

Abstract
Obstructive Sleep Apnea (OSA) syndrome has been recognized as a highly prevalent public health problem and is associated with major neurobehavioral morbidity. Chronic intermittent hypoxia (CIH), a major pathological component of OSA, increases oxidative damage to the brain cortex and decreases neurocognitive function in rodent models resembling human OSA. We employed in vitro and in vivo approaches to identify the specific phases and subcellular compartments in which enhanced reactive oxygen species (ROS) are generated during CIH. In addition, we utilized the cell culture and animal models to analyze the consequences of enhanced production of ROS on cortical neuronal cell damage and neurocognitive dysfunction. In a primary cortical neuron culture system, we demonstrated that the transition phase from hypoxia to normoxia (NOX) during CIH generates more ROS than the transition phase from NOX to hypoxia or hypoxia alone, all of which generate more ROS than NOX. Using selective inhibitors of the major pathways underlying ROS generation in the cell membrane, cytosol, and mitochondria, we showed that the mitochondria are the predominant source of enhanced ROS generation during CIH in mouse cortical neuronal cells. Furthermore, in both cell culture and transgenic mice, we demonstrated that overexpression of MnSOD decreased CIH-mediated cortical neuronal apoptosis, and reduced spatial learning deficits measured with the Morris water maze assay. Together, the data from the in vitro and in vivo experiments indicate that CIH-mediated mitochondrial oxidative stress may play a major role in the neuronal cell loss and neurocognitive dysfunction in OSA. Thus, therapeutic strategies aiming at reducing ROS generation from mitochondria may improve the neurobehavioral morbidity in OSA.
Introduction
Obstructive sleep apnea (OSA) syndrome, characterized by episodic cessation of airflow during sleep, has been recognized as a highly prevalent public health problem affecting 2% of women and 4% of men in the general US population. Chronic intermittent hypoxia (CIH) is a major pathological element of OSA that Consequently decreases neurocognitive function and increases cardiovascular morbidity in patients with OSA (Young et al., 2002a; Young et al., 2002b; Naegele et al., 1995). More recently, brain structural abnormalities have been detected in OSA patients using functional MRI approaches (Macey et al., 2002; Macey et al., 2005). The significant brain gray matter loss found in OSA patients is apparently associated with alterations in neural circuitry and neurocognitive function (Macey and Harper, 2005; Woo et al., 2005). Similarly, animal models mimicking the intermittent hypoxic episodes of OSA elicit hippocampal and cortical neuronal cell damage, and consequently result in deficits in spatial task learning and working memory (Xu et al., 2004; Gozal et al., 2003; Gozal et al., 2001). However, the cellular and molecular processes underlying the neuronal cell loss and consequent neurocognitive dysfunction remain incompletely delineated.
Reactive oxygen species (ROS) including superoxide, hydroxyl radicals, hydrogen peroxide, and peroxynitrite are the oxygen-containing molecules that are highly reactive with proteins, lipids and nucleic acids. They have been identified as perpetrators of cell death, tissue damage and functional alterations in human neurodegenerative diseases. Evidence has accumulated in recent years to suggest that increased oxidative stress is associated with neurocognitive and cardiovascular dysfunction in human OSA (Lavie et al., 2004; El Solh et al., 2006; Grebe et al., 2006; Carpagnano et al., 2003). The oscillations in oxygen concentration during CIH are reminiscent of the process of ischemia (hypoxia)/re-oxygenation, and may contribute to the increased production of ROS (Prabhakar et al., 2006; Prabhakar and Kumar, 2004; Lavie, 2005; Lavie, 2003). In support of this assumption, we have previously shown in rats that systemic administration of an electron spin trapper abrogated CIH-induced deficits in the acquisition of a spatial task in the water maze (Row et al., 2003). Furthermore, the excessive somnolence that develops following CIH exposures in mice appears to be mediated at least in part by oxidative stress-induced cellular losses within the locus coeruleus, and is markedly reduced in NADPH oxidase deficient mice (Zhan et al., 2005; Veasey et al., 2004). Although increased oxidative stress has been causally linked to neuronal cell losses in a variety of neurodegenerative diseases, the cellular and molecular mechanisms of CIH-induced brain structural changes and consequent neurophysiological alterations remain largely undefined. In addition, the specific phase(s) and sub-cellular compartment(s) where increased ROS production occurs remain to be determined. To this end, we utilized cell culture and transgenic mouse models to analyze CIH-mediated oxidant generation, CIH-induced neuronal apoptosis, and CIH-mediated neurocognitive dysfunction.
Methods
Chemicals and reagents
Rotenone, allopurinol, apocynin, and 5,5-dimethyl pyrroline-N-oxide (DMPO) were purchased from Sigma Chemicals (St. Louis, MO). 2′,7′-dicholorodihydrofluorescein diacetate (DCF), and di-hydroethidine (DHE) were purchased from Molecular Probes (Eugene, OR).
Neurobasal medium, fetal bovine serum (FBS), B27 and glutamine were purchased from Invitrogen Inc. (Carlsbad, CA).
Animals
C57BL/6J mice (Stock # 000664) (Male, 12 weeks of age, body weight 22–25g) were purchased from Jackson Laboratories (Bar Harbor, ME). Transgenic mice overexpressing MnSOD were provided by Dr. Daret St. Clair (University of Kentucky) (Yen et al., 1996; Yen et al., 1999). The experimental protocols used for breeding and CIH experiments were approved by the Institutional Animal Use and Care Committee and are in close agreement with the National Institutes of Health guidelines for the care and use of laboratory animals. All efforts were made to minimize animal stress and to reduce the number of animals used.
Primary cortical neuronal cell culture
Primary cortical neuronal cultures were prepared and maintained according to a previously described method with minor modifications (Brewer, 1995). Briefly, the cerebral cortical regions from embryonic day 15.5 (E15.5) mouse fetuses were dissected out and rinsed twice with ice cold Ca2+/Mg2+-free Hanks’ salt solution (HSS), pH 7.5. After removal of meninges, cells were dissociated by trituration through a 5-ml pipette first and then a flame-polished Pasteur pipette. Cells were spun down by centrifugation and resuspended in Neurobasal medium supplemented with 0.2% heat inactivated fetal bovine serum (FBS), 2% B27, 1.2 mM glutamine and penicillin/streptomycin (100 U/ml and 100 μg/ml, respectively). Dissociated cells were then either plated 35 mm dishes or chamber slides (Corning Life Sciences, NY) that were coated with poly-D-lysine (0.1 mg/ml). The neurons were grown in a humidified atmosphere of 5% CO2–95% air at 37°C for 3–4 days and were used for experiments in Neurobasal medium containing 2% B27 and 0.2% FBS.
Chronic intermittent hypoxic conditions for primary cortical neurons
Cultured cortical neurons were either exposed to normoxia (NOX: 21% O2, 5% CO2, and balance N2), or to CIH (Episodic cycles of NOX: 21% O2, 5% CO2, and for 35 min), balance N2 for 25 min; and hypoxia: 0.1% O2, 5% CO2, and balance N2 using a custom-designed, computer-controlled incubation chamber attached to an external O2-CO2 computer-driven controller (Biospherix, Redfield, NY). Chamber O2, levels were continuously monitored and adjusted according to the desired N2, and CO2 programmed profile. In addition, O2 content in the medium was monitored with a fiber optic sensor (Ocean Optics, Dunedin, FL) placed 1 mm above the cell layer to O2 ensure the specific experimental profile implementation. O2 levels in the air phase as well as in the medium were continuously sensor during CIH (Ocean monitored O2 Optics, Dunedin, FL).
Chronic intermittent hypoxic conditions for experimental mice
Exposures of mice to CIH were carried out as previously described (Xu et al., 2004). Briefly, animals were housed in a chamber (30″×20″×20″) and were operated under 12 hr light-dark cycle (Oxycycler model A44XO, Biospherix, Redfield, NY). Gas was circulated around the chamber at 60 L.min−1 (i.e., one complete change per 10 seconds). The O2 concentration was continuously measured by an O2 analyzer, and was changed throughout the 12 hr of light time (06:00 a.m.– 6:00 p.m.) by a computerized system controlling the gas valve outlets, such that the moment to moment desired oxygen concentration of the chamber was adjusted automatically. This system permitted delineation of oxygenation profiles in the mouse model that mimic those of patients with OSA. For all of the experiments, the CIH profiles consisted of alternating NOX (21% O2) and hypoxia (5.7% O2) every 90 seconds. Deviations from the desired O2 concentrations were supplied by addition of N2 or room air through solenoid valves. Ambient CO2 in the chamber was periodically monitored and maintained at 0.03% by modifying the ventilation of the chamber. Humidity was measured and maintained at 40–50% and temperature was kept at 22–24°C. The selection of this CIH profile emanated from a series of preliminary experiments in which the magnitude of hypoxia, and length of hypoxia and NOX were tested and confirmed to reproduce the respiratory patterns and neurocognitive deficits in the mouse model (Tagaito et al., 2001; Polotsky et al., 2006). The durations of CIH exposures were from 1 day to 30 days. NOX control mice were placed in neighboring chambers but were exposed to normoxic gas. At least three mice per group were used for the specific studies.
At the end of the designated duration of CIH exposures, mice were anesthetized with pentobarbital (50 mg/kg) and perfused with 0.9% saline buffer. Mouse brain cortical regions were dissected out, frozen in liquid nitrogen immediately and stored in a −80 °C freezer until analysis.
Dicholorodihydrofluorescein (DCF) and di-hydroethidine (DHE) oxidation assays of CIH-mediated ROS production in primary cortical neurons
The production of ROS was measured spectrofluorometrically by using the probe of 2′,7′-dicholorodihydrofluorescein diacetate (DCF), or di-hydroethidine (DHE). Briefly, 6×104 cortical neurons (final number of neuronal cells in each well) were cultured in 48-well plate in triplicate and were preincubated with 50 μM of DCF or 75 μM of DHE (Molecular Probes, Eugene, OR) for 10 min. Cells were either exposed to NOX (21% O2, 5% CO2, balanced N2) or CIH (episodic cycles of NOX: 21% O2, 5% for 35 CO2, and balance N2 for 25 min; and hypoxia: 0.1% O2, 5% CO2, and balance N2 min) for 1 to 8 hr. Then, cells were measured for cumulative ROS generation by assaying DCF or DHE oxidation in a spectrofluorometer (Molecular Devices, Sunnyvale, CA) at the specific excitation and emission wavelengths (For DCF: 485 nm and 530 nm respectively; For DHE: 475 nm and 610 nm respectively).
To define ROS generation in specific phases, we extended the intermittent hypoxia into a 120 min of hypoxic phase (0.1% O2, 5% CO2, balanced N2) and a 120 min NOX phase (21% O2, 5% CO2, balanced N2) respectively. The same DCF and DHE assays were used to measure ROS generation as described above.
To determine ROS generation in the specific subcellular compartments under CIH conditions, we preincubated mouse cortical neurons with 75 μM of Allopurinol, 50 μM of Apocynin, or 50 μM of Rotenone respectively with specific vehicle controls prior to exposure to CIH or NOX. Subsequently, DCF and DHE assays were used to measure ROS generation as described above. In addition, primary cortical neuronal cells from MnSOD transgenic mice were also used to verify and confirm mitochondrial ROS production.
Electron paramagnetic resonance (EPR) assay
The production of ROS was also measured with electron paramagnetic resonance (EPR) according to previously described methods with minor modifications (Liu et al., 2000). Briefly, 1×106 cortical neurons cultured in 6-well plate in triplicates were preincubated with 50 mM 5,5-dimethyl-1-pyrroline N-oxide (DMPO) for 10 min. Cells were then exposed to NOX (21% O2, 5% CO2, balanced N2) or CIH (Episodic cycles of NOX: 21% O2, 5% CO2, and balance N2 for 25 min; and hypoxia: 0.1% O2, 5% for 35 min) for 6 hr. Cells were scraped off the wells with a CO2, and balance N2 rubber scraper at the end of exposure and frozen immediately in liquid nitrogen. EPR spectroscopy was performed in 500 μl samples in a TM flat corvette using a Bruker EPR spectrometer (Bruker Instruments, Germany). Instrument settings were as follows: Microwave power, 10 mW; Klystron frequency, 9.764 GHz; Modulation amplitude, 1.05Gauss; Scan rate, 80G/84s; and Time constant, 0.16s.
Immunohistochemical staining
Cortical neurons were grown in pre-coated chamber slides and exposed to NOX or CIH as described in the previous sections. At the end of NOX and CIH exposures, cortical neurons were fixed with 4% paraformaldehyde (PFA) for 30 min followed by incubating with 0.1% triton X-100 in 1×PBS (pH7.5) for 15 min to permeabilize cells. After blocking with 10% goat serum in 1×PBS containing 0.1% NP-40 for 1hr, cells were incubated with antibody overnight at 4 °C (primary antibodies: anti-MDA at 1:500; anti-Bcl2 at 1:400; anti-AIF at 1:600: anti-cleaved caspase-3 at 1:400; Chemicon Inc.). Cells were washed with 0.1% triton X-100 in 1×PBS for 5 min each for 5 times and then incubated with specific secondary anti-body conjugated with fluorescein for 2 hr at room temperature in the dark. After extensive washes, slides were mounted with anti-fade medium and examined under a fluorescent microscope. All images were collected and analyzed with a Nikon fluorescent microscope (80I Eclipse Model, Nikon, Japan) equipped with the Spot digital camera (Diagnostic Instruments, Sterling Heights, MI) and Photoshop software (Adobe Systems, San Jose, CA).
Analysis of CIH-mediated protein oxidation
Oxidized proteins can be derivatized with 2,4-dinitropenylhydrazine (DNPH) (Levine et al., 2000; Levine et al., 1994). Both Western blotting and spectrophotometric approaches were used to analyze CIH-mediated protein oxidation in cortical tissues and cortical neurons compared to that of NOX controls. For Western analysis, total 20 μg protein from samples was reacted with DNPH according to manufacturer’s instruction (Chemicon Inc, Temecula, CA). After neutralization, oxidized protein adducts were separated on a 12.5% SDS PAGE gel and transferred onto nitrocellulose membrane. The membrane was blocked with 5% nonfat milk dissolved in TBST (10 mM Tris-HCl, pH 7.5, 150 mM NaCl, 0.1% Tween-20) for 1 hr at room temperature on a shaker plate and then incubated with the specific anti-DNPH antibody overnight at 4°C. The membrane was washed three times for 5 min each time with TBST at room temperature and then incubated with horseradish peroxidase (HRP)-conjugated secondary antibody (KPL, Gaithersburg, MD) for 1 hr at room temperature. The membrane was washed three times with TBST for 5 min each and visualized in ECL reagents (Amersham Bioscience, Piscataway, NJ).
For spectrophotometric analyses, samples were homogenized in 20 mM phosphate buffer (pH 6.5) and treated with 1× DNPH (Chemicon Inc., Temecula, CA) in 2.0 M HCl at room temperature for 20 min. After precipitation with 10% trichloroacetic acid, the pellets were washed three times with ethanol/ethyl acetate (1:1). The final pellets were dried and dissolved in 6 M guanidine HCl in 20 mM sodium phosphate buffer (pH6.5). Insoluble debris was removed by centrifugation at 12,000g for 5 min at 4 °C. The spectrum of the DNPH-derived samples was measured at 375 nm (Levine et al., 2000; Levine et al., 1994; Oliver et al., 1990; Ahn et al., 1987). The relative protein oxidation was then expressed in fold-increase in the CIH-exposed cortical tissues or cortical neurons compared with that of NOX controls.
Analysis of CIH-mediated lipid peroxidation
The relative levels of lipid peroxidation in the cortical tissues were measured by the generation of malondialdehyde (MDA), a product of lipid peroxidation, according to the manufacturer’s instructions (OxisResearch, Portland, OR) with minor modifications (Dei et al., 2002; Hall et al., 1996; Row et al., 2003; Xu et al., 2004). Briefly, cortical tissues from control and CIH-exposed mice were homogenized in 20 mM phosphate buffer (pH7.4) containing 0.5M butylated hydroxytolene to prevent sample oxidation. After protein concentration measurements, equal amounts of protein (2.0 to 2.5 mg protein from each sample in triplicates) were incubated with the chromogenic reagents at 45 °C in 500 μl buffer for 1–2 hr. The samples were then centrifuged at 8000g for 5 min and the clear supernatants were measured at 586 nm. The levels of MDA production were calculated with the standard curve according to the manufacturer’s instruction (OxisResearch, Portland, OR). The relative levels of lipid peroxidation were expressed in fold-increase in MDA production in CIH-exposed cortical tissues vs. that of NOX controls.
Analysis of Apoptosis
Apoptosis assays of cortical neurons or cortical tissues exposed to NOX and CIH were carried out according the manufacturer’s instructions to detect histone-associated DNA fragments (Roche Biochemicals, Indianapolis, IN). Briefly, for cell culture system, control (NOX) and CIH-exposed cortical neurons (2×105 cells/well in 6-well plate) were lysed with 200 μl lysis buffer for 30 min at room temperature. After centrifugation at 200×g for 10 min, the supernatant was carefully transferred to a new tube and protein concentration was adjusted to 10 μg/ml. A total of 20 μl (total 200 ng protein) was used for apoptotic assay as defined by manufacturer’s instructions. Similarly, mouse brain cortical tissues were homogenized with a Dounce glass homogenizer in lysis buffer to release histone-associated DNA complexes. After centrifugation and determination of protein concentration, apoptotic assays were performed in the supernatant in the same way as described above.
Morris water maze analysis
Morris water maze analysis was performed in 12 male 12-week old MnSOD transgenic mice and age-matched normal littermate controls. Animals were housed in groups of four in standard clear polycarbonate cages with food and water available ad libitum. Mice were randomly assigned to be exposed during daylight to either CIH (n=12), or normal room air (NOX, n=12) for a period of 14 and 21 days. Animals were kept on a 12-hour light/dark cycle (6:00 am–6:00 pm), and Morris water maze analysis was conducted during the light phase at the end of CIH exposure (Morris, 1984). Briefly, the maze consisted of a white circular pool, 1.8m in diameter and 0.6m in height, filled to a level of 35cm with water maintained at a temperature of 22°C. Pool water was made opaque by addition of 100ml of non-toxic white tempera paint. A Plexiglas escape platform (10cm in diameter) was positioned 1cm below the water surface and could be placed at various locations throughout the pool. Extramaze cues surrounding the maze were located at fixed locations, and visible to the mice while in the maze. Maze performance was recorded by a video camera suspended above the maze and interfaced with a video tracking system (HVS Imaging, Hampton, UK). One day prior to water maze testing, mice were habituated to the water maze during a swim training. Place learning was then assessed using a modified space training regimen that has been demonstrated to elicit optimal learning in mice (Gerlai and Clayton, 1999). During the water maze test, mice were placed into the pool from a specific point and allowed a maximum of 90 seconds to escape to the platform where the mice were allowed to stay for 15 seconds. Mice that failed to escape were led to the platform. The position of the platform remained constant during the trials. The escape latency and pathlength of a total of five trials were averaged in the water maze testing.
Statistical analysis
Results were expressed as Mean ± SE. Comparisons between normal control (NOX) and CIH-exposed brain cortical samples were analyzed by a Student t-test. To assess the spatial learning and memory in the water maze, mean escape latencies, and swim distance were analyzed by analysis of variance (ANOVA). Student-Newman-Keuls’ post-hoc tests were used when significant differences were found. A p-value less than 0.05 was considered statistically significant.
Results
CIH increased ROS production in mouse brain cortical neurons in the primary culture system
After a series of preliminary experiments, we established an adequate cell culture profile to study the effect of CIH on neuronal cell toxicity in vitro in the cell culture system (Fig. 1A). The actual hypoxic and normoxic exposure duration to cortical neuronal cells in each cycle is 35 min and 25 min respectively. Under these conditions, we applied the most frequently used DCF fluorescent oxidation approach to analyze CIH-mediated ROS production in the cortical neurons. Cells exposed to CIH significantly increased DCF oxidation compared to the NOX controls, indicating there was an increase of ROS production by CIH (Fig. 1B). The increased CIH-mediated ROS generation was subsequently confirmed with di-hydroethidine (DHE) oxidation and spin trapping electron paramagnetic resonance (EPR) assays (Fig. 1C and 1D).
Figure 1. CIH increased ROS generation in mouse brain cortical neurons.

A.The experimental profile for primary brain cortical neurons exposed to chronic intermittent hypoxia (NOX: normoxia control; CIH: chronic intermittent hypoxia). B. DCF fluorescent analysis of ROS generation in cortical neurons exposed to CIH (n=3; * indicates significant compared to NOX control, p < 0.05). C. DHE fluorescent analysis of ROS generation in cortical neurons exposed to CIH (n=3; * indicates significant compared to NOX control, p < 0.05). D. Representative spin trapping EPR analysis of ROS generation in cortical neurons exposed to CIH (n=3).
ROS generation at different phases of intermittent hypoxia in the brain cortical neuronal cells
CIH consists of multiple phases including NOX, hypoxia, transition phase from NOX to hypoxia, and transition phase from hypoxia to NOX (Fig. 1A). We extended the intermittent hypoxia profile to a 120 min interval at different phases including NOX, hypoxia, and transition phases in order to measure ROS generation accurately (Fig. 2A). We demonstrated that transition phase from hypoxia to NOX generated more ROS than that of the hypoxia, or transition phase from NOX to hypoxia, all of which produced more ROS than NOX controls, as measured by the DCF (Fig. 2B) and DHE assays (Fig. 2C). The increased ROS production at transition phase from hypoxia to NOX, hypoxia phase, or transition phase from NOX to hypoxia was continuously increased after shifting to NOX for 20 min, as measured by kinetic DCF assay (Fig. 2D).
Figure 2. ROS generation at different phases of intermittent hypoxia in mouse brain cortical neurons during CIH.

A. The experimental profile to analyze ROS generation in mouse brain cortical neurons at the specific phases of CIH (NOX: normoxia; HP: hypoxia). B. DCF fluorescent analysis of ROS generation in the cortical neurons exposed to CIH at the specific phases of CIH (n=3; * indicates significant compared to NOX control, p < 0.05). C. DHE fluorescent analysis of ROS generation in the cortical neurons exposed to CIH at the specific phases of CIH (n=3; * indicates significant compared to NOX control, p < 0.05). D. A kinetic representation of DCF fluorescent assay of ROS production in the cortical neurons after exposure of cells to NOX from the specific phases of CIH.
CIH-mediated ROS generation at different intracellular compartments of mouse brain cortical neurons
The production of ROS can be derived from several intracellular compartments, i.e., mitochondria, cytosol or membrane. We applied selective inhibitors that target different pathways of ROS generation to define the specific sources of enhanced ROS production in response to CIH in mouse brain cortical neurons. We first defined the optimal doses of the specific inhibitors that transiently inhibit specific cellular pathway of ROS generation without significantly affecting cell viability (Fig. 3A–3C), which are 75μM of Allopurinol, 50μM of Apocynin, and 50μM of Rotenone respectively. Subsequently, we used the optimal doses to identify compartment-specific ROS generation by CIH effects. We demonstrated that mitochondria generated more ROS during CIH than cytosol (cytosolic xanthine oxidase) or cell membrane (NADPH oxidase), as measured by DCF (Fig. 3D) and DHE assays (Fig. 3E).
Figure 3. Mitochondria are the predominate source of the enhanced ROS generation by CIH in the mouse brain cortical neurons.

A. Effects of Apocynin (a NADPH oxidase inhibitor) on ROS generation and cell viability in the cortical neurons under NOX conditions. B. Effects of Rotenone (a mitochondrial oxidase inhibitor) on ROS generation and cell viability in the cortical neurons under NOX conditions. C. Effects of Allopurinol (a xanthine oxidase inhibitor) on ROS generation and cell viability in the cortical neurons under NOX conditions. D. DCF fluorescent analysis of ROS generation in cortical neurons exposed to CIH in the presence of Apocynin, Rotenone and Allopurinol respectively (n=3; * indicates significant compared to CIH, p < 0.05). E. DHE fluorescent analysis of ROS generation in cortical neurons exposed to CIH in the presence of Apocynin, Rotenone and Allopurinol respectively (n=3; * indicates significant compared to CIH, p < 0.05).
Overexpression of MnSOD reduced CIH-mediated mouse brain cortical neuronal apoptosis in the primary cell culture system
Mouse brain cortical neurons exposed to CIH increased oxidative stress as detected with the lipid peroxidation marker, MDA (Fig. 4A), and the protein oxidation marker, carbonyl protein production (data not shown). In addition, CIH increased the dissociation of Bcl2 from the mitochondrial membrane (Fig. 4B), translocation of apoptosis inducing factor (AIF) from mitochondria to nucleus (Fig. 4C), and activation of caspase 3 in cortical neuronal cells (Fig. 4D). Consequently, CIH promoted time-dependent neuronal apoptosis (Fig. 4E). In contrast, overexpression of MnSOD significantly decreased CIH-mediated neuronal apoptosis in cell culture (Fig. 4E).
Figure 4. Overexpression of MnSOD reduced CIH-mediated oxidative stress and neuronal apoptosis in cell culture system.
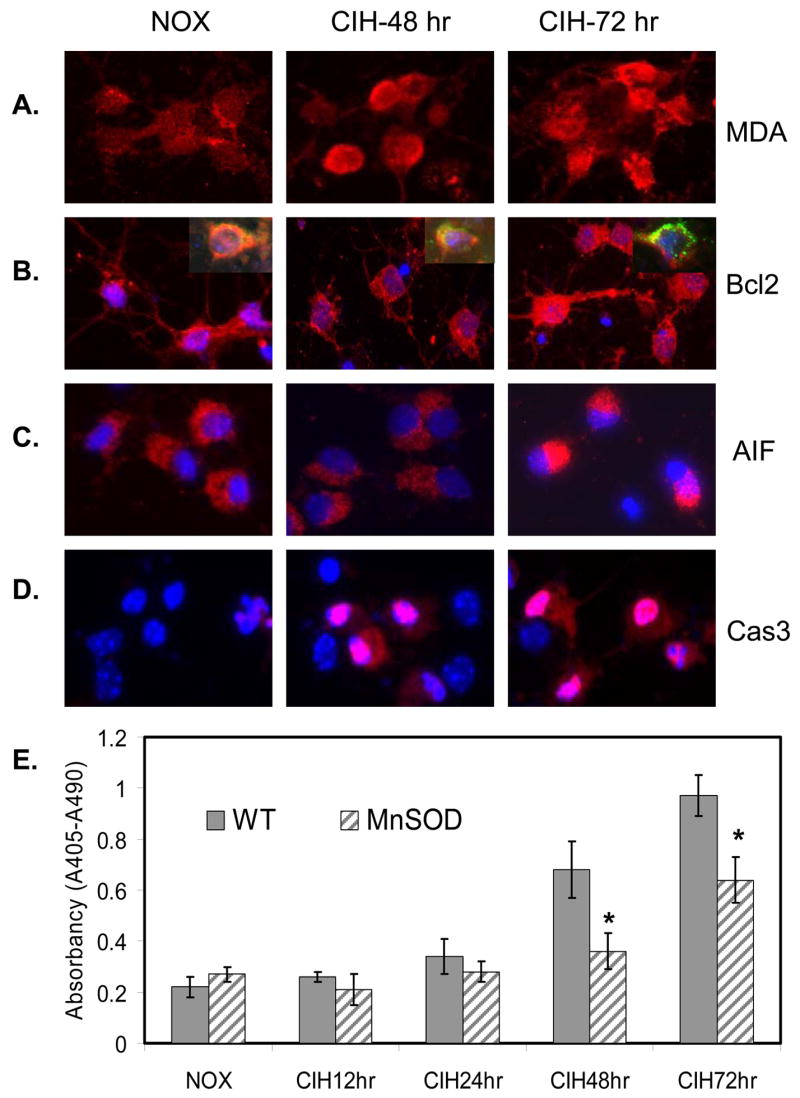
A. Representative immunohistochemical (IHC) images of MDA production in mouse cortical neurons exposed to CIH compared to NOX control. B. Representative IHC images of Bcl2 dissociation from mitochondria in mouse cortical neurons exposed to CIH compared to NOX control. Inserts show the co-localization of Mito-tracker (green) and Bcl2 (red) along with DAPI (blue) staining of nuclei. C. Representative IHC images of AIF nuclear translocation in mouse cortical neurons exposed to CIH compared to NOX control [DAPI (blue) staining of nuclei]. D. Representative IHC images of caspase 3 activation in mouse cortical neurons exposed to CIH compared to NOX control [DAPI (blue) staining of nuclei]. E. Overexpression of MnSOD reduced CIH-mediated neuronal apoptosis (n=3; * indicates significant compared to WT-control, p < 0.05).
Transgenic mice overexpressing MnSOD reduced CIH-mediated oxidative damage on mouse brain cortical cells
We have established a mouse model that mimics the episodic event of hypoxia and normoxia in OSA to study the effects of CIH on neuronal cell degeneration and neurocognitive dysfunction (Xu et al., 2004). Wild-type (WT) and MnSOD transgenic mice were exposed to NOX or CIH conditions as defined in Fig. 5A. At different time points of exposure, mouse cortical tissues were collected, and analyzed with Western blotting and spectrophotometric assays to determine protein oxidation and lipid peroxidation. Compared with NOX controls, CIH significantly increased cortical protein oxidation (Fig. 5B, and 5C) and lipid peroxidation (Fig. 5D). Overexpression of MnSOD decreased protein oxidation and lipid peroxidation (Fig. 5C and 5D), and reduced cortical cell apoptosis (Fig. 5E).
Figure 5. Overexpression of MnSOD reduced CIH-mediated oxidative damage and neuronal apoptosis in the mouse model.
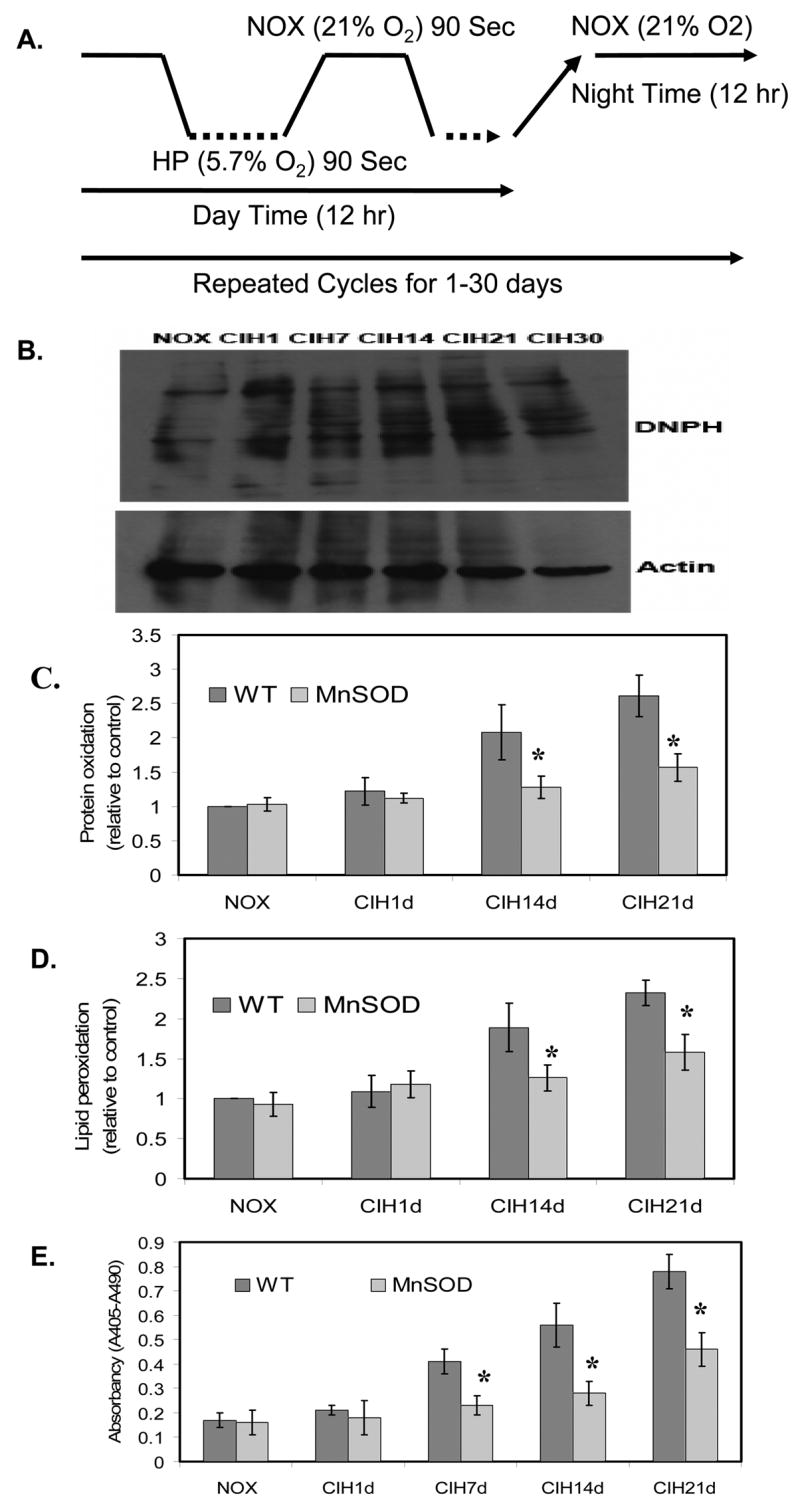
A. An experimental profile for mice exposed to chronic intermittent hypoxia (CIH). For CIH, mice were underwent the alterations of 90 seconds of room air (21% oxygen) and 90 seconds of hypoxia (5.7% oxygen) during day time (6:00Am to 6:00 Pm) for 1 day to 30 days respectively. For NOX control, mice were underwent normal room air (21% oxygen) all the time in the same environment. B. A representation of western blotting analysis demonstrating protein oxidation in mouse brain cortex during CIH compared to that of NOX control. The upper panel is shown the DNPH-derived protein oxidation and the lower panel is shown the actin for loading calibration. C. Overexpression of MnSOD reduced CIH-mediated protein oxidation as detected by the carbonyl contents in mouse brain cortex compared to that of WT-mice (n=3; * indicates p< 0.05). D. Overexpression of MnSOD reduced CIH-mediated lipid peroxidation as detected by MDA contents in mouse brain cortex compared to that of WT-mice (n=3; * indicates p< 0.05). E. Overexpression of MnSOD significantly reduced CIH-mediated mouse brain cortical cell apoptosis compared to that of WT-mice (n=3; * indicates significant compared to WT-mice, p < 0.05).
Transgenic mice overexpressing MnSOD improved CIH-mediated neurocognitive dysfunction
Long-term exposure of mice to CIH (14 and 21 days) that recapitulates the episodic event in OSA resulted in neurocognitive dysfunction as measured with the spatial learning and memory task in the Morris water maze (Fig. 6A and 6B). On the other hand, transgenic mice overexpressing MnSOD had significant preservation of their spatial learning and memory function compared with WT-control mice under same CIH conditions (Fig. 6A and 6B).
Figure 6. Overexpression of MnSOD improved CIH-induced neurocognitive dysfunction as determined by Morris water maze assay.

A. Mean time in seconds to locate the target platform in mice exposed to 14 and 21 days of CIH and NOX. WT-mice exposed to CIH for 14 and 21 days significantly increased the time to find the target compared to that of NOX controls respectively (n=12 in each group, * indicates significant compared CIH exposed WT-mice to NOX controls, p < 0.05). Transgenic mice overexpressing MnSOD exposed to CIH significantly decreased the time to find the target, compared to that of WT-mice exposed to CIH (n=12 in each group, ** indicates significant compared to WT-mice, p < 0.05). B. Mean pathlength in meters to locate the target platform in mice exposed to 14 and 21 days of CIH and NOX. WT-mice exposed to CIH significantly increased the pathlength to find the target compared to that of NOX controls (n=12 in each group, * indicates significant compared CIH exposed WT-mice to NOX controls, p < 0.05). Transgenic mice overexpressing MnSOD exposed to CIH significantly decreased the pathlength to find the target, compared to that of WT-mice (n=12 in each group, ** indicates significant compared to WT-mice, p < 0.05).
Discussion
Both clinical investigations (Younes et al., 2001; Beebe and Gozal, 2002; Hayakawa et al., 1996; Balfors and Franklin, 1994; Gozal et al., 2004) and animal model studies (Xu et al., 2004; Gozal et al., 2003; Row et al., 2003; Zhan et al., 2005; Veasey et al., 2004; Prabhakar and Kumar 2004) have demonstrated that CIH, one of the major components of OSA, leads to brain structural, functional and physiological alterations. Nevertheless, the cellular and molecular processes of CIH-mediated neuronal damage and neurocognitive dysfunction remain unclear. The present study using a primary cell culture system and a mouse model in conjunction with CIH paradigms revealed four major findings: 1). The episodic oscillations of O2 concentration during CIH significantly increased ROS generation in mouse brain cortical neurons in vitro and in vivo; 2). The transition phase from hypoxia to NOX generated more ROS than that of the transition phase from NOX to hypoxia or hypoxia alone, all of which produced more ROS than NOX; 3). Mitochondria were the major source of CIH-mediated ROS generation; 4). Overexpression of MnSOD reduced CIH-mediated oxidative damage, decreased CIH-induced cortical neuronal apoptosis, and improved CIH-mediated neurocognitive dysfunction.
The repeated episodes of oscillatory behaviors of oxygen blood content in the OSA patients during sleep may enhance ROS generation, similar to the events that occur during hypoxia/ischemia and re-oxygenation processes. Indeed, higher circulating levels of thiobarbituric acid-reactive substances (TBARS), 8-isoprostane, and other oxidative stress markers have been found in OSA patients when compared with controls (Barcelo et al., 2000), suggesting that increased oxidative stress plays an important role in the pathophysiology of OSA-induced tissue and organ injury. To validate and identify the cellular processes of ROS-mediated neuropathology, we developed a mouse model that recapitulates the alterations of oxygenation levels in OSA. We demonstrated that exposure of mice to CIH significantly increased the production of oxidative stress markers in cortical tissues (Xu et al., 2004). More importantly, the increased oxidative damage is associated with a decline in neurocognitive function (Xu et al., 2004; Gozal et al., 2003; Row et al., 2003). These observations in animal models are remarkably similar to and supportive of the clinical findings in human OSA patients (Lavie et al., 2004; El Solh et al., 2006; Grebe et al., 2006; Carpagnano et al., 2003; Hayakawa et al.,1996; Barcelo et al., 2000). However, not all studies in human OSA have confirmed the presence of increased oxidative stress-related markers (Svatikova et al., 2005), indicating that sampling issues and patient selection factors (such as co-existence of other disease states) may markedly affect findings. To this end, we tested and selected the CIH paradigms that readily eliminated other potential confounders to discriminately analyze the effect of episodic hypoxia on the generation of ROS in vitro and in vivo in the current study setting. We further employed several well-established and accepted techniques to measure ROS production, and demonstrated that CIH increases ROS generation in cortical neuronal cells. The increased DHE oxidation and EPR signals suggest that enhanced superoxide production is most likely the early and dominant molecules in cortical neuronal cells during CIH (Chen et al., 2005; Buettner, 1990; Liu et al., 2000).
Since intermittent hypoxia includes multiple phases, we were interested in which of these phases, would predominate generation of ROS during CIH. We expanded the NOX phase and hypoxic phase of intermittent hypoxia to 120 min each, so that, we can measure the relative ROS generation accurately. The transition phase from NOX to hypoxia, and transition phase from hypoxia to NOX was 240 min respectively (Fig. 2A). Under these experimental conditions, we demonstrated a phase dependent ROS production that is in agreement with previous literature on hypoxia-ischemia/reoxygenation events (Zwicker et al., 1998; Weissmann et al., 2006). To further define the specific subcellular compartments where the enhanced ROS are generated, we selected the specific inhibitors that inhibit the pathways of ROS production, and found that mitochondria were the major source for the enhanced ROS generation by CIH in mouse cortical neurons (Fig. 3). CIH-mediated mitochondrial ROS production was further confirmed with primary cortical neuronal cell cultures obtained from MnSOD transgenic mice (Fig. 4 and data not shown). Identification of the specific phases and specific subcellular compartments that are associated with CIH-mediated ROS generation is important for target-specific design of interventions to reduce or prevent oxidative damage in the context of disorders characterized by CIH. To the best of our knowledge, this is the first report to analyze phase-specific and subcellar compartment-specific ROS generation by CIH. More recently, Zhan et al. (2005) showed that long-term of intermittent hypoxia increased NADPH oxidase gene and protein response in wake-active brain regions. It appears that NADPH oxidase-mediated ROS generation was associated with hypersomnolence related brain oxidative injury. On the other hand, CIH-mediated mitochondrial ROS production seemed to induce cortical neuronal cell death and increased neurocognitive dysfunction (Fig. 4E, Fig. 5E and Fig. 6). Collectively, these findings rather than contradictory, are probably complementary and suggest that CIH-induced ROS production mediates different neurological alterations at different neuroanatomical regions (Zhan et al., 2005). Thus, decrease or removal of one of the major sources of ROS production (as might be anticipated to occur in NADPH oxidase knock out mice or MnSOD overexpression transgenic mice), will reduce specific initiation of cell death mechanisms.
Based on our findings that CIH-mediated mitochondrial ROS generation induced oxidative damage and neuronal apoptosis (Fig. 4 and Fig. 5), it is reasonable to hypothesize that an antioxidant enzyme that inhibits mitochondrial ROS generation, or reduces ROS accumulation and propagation should protect neurons from CIH damage and improve neurocognitive function. To this end, transgenic mice overexpressing MnSOD were used and confirmed this assumption. Overexpression of MnSOD significantly reduced protein oxidation and lipid peroxidation, and improved CIH-mediated neurocognitive dysfunction (Fig. 5 and Fig. 6). Taken together, the data suggest that enhanced production of ROS from mitochondria by CIH is an important pathogenic factor that contributes to neuronal cell death, and that pharmacological approaches aiming to reduce mitochondrial ROS generation and oxidative stress propagation may provide effective therapy to reduce or improve OSA-mediated neurocognitive dysfunction.
In conclusion, the present studies combined cell culture and transgenic mouse models demonstrated that mitochondrial ROS production constitutes a critically important element in neuronal cell injury induced by CIH. More importantly, overexpression of mitochondrial MnSOD significantly reduced neuronal cell death and improved CIH-mediated neurocognitive dysfunction. These findings identify mitochondria as a potential target for therapy of neurocognitive morbidity in human OSA patients.
Acknowledgments
We thank Dr. St. Clair for proving the MnSOD transgenic mice. This study was supported by National Institutes of Health Grants NS45829 and HL75034.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Reference List
- Ahn B, Rhee SG, Stadtman ER. Use of fluorescein hydrazide and fluorescein thiosemicarbazide reagents for the fluorometric determination of protein carbonyl groups and for the detection of oxidized protein on polyacrylamide gels. Anal Biochem. 1987;161:245–257. doi: 10.1016/0003-2697(87)90448-9. [DOI] [PubMed] [Google Scholar]
- Balfors EM, Franklin KA. Impairment of cerebral perfusion during obstructive sleep apneas. Am J Respir Crit Care Med. 1994;150:1587–1591. doi: 10.1164/ajrccm.150.6.7952619. [DOI] [PubMed] [Google Scholar]
- Barcelo A, Miralles C, Barbe F, Vila M, Pons S, Agusti AG. Abnormal lipid peroxidation in patients with sleep apnoea. Eur Respir J. 2000;16:644–647. doi: 10.1034/j.1399-3003.2000.16d13.x. [DOI] [PubMed] [Google Scholar]
- Beebe DW, Gozal D. Obstructive sleep apnea and the prefrontal cortex: towards a comprehensive model linking nocturnal upper airway obstruction to daytime cognitive and behavioral deficits. J Sleep Res. 2002;11:1–16. doi: 10.1046/j.1365-2869.2002.00289.x. [DOI] [PubMed] [Google Scholar]
- Brewer GJ. Serum-free B27/neurobasal medium supports differentiated growth of neurons from the striatum, substantia nigra, septum, cerebral cortex, cerebellum, and dentate gyrus. J Neurosci Res. 1995;42:674–683. doi: 10.1002/jnr.490420510. [DOI] [PubMed] [Google Scholar]
- Buettner GR. On the reaction of superoxide with DMPO/.OOH. Free Radic Res Commun. 1990;10:11–15. doi: 10.3109/10715769009145928. [DOI] [PubMed] [Google Scholar]
- Carpagnano GE, Kharitonov SA, Resta O, Foschino-Barbaro MP, Gramiccioni E, Barnes PJ. 8-Isoprostane, a marker of oxidative stress, is increased in exhaled breath condensate of patients with obstructive sleep apnea after night and is reduced by continuous positive airway pressure therapy. Chest. 2003;124:1386–1392. doi: 10.1378/chest.124.4.1386. [DOI] [PubMed] [Google Scholar]
- Chen YR, Chen CL, Zhang L, Green-Church KB, Zweier JL. Superoxide generation from mitochondrial NADH dehydrogenase induces self-inactivation with specific protein radical formation. J Biol Chem. 2005;280:37339–37348. doi: 10.1074/jbc.M503936200. [DOI] [PubMed] [Google Scholar]
- Dei R, Takeda A, Niwa H, Li M, Nakagomi Y, Watanabe M, Inagaki T, Washimi Y, Yasuda Y, Horie K, Miyata T, Sobue G. Lipid peroxidation and advanced glycation end products in the brain in normal aging and in Alzheimer’s disease. Acta Neuropathol (Berl) 2002;104:113–122. doi: 10.1007/s00401-002-0523-y. [DOI] [PubMed] [Google Scholar]
- El Solh AA, Saliba R, Bosinski T, Grant BJ, Berbary E, Miller N. Allopurinol improves endothelial function in sleep apnoea: a randomised controlled study. Eur Respir J. 2006;27:997–1002. doi: 10.1183/09031936.06.00101005. [DOI] [PubMed] [Google Scholar]
- Gerlai R, Clayton NS. Analysing hippocampal function in transgenic mice: an ethological perspective. Trends Neurosci. 1999;22:47–51. doi: 10.1016/s0166-2236(98)01346-0. [DOI] [PubMed] [Google Scholar]
- Gozal D, Daniel JM, Dohanich GP. Behavioral and anatomical correlates of chronic episodic hypoxia during sleep in the rat. J Neurosci. 2001;21:2442–2450. doi: 10.1523/JNEUROSCI.21-07-02442.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gozal D, O’Brien L, Row BW. Consequences of snoring and sleep disordered breathing in children. Pediatr Pulmonol Suppl. 2004;26:166–168. doi: 10.1002/ppul.70094. [DOI] [PubMed] [Google Scholar]
- Gozal D, Row BW, Kheirandish L, Liu R, Guo SZ, Qiang F, Brittian KR. Increased susceptibility to intermittent hypoxia in aging rats: changes in proteasomal activity, neuronal apoptosis and spatial function. J Neurochem. 2003;86:1545–1552. doi: 10.1046/j.1471-4159.2003.01973.x. [DOI] [PubMed] [Google Scholar]
- Grebe M, Eisele HJ, Weissmann N, Schaefer C, Tillmanns H, Seeger W, Schulz R. Antioxidant vitamin C improves endothelial function in obstructive sleep apnea. Am J Respir Crit Care Med. 2006;173:897–901. doi: 10.1164/rccm.200508-1223OC. [DOI] [PubMed] [Google Scholar]
- Hall ED, Smith SL, Oostveen JA. Inhibition of lipid peroxidation attenuates axotomy-induced apoptotic degeneration of facial motor neurons in neonatal rats. J Neurosci Res. 1996;44:293–299. doi: 10.1002/(SICI)1097-4547(19960501)44:3<293::AID-JNR10>3.0.CO;2-6. [DOI] [PubMed] [Google Scholar]
- Hayakawa T, Terashima M, Kayukawa Y, Ohta T, Okada T. Changes in cerebral oxygenation and hemodynamics during obstructive sleep apneas. Chest. 1996;109:916–921. doi: 10.1378/chest.109.4.916. [DOI] [PubMed] [Google Scholar]
- Lavie L. Sleep-disordered breathing and cerebrovascular disease: a mechanistic approach. Neurol Clin. 2005;23:1059–1075. doi: 10.1016/j.ncl.2005.05.005. [DOI] [PubMed] [Google Scholar]
- Lavie L. Obstructive sleep apnoea syndrome--an oxidative stress disorder. Sleep Med Rev. 2003;7:35–51. doi: 10.1053/smrv.2002.0261. [DOI] [PubMed] [Google Scholar]
- Lavie L, Vishnevsky A, Lavie P. Evidence for lipid peroxidation in obstructive sleep apnea. Sleep. 2004;27:123–128. [PubMed] [Google Scholar]
- Levine RL, Wehr N, Williams JA, Stadtman ER, Shacter E. Determination of carbonyl groups in oxidized proteins. Methods Mol Biol. 2000;99:15–24. doi: 10.1385/1-59259-054-3:15. [DOI] [PubMed] [Google Scholar]
- Levine RL, Williams JA, Stadtman ER, Shacter E. Carbonyl assays for determination of oxidatively modified proteins. Methods Enzymol. 1994;233:346–357. doi: 10.1016/s0076-6879(94)33040-9. [DOI] [PubMed] [Google Scholar]
- Liu R, Buettner GR, Oberley LW. Oxygen free radicals mediate the induction of manganese superoxide dismutase gene expression by TNF-alpha. Free Radic Biol Med. 2000;28:1197–1205. doi: 10.1016/s0891-5849(00)00237-9. [DOI] [PubMed] [Google Scholar]
- Macey KE, Macey PM, Woo MA, Henderson LA, Frysinger RC, Harper RK, Alger JR, Yan-Go F, Harper RM. Inspiratory loading elicits aberrant fMRI signal changes in obstructive sleep apnea. Respir Physiol Neurobiol. 2005 doi: 10.1016/j.resp.2005.05.024. [DOI] [PubMed] [Google Scholar]
- Macey PM, Harper RM. OSA brain morphology differences: magnitude of loss approximates age-related effects. Am J Respir Crit Care Med. 2005;172:1056–1057. doi: 10.1164/ajrccm.172.8.954. [DOI] [PubMed] [Google Scholar]
- Macey PM, Henderson LA, Macey KE, Alger JR, Frysinger RC, Woo MA, Harper RK, Yan-Go FL, Harper RM. Brain morphology associated with obstructive sleep apnea. Am J Respir Crit Care Med. 2002;166:1382–1387. doi: 10.1164/rccm.200201-050OC. [DOI] [PubMed] [Google Scholar]
- Morris R. Developments of a water-maze procedure for studying spatial learning in the rat. J Neurosci Methods. 1984;11:47–60. doi: 10.1016/0165-0270(84)90007-4. [DOI] [PubMed] [Google Scholar]
- Naegele B, Thouvard V, Pepin JL, Levy P, Bonnet C, Perret JE, Pellat J, Feuerstein C. Deficits of cognitive executive functions in patients with sleep apnea syndrome. Sleep. 1995;18:43–52. [PubMed] [Google Scholar]
- Oliver CN, Starke-Reed PE, Stadtman ER, Liu GJ, Carney JM, Floyd RA. Oxidative damage to brain proteins, loss of glutamine synthetase activity, and production of free radicals during ischemia/reperfusion-induced injury to gerbil brain. Proc Natl Acad Sci U S A. 1990;87:5144–5147. doi: 10.1073/pnas.87.13.5144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Polotsky VY, Rubin AE, Balbir A, Dean T, Smith PL, Schwartz AR, O’Donnell CP. Intermittent hypoxia causes REM sleep deficits and decreases EEG delta power in NREM sleep in the C57BL/6J mouse. Sleep Med. 2006;7:7–16. doi: 10.1016/j.sleep.2005.06.006. [DOI] [PubMed] [Google Scholar]
- Prabhakar NR, Kumar GK. Oxidative stress in the systemic and cellular responses to intermittent hypoxia. Biol Chem. 2004;385:217–221. doi: 10.1515/BC.2004.015. [DOI] [PubMed] [Google Scholar]
- Prabhakar NR, Peng YJ, Yuan G, Kumar GK. Reactive oxygen species facilitate oxygen sensing. Novartis Found Symp. 2006;272:95–99. [PubMed] [Google Scholar]
- Row BW, Liu R, Xu W, Kheirandish L, Gozal D. Intermittent hypoxia is associated with oxidative stress and spatial learning deficits in the rat. Am J Respir Crit Care Med. 2003;167:1548–1553. doi: 10.1164/rccm.200209-1050OC. [DOI] [PubMed] [Google Scholar]
- Svatikova A, Wolk R, Lerman LO, Juncos LA, Greene EL, McConnell JP, Somers VK. Oxidative stress in obstructive sleep apnoea. Eur Heart J. 2005;26:2435–2439. doi: 10.1093/eurheartj/ehi440. [DOI] [PubMed] [Google Scholar]
- Tagaito Y, Polotsky VY, Campen MJ, Wilson JA, Balbir A, Smith PL, Schwartz AR, O’Donnell CP. A model of sleep-disordered breathing in the C57BL/6J mouse. J Appl Physiol. 2001;91:2758–2766. doi: 10.1152/jappl.2001.91.6.2758. [DOI] [PubMed] [Google Scholar]
- Veasey SC, Davis CW, Fenik P, Zhan G, Hsu YJ, Pratico D, Gow A. Long-term intermittent hypoxia in mice: protracted hypersomnolence with oxidative injury to sleep-wake brain regions. Sleep. 2004;27:194–201. doi: 10.1093/sleep/27.2.194. [DOI] [PubMed] [Google Scholar]
- Weissmann N, Zeller S, Schafer RU, Turowski C, Ay M, Quanz K, Ghofrani HA, Schermuly RT, Fink L, Seeger W, Grimminger F. Impact of mitochondria and NADPH oxidases on acute and sustained hypoxic pulmonary vasoconstriction. Am J Respir Cell Mol Biol. 2006;34:505–513. doi: 10.1165/rcmb.2005-0337OC. [DOI] [PubMed] [Google Scholar]
- Woo MA, Macey PM, Keens PT, Kumar R, Fonarow GC, Hamilton MA, Harper RM. Functional abnormalities in brain areas that mediate autonomic nervous system control in advanced heart failure. J Card Fail. 2005;11:437–446. doi: 10.1016/j.cardfail.2005.02.003. [DOI] [PubMed] [Google Scholar]
- Xu W, Chi L, Row BW, Xu R, Ke Y, Xu B, Luo C, Kheirandish L, Gozal D, Liu R. Increased oxidative stress is associated with chronic intermittent hypoxia-mediated brain cortical neuronal cell apoptosis in a mouse model of sleep apnea. Neuroscience. 2004;126:313–323. doi: 10.1016/j.neuroscience.2004.03.055. [DOI] [PubMed] [Google Scholar]
- Yen HC, Oberley TD, Gairola CG, Szweda LI, St Clair DK. Manganese superoxide dismutase protects mitochondrial complex I against adriamycin-induced cardiomyopathy in transgenic mice. Arch Biochem Biophys. 1999;362:59–66. doi: 10.1006/abbi.1998.1011. [DOI] [PubMed] [Google Scholar]
- Yen HC, Oberley TD, Vichitbandha S, Ho YS, St Clair DK. The protective role of manganese superoxide dismutase against adriamycin-induced acute cardiac toxicity in transgenic mice. J Clin Invest. 1996;98:1253–1260. doi: 10.1172/JCI118909. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Younes M, Ostrowski M, Thompson W, Leslie C, Shewchuk W. Chemical control stability in patients with obstructive sleep apnea. Am J Respir Crit Care Med. 2001;163:1181–1190. doi: 10.1164/ajrccm.163.5.2007013. [DOI] [PubMed] [Google Scholar]
- Young T, Peppard PE, Gottlieb DJ. Epidemiology of obstructive sleep apnea: a population health perspective. Am J Respir Crit Care Med. 2002a;165:1217–1239. doi: 10.1164/rccm.2109080. [DOI] [PubMed] [Google Scholar]
- Young T, Shahar E, Nieto FJ, Redline S, Newman AB, Gottlieb DJ, Walsleben JA, Finn L, Enright P, Samet JM. Predictors of sleep-disordered breathing in community-dwelling adults: the Sleep Heart Health Study. Arch Intern Med. 2002b;162:893–900. doi: 10.1001/archinte.162.8.893. [DOI] [PubMed] [Google Scholar]
- Zhan G, Serrano F, Fenik P, Hsu R, Kong L, Pratico D, Klann E, Veasey SC. NADPH oxidase mediates hypersomnolence and brain oxidative injury in a murine model of sleep apnea. Am J Respir Crit Care Med. 2005;172:921–929. doi: 10.1164/rccm.200504-581OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zwicker K, Dikalov S, Matuschka S, Mainka L, Hofmann M, Khramtsov V, Zimmer G. Oxygen radical generation and enzymatic properties of mitochondria in hypoxia/reoxygenation. Arzneimittelforschung. 1998;48:629–636. [PubMed] [Google Scholar]