

Abstract
This study defines the in vitro phenomenon of ciliated bovine bronchial epithelial cell (BBEC) detachment from the basal epithelium and regulation of cilia motility mediated through protein kinase C epsilon (PKCε). The authors determined the time course of activation and downregulation of PKCε by the known PKC activator phorbol 12-myristate 13-acetate (PMA) and demonstrate that chemical inhibition of PKC by calphostin C or the novel PKC isoform inhibitor Ro 31-8220 induced striking detachment of ciliated BBECs from the basal cell monolayer within 1 hour, independent of apoptosis or necrotic cell death. The results of this study support a possible novel PKCε-mediated signaling pathway through which ciliated cell attachment is maintained.
Keywords: airway epithelium, bronchial epithelial cell, cell detachment, cilia, kinase, protein kinase C epsilon
The protein kinase C (PKC) family of serine/threonine kinases transmits cellular signals via phosphorylation in response to diverse stimuli, regulating biological processes such as proliferation and differentiation. PKCε has been shown in multiple cell types to be activated by second messengers such as diacylglyercerol (DAG), fatty acids, and phosphatidylinositol 3,4,5,-triphosphate (PIP3). The activated kinase translocates from the cytoplasm to the membrane or cytoskeleton in response to DAG or PIP3, whereas binding of certain fatty acids causes translocation to the Golgi network [1]. Specific subcellular localization of PKCε is also enhanced by association with the selective adaptor protein beta’-COP (also called RACK2) within the Golgi apparatus [2], whereas binding to the scaffolding protein receptor of activated C kinase 1 (RACK1) at the cellular membrane can enhance focal adhesion assembly and induce migration and spreading of certain mammalian cell types [3, 4]. Phosphorylation by PKCε has been implicated in the regulation of several diverse mammalian systems such as (1) modulation of stress signals via heat-shock proteins [5]; (2) induction of neurite outgrowth [6]; (3) regulation of sensitivity of gamma aminobutyrate type A receptors to ethanol and other allosteric modulators [7, 8]; (3) cardioprotection against ischemic heart disease [9]; (5) early-stage lipo-polysaccharide (LPS)-mediated macrophage activation [10] and LPS-induced, tumor necrosis factor-α (TNF-α)–stimulated apoptosis [11]; and (6) positive regulation of cell spreading and adhesion [3, 4, 12]. PKCε has also been implicated in the loss of cisplatin-induced cell-cell contacts in the renal epithelial cell line LLC-PK1 [13].
Few specific PKCε-activating proteins within the airway and lung have been identified [14] and the role PKCε plays in inflammatory lung diseases remains unclear. This novel isozyme is likely important because Western blot protein studies demonstrate ubiquitous expression of PKCε in the lung, including bronchial [15] and alveolar epithelial cells [16]. In this context, we hypothesize that PKCε signaling is required for ciliated epithelial cell attachment to the airway epithelium. This study confirms that PKCε is present and active in airway cells and demonstrates that selective chemical inhibition of PKCε causes detachment of clump, ciliated bronchial epithelial cells (BBECs) from the basal epithelial cell monolayer in primary, cultured cells. Utilizing specialized equipment to measure ciliary motility developed in our laboratory (the Sisson-Ammons Video Analysis system or SAVA) [17]. The results of this study support a possible novel PKCε-mediated signaling pathway through which ciliated cell attachment is maintained.
MATERIALS AND METHODS
Cell Culture and Treatments
Primary bovine bronchial epithelial cells (BBECs) were prepared from bovine lung as described previously [18]. Briefly, bronchi were dissected from the lung and digested overnight at 4°C in 0.1% bacterial protease type IV in minimum essential medium (M199 with Earl’s salts; Gibco, Carlsbad, CA). The following day, the bronchi were repeatedly rinsed in M199 containing 10% fetal bovine serum (FBS; Gibco) to collect a mixture >95% viable ciliated and basal epithelial cells lining the lumen. These cells were then washed in M199 medium, counted with a hemacytometer, and 3×106 cells were plated on 60-mm tissue culture dishes coated with 1% type I collagen (Vitrogen; Cohesion, Palo Alto, CA). BBECs were grown in M199 complete medium containing 10% FBS, 50 U/mL penicillin and streptomycin (Gibco), and 2 μg/mL fungizone (Gibco) in a humidified 95% air/5% CO2 incubator at 37°C. Confluent monolayers of primary BBECs (a mixed cell population of clumped, ciliated cells attached to the basal cell monolayer) were obtained in 3 days and treated as described below.
To assess the effect of chemical inhibition of various isoforms of PKC, confluent, submerged monolayers of BBECs were treated with the general PKC inhibitor, calphostin C (1 μM); PKCα inhibitor, Gö 6976 (1 μM); PKCα and PKCγ inhibitor, HBDDE (5 μM); PKCδ inhibitor, rottlerin (20 μM); PKCβ1 inhibitor, hispidin (10 μM); PKCζ inhibitor, myristylated PKCζ(1 μM); PKCβ2 inhibitor, LY 379196 (20 μM), or medium only as a control. In experiments involving the PKC novel isoform inhibitor, Ro 31-8220 (BioMol, Plymouth Meeting, PA), concentrations from 10 nM to 10 μM were tested for effective inhibition of kinase activity and induction of ciliated cell detachment. Optimal PKCε activity inhibition occurred at 10 μM Ro 31-8220 and this concentration of inhibitor was used for subsequent experiments. In some experiments, 100 ng/mL of phorbol 12-myristate 13-acetate (PMA) was added to BBECs to activate PKC. All other reagents not specified were purchased from Sigma.
Fluorescent Localization of PKCε and Acetylated α-Tubulin
Primary BBECs were grown on collagen-coated glass coverslips in 35-mm dishes and fixed using the following method: coverslips were rinsed three times with ice-cold phosphate-buffered saline, pH 7.4 (PBS), fixed with 1% fresh paraformaldehyde in PBS for 15 minutes at 4°C, rinsed with PBS, permeabilized with 0.1% Triton X-100 in PBS for 15 minutes at 4°C, and then rinsed thoroughly with PBS. Primary rabbit anti-PKCε antibodies (Sigma) were diluted in 1% bovine serum albumin (BSA)/PBS (at 1:200) and carefully applied to coverslips. After incubating with the primary antibody overnight at 4°C, coverslips were rinsed with PBS and then incubated with secondary antibodies (purified anti-rabbit immunoglobulin G [IgG] conjugated to fluorescein isothiocyanate [FITC] for the polyclonal antibodies) diluted 1:500 in 1% BSA/PBS for 1 hour at room temperature. Subsequently, IgG-treated coverslips were rinsed 5 times with PBS, mounted on slides, and viewed with a Nikon Diaphot epifluorescence microscope equipped with appropriate filters to discern FITC. Digital micrographs of numerous representative fields were recorded.
To demonstrate that detached BBECs were preferentially ciliated, cells were collected following a 2-hour, 10 μM Ro 31-8220 treatment, cytospun, fixed in methanol, and stained with a monoclonal primary antibody to acetylated α-tubulin (a cilia-specific marker) and a secondary antibody labeled with Alexa488 fluor. Cells were imaged following laser scanning confocal microscopy.
Quantification of Ciliated Cell Detachment
Detached ciliated cells were quantified in BBECs by washing monolayers twice with warmed, serum-free M199. Cells were incubated with Ro 31-8220 (10 μM) for 3 hours at 37°C, followed by collection of the medium and a gentle wash of the cells with 2 mL M199, pooling the supernatants. The cells in the supernatant were spun at 100×g and cells were resuspended and counted using a hemocytometer or Coulter counter. Our ciliated BBEC preparation consists of clump, ciliated primary cells attached to a confluent basal monolayer grown on tissue culture dishes. This preparation resembles the ciliated, goblet, and basal cells that populate the normal airway. The ciliated and basal BBECs in vitro can clearly be distinguished by motility using SAVA.
PKCε Activity Assay
PKCε activity was determined in crude whole cell cytosolic and particulate fractions from BBECs similar to methods described previously [18, 19]. Cell monolayers were flash-frozen in liquid N2 in cell lysis buffer containing 50 mM Tris-HCl (pH 7.4) and 1 μg/mL each of leupeptin A, phenylmethyl-sulphonylfluoride, and aprotinin. The cells were scraped with a cell lifter, sonicated, and centrifuged at 10,000×g for 30 minutes at 4°C. The supernatant was removed (cytosolic fraction) and the pellet was resuspended in cell lysis buffer containing 0.01% Triton X-100 and sonicated again (particulate fraction). To measure PKCε activity specifically, 24 μg/mL PMA, 30 mM dithiotreitol, 150 μM ATP, 45 mM Mg-acetate, the PKCε-specific substrate peptide (ERMRPRKRQGSVRRRV; Calbiochem, San Diego, CA), and 10 μCi/mL [γ-32P]ATP were mixed in a Tris-HCl buffer (pH 7.5). Chilled (4°C) cell lysate (cytosolic or particulate) samples (20 μL) were added to 40 μL of the reaction mix and incubated for 15 minutes at 30°C. This mixture (60 μL) was then spotted onto P-81 phosphocellulose papers (Whatman, Clinton, NJ) to halt incubation, and papers were subsequently washed 5 times in 75 mM phosphoric acid for 5 minutes, washed once in 100% ethanol for 1 minute, dried, and counted in nonaqueous scintillant (National Diagnostics, Atlanta GA). PKCε activity was expressed in relation to the total amount of cellular protein assayed as picomoles of phosphate incorporated per minutes per milligram.
Cell Viability Assay
Medium supernatant (50 μL) from cell monolayers treated with PKC inhibitors for 2 hours or medium alone was sampled for cell viability assays. In addition, confluent 60-mm dishes of cells were lysed as a positive control for lactate dehydrogenase (LDH) release. Cell viability was determined by a commercially available kit (Sigma) to measure LDH release, according to the manufacturer’s instructions.
Apoptosis Quantitation
Briefly, BBECs were treated with 10 μM Ro 31-8220, 1 μM calphostin C, or 1 μM positive control tumor necrosis factor-α (TNF-α) for 2 hours and the supernatant was collected into 15-mL conical tubes. This was spun at 100×g for 5 minutes to concentrate cells followed by resuspension of cells at concentration of 2×105 cells/mL in 1% paraformaldehyde. Cells (100 μL) were cytospun (Wescor) onto each slide. Apoptosis of the BBECs following treatments was determined with ApopTag In Situ Apoptosis Detection Kit (Intergen, Purchase, NY) following the peroxidase staining protocol for the cells. The cells were analyzed and 6 fields of view of 100 cells were counted on an Olympus IMT-2 Microscope.
Ciliary Beat Frequency Measurement
A detailed description and characterization of the Sisson-Ammons Video Analysis (SAVA) system for measurement of ciliary beat frequency (CBF) is provided in Sisson and Colleagues [17]. Actively beating ciliated cells were observed and their motion quantified by whole field analysis using SAVA, which combines phase contrast microscopy and computerized frequency spectrum analysis [17]. The number of motile points for each 3-second digital video image was determined using a software algorithm in SAVA. The algorithm assesses if a change in light intensity occurs in a 16-pixel zone in which each zone represents a 4×4-pixel area. For every 640×480-pixel video image, the number of motile zones are calculated from a possible 19,200 total zones. As cilia stop beating or ciliated cells detach the number of motile points decrease over time. CBF was determined using Fourier analysis of the field of view and all frequencies represent the mean ± 1 standard error of the mean (SEM) from 6 separate cell groups or fields.
Statistical Analysis
All quantitative experiments were performed in triplicate. All of our data were analyzed using GraphPad Prism (version 4.00 for Windows; GraphPad Software, San Diego CA) and represented as mean ± standard error. Data were analyzed for statistical significance using analysis of variance (ANOVA).
RESULTS
PKCε is Present in Airway Epithelial Cells and is Activated by PMA
We have shown previously by Western blot that mammalian airway epithelial cells express the novel PKCε isoform [15]. However, the specific cellular localization of PKCε has not been characterized in resting and stimulated airway cells. We utilized immunofluorescent secondary antibodies directed against native PKCε primary antibodies to localize this kinase in bovine bronchial epithelial cells (BBECs). Resting airway cells display bright, punctate staining in the perinuclear region (Figure 1a). However, when 100 ng/mL of the known PKC activator PMA is applied to BBECs, the cells initially flatten out and display a diffuse staining pattern within 15 minutes (Figure 1b), as the activated kinase translocates from the cytoplasm to the membrane. Further timepoints demonstrate that activation of PKCε by PMA induces autodownregulation of the kinase, as the staining at 45 and 60 minutes is significantly decreased (Figure 1c, d).
FIGURE 1.
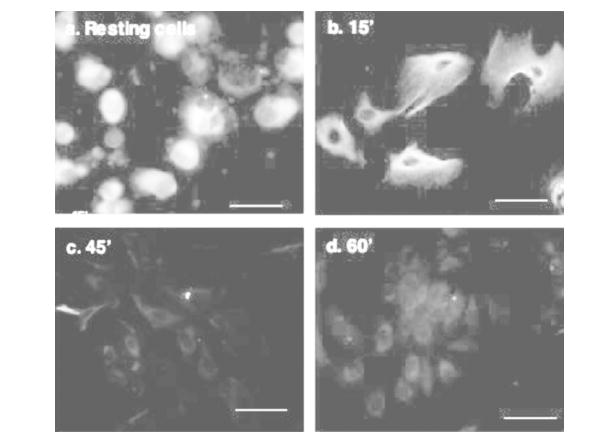
Localization of PKCε in resting and activated primary bovine bronchial epithelial cells. Unstimulated BBECs (a) or BBECs activated with 100 ng/mL PMA for 15 (b), 45 (c) or 60 (d) minutes were incubated with primary antibody overnight at 4°C, coverslips were rinsed with PBS, and then incubated with secondary antibodies (purified anti-mouse IgG conjugated to rhodamine [TRITC] or purified anti-rabbit IgG conjugated to fluorescein [FITC]. Subsequently, IgG-treated coverslips were rinsed 5 times with PBS, mounted on slides, and viewed with a Nikon epifluorescence microscope equipped with appropriate filters to discern TRITC or FITC. Digital micrographs were recorded of multiple representative fields. The size bar equals 25 μm.
The Effect of PMA and Ro 31-8220 on PKCε Kinase Activity
To demonstrate that activated translocation of PKCε by PMA in BBECs correlates with measurable changes in PKCε activity, we performed an in vitro radioactive kinase assay utilizing a peptide substrate recognized specifically by the PKCε isoform. As shown in Figure 2, a significant 3-fold increase in PKCε activity over the medium-treated control occurred after 15 minutes of treatment with 100 ng/mL PMA. By 45 minutes, the PKCε activity in PMA-treated BBECs dropped to below control levels, consistent with the autodownregulation of the kinase represented by the noticeably fainter PKCε fluorescence in Figure 1c. If BBECs were pretreated for 1 hour with 10 μM of the selective chemical inhibitor Ro 31-8220 [20] then stimulated with PMA (Figure 2; grey bars), the PKCε activity remained at baseline levels through 15 minutes and dropped below the control within 45 to 60 minutes, also consistent with significant chemical inhibition of the PKCε isoform.
FIGURE 2.
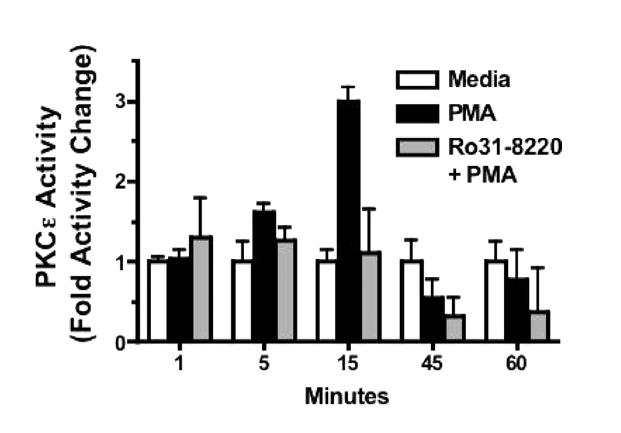
The effect of PMA and Ro 31-8220 on PKCε kinase activity in BBECs. The effect of PMA and Ro 31-8220 on BBECs was directly measured using a PKCε-specific in vitro radioactive kinase assay utilizing a peptide substrate recognized specifically by the PKCε isoform. Cells were treated with medium only (white bars), 100 ng/mL PMA (black bars), or were pretreated with the selective chemical inhibitor Ro 31-8220 (10 μM) then stimulated with PMA (grey bars). Results are presented as fold activity change.
Figure 3A, B demonstrates the inhibition of total PKCε activity in the BBEC cytosolic and particulate cell lysate fractions at various concentrations of Ro 31-8220. Significantly higher PKCε activity can be detected in the particulate fraction compared to the cytosolic fraction at all concentrations of the inhibitor or the medium control, which is consistent with activation and translocation of the kinase to the membrane. Effective kinase inhibition occurred within 1 hour in both particulate and cytosolic fractions at a Ro 31-8220 concentration of 1 μM. Though there was little difference in inhibition of kinase activity between 1 μM and 10 μM, all subsequent experiments were performed with 10 μM of Ro 31-8220.
FIGURE 3.
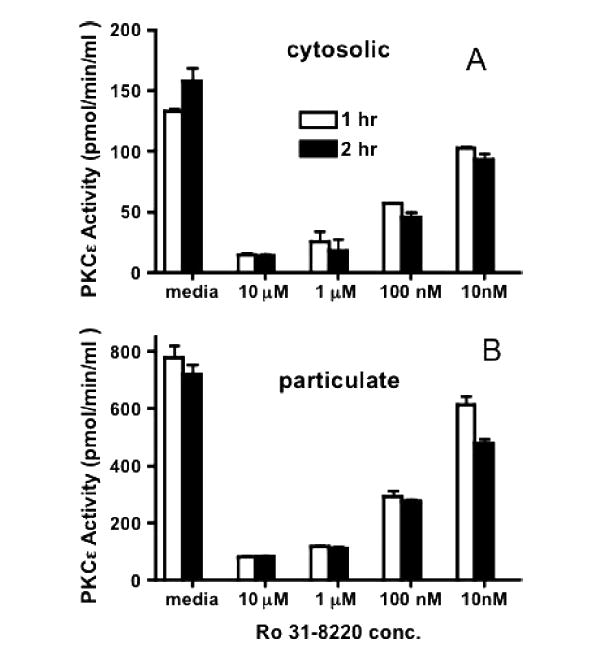
The inhibition of in vitro PKCε kinase activity by Ro 31-8220. Direct PKCε activity was measured in vitro in BBEC cytosolic (A) and particulate (B) cell lysate fraction at increasing concentrations of Ro 31-8220 for 1 (white bars) or 2 (black bars) hours.
Selective Chemical Inhibition of PKCε Causes Ciliated Cell Detachment
Coincident with PKCε inhibition by Ro 31-8220, we observed the detachment of ciliated BBECs. To determine the isoform specificity of the detached response, selective chemical inhibitors of PKC isoforms were added to submerged cultures of ciliated BBECs for 1 hour: general PKC inhibitor, calphostin C (1 μM); PKCα inhibitor, Gö6976 (1 μM); PKCα and PKCγ inhibitor, HBDDE (5 μM); PKCδ inhibitor, rottlerin (20 μM); PKCβ1 inhibitor, hispidin (10 μM); PKCζ inhibitor, myristylated PKCζ(1 μM); PKCβ2 inhibitor, LY 379196 (20 μM); or medium only as a control. As shown in Figure 4A, calphostin C caused a significant increase in the detachment of ciliated BBECs from the basal cell monolayer as measured by direct cell count, compared to a medium-only control. Utilizing the SAVA system to measure cilia motility in real time, we determined that this effect was specific to the ciliated BBECs; basal epithelial cells were not affected and did not detach from the dish. None of the other PKC inhibitors alone induced this specific ciliated cell detachment. PKA and PKG selective inhibitors were also investigated and no ciliated cell detachment was noted (data not shown).
FIGURE 4.
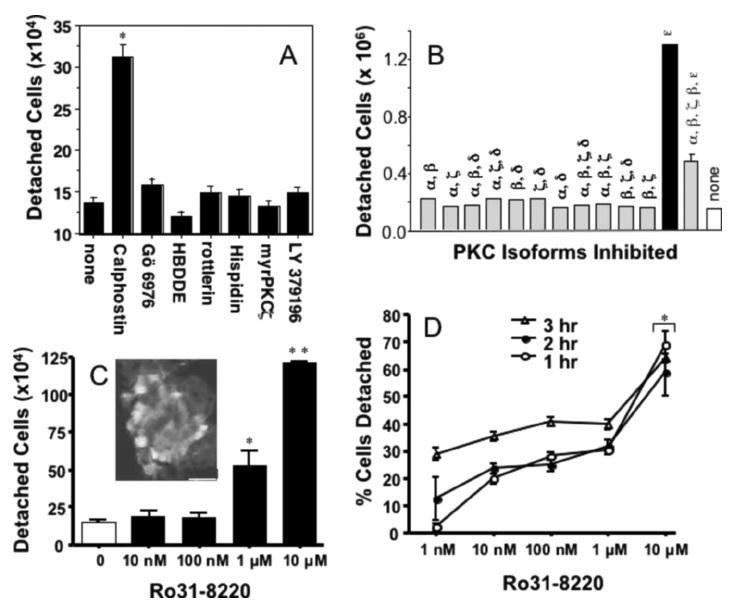
Inhibition of PKCε causes ciliated BBEC detachment. A, Following 1 hour of treatment of ciliated primary BBECs with selective chemical inhibitors of PKC isoforms: general PKC inhibitor, calphostin C (1 μM); PKCα inhibitor, Gö6976 (1 μM); PKCα and PKCγ inhibitor, HBDDE (5 μM); PKCδ inhibitor, rottlerin (20 μM); PKCβ1 inhibitor, hispidin (10 μM); PKCζ inhibitor, myristylated PKCζ(1 μM); PKCβ2 inhibitor, LY 379196 (20 μM); or medium only as a control, ciliated cell detachment was quantitated by direct cell count. B, Multiple combinations of PKC inhibitors, including 10 μM PKC inhibitor Ro 31-8220, were tested to determine which isoform caused ciliated cell detachment. C, A 1-hour Ro 31-8220 dose-response curve indicates that significant ciliated BBEC detachment compared to control occurs at 1 μM (P≤.05), and even greater detachment occurs at 10 μM Ro 31-8220 (P≤.01). The inset picture of fixed BBECs stained with a primary antibody to acetylated α-tubulin following 2 hours, of 10 μM Ro 31-8220 treatment demonstrates that the detached cells are ciliated. Size bar equals 25 μm. D, During 1 to 3 hours, 1 μM Ro 31-8220 causes approximately 35% of the ciliated BBECs to detach within 3 hours and 10 μM Ro 31-8220 causes extremely rapid detachment within 1 hour (P≤.05 compared to medium only control).
To identify the specificity of these chemical inhibitors in the detachment of ciliated cells, we utilized multiple combinations of PKC inhibitors for various isoforms (Figure 4B). As shown in Figure 4A, after 1 hour of treatment, the broad spectrum PKC inhibitor calphostin C causes the detachment of approximately 3–5×105 ciliated BBECs. However, the addition of the selective novel chemical PKCε inhibitor Ro 31-8220 caused the detachment of >106 ciliated BBECs. Without the inhibition of PKCε, no other combination of PKC inhibitors, including the PKCδ specific inhibitor, rottlerin, reproduced this effect (Figure 4B). It is likely that the effect of calphostin C and Ro 31-8220 on the novel PKCε isoform is causing specific ciliated cell detachment. A 1-hour Ro 31-8220 dose response experiment (Figure 4C) shows that significant ciliated BBEC detachment compared to control (6×105 to 106 cells) occurred at concentrations ranging from 1 to 10 μM, which reflects the nearly complete inhibition of PKCε kinase activity (Figure 3A, B). The inset picture of fixed, detached BBECs stained with a primary antibody against acetylated α-tubulin (a cilia-specific marker) following a 2-hour, 10 μM Ro 31-8220 treatment demonstrates that the detached cells are preferentially ciliated. Medium alone did not induce significant detachment of ciliated cells (data not shown). It was also noted that PMA treatment did not cause ciliated cell detachment (data not shown). PMA activates several isoforms of PKC, not only PKCε, in addition to causing several phenotypic changes in cells [21] including increased cell adhesion. Although PMA is able to activate and downregulate PKCε, it is not selective for this isoform and therefore is not a specific initiator of ciliated cell detachment.
At 1 μM Ro 31-8220, approximately 35% of the ciliated BBECs were detached within 3 hours. At 10 μM Ro 31-8220, extremely rapid detachment takes place and >65% of the ciliated BBECs were detached within 1 hour (Figure 4D). As cell detachment occurred, cilia were noticeably beating (and could be measured by SAVA) and cells appeared alive and floating in the media despite having detached from the basal epithelium. To demonstrate that PKC inhibition does not cause BBEC death, lactate dehydrogenase (LDH) release was calculated using media from BBECs that had been treated with Ro 31-8220 (Figure 5A) as well as other PKC inhibitors for 2 hours. Even though 1 μM Calphostin C and Ro 31-8220 induced ciliated cell detachment (Figure 4), no LDH release was measured from these cells (Figure 5A). We also confirmed using the commercially available ApopTag kit, that the adherent and detached BBECs (treated with Calphostin C or Ro 31-8220 for 2 hours) were not apoptotic (Figure 5B) compared to the positive control (cells treated with 1 μM TNF-α), which displayed hallmarks of apoptosis within 2 hours.
FIGURE 5.
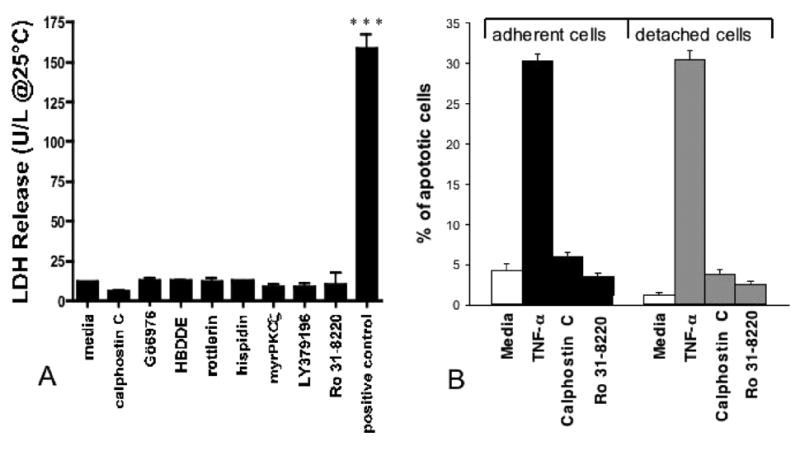
Ciliated BBECs are viable following detachment. A, A standard lactate dehydrogenase assay was utilized to measure cell viability in detached ciliated cells following 2-hour treatments with various PKC inhibitors or medium only. The positive control (lysed BBECs) caused significant (P≤.001) LDH release. B, The ApopTag kit was used to measure hallmarks of apoptosis in adherent (black bars) or detached (grey bars) BBECs treated with 1 μM calphostin C or 10 μM Ro 31-8220 compared to the positive control (1 μM TNF-α).
Inhibition of PKCε and Ciliary Beat Frequency
Our laboratory has demonstrated the capacity to effectively measure CBF in ciliated BBECs using the SAVA system [17]. In the control cells, CBF averaged approximately 10 Hz and the number of motile points per measured field was consistently >3000 (Figure 6A), which corresponds to ~ 15% of the entire field showing motile cilia. In the ciliated cells treated with Ro 31-8220, the number of motile points dropped steeply from 3000 to undetectable within 3 hours, whereas CBF decreased from approximately 10 Hz to 3 Hz in the same time frame and leveled off to 2 Hz from 3 to 6 hours (Figure 6B). The number of motile points is indicative of attached ciliated cells and after treatment with Ro 31-8220, CBF was rapidly decreased. After 3 hours, ciliated cells were nearly completely detached and beating had ceased.
FIGURE 6.
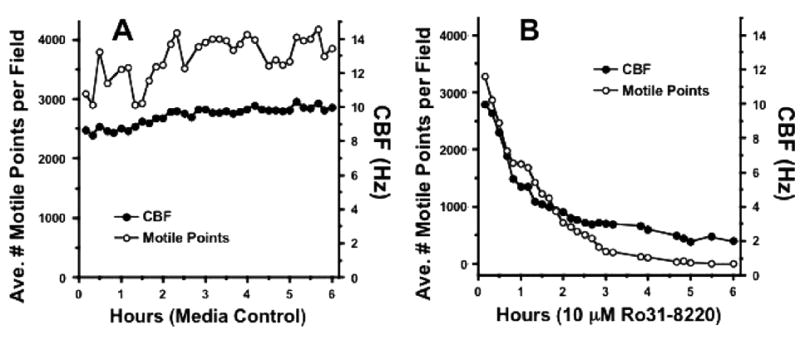
Inhibition of PKCε decreases ciliary motility. In order to verify that inhibition of PKCε was responsible for the decrease in CBF, a 6-hour time course was performed using ciliated BBEC controls in medium only (A) and 10 μM PKCε inhibitor Ro 31-8220 (B), measuring both CBF and the number of motile points using SAVA.
DISCUSSION
The role PKCε plays in mucociliary clearance and inflammatory lung diseases has not been well characterized. In this study, we defined the time-course of activation and downregulation of PKCε by PMA and demonstrated that selective inhibition of PKCε induces ciliated cell detachment. Using an antibody against the native kinase, PKCε was localized to the peri-nuclear region of the cytoplasm in resting bronchial epithelial cells. Following PMA treatment, PKCε translocated to the cellular membrane, which paralleled a significantly increased in vitro kinase activity in the particulate fraction within 15 minutes. However, within 1 hour, kinase activity dropped below baseline, consistent with autodownregulation of PKCε. To further define the downstream effects of PKC signaling on ciliated bronchial epithelial cells, BBECs were incubated with various inhibitors of specific iso-forms. The broadspectrum PKC inhibitor Calphostin C induced rapid detachment of ciliated BBECs from the basal epithelium and selective inhibition of the PKCε isoform by 10 μM Ro 31-8220 caused the detachment of > 65% of the ciliated BBECs within 1 hour. Viability assays determined that ciliated cells were alive and did not appear to be apoptotic. In fact, detached cells continued to beat for a short time after detachment; however, within 3 hours following treatment with Ro 31-8220, nearly all ciliated cells had detached and beating had stopped. Detachment does not appear to be caused by apoptosis or necrosis, as the ciliated cells remain alive and cilia continue beating.
Although several in vivo environmental insults can cause stasis of cilia motility, widespread cilia shedding and detachment of ciliated cells from the basal epithelium, one example which implicates PKC signaling in ciliated cell detachment is respiratory syncytial virus (RSV) viral infection within the upper airway [22, 23]. Our laboratory has previously studied this effect in bovine bronchial epithelial cells infected with bovine RSV [23]. The in vitro ciliated cell detachment phenomenon described in this paper parallels the detachment of ciliated cells following viral infection and may elucidate a novel PKCε-mediated signaling pathway to regulate ciliated cell attachment and to modulate cilia motility. Though the molecular pathogenesis of RSV is not clearly understood, RSV infection has been shown to activate and induce the translocation of PKCε and other novel PKC isoforms at 24 to 48 hours post infection [16]. We have shown that sustained activation of the kinase can autodownregulate PKCε, and we propose that effective inhibition of PKCε induces ciliated cell detachment. Several other in vitro treatments, including incubation of ciliated cells with sodium metabisulfite, can also slow ciliary beat and induce ciliated cell detachment at high concentrations [24]. Furthermore, neuropeptide Y was found to inhibit CBF in cultured human BECs, presumably mediated through a novel PKC isoform–mediated pathway [25]. These examples from the literature and our own experiments provide evidence that a novel PKC, possibly PKCε, can modulate ciliated cell slowing in response to various stimuli. The mechanism of PKC regulation of the cilia motility is still under investigation, though preliminary localization data from our laboratory suggests that in ciliated airway cells, PKCε localizes to the cytoplasm as well as to the apical region of cell, near the axoneme, perhaps targeted to specific substrates which modulate cilia motility (data not shown).
In order to understand the role of PKCε signaling in cell detachment, an area of future study to determine if PKCε substrate targets are localized to specific cell attachment molecules. Few PKCε substrates have been identified in the airway and the specific downstream events following the translocation of the kinase to the membrane are unknown. In other mammalian cell types, activated PKCε binds to the scaffolding protein RACK1 at the membrane [1]. RACK1 can also interact with the integrin β chain, resulting in increased focal adhesion formation, adhesion, and motility [4]. Whether this series of events is also occurring in bronchial epithelial cells and mediating ciliated cell attachment is the subject of future investigation.
Acknowledgments
This research was supported by VA Merit Review (TAW), NRSA Institutional Fellowship 5T32AA007582 (RES), and NIH R01 AA008769 (JHS). The authors wish to thank Lisa Chudomelka for manuscript preparation and Jan Taylor of the UNMC Laser Scanning Microscope Core Facility for operation of the confocal microscope.
References
- 1.Akita Y. Protein kinase C-epsilon (PKC-epsilon): its unique structure and function. J Biochem (Tokyo) 2002;132:847–852. doi: 10.1093/oxfordjournals.jbchem.a003296. [DOI] [PubMed] [Google Scholar]
- 2.Csukai M, et al. The coatomer protein beta’-COP, a selective binding protein (RACK) for protein kinase Cepsilon. J Biol Chem. 1997;272:29200–29206. doi: 10.1074/jbc.272.46.29200. [DOI] [PubMed] [Google Scholar]
- 3.Berrier AL, et al. Activated R-ras, Rac1, PI 3-kinase and PKCepsilon can each restore cell spreading inhibited by isolated integrin beta1 cytoplasmic domains. J Cell Biol. 2000;151:1549–1560. doi: 10.1083/jcb.151.7.1549. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Besson A, Wilson TL, Yong VW. The anchoring protein RACK1 links protein kinase Cepsilon to integrin beta chains. Requirements for adhesion and motility. J Biol Chem. 2002;277:22073–22084. doi: 10.1074/jbc.M111644200. [DOI] [PubMed] [Google Scholar]
- 5.Wu JM, et al. PKC epsilon is a unique regulator for hsp90 beta gene in heat shock response. J Biol Chem. 2003;278:51143–51149. doi: 10.1074/jbc.M305537200. [DOI] [PubMed] [Google Scholar]
- 6.Zeidman R, et al. PKCepsilon, via its regulatory domain and independently of its catalytic domain, induces neurite-like processes in neuroblastoma cells. J Cell Biol. 1999;145:713–726. doi: 10.1083/jcb.145.4.713. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Hodge CW, et al. Supersensitivity to allosteric GABA(A) receptor modulators and alcohol in mice lacking PKCepsilon. Nat Neurosci. 1999;2:997–1002. doi: 10.1038/14795. [DOI] [PubMed] [Google Scholar]
- 8.Hodge CW, et al. Decreased anxiety-like behavior, reduced stress hormones, and neurosteroid supersensitivity in mice lacking protein kinase C epsilon. J Clin Invest. 2002;110:1003–1010. doi: 10.1172/JCI15903. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Murriel CL, Mochly-Rosen D. Opposing roles of delta and epsilonPKC in cardiac ischemia and reperfusion: targeting the apoptotic machinery. Arch Biochem Biophys. 2003;420:246–254. doi: 10.1016/j.abb.2003.08.038. [DOI] [PubMed] [Google Scholar]
- 10.Castrillo A, et al. Protein kinase Cepsilon is required for macrophage activation and defense against bacterial infection. J Exp Med. 2001;194:1231–1242. doi: 10.1084/jem.194.9.1231. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Comalada M, et al. PKC epsilon is involved in JNK activation that mediates LPS-induced TNF-alpha, which induces apoptosis in macrophages. Am J Physiol Cell Physiol. 2003;285:C1235–C1245. doi: 10.1152/ajpcell.00228.2003. [DOI] [PubMed] [Google Scholar]
- 12.Chun JS, Ha MJ, Jacobson BS. Differential translocation of protein kinase C epsilon during HeLa cell adhesion to a gelatin substratum. J Biol Chem. 1996;271:13008–13012. doi: 10.1074/jbc.271.22.13008. [DOI] [PubMed] [Google Scholar]
- 13.Imamdi R, de Graauw M, van de Water B. Protein kinase C mediates cisplatin-induced loss of adherens junctions followed by apoptosis of renal proximal tubular epithelial cells. J Pharmacol Exp Ther. 2004;311:892–903. doi: 10.1124/jpet.104.072678. [DOI] [PubMed] [Google Scholar]
- 14.Wang W, et al. Endogenous calcitonin gene-related peptide protects human alveolar epithelial cells through protein kinase Cepsilon and heat shock protein. J Biol Chem. 2005;280:20325–20330. doi: 10.1074/jbc.M413864200. [DOI] [PubMed] [Google Scholar]
- 15.Wyatt TA, et al. Stimulation of protein kinase C activity by tumor necrosis factor-alpha in bovine bronchial epithelial cells. Am J Physiol. 1997;273(5 Pt 1):L1007–L1012. doi: 10.1152/ajplung.1997.273.5.L1007. [DOI] [PubMed] [Google Scholar]
- 16.Monick M, et al. Respiratory syncytial virus infection results in activation of multiple protein kinase C isoforms leading to activation of mitogen-activated protein kinase. J Immunol. 2001;166:2681–2687. doi: 10.4049/jimmunol.166.4.2681. [DOI] [PubMed] [Google Scholar]
- 17.Sisson JH, et al. All-digital image capture and whole-field analysis of ciliary beat frequency. J Microsc. 2003;211(Pt 2):103–111. doi: 10.1046/j.1365-2818.2003.01209.x. [DOI] [PubMed] [Google Scholar]
- 18.Wyatt TA, Forget MA, Sisson JH. Ethanol stimulates ciliary beating by dual cyclic nucleotide kinase activation in bovine bronchial epithelial cells. Am J Pathol. 2003;163:1157–1166. doi: 10.1016/S0002-9440(10)63475-X. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Wyatt TA, et al. Acetaldehyde-stimulated PKC activity in airway epithelial cells treated with smoke extract from normal and smokeless cigarettes. Proc Soc Exp Biol Med. 2000;225:91–97. doi: 10.1046/j.1525-1373.2000.22511.x. [DOI] [PubMed] [Google Scholar]
- 20.Chen CS, Poenie M. New fluorescent probes for protein kinase C. J Biol Chem. 1993;21:15812–15822. [PubMed] [Google Scholar]
- 21.Liu WS, Heckman CA. The sevenfold way of PKC regulation. Cell Signal. 1998;10:529–542. doi: 10.1016/s0898-6568(98)00012-6. [DOI] [PubMed] [Google Scholar]
- 22.Ogra PL. Respiratory syncytial virus: the virus, the disease and the immune response. Paediatr Respir Rev. 2004;5(Suppl A):S119–S126. doi: 10.1016/S1526-0542(04)90023-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Sisson JH, et al. Smoke and viral infection cause cilia loss detectable by bronchoalveolar lavage cytology and dynein ELISA. Am J Respir Crit Care Med. 1994;149:205–213. doi: 10.1164/ajrccm.149.1.8111584. [DOI] [PubMed] [Google Scholar]
- 24.O’Brien DW, et al. A mechanism of airway injury in an epithelial model of mucociliary clearance. Respir Res. 2004;5:10. doi: 10.1186/1465-9921-5-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Wong LB, Park CL, Yeates DB. Neuropeptide Y inhibits ciliary beat frequency in human ciliated cells via nPKC, independently of PKA. Am J Physiol. 1998;275(2 Pt 1):C440–C448. doi: 10.1152/ajpcell.1998.275.2.C440. [DOI] [PubMed] [Google Scholar]