu231 main chain carbonyl into the binding pocket enabling it to interact with the presumably protonated and thus positively charged amino methyl group of preQ1 (Figure 3(b)). It should be noted that in this scenario the charges of the Glu235 carboxylate and the protonated preQ1 amino methyl group compensate for each other.
In the Tgt·preQ0 complex the Leu231/Ala232 peptide bond is flipped compared to the preQ1 bound situation, i.e. the Ala232 main chain amide is facing the binding pocket, where it H-bonds the ligand’s acceptor nitrile group. As mentioned above, this conformation of the Leu231/Ala232 peptide bond exactly corresponds to the one observed upon binding of the guanine analogue inhibitor BIH. However, the way this conformation is stabilised by the Glu235 side chain carboxyl, is significantly different. Indeed, the Glu235 carboxyl function does not H-bond in its protonated state the backwards oriented Leu231 carbonyl. It rather stacks with its hydrophobic π face between the Leu231/Ala232 peptide bond and the aromatic ring system of the Tyr161 side chain. This stacking interaction is stabilised by H-bonds formed by the Glu235 carboxyl group to the main chain amide of Val233 (3.1 Å) and to the carbonyl oxygen of Gly230 (2.8 Å). The latter H-bond clearly indicates that also here the Glu235 carboxyl function is present in a protonated state. A shift of the Glu235 side chain by ca. 1.5 Å towards the binding pocket is prerequisite for the stacking interaction. This shift is rendered possible by a Glu235/Gly236 peptide bond flip which so far had not been observed in any other Tgt crystal structure18 (Figures 3(c),(e)).
The present work was aimed to confirm and further analyse the role of Glu235 in the Leu231/Ala232 peptide switch and in substrate promiscuity of bacterial Tgt. In order to mimic a permanently protonated state of the Glu235 carboxyl function we introduced a conservative Glu235Gln mutation into the Z. mobilis Tgt enzyme. This mutation was expected to firmly fix the peptide switch in its NH exposing form independent of the applied pH or any bound substrate. This article describes the consequences of this mutation on substrate specificity of Z. mobilis Tgt as studied by means of enzyme kinetics and crystal structure analyses.
Results
Crystal structure of wild type Z. mobilis Tgt in complex with guanine
The crystal structure of Z. mobilis Tgt in complex with the inhibitor 2-butyl-1H-imidazole-4,5-dicarboxylic acid hydrazide (BIH) had shown, for the first time, a conformation of the Leu231/Ala232 peptide bond, in which the main chain amide of Ala232 was exposed to the binding pocket.19 This had been in contrast to all other crystal structures of Z. mobilis Tgt determined previously, where this peptide bond was oriented such that the main chain carbonyl of Leu231 was facing the binding pocket. Since guanine just like BIH lacks the exocyclic aminomethyl group of preQ1, it had immediately been postulated that binding of guanine to Tgt resulted in the same conformation of the Leu231/Ala232 peptide bond as binding of BIH. Due to the lack of a high resolution crystal structure of guanine-bound Tgt, this assumption had not yet been proven so far. Thus, in order to figure out the exact geometry of the Tgt binding pocket with guanine bound, we crystallised Z. mobilis Tgt in the presence of guanine hydrochloride and determined the complex structure at a resolution of 1.77 Å (Table 2). In the electron density map the bound ligand was clearly defined. As observed for the Tgt·BIH complex, the Leu231/Ala232 peptide bond was present in the NH exposing conformation stabilised by a strong H-bond (2.4 Å) which was formed between the backwards oriented Leu231 carbonyl oxygen and the protonated carboxyl group of Glu235. Also in absolute accordance with the situation in the Tgt·BIH complex an interstitial water was bound H-bonding both the Ala232 main chain amide and the guanine N7 (Figure 3(a)). Thus, the presented structure confirmed that binding of guanine by Tgt leads to the same Leu231/Ala232 peptide bond orientation and Glu235 conformation as binding of the BIH inhibitor.
Table 2.
Crystallographic data collection and refinement statistics
| Crystal data | wild type·guanine (2PWU) | Glu235Gln pH 5.5 (2OKO) | Glu235Gln pH 8.5 (2Z1V) | Glu235Gln·guanine (2POT) |
|---|---|---|---|---|
| A. Data processing | ||||
| Space group | C2 | C2 | C2 | C2 |
| a, b, c (Å) | 90.2, 64.5, 70.8 | 90.4, 65.2, 70.5 | 90.8, 65.2, 70.5 | 89.1, 63.7, 71.1 |
| β (deg.) | 92.8 | 96.4 | 96.1 | 92.9 |
| Resolution rangea (Å) | 50-1.77 (1.80 -1.77) | 50-1.50 (1.53-1.50) | 20–1.55 (1.58–1.55) | 50-1.80 (1.83-1.80) |
| Total no. of reflections | 105,401 | 210,241 | 132,929 | 90,611 |
| No. of unique reflections | 36,424 | 59,104 | 56,093 | 33,979 |
| Completenessa (%) | 94.8 (83.9) | 91.5 (85.1) | 95.6 (78.5) | 97.6 (83.4) |
| Redundancy | 2.8 | 3.5 | 2.3 | 2.5 |
| R(I)syma,b (%) | 4.4 (38.9) | 3.2 (15.9) | 5.8 (23.9) | 4.1 (46.7) |
| I/σ (I) a | 22 (2.3) | 33 (8.0) | 16 (3.8) | 21 (1.5) |
| B. Refinement | ||||
| Rworkc/Rfreed (%) | 20.0/27.1 | 12.5/17.4 | 14.3/20.3 | 21.9/28.6 |
| No. of atoms/residues (molecules) | ||||
| Protein | 2,702/358 | 2,824/365 | 2,860/369 | 2,688/355 |
| Water | 101 | 346 | 305 | 86 |
| Glycerol (cryo buffer) | 18/3 | 96/16 | 24/4 | 42/7 |
| Ligand | 14/1 | --- | --- | 11/1 |
| Mean B-factors (Å2) | ||||
| Protein | 32.8 | 21.8 | 21.6 | 39.0 |
| Water | 34.0 | 34.8 | 31.3 | 40.4 |
| Glycerol (cryo buffer) | 52.6 | 48.1 | 39.4 | 62.6 |
| Ligand | 60.4 | --- | --- | 53.5 |
| rmsd angle (°) | 2.0 | 2.2 | 2.2 | 2.0 |
| rmsd bond (Å) | 0.007 | 0.011 | 0.010 | 0.006 |
| (An)isotropic refinement | isotropic | anisotropic | anisotropic | isotropic |
| Crystal data | Glu235Gln·BIH (2Z1W) | Glu235Gln·preQ0 (2PWV) | Glu235Gln·preQ1 (2Z1X) | |
|
| ||||
| A. Data processing | ||||
| Space group | C2 | C2 | C2 | |
| a, b, c (Å) | 90.9, 65.3, 70.6 | 90.5, 65.2, 70.4 | 89.0, 64.2, 70.6 | |
| β (deg.) | 96.7 | 96.3 | 93.1 | |
| Resolution rangea (Å) | 50-1.63 (1.66-1.63) | 20–1.70 (1.73–1.70) | 50-1.63 (1.66-1.63) | |
| Total no. of reflections | 170,346 | 124,483 | 103,473 | |
| No. of unique reflections | 47,068 | 42,679 | 42,180 | |
| Completenessa (%) | 92.5 (83.9) | 98.3 (96.9) | 89.8 (93.1) | |
| Redundancy | 3.6 | 2.8 | 2.3 | |
| R(I)syma,b (%) | 3.2 (17.2) | 5.9 (49.1) | 7.8 (31.6) | |
| I/σ(I) a | 33 (8.3) | 12 (2.2) | 24 (2.0) | |
| B. Refinement | ||||
| Rworkc/Rfreed (%) | 13.2/19.4 | 20.1/24.5 | 15.3/21.6 | |
| No. of atoms/residues (molecules) | ||||
| Protein | 2,801/362 | 2,799/361 | 2,790/364 | |
| Water | 298 | 228 | 172 | |
| Glycerol (cryo buffer) | 60/10 | 36/6 | 48/8 | |
| Ligand | 15/1 | 13/1 | 13/1 | |
| Mean B-factors (Å2) | ||||
| Protein | 23.5 | 22.8 | 25.7 | |
| Water | 34.0 | 28.6 | 32.9 | |
| Glycerol (cryo buffer) | 54.8 | 48.5 | 53.0 | |
| Ligand | 22.2 | 24.9 | 25.2 | |
| rmsd angle (°) | 2.1 | 2.1 | 2.1 | |
| rmsd bond (Å) | 0.010 | 0.009 | 0.008 | |
| (An)isotropic refinement | anisotropic | isotropic | anisotropic | |
number in parentheses is for highest resolution shell
R(I)sym = Σ |I −〈I〉|/ΣI with I representing the observed intensities and 〈I〉 representing the mean observed intensity
Rwork = Σhkl |Fo − Fc|/Σhkl |Fo|.
Rfree was calculated as for Rwork but on 5 % of the data excluded from the refinement
Enzymatic characterisation of wild type Z. mobilis Tgt
Since no kinetic data were available for the Z. mobilis Tgt enzyme regarding preQ0 and preQ1 we determined and accordingly redetermined kcat and KM concerning guanine, preQ0, preQ1 and tRNA. Redetermination of the Michaelis/Menten parameters for guanine and tRNA was done by means of a well established assay monitoring the incorporation of radio-labelled guanine into tRNA.21 Since no radio-labelled preQ0 and preQ1 is commercially available, we used for these substrates a guanine washout assay which in essence was first described by Hoops et al. 20 Here, the decrease of radioactivity in tRNA labelled in position 34 with [8-3H]-guanine due to the incorporation of the respective substrate is monitored. The kinetic parameters for all four substrates are summarised in Table 3. With respect to preQ0 and preQ1 the six-fold difference in KM reported for the E. coli enzyme was not observed for Z. mobilis Tgt. Within the experimental errors the KM-values of preQ0 and preQ1 were virtually identical and did not differ from that measured for guanine. Instead, the kcat-values of preQ0 and preQ1 differed significantly by a factor of almost 10. While, compared to guanine, preQ1 was incorporated into tRNA with a higher rate, the incorporation rate of preQ0 was substantially lower than that of guanine. Obviously, in contrast to E. coli Tgt, in Z. mobilis Tgt the preferred insertion of preQ1 into tRNA is not achieved by a higher affinity of preQ1 to the enzyme compared to preQ0, but by a clearly higher turnover number.
Table 3.
Kinetic parameters for wild type Tgt and Tgt(Glu235Gln)
| wild type Tgt | tRNATyr* | [3H]-guanine | preQ1 | preQ0 |
|---|---|---|---|---|
| KM [μM] | 0.9 ± 0.2 | 1.2 ± 0.2 | 0.7 ± 0.2 | 0.9 ± 0.2 |
| kcat [s−1] ** | 5.4 · 10−2 | 5.6 · 10−2 | 10.2 · 10−2 | 1.2 · 10−2 |
| kcat/KM [μM−1s−1] | 6.0 · 10−2 | 4.6 · 10−2 | 14.6 · 10−2 | 1.3 · 10−2 |
|
| ||||
| Tgt(Glu235Gln) | tRNATyr * | [3H]-guanine | preQ1 | preQ0 |
|
| ||||
| KM [μM] | 1.0 ± 0.1 | 3.3 ± 0.3 | 32 ± 7 | < 0.5 |
| kcat [s−1] ** | 7.0 ·10−2 | 7.6 ·10−2 | 10.0 ·10−2 | 0.6 ·10−2 |
| kcat/KM [μM−1s−1] | 7.0 ·10−2 | 2.4 ·10−2 | 0.3 ·10−2 | > 1.2 ·10−2 |
Construction and enzymatic characterisation of Z. mobilis Tgt(Glu235Gln)
To further investigate the influence of the Glu235 carboxyl on the Leu231/Ala232 peptide switch and thus on substrate selectivity we introduced a Glu235Gln mutation into Z. mobilis Tgt by site directed mutagenesis. The carboxamide of the mutated residue was intended to mimic a permanently protonated and electrically neutral Glu235 side chain carboxyl. Subsequently, we determined Michaelis/Menten parameters for the mutated enzyme concerning guanine, preQ0, preQ1 and tRNA. The results are summarised in Table 3. As expected, the mutation had no significant influence on the affinity of the tRNA substrate reflected by a virtually unchanged KM compared to wild type Tgt. A slight increase of kcat (7.0· 10−2 s−1 vs. 5.4 · 10−2 s−1 measured for the wild type enzyme) using guanine as second substrate was not considered significant with respect to the error range of the assay. Alike, the Glu235Gln mutation obviously had no significant influence on the turnover numbers for any of the three substrate bases. The kcat values regarding guanine, preQ0 and preQ1 determined for the mutated enzyme agreed well with those determined for the wild-type enzyme. In contrast, however, the mutation caused a measurable change in KM with respect to the substrate bases. While KM(guanine) was only marginally changed, the most drastical impact of the mutation was observed for KM(preQ1). Although this base was still accepted as a substrate, KM was increased by a factor of almost 50 suggesting a noticeable decrease in affinity. Unlike guanine and preQ1, preQ0 seemed to bind with an increased affinity to the mutated enzyme as KM for this substrate was, compared to wild type Tgt, lowered from 0.9 μM to less than 0.5 μM. Due to the slow turnover rate achieved by Tgt, in particular with respect to this substrate, an exact determination of KM was not possible. To assure an excess of substrate even at low concentrations the amount of enzyme used in the assay would have had to be kept too low to obtain a reasonable signal to noise ratio.
In summary, while the Glu235Gln mutation had no significant influence on the turnover number for any of the three substrate bases, KM was drastically increased for preQ1, marginally elevated for guanine and obviously lowered (even though to an unknown extent) for preQ0. Thus, regarding kcat/KM as a measure of catalytic efficiency, the Glu235Gln mutation resulted in a clearing or even inversion of substrate preference from preQ1 to preQ0 (see Table 3). This agrees well with the hypothesis, that the Leu231/Ala232 peptide conformation necessary for guanine and preQ0 accommodation is in wild type Tgt stabilised by the protonated state of Glu235, which was mimicked by the Glu235Gln mutation.
Crystal structure analyses of Tgt(Glu235Gln)
To allow the interpretation of the measured enzyme kinetic data at a structural level, we crystallised the mutated enzyme in its apo form as well as in complex with its substrates and the guanine analogous inhibitor BIH (Table 2).
Two structures of ligand-free Tgt(Glu235Gln) derived from crystals grown at pH 5.5 and pH 8.5, respectively, were determined at nominal resolutions of better than 1.6 Å. In the electron densities of both structures the residues of and near the active site were excellently defined. As expected, in contrast to wild type Tgt no pH-dependent conformational change was observed for the Leu231/Ala232 peptide bond. Independently whether crystallisation had been performed at pH 5.5 or 8.5, the carbonyl of this peptide bond was oriented backwards while the amide was exposed into the binding pocket. Surprisingly, however, the way this conformation was stabilised by the Gln235 carboxamide did not correspond to the way it is stabilised by the protonated Glu235 carboxyl in wild type apo Tgt crystallised at pH 5.5. Rather, the geometry of the binding pocket was virtually identical to the one observed in the structure of wild type Tgt with preQ0 bound. No H-bond was formed between the Gln235 carboxamide and the Leu231 carbonyl oxygen. Instead, the carboxamide stacked between the Leu231/Ala232 peptide bond and the side chain aromatic ring system of Tyr161. As a consequence, the Gln235 side chain was shifted towards the binding pocket concomitant with a Gln235/Gly236 peptide flip so far only observed in preQ0 bound wild type Tgt (with Gln235 being replaced by Glu235). To investigate the influence of a bound substrate on the active-site geometry of Tgt(Glu235Gln), we determined the crystal structures of the mutated enzyme in complex with preQ0, guanine, the BIH inhibitor, and preQ1, respectively.
The Tgt(Glu235Gln) · preQ0 complex structure was determined at a resolution of 1.70 Å. The electron density for the preQ0 ligand was clearly defined. Superposition of the Tgt(Glu235Gln) · preQ0 complex structure and the structures of the ligand-free mutated enzyme revealed that the binding pocket geometry was virtually identical in all three structures (Figure 4(a)) and exactly corresponded to the one observed in preQ0 bound wild type Tgt (Figure 3(e)). The nitrile nitrogen of preQ0 was present at a position occupied by a glycerol oxygen picked up from the cryo buffer in the structures of uncomplexed mutated Tgt (Figure 4(a)).
Figure 4. Crystal structures of Tgt(Glu235Gln).
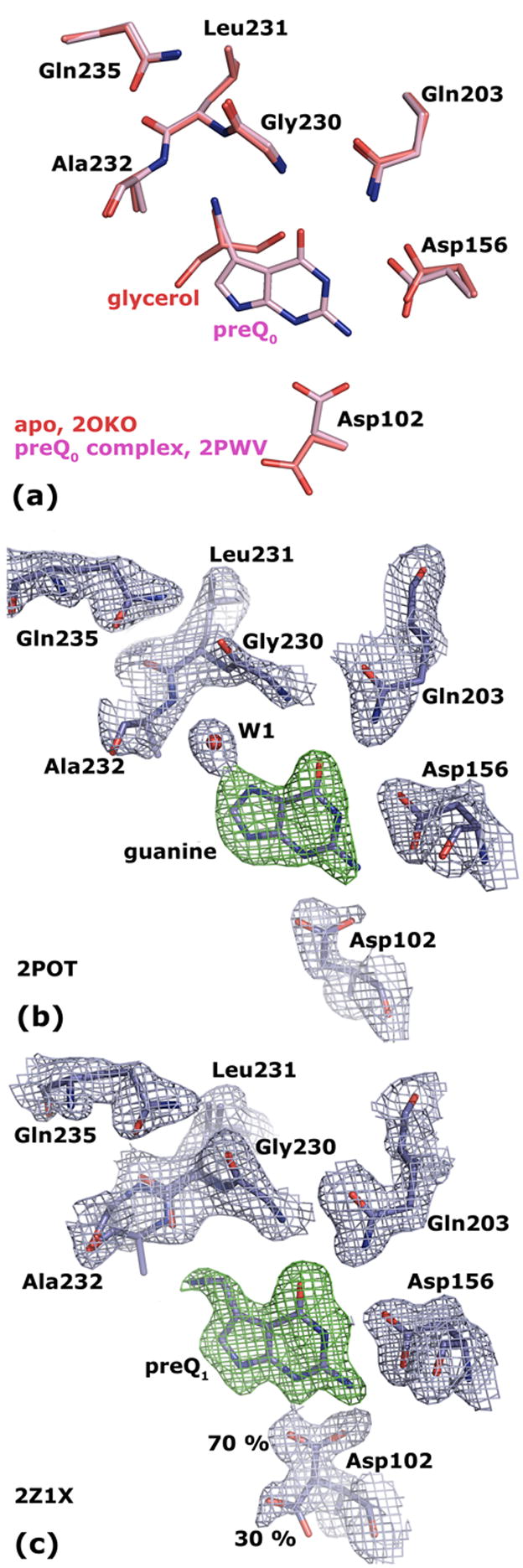
(a) Superposition of Tgt(Glu235Gln) in its apo form (crystallised at pH 5.5; the structure obtained at pH 8.5 looks identical) and in complex with preQ0 reveals equal binding pocket geometries. (b) 2 |Fo| − |Fc| electron density map of Tgt(Glu235Gln) in complex with guanine contoured at 1.5 σ (blue). The green density represents an |Fo| − |Fc| map with the ligand omitted from the calculation contoured at 3.0 σ. (c) 2 |Fo| − |Fc| electron density map of Tgt(Glu235Gln) in complex with preQ1 contoured at 1.5σ (blue). The green density represents an |Fo| − |Fc| map with the ligand omitted from the calculation contoured at 3.0 σ. The electron density reveals a split conformation for the Asp102 side chain indicating an uncomplete occupancy of the binding pocket with the substrate base. The Leu231/Ala232 peptide bond is present in two different conformations. The occupancy of the ligand and of the predominant Leu231/Ala232 and Asp102 conformers refine to 0.7.
The crystal structures of Tgt(Glu235Gln) in complex with guanine and with the guanine analogous inhibitor BIH were determined at a resolution of 1.80 Å and 1.63 Å, respectively. In both structures the electron densities for the ligands as well as for the residues constituting the binding pockets were excellently defined (for the guanine complex see Figure 4(b)). It was clearly visible that also binding of guanine or BIH did, compared to the apo-form, not result in any conformational change of the Leu231/Ala232 peptide bond or of Gln235. As in guanine or BIH-bound wild-type Tgt, the exposed Ala232 main chain amide formed an H bond to an interstitial water molecule, which was further H-bonded to N7 of guanine or the corresponding nitrogen of BIH, respectively. In contrast to wild type Tgt, however, this conformation was not stabilised by an H-bond formed between the Leu231 carbonyl and the carboxamide of Gln235 but by a stacking interaction between the Leu231/Ala232 peptide bond and the Gln235 side chain.
In order to understand the way preQ1 was bound by the mutated Tgt we determined the crystal structure of the Tgt(Glu235Gln) · preQ1 complex at a resolution of 1.63 Å. Also in this structure the electron density attributable to the ligand was very well defined. A split conformation of the Asp102 side chain, however, indicated an incomplete occupancy of the binding pocket with preQ1 (Figure 4(c)). In apo structures of Tgt the Asp102 side chain carboxylate is oriented towards the outside of the binding pocket H-bonding the side chain amide of Asn70 as well as the main chain amide of Thr71. Once a substrate base has bound the Asp102 side chain rotates into the binding pocket towards the ligand in order to H-bond its exocyclic amine at position 3.22 In the Tgt(Glu235Gln) · preQ1 complex structure ca. 30 % of Asp102 exhibited a side chain conformation pointing towards the outside of the binding pocket suggesting a portion of the binding pocket being unoccupied by the ligand. This was in line with the fact that KM(preQ1) was drastically increased for Tgt(Glu235Gln). As in all other structures of Tgt(Glu235Gln) the Gln235 side chain stacked on top of the Leu231/Ala232 peptide bond. Accordingly, the conformation of the Gln235/Gly236 peptide bond remained also here unchanged compared to all other Tgt(Glu235Gln) structures as well. Interestingly, however, the electron density attributable to the Leu231/Ala232 peptide bond could only been interpreted such that it was present in two different conformations. One conformer corresponded exactly to the one observed in the remaining Tgt(Glu235Gln) structures with the main chain amide of Ala232 being exposed to the binding pocket. Indeed, this conformation was hardly compatible with a bound preQ1 molecule due to unfavourable interactions between the Ala232 main chain amide and the amino methyl group of preQ1. The fact that this conformation could be refined to an occupancy of ca. 30 % agreed well with the assumption that a portion of the binding pocket was devoid of ligand. In the second conformer the Leu231/Ala232 peptide bond was flipped enabling the exposed Leu231 carbonyl oxygen to H-bond the amino methyl group of preQ1. This geometry combining the exposed Leu231 carbonyl with the stacking conformation of Gln235 (or Glu235) had never been observed in any Tgt crystal structure before. As a consequence, exposure of the Leu231 carbonyl into the binding pocket does not necessarily require the deprotonated carboxyl of Glu235 H-bonding the backwards oriented Ala232 main chain amide. It is also compatible with a stacking interaction of the Gln235 side chain, although this mode of stabilisation is obviously energetically less favourable as reflected by the drastically increased KM(preQ1) measured for Tgt(Glu235Gln).
Discussion
In this study we have confirmed and further analysed the role of Glu235 as a general acid/base not directly involved in catalysis but facilitating the binding of various substrates with different H-donor and -acceptor properties. The high resolution crystal structure of Z. mobilis Tgt in complex with guanine presented in this work gives the final proof that binding of each of the three substrate bases (guanine, preQ0 and preQ1) triggers a different geometry of the Tgt binding pocket. Prerequisite of the substrate promiscuity observed in Tgt is the ability of the Leu231/Ala232 peptide bond to flip and thus to change its orientation depending on the bound substrate. Remarkably, the bound substrate not only influences the orientation of this peptide bond but, moreover, the way it is stabilised by the strictly conserved Glu235. Obviously, the highly flexible interplay of the Leu231/Ala232 peptide bond and the general acid/base Glu235 facilitates an optimal adaptation of the Tgt binding pocket to each of its substrate bases. This is reflected by the virtually identical KM values of Z. mobilis Tgt for guanine, preQ0 and preQ1 as determined in this study. Moreover, our results show that the preferred incorporation of the “correct” substrate preQ1 into tRNA over its biosynthetic precursor preQ0 is achieved by a ca. 10 fold faster turnover rate. It should be noted, however, that to the best of our knowledge it has not been investigated so far, if QueF, the nitrile reductase reducing preQ0 to preQ1, is also acting on preQ0 incorporated into tRNA. In this case preQ0 could represent an in vivo substrate of Tgt as well. The observed differences in kcat for the three substrate bases may hardly be explained by their binding modes observed in the corresponding crystal structures, but possibly may be deduced from their chemical properties. In each case nitrogen N9 of the respective base nucleophilically attacks C1 of the covalently bound tRNA ribose34 during catalysis (Figure 2(c) and Table 1). In preQ1 this nitrogen is the most potent nucleophile, as it is in no electronic conjugation with the exocylic amino methyl group. In guanine, N9 is less nucleophilic due to an electron withdrawing effect of N7 being in conjugation with N9. The preQ0 substrate is the least reactive base in this series. Here, the exocyclic nitrile function is conjugated to the endocyclic nitrogen, resulting in an even more pronounced electron withdrawing effect. This is fully consistent with the observed decrease in turnover in the order preQ1 > guanine > preQ0. The virtually identical KM-values for the three substrate bases indicate that the conformation of the Leu231/Ala232 peptide bond as well as the way it is stabilised by the Glu235 side chain carboxyl may have little influence on the energetic state of the substrate binding pocket.
With intent to create a bacterial Tgt with altered substrate specificity Glu235 was mutated to Gln in an attempt to mimic a permanently protonated Glu235 side chain carboxyl. Since the Leu231/Ala232 peptide bond conformation required for the accommodation of preQ1 involves stabilisation by a deprotonated Glu235 the mutation was expected to strongly disfavour or even disable preQ1 binding while leaving unaffected or even improving the binding of guanine and preQ0. Kinetic characterisation of Tgt(Glu235Gln) showed that the change of KM(guanine) caused by the mutation was at the border of significance, while KM(preQ0) seemed to be (to an unknown extent) decreased compared to wild type Tgt. Although KM(preQ1) of the mutated enzyme was increased by a factor of almost 50, the remaining affinity to this substrate was still unexpectedly high. The crystal structures of Tgt(Glu235Gln) in its apo-form as well as in complex with various ligands provided an explanation to the observed kinetic data. The same geometry of the binding pocket was observed, no matter if the mutated enzyme was crystallised in its apo-form (independent of the applied pH) or in complex with guanine, BIH, or preQ0. In each case, the Ala232 main chain amide was exposed to the binding pocket, stabilised by a stacking interaction with the Gln235 side chain carboxamide. This conformation was analogous to that observed in wild 231 type Tgt bound to preQ0. Seemingly, a surprisingly strong H-bond formed between the Leucarbonyl and the protonated Glu235 side chain carboxyl (2.4 Å) plays an important role in stabilising the binding pocket geometry observed in wild type Tgt crystallised at pH 5.5 or in complex with guanine/BIH. Due to its less electronegative nitrogen and the presence of two hydrogens over which the positive partial charge is distributed the carboxamide of Gln235 is obviously not able to form as strong an H-bond to the Leu231 carbonyl rendering the stacking interaction energetically more favourable. Hence, the binding pocket of Tgt(Glu235Gln) seems predestined for the accommodation of preQ0, since independent of any pH no structural rearrangement is required for its binding. A priori the binding pocket is present in a geometry identical to the one induced upon preQ0 binding in wild-type Tgt. This finding is supported by the (even though to an unknown extent) reduced KM(preQ0) determined for Tgt(Glu235Gln). As a surprise, the crystal structure of Tgt(Glu235Gln) in complex with preQ1 revealed a binding pocket geometry featuring an exposed Leu231 carbonyl stabilised by a Gln235 stacking interaction. This unexpected geometry showed that the stacking interaction still allows the Leu231/Ala232 peptide flip. As a result, the substrate specificity achieved by the Glu235Gln mutation was not as strict as that observed for archaeal Tgt which is absolutely unable to bind preQ1. It permanently exposes the amide group of the Val197/Val198 peptide bond (corresponding to Leu231/Ala232 in bacterial Tgt) allowing both the binding of guanine and of preQ0 but not of preQ1. The peptide flip necessary to accommodate this base is rendered impossible, since the opposing carbonyl of this peptide bond is involved in an invariant backbone H-bond network which does not imply the carboxyl group of an acidic amino acid1,23 (Figure 3(f)).
In summary, the mutated Tgt(Glu235Gln) investigated in this study behaved in several details different from what was expected, although a more conservative exchange than glutamic acid/glutamine can hardly be performed. The present work clearly shows the indispensability of crystal structure analyses for the correct interpretation of kinetic data obtained from mutated proteins.
Furthermore, this study provides an example that mutation of a residue which does not even directly interact with any ligand enables the inversion of substrate selectivity of an enzyme. This may generally underline the importance to consider residues in the “second sphere” of binding pockets during attempts of creating enzymes with new binding properties.
The successful modulation of selectivity is an important step towards the understanding of selectivity and specificity determining features in Tgts. In E. coli Tgt replacement of Asp156 (Z. mobilis numbering) by Asn altered the specificity towards xanthine even though at the expense of a reduced catalytic activity.24 In this context it might be interesting to study mutational exchanges in the close neighbourhood of the attacking nucleophile Asp280. In bacterial Tgt this residue is tightly kept in position by H-bonds formed with Gly261 and Tyr258.17 In archaeal Tgt the tyrosine is replaced by a strictly conserved histidine.1 Likely, this exchange will have pronounced influence on the catalytic properties.
These considerations provide a perspective towards an ambitious goal: the modification of substrate specificity towards bases other than preQ0 or preQ1 and their efficient incorporation into the wobble position of tRNAs. This would allow to study the translational process in more detail via well designed modulation of the accuracy of the wobble base pairing.
Materials and Methods
Cloning and Tgt preparation
The QuikChange™ site-directed mutagenesis kit (Stratagene) was used to introduce a Glu235Gln mutation into the wild-type tgt expression plasmid pET9d-ZM425 following the vendor’s protocol. The primers used for mutagenesis were E235Q-s (5′-GGG GGA TTG GCT GTG GGT CAA GGA CAG GAT GAA ATG-3′) and E235Q-a (5′-CAT TTC ATC CTG TCC TTG ACC CAC AGC CAA TCC CCC-3′) (mutation underlined). Sequencing of the entire tgt gene (MWG Biotech, Ebersberg) confirmed the presence of the desired mutation as well as the absence of any further unwanted mutation. Subsequently, the mutated plasmid pET9d-ZM4-E235Q was transformed into E. coli BL21(DE3) pLysS cells. These cells were used for the preparation of the Tgt enzyme following the method described by Romier et al. .26
Preparation of tRNATyr
Preparation of E. coli tRNATyr (ECY2) via in vitro transcription was done following the method described by Curnow et al. .27
Kinetic parameters
Kinetic parameters for Tgt and Tgt(Glu235Gln) have been determined for the different substrates essentially as described in Meyer et al. .21
Michaelis-Menten parameters for tRNA and guanine were determined monitoring incorporation of radioactively labelled guanine into tRNATyr in position 34. Kinetic data for both substrates were determined separately in triplicate and average values were calculated. Kinetic parameters for guanine were measured using 150 nM Tgt, 15 μM unlabelled tRNATyr, and variable concentrations of [8-3H]-guanine (0.9 Ci/mmol; Hartmann Analytic) in the range of 0.5 – 20 μM in buffer solution (200 mM HEPES pH 7.3, 20 mM MgCl2) and 2.95 μM (≡ 5 % of critical micellar concentration) Tween 20 (Roth). Kinetic parameters for tRNA were measured using 150 nM Tgt, 20 μM [8-3H]-guanine, and variable concentrations of tRNATyr (0.25 – 15 μM). Initial velocities of the base exchange reaction in counts per minute were converted to [μM/min] using a calibration constant derived from liquid scintillation counting of [8-3H]-guanine solutions with variable concentrations. Kinetic parameters were determined via double-reciprocal linearization using the method of Eadie-Hofstee and linear regression using GraFit.28
Michaelis-Menten parameters for preQ0 and preQ1 were calculated via monitoring the loss of [8-3H]-guanine from tRNATyr radio-labelled in position 34. To produce radioactively labelled tRNA 50 μM unmodified tRNATyr was incubated with 0.5 μM Tgt and 10 μM [8-3H]-guanine (11.8 Ci/mmol; Hartmann Analytic) in Tgt assay buffer for 2 h. Tgt and free guanine/[8-3H]-guanine were extracted from the reaction mixture by the addition of equal volumes of Roti-Phenol (Roth) and chloroform : isoamylalcohol (24:1). The aqueous supernatant was once more treated with an equal volume of chloroform : isoamylalcohol (24:1). From the aqueous supernatant containing the radioactively labelled tRNA samples were retained to determine the specific activity of the radio-labelled tRNA. An additional purification step was performed via gel filtration using NAP-columns (GE Healthcare, Life Science) and Tgt assay buffer. From the eluted solution again samples were retained to derive the final concentration of the radio-labelled tRNA from its specific activity. For each preparation it amounted to ca. 20 μM. Kinetic parameters for preQ0 and preQ1 were measured using 150 nM wild type Tgt or Tgt(Glu235Gln) and 8 μM radio-labelled tRNATyr. The concentration of preQ0 was varied in a range of 0.5 – 15 μM. For preQ1 variable concentrations in a range of 0.5 – 15 μM for wild type Tgt and 2 – 80 μM for Tgt(Glu235Gln) were applied. Initial velocities in counts per minute were calculated from the decreasing tritium labelling level of tRNA due to the incorporation of the respective substrate bases. Initial velocities in counts per minute were converted to [μM/min] using a calibration constant derived from liquid scintillation counting of guanine/[8-3H]-guanine solutions with variable concentrations. Kinetic parameters were determined via double-reciprocal linearization using the method of Eadie-Hofstee and linear regression using GraFit.28
Crystal structure analyses
Tgt(Glu235Gln) crystals suitable for ligand soaking were produced in a two step procedure. Droplets were prepared by mixing 2 μL of concentrated protein solution (14 mg/mL) with 2 μL reservoir solution [100 mM MES, pH 5.5, 1 mM DTT, 13 % (w/v) PEG 8,000, 10 % (v/v) DMSO]. Micro-crystals were grown at 291 K using the hanging-drop vapour diffusion method in the presence of 1.0 mL of reservoir solution of the respective seeding buffer. Micro crystals grew within two weeks.
Subsequently, macro-seeding was performed under similar conditions to obtain crystals of the uncomplexed protein. Again droplets were prepared by mixing 2 μL of concentrated protein solution with 2 μL macro-seeding reservoir solution [100 mM MES, pH 5.5, 1 mM DTT, 8 % (w/v) PEG 8,000, 10 % (v/v) DMSO]. One micro-crystal was transferred into this solution. Single crystals with a size of approximately 0.7 × 0.7 × 0.2 mm3 grew within two to four weeks.
To obtain crystals of apo-Tgt(Glu235Gln) at pH 8.5, buffers were used identical to those described above but with 100 mM TrisHCl pH 8.5 instead of 100 mM MES pH 5.5 as buffer system.
To allow cocrystallization of the mutated Tgt in complex with preQ0 the compound was dissolved in DMSO and added to the macro-seeding droplet to a final concentration of 2 mM. Crystals of mutated Tgt complexed to preQ1, BIH and guanine as well as crystals of wild type Tgt bound to guanine were produced by a soaking procedure. The compounds were dissolved in DMSO and added to 2 μL of a crystallization droplet to a final concentration of 10 to 20 mM. Finally, a single Tgt crystal was transferred into the droplet and soaked for 30 min.
For data collection, crystals were cryoprotected using glycerol. The glycerol and PEG 8,000 concentrations of the macro-seeding buffers were increased stepwise by transferring the crystals to six different 2 μL cryo-droplets each with 5 to 30 min incubation times [glycerol concentrations (v/v): 5 % → 10 % → 15 % → 20 % → 25 % → 30 %; and PEG 8,000 concentrations (w/v): 5.0 % → 6.3 % → 7.5 % → 8.0 % → 8.8 % → 9.8 %, respectively]. These droplets also contained the ligands at equivalent concentrations compared to soaking and co-crystallization conditions. The cryo-soaked crystals were flash-frozen in liquid N2. Data sets were collected at cryo conditions (100 K) with CuKα radiation (λ = 1.5418 Å) using a Rigaku RU-300 rotating-anode generator at 50 kV and 90 mA equipped with Xenocs focussing optics and an R-AXIS IV detector. All crystals tested exhibited monoclinic symmetry in space group C2 containing one monomer per asymmetric unit with Matthews coefficients of 2.3 – 2.4. Data processing and scaling was performed using the HKL2000 package.29 For all refined structures unit cell dimensions for the crystals, data collection and processing statistics are given in Table 2.
For crystals grown at pH 5.5 coordinates of the apo Tgt crystal structure grown at pH 5.5 (PDB-code: 1P0D) were directly applied for initial rigid-body refinement of the protein molecule followed by repeated cycles of conjugate gradient energy minimisation, simulated annealing and B-factor refinement using the CNS program package.30 For the crystal grown at pH 8.5 the coordinates of the apo Tgt crystal structure grown at pH 8.5 (PDB-code: 1PUD) were applied. Refinement at the later stages for all other structures was performed with SHELXL.31 Here, up to 50 cycles of conjugate gradient minimisation were performed with default restraints on bonding geometry and B-values: 5 % of all data were used for Rfree calculation. Amino acid side chains were fit to σA-weighted 2 |Fo|−|Fc| and |Fo|−|Fc| electron density maps using Coot.32 Water, glycerol molecules, and ligands were located in the difference electron density and added to the model for further refinement cycles. Anisotropic conjugate gradient refinement resulted in a significant improvement of the models built for apo Tgt(Glu235Gln) (pH 5.5 and 8.5), Tgt(Glu235Gln)·BIH, and Tgt(Glu235Gln)·preQ1. During the last refinement cycles, riding H-atoms were introduced for the protein residues (not for the ligand) without using additional parameters. All final models were validated using PROCHECK.33 Data refinement statistics are given in Table 2.
Protein Data Bank Accession Codes
The following Protein Data Bank (PDB) accession codes were allocated to the crystal structures determined during this work:
Tgt·guanine: 2PWU; Tgt(Glu235Gln) pH 5.5: 2OKO; Tgt(Glu235Gln) pH 8.5 2Z1V; Tgt(Glu235Gln) · guanine 2POT; Tgt(Glu235Gln) · BIH 2Z1W; Tgt(Glu235Gln) · preQ0 2PWV; Tgt(Glu235Gln) · preQ1: 2Z1X (also given in Table 2).
Alignment and Figures
Alignment of structures with similar or identical sequences was performed with the alignment function implemented in Pymol (http://www.pymol.org).
Figures were prepared using Isis Draw (MDL, San Leandro, USA) and Pymol.
Acknowledgments
This research was supported by the NIH (Grant GM065489) and by the Deutsche Forschungsgemeinschaft (Grants KL1204/1-3, KL1204/9-1 and the Graduiertenkolleg “Protein Function at the Atomic Level”). We thank Christian Sohn for his support during X-ray data collection, Hans-Dieter Gerber for synthesising preQ1 and Andreas Blum for helpful discussions.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Stengl B, Reuter K, Klebe G. Mechanism and substrate specificity of tRNA-guanine transglycosylases (Tgts): tRNA modifying enzymes from the three different kingdoms of life share a common catalytic mechanism. ChemBioChem. 2005;6:1–15. doi: 10.1002/cbic.200500063. [DOI] [PubMed] [Google Scholar]
- 2.Iwata-Reuyl D. Biosynthesis of the 7-deazaguanosine hypermodified nucleosides of transfer RNA. Bioorg Chem. 2003;31:24–43. doi: 10.1016/s0045-2068(02)00513-8. [DOI] [PubMed] [Google Scholar]
- 3.Watanabe M, Matsuo M, Tanaka S, Akimoto H, Asahi S, Nishimura S, Katze JR, Hashizume T, Crain PF, McCloskey JA, Okada N. Biosynthesis of archaeosine, a novel derivative of 7-deazaguanosine specific to archaeal tRNA, proceeds via a pathway involving base replacement on the tRNA polynucleotide chain. J Biol Chem. 1997;272:20146–20151. doi: 10.1074/jbc.272.32.20146. [DOI] [PubMed] [Google Scholar]
- 4.Okada N, Nishimura S. Isolation and characterization of a guanine insertion enzyme, a specific tRNA transglycosylase, from Escherichia coli. J Biol Chem. 1979;254:3061–3066. [PubMed] [Google Scholar]
- 5.Nakanishi S, Ueda T, Hori H, Yamazaki N, Okada N, Watanabe K. A UGU sequence in the anticodon loop is a minimum requirement for recognition by Escherichia coli tRNA-guanine transglycosylase. J Biol Chem. 1994;269:32221–32225. [PubMed] [Google Scholar]
- 6.Curnow AW, Garcia GA. tRNA-guanine transglycosylase from Escherichia coli. Minimal tRNA structure and sequence requirements for recognition. J Biol Chem. 1995;270:17264–17267. doi: 10.1074/jbc.270.29.17264. [DOI] [PubMed] [Google Scholar]
- 7.Kuchino Y, Kasai H, Nihei S, Nishimura S. Biosynthesis of the modified nucleoside Q in transfer RNA. Nucleic Acids Res. 1976;3:393–398. doi: 10.1093/nar/3.2.393. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Reader JS, Metzgar D, Schimmel P, de Crécy-Lagard V. Identification of four genes necessary for biosynthesis of the modified nucleoside queuosine. J Biol Chem. 2004;279:6280–6285. doi: 10.1074/jbc.M310858200. [DOI] [PubMed] [Google Scholar]
- 9.Gaur R, Varshney U. Genetic analysis identifies a function for the queC (ybaX) gene product at an initial step in the queuosine biosynthetic pathway in Escherichia coli. J Bacteriol. 2005;187:6893–6901. doi: 10.1128/JB.187.20.6893-6901.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Van Lanen SG, Reader JS, Swairjo MA, de Crécy-Lagard V, Lee B, Iwata-Reuyl D. From cyclohydrolase to oxidoreductase: discovery of nitrile reductase activity in a common fold. Proc Natl Acad Sci U S A. 2005;102:4264–4269. doi: 10.1073/pnas.0408056102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Van Lanen SG, Kinzie SD, Matthieu S, Link T, Culp J, Iwata-Reuyl D. tRNA modification by S-adenosylmethionine:tRNA ribosyltransferase-isomerase. Assay development and characterization of the recombinant enzyme. J Biol Chem. 2003;278:10491–10499. doi: 10.1074/jbc.M207727200. [DOI] [PubMed] [Google Scholar]
- 12.Mathews I, Schwarzenbacher R, McMullan D, Abdubek P, Ambing E, Axelrod H, et al. Crystal structure of S-adenosylmethionine:tRNA ribosyltransferase-isomerase (QueA) from Thermotoga maritima at 2.0 Å resolution reveals a new fold. Proteins. 2005;59:869–874. doi: 10.1002/prot.20419. [DOI] [PubMed] [Google Scholar]
- 13.Grimm C, Ficner R, Sgraja T, Haebel P, Klebe G, Reuter K. Crystal structure of Bacillus subtilis S-adenosylmethionine:tRNA ribosyltransferase-isomerase. Biochem Biophys Res Comm. 2006;351:695–701. doi: 10.1016/j.bbrc.2006.10.096. [DOI] [PubMed] [Google Scholar]
- 14.Frey B, McCloskey J, Kersten W, Kersten H. New function of vitamin B12: cobamide-dependent reduction of epoxyqueuosine to queuosine in tRNAs of Escherichia coli and Salmonella typhimurium. J Bacteriol. 1988;170:2078–2082. doi: 10.1128/jb.170.5.2078-2082.1988. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Goodenough-Lashua DM, Garcia GA. tRNA-guanine transglycosylase from E. coli: a ping-pong kinetic mechanism is consistent with nucleophilic catalysis. Bioorg Chem. 2003;31:331–344. doi: 10.1016/S0045-2068(03)00069-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Romier C, Reuter K, Suck D, Ficner R. Crystal structure of tRNA-guanine transglycosylase: RNA modification by base exchange. EMBO J. 1996;15:2850–2857. [PMC free article] [PubMed] [Google Scholar]
- 17.Xie W, Liu X, Huang RH. Chemical trapping and crystal structure of a catalytic tRNA guanine transglycosylase covalent intermediate. Nat Struct Biol. 2003;10:781–788. doi: 10.1038/nsb976. [DOI] [PubMed] [Google Scholar]
- 18.Brenk R, Stubbs MT, Heine A, Reuter K, Klebe G. Flexible adaptations in the structure of the tRNA-modifying enzyme tRNA-guanine transglycosylase and their implications for substrate selectivity, reaction mechanism and structure-based drug design. ChemBioChem. 2003;4:1066–1077. doi: 10.1002/cbic.200300644. [DOI] [PubMed] [Google Scholar]
- 19.Brenk R, Naerum L, Gradler U, Gerber HD, Garcia GA, Reuter K, Stubbs MT, Klebe G. Virtual screening for submicromolar leads of tRNA-guanine transglycosylase based on a new unexpected binding mode detected by crystal structure analysis. J Med Chem. 2003;46:1133–1143. doi: 10.1021/jm0209937. [DOI] [PubMed] [Google Scholar]
- 20.Hoops GC, Townsend LB, Garcia GA. tRNA-guanine transglycosylase from Escherichia coli: structure-activity studies investigating the role of the aminomethyl substituent of the heterocyclic substrate preQ1. Biochemistry. 1995;34:15381–15387. doi: 10.1021/bi00046a047. [DOI] [PubMed] [Google Scholar]
- 21.Meyer EA, Donati N, Guillot M, Schweizer WB, Diederich F, Stengl B, Brenk R, Reuter K, Klebe G. Synthesis, biological evaluation, and crystallographic studies of extended guanine based (lin-benzoguanine) inhibitors for tRNA-guanine transglycosylase (Tgt) Helv Chim Acta. 2006;89:573–597. [Google Scholar]
- 22.Brenk R, Meyer EA, Reuter K, Stubbs MT, Garcia GA, Diederich F, Klebe G. Crystallographic study of inhibitors of tRNA-guanine transglycosylase suggests a new structure-based pharmacophore for virtual screening. J Mol Biol. 2004;338:55–75. doi: 10.1016/j.jmb.2004.02.019. [DOI] [PubMed] [Google Scholar]
- 23.Ishitani R, Nureki O, Fukai S, Kijimoto T, Nameki N, Watanabe M, Kondo H, Sekine M, Okada N, Nishimura S, Yokoyama S. Crystal structure of archaeosine tRNA-guanine transglycosylase. J Mol Biol. 2002;318:665–677. doi: 10.1016/S0022-2836(02)00090-6. [DOI] [PubMed] [Google Scholar]
- 24.Todorov KA, Garcia GA. Role of aspartate 143 in Escherichia coli tRNA-guanine transglycosylase: alteration of heterocyclic substrate specificity. Biochemistry. 2006;45:617–625. doi: 10.1021/bi051863d. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Reuter K, Ficner R. Sequence analysis and overexpression of the Zymomonas mobilis tgt gene encoding tRNA-guanine transglycosylase: purification and biochemical characterization of the enzyme. J Bacteriol. 1995;177:5284–5288. doi: 10.1128/jb.177.18.5284-5288.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Romier C, Ficner R, Reuter K, Suck D. Purification, crystallization, and preliminary X-ray diffraction studies of tRNA-guanine transglycosylase from Zymomonas mobilis. Proteins. 1996;24:516–519. doi: 10.1002/(SICI)1097-0134(199604)24:4<516::AID-PROT11>3.0.CO;2-O. [DOI] [PubMed] [Google Scholar]
- 27.Curnow AW, Kung FL, Koch KA, Garcia GA. tRNA-guanine transglycosylase from Escherichia coli: gross tRNA structural requirements for recognition. Biochemistry. 1993;32:5239–5246. doi: 10.1021/bi00070a036. [DOI] [PubMed] [Google Scholar]
- 28.Leatherbarrow R. GraFit 4.09 edit. Erithacus Software Limited; USA: 1999. [Google Scholar]
- 29.Otwinowski Z, Minor W. Processing of X-ray diffraction data collected in oscillation mode. Methods Enzymol. 1997;276:307–326. doi: 10.1016/S0076-6879(97)76066-X. [DOI] [PubMed] [Google Scholar]
- 30.Brunger AT, Adams PD, Clore GM, DeLano WL, Gros P, Grosse-Kunstleve RW, Jiang JS, Kuszewski J, Nilges M, Pannu NS, Read RJ, Rice LM, Simonson T, Warren GL. Crystallography and NMR system: A new software suite for macromolecular structure determination. Acta Crystallogr sect D. 1998;54:905–921. doi: 10.1107/s0907444998003254. [DOI] [PubMed] [Google Scholar]
- 31.Sheldrick GM, Schneider TR. SHELXL: high-resolution refinement. Methods Enzymol. 1997;277b:319–343. [PubMed] [Google Scholar]
- 32.Emsley P, Cowtan K. Coot: model-building tools for molecular graphics. Acta Crystallogr sect D. 2004;60:2126–2132. doi: 10.1107/S0907444904019158. [DOI] [PubMed] [Google Scholar]
- 33.Laskowski RA, MacArthur MW, Moss DS, Thornton JM. PROCHECK: a program to check the stereochemical quality of protein structures. J Appl Crystallogr. 1993;26:283–291. [Google Scholar]
- 34.Stengl B, Meyer EA, Heine A, Brenk R, Diederich F, Klebe G. Crystal structures of tRNA-guanine transglycosylase (TGT) in complex with novel and potent inhibitors unravel pronounced induced-fit adaptations and suggest dimer formation upon substrate binding. J Mol Biol. 2007;370:492–511. doi: 10.1016/j.jmb.2007.04.008. [DOI] [PubMed] [Google Scholar]