

Abstract
Background/Aims
Sepsis remains a leading cause of death in critically ill patients. Because endotoxemia is viewed as a key mediator of sepsis-induced inflammation, administration of bacterial endotoxin (LPS) is often used to simulate sepsis in experimental animals. This study tests the hypothesis that LPS is a critical determinant of the hepatic microvascular dysfunction in mice made septic by cecal ligation and puncture (CLP).
Methods
Intravital videomicroscopy was used to quantify sinusoidal perfusion, and platelet and leukocyte adhesion in terminal hepatic venules (THV) and sinusoids in LPS-sensitive and LPS-insensitive mice subjected to CLP or LPS (i.p.). mRNA expression of TLR-2, TLR-4, MyD-88 and Ly-96 was also assessed.
Results
While LPS-sensitive mice responded to both CLP and LPS challenges with elevated leukocyte and platelet adhesion in THV and sinusoids, and a reduced sinusoidal perfusion density, LPS-insensitive mice exhibited comparable blood cell adhesion and sinusoidal malperfusion following CLP, but not LPS. Hepatic mRNA of MyD-88 and TLR-2 were elevated in the CLP and LPS groups. Endotoxin was not detectable in the blood of LPS-sensitive mice after CLP, but was elevated after LPS administration.
Conclusion
These findings do not support a major role for LPS in the hepatic microvascular disturbances associated with polymicrobial sepsis.
Keywords: LPS, platelets, sepsis, intravital fluorescence microscopy, liver injury, TLR-4
Introduction
Sepsis remains a major clinical problem, with an incidence of about 3 cases per 1000 population and a mortality rate of ~30%. (1) Multiple organ failure derived from sepsis is associated with an even higher mortality that increases substantially with the number of organs involved. (2) The liver is the second most commonly affected organ in sepsis/multiple organ failure and the development of hepatic dysfunction during the multiple organ dysfunction syndrome is usually fatal. (3) Microcirculatory failure resulting from the recruitment and activation of neutrophils is considered to be a critical event in the development of liver injury during sepsis. (4) Although neutrophils make an important contribution to the elimination of invading microorganisms, these phagocytic cells can also cause tissue damage that results from the overproduction of reactive oxygen metabolites and inflammatory mediators. (5) There is also growing evidence for the involvement of other blood cells, such as platelets in the development of the microcirculatory dysfunction during sepsis. (6) However, the mechanisms that are responsible for promoting the participation of leukocytes and platelets in the hepatic microcirculatory failure and hepatocellular dysfunction during sepsis remain unclear.
Several methods have been used to induce and study sepsis in animal models. These include soft tissue infection, intravascular infusion of live bacteria, administration of endotoxin/lipopolysaccharide (LPS) and cecal ligation and puncture (CLP), with the latter two models representing the most commonly used. (7) There are prominent similarities and differences between the LPS and CLP models of experimental sepsis. In mice, the two models are associated with similar behavioral deficits, mortality rates, and reductions in leukocyte counts. (8) A major difference between the models is that LPS simulates gram-negative sepsis whereas CLP simulates the clinical situation of polymicrobial sepsis. There are also significant qualitative and quantitative differences in the cytokine profiles in serum and organ homogenates between the two models. (9) While endotoxemia and CLP are sometimes used interchangeably (10) and it is often assumed that LPS-mediated events contribute to the pathogenesis of sepsis (11, (12), the differences between the models noted above, coupled to reports describing the involvement of LPS-independent apoptotic pathways in thymus, spleen, lung and gut (13), as well as LPS-independent mechanisms in the increased endothelial cell adhesion molecules expression associated with CLP (14) place the validity of this assumption in question.
Toll-like receptors (TLRs) are receptors of the innate immune system recognizing pathogen associated molecular patterns. TLR-2 is the major signaling receptor for peptidoglycan, a major part of the cell wall of gram-positive bacteria, whereas the major signaling receptor of LPS/endotoxin (part of the cell wall of gram-negative bacteria) is TLR-4. (15) Stimulation of the extracellular domain of a TLR triggers the intracellular association of myeloid differentiation factor 88 (MyD-88) with its cytosolic domain. (16) MyD-88 therefore serves as a key adaptor protein for both TLR-2 and TLR-4, linking the receptors to downstream kinases. (15) Another protein required for functional TLR-4 signaling is the lymphocyte antigen 96 (Ly96) also called myeloid differentiation-2 (MD-2). (17) This protein is associated with the extracellular domain of TLR-4 and is required for LPS signaling through TLR-4, which is confirmed by studies showing that Ly96 null mice do not respond to LPS. (18)
The overall objective of this study was to determine whether LPS contributes to the hepatic microvascular dysfunction induced by CLP. This objective was achieved by comparing the responses of blood cell-endothelial interactions and perfused sinusoidal density following CLP or LPS challenge in LPS-sensitive (C3HeB/FeJ) and LPS-insensitive (C3H/HeJ) mice. To further elucidate the role of LPS in CLP-induced hepatic microvascular dysfunction, we monitored expression of TLR-2, TLR-4 (the main receptor for LPS signaling), MyD88 and Ly96 using RT-PCR in livers of LPS-sensitive mice following CLP and compared the results to values obtained from sham-operated animals and mice injected with 50 and 500 μg/kg LPS, respectively. LPS concentrations were also measured in the portal vein and the inferior vena cava of LPS-sensitive mice after sham operation, CLP or LPS administration.
Material and Methods
Animals
Male C3H/HeJ (LPS-insensitive) or C3HeB/FeJ (LPS-sensitive) mice (6–8 weeks; 26.0g ± 0.5g) were obtained from Jackson Laboratory (Bar Harbor, ME). Animal handling procedures were approved by the LSU Health Sciences Center Institutional Animal Care and Use Committee and were in accordance with the guidelines of the American Physiological Society.
Experimental protocols
In the first set of experiments, the hepatic microcirculatory responses to CLP or 50 or 500 μg/kg LPS were compared between LPS-sensitive or LPS-insensitive animals. At the end of each experiment, blood was taken from the heart for blood cell counts and measurement of ALT. In a second set of experiments, liver was harvested at 6 hours following CLP or LPS, and RT-PCR was performed for measurement of hepatic expression of TLR-2, TLR-4, My-D88 and Ly96. Additionally blood was taken from the portal vein and the inferior vena cava for assessment of endotoxin concentration.
Cecal ligation and perforation (CLP)
To induce sepsis, the CLP procedure was used as described previously. (19) Briefly, animals were anesthetized with ketamine hydrochloride (150 mg/kg i.m.) and xylazine (7.5 mg/kg i.m.). A midline laparatomy was performed, the cecum was exteriorized and ligated distal of the ileocecal valve without causing intestinal obstruction. Then, the cecum was perforated three times using a 20-gauge needle (top, middle and bottom third) and squeezed to extrude fecal contents that were spread around the cecum using a cotton swab. The incision was closed and each mouse received 1 ml of normal saline subcutaneously for fluid resuscitation. The animals were allowed free access to standard chow and water after induction of sepsis. In sham animals, the cecum was exteriorized without ligation and puncture, whereas control mice did not undergo any surgery at all. These groups yielded comparable results and therefore were combined as the control group for the CLP data in Figures 1–3.
Figure 1.
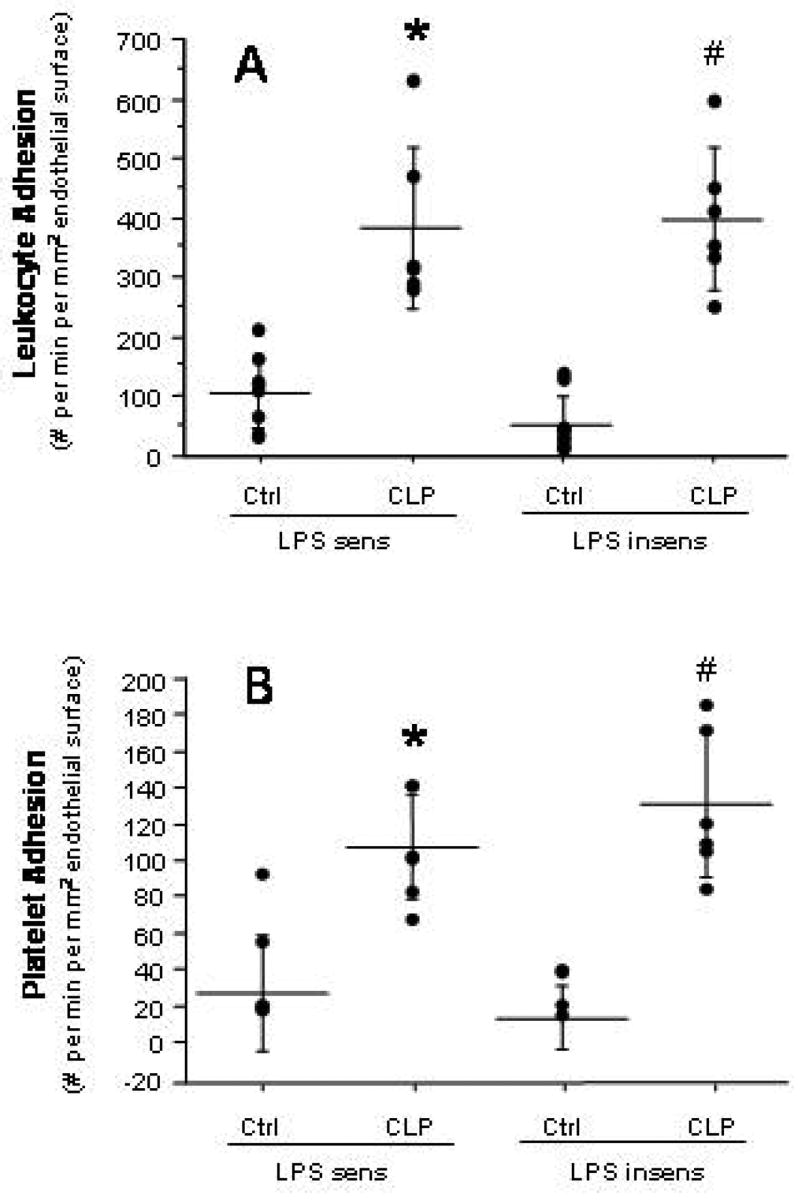
Firm adhesion of leukocytes (panel A) and platelets (panel B) in terminal hepatic venules following cecal ligation and puncture (CLP) in LPS-sensitive and LPS-insensitive mice. * p < 0.005 versus LPS sens Ctrl; # p < 0.0005 versus LPS insens Ctrl. n=8 for LPS sens Ctrl, n=6 for LPS sens CLP, n=9 for LPS insens Ctrl, n=6 for LPS insens CLP.
Figure 3.
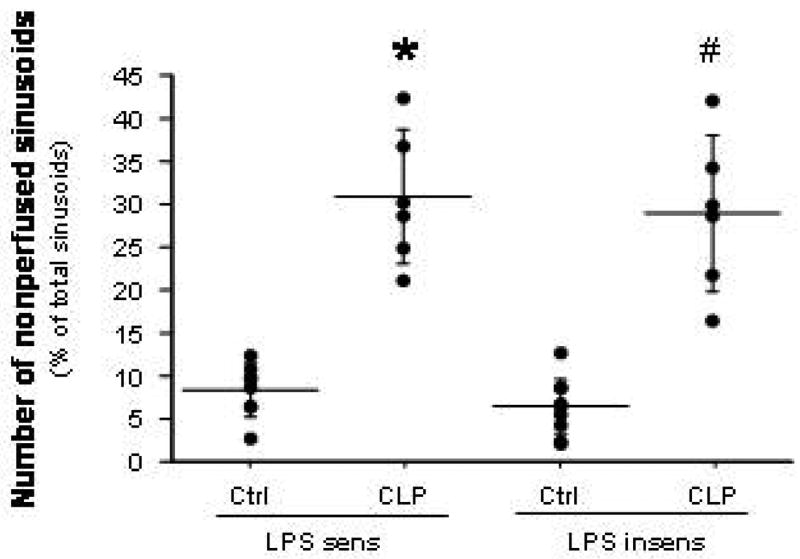
Number of nonperfused sinusoids (NPS) in LPS-sensitive and LPS-insensitive mice after cecal ligation and puncture (CLP). * p < 0.001 versus LPS sens Ctrl; # p < 0.0005 versus LPS insens Ctrl. n=8 for LPS sens Ctrl, n=6 for LPS sens CLP, n=9 for LPS insens Ctrl, n=6 for LPS insens CLP.
LPS-administration
LPS-sensitive or LPS-insensitive mice were anesthetized as above and injected with 50 or 500 μg/kg LPS from E. coli 0111:B4 (Sigma, St. Louis, MO) i.p. After the injection, each mouse received 1 ml of normal saline subcutaneously for fluid resuscitation.
Isolation of platelets
Platelets were obtained from the blood of corresponding donor mice and labeled with the fluorchrome carboxyfluorescein diacetate succinimudyl ester (CFDASE; Molecular Probes, Eugene, OR) as previously described. This isolation process does not cause activation of platelets as determined by P-selectin expression. (20)
Intravital microscopy
The animals were re-anesthetized 6 hours after the induction of CLP or LPS administration, and intravital videomicrosopy was performed as described in detail elsewhere (21). The liver surface was scanned for 3 – 5 venules, each of which was recorded for 1 minute. 100 μm segments of terminal hepatic venules (THV) (diameter=15–45 μm) were observed. Leukocytes and platelets were classified as firmly adherent cells if they remained stationary on the vessel wall for >10 seconds and were quantified as #/mm2 venular wall (calculated assuming cylindrical vessel geometry). (22) Within sinusoids, blood cells were considered stationary if they did not move for the entire one minute observation period and expressed as #/mm2 liver surface. Sinusoidal perfusion failure was quantified by the number of nonperfused sinusoids (expressed as nonperfused:total sinusoids per field of view). A sinusoid was considered nonperfused if no white blood cells were observed flowing through it.
Blood cell counts and Alanine aminotransferase (ALT) activity
Blood was collected via cardiac puncture at the end of the experiments. Leukocyte and platelet counts were performed with a hemocytometer, and serum ALT was measured using a kit from ThermoDMA (Louisville, CO). ALT data are presented as units per liter at 37°C.
TLR-4 expression
Real-time PCR using standard protocols was employed to measure the expression of TLR-2, TLR-4, MyD-88 and Ly96 in liver samples from LPS-sensitive mice following either CLP or LPS-administration. Gene expression was determined using a comparative critical threshold (CT) method. The content of the relevant gene in livers of mice exposed to CLP or LPS was normalized to 18s content and expressed relative to the sham-operated group.
Endotoxin measurement
At the end of each experiment, a heparinized blood sample was collected directly from the portal vein and the inferior vena cava. The platelet-rich plasma was prepared as described previously. (23) Endotoxin was determined using a kinetic chromogenic assay with a sensitivity range=0.005–50 EU/ml (Cambrex; Cottonwood, Az).
Statistical analysis
The Mann Whitney non-parametric test was used to compare two groups, and the Kruskal Wallis test with Dunns post-hoc test was used to compare three or more groups, as appropriate. The values are expressed as means ± standard deviation.
Results
CLP reduced platelet counts in both LPS-sensitive and –insensitive mouse strains (not significant) (Table 1). In contrast, either 50 or 500 μg/kg LPS significantly reduced blood leukocyte, but not platelet, count in LPS-sensitive mice, while neither blood cell was affected by LPS in LPS-insensitive mice.
Table 1.
Changes in blood cell counts following CLP or LPS treatment in LPS-sensitive and LPS-insensitive mice.
| Leukocytes (#/μl) | Platelets (# x 103/μl) | ALT (IU/l) | |
|---|---|---|---|
| LPS-sensitive | |||
| Control | 2680 ± 460 | 1100 ± 83 | 32 ± 5 |
| Sham | 3340 ± 860 | 1089 ± 114 | 62 ± 6 |
| CLP | 2910 ± 360 | 841 ± 56 | 54 ± 4 |
| 50 μg/kg LPS | 1520 ± 180 * # | 621 ± 73 * # | 44 ± 6 |
| 500 μg/kg LPS | 1060 ± 120 * # | 784 ± 65 * | 81 ± 10 * |
|
| |||
| LPS-insensitive | |||
| Control | 4330 ± 740 | 1145 ± 70 | 35 ± 9 |
| Sham | 4900 ± 570 | 985 ± 55 | 56 ± 7 |
| CLP | 2970 ± 340 | 794 ± 67 | 64 ± 5 |
| 50 μg/kg LPS | 5450 ± 980 | 998 ± 104 | 65 ± 10 |
| 500 μg/kg LPS | 4180 ± 1140 | 1017 ± 19 | 56 ± 7 |
p < 0.05 versus corresponding controls
p < 0.05 versus corresponding dose in LPS-insensitive mice.
CLP experiments
Compared to their respective control mice, both LPS-sensitive and LPS-insensitive mice exhibited significantly elevated yet comparable levels of leukocyte (panel A) and platelet (panel B) adhesion in terminal hepatic venules (THV) at 6 hrs following induction of sepsis by CLP (Figure 1). A similar pattern of leukocyte and platelet recruitment responses was noted in hepatic sinusoids (Figure 2) of the two mouse strains after CLP. The blood cell recruitment responses in liver sinusoids were accompanied by a significant reduction in the density of perfused sinusoids (Figure 3) in both LPS-sensitive and LPS-insensitive mice. Hence, for all measured variables related to blood cell recruitment and sinusoidal perfusion, no significant differences were noted in the liver microcirculations of LPS-sensitive and LPS-insensitive mice in response to CLP.
Figure 2.
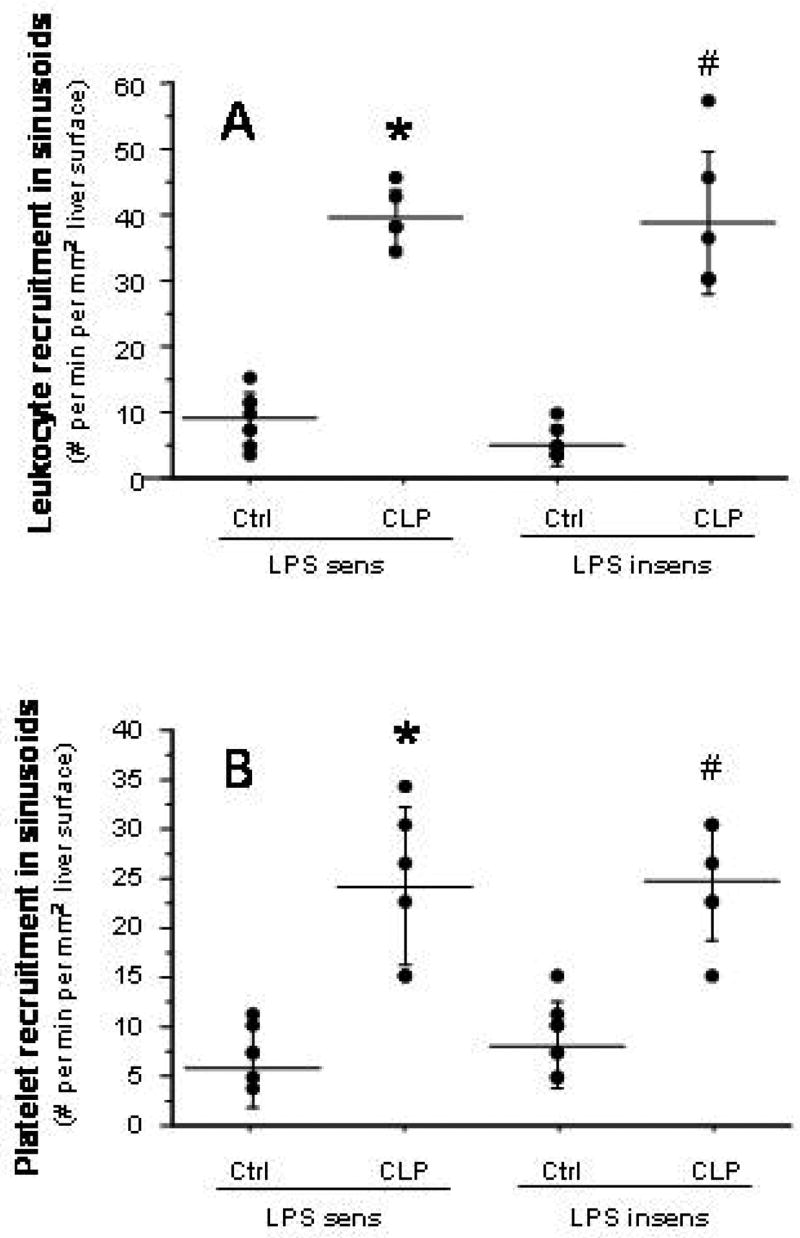
Leukocyte (panel A) and platelet (panel B) recruitment in hepatic sinusoids of LPS-sensitive and LPS-insensitive mice after cecal ligation and puncture (CLP). * p < 0.001 versus LPS sens Ctrl; # p < 0.0005 versus LPS insens Ctrl. n=8 for LPS sens Ctrl, n=6 for LPS sens CLP, n=9 for LPS insens Ctrl, n=6 for LPS insens CLP.
Serum-ALT levels (an indicator of hepatocellular injury) were not altered 6 hours following the CLP-procedure in both strains of mice when compared with control or sham-operated mice (Table 1).
LPS experiments
Treatment of LPS-sensitive mice with LPS resulted in significant increases in both leukocyte and platelet adhesion in THV of LPS-sensitive mice at 500 μg/kg, but not at 50 μg/kg LPS (Figure 4). In LPS-insensitive mice neither dose of LPS affected leukocyte or platelet adhesion in THV.
Figure 4.
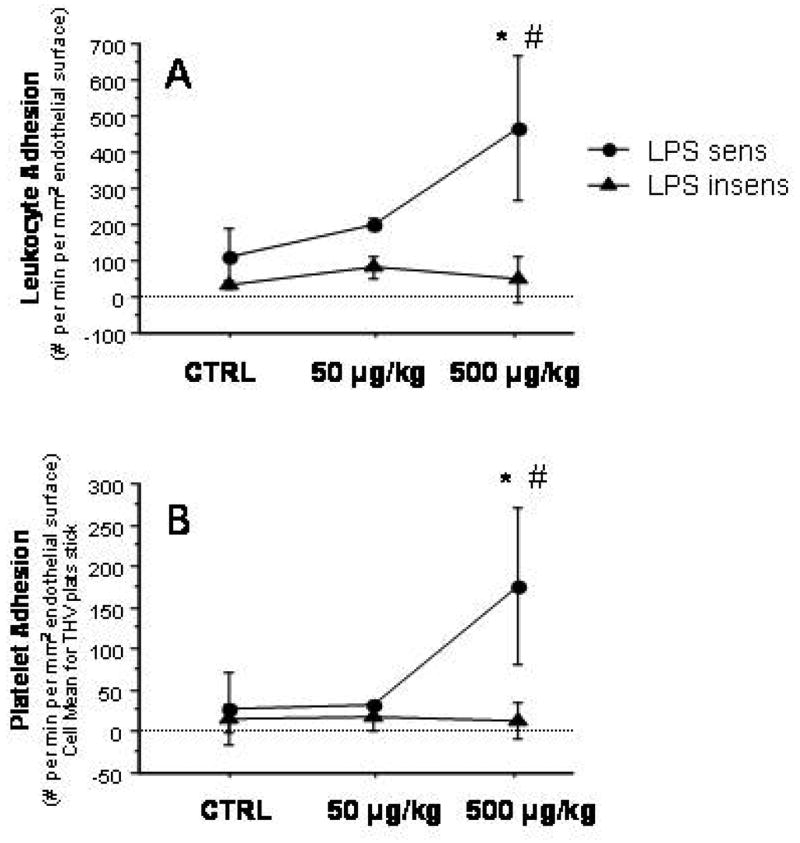
Leukocyte (panel A) and platelet adhesion (panel B) in terminal hepatic venules of LPS-sensitive and LPS-insensitive mice after graded doses of LPS (50 and 500 μg/kg, respectively) and controls. * p < 0.05 versus corresponding Ctrl; # p < 0.05 versus corresponding LPS.insens group. n=4 for LPS sens Ctrl, n=3 for LPS sens 50g/kg, n=6 for LPS sens 500 g/kg, n=5 for LPS insens Ctrl, n=3 for LPS insens 50g/kg, n=3 for LPS insens 500 g/kg.
Injection of LPS in LPS-sensitive mice resulted in the recruitment of both leukocytes and platelets in hepatic sinusoids (Figure 5). However, these blood cell adhesion responses were not elicited by LPS in the liver sinusoids of LPS-insensitive mice. LPS administration also caused an impairment of the sinusoidal perfusion, as reflected by the nearly 3-fold increase in the number of nonperfused sinusoids observed in livers of LPS-sensitive mice receiving 500 μg/kg LPS (Figure 6). No changes in the density of perfused sinusoids were noted after either dose of LPS in LPS-insensitive mice.
Figure 5.
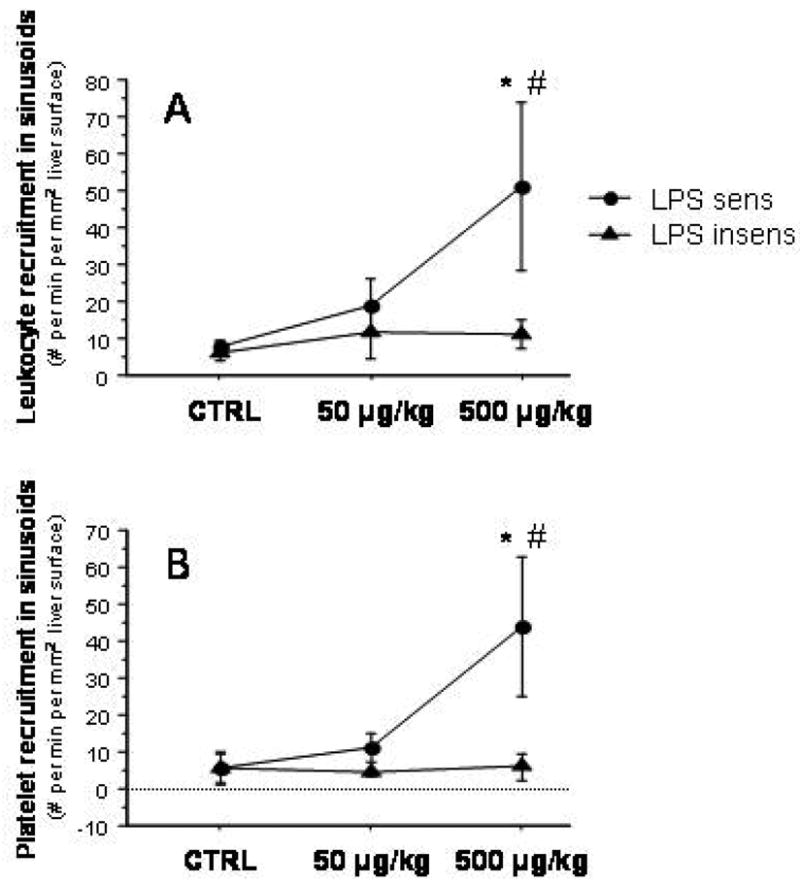
Leukocyte (panel A) and platelet (panel B) recruitment in hepatic sinusoids following injection of 50 and 500 μg/kg LPS in LPS-sensitive and LPS-insensitive mice. * p < 0.01 versus corresponding Ctrl; # p < 0.05 versus corresponding LPS insens group. n=4 for LPS sens Ctrl, n=3 for LPS sens 50g/kg, n=6 for LPS sens 500 g/kg, n=5 for LPS insens Ctrl, n=3 for LPS insens 50g/kg, n=3 for LPS insens 500 g/kg.
Figure 6.
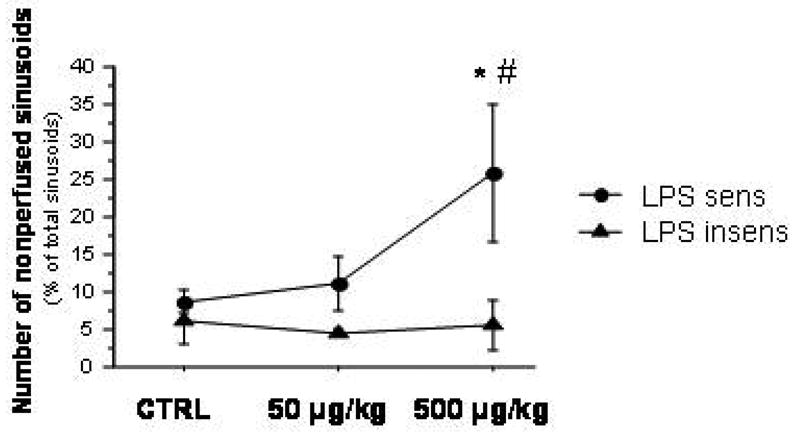
Sinusoidal perfusion failure (number of nonperfused sinusoids) in LPS-sensitive and LPS-insensitive animals after treatment with 50 and 500 μg/kg LPS. * p < 0.05 versus corresponding Ctrl; # p < 0.05 versus corresponding LPS insens group. n=4 for LPS sens Ctrl, n=3 for LPS sens 50g/kg, n=6 for LPS sens 500 g/kg, n=5 for LPS insens Ctrl, n=3 for LPS insens 50g/kg, n=3 for LPS insens 500 g/kg.
LPS administration (at both doses) in LPS-insensitive mice did not alter the serum ALT levels. However, ALT levels were significantly increased by 500 μg/kg in LPS-sensitive mice (Table 1).
Endotoxin concentrations
While LPS/endotoxin was not detected in either sham or CLP mice, i.p. injection of LPS increased the values of LPS in both LPS treated groups reaching statistical significance at a dose of 500 μg/kg LPS (Table 2).
Table 2.
Endotoxin-Concentrations in Portal Vein and V. cava inferior in LPS-sensitive mice after sham operation, CLP and graded doses of LPS-injection (50 and 500 μg/kg, respectively).
| LPS in Portal Vein (EU/ml) | LPS in V. cava inferior (EU/ml) | |
|---|---|---|
| Sham | n.d. (n=3) | n.d. (n=3) |
| CLP | n.d. (n=3) | n.d. (n=3) |
| LPS 50 μg/kg | 18.9 ± 4.8(n=4) | 49.3 ± 26.1 (n=4) |
| LPS 500 μg/kg | 653.7 ± 175.7 * (n=5) | 561.4 ± 139.1 * (n=5) |
n.d. not detectable
p < 0.05 versus sham, CLP, LPS 50 μg/kg
Hepatic mRNA expression
CLP and both LPS doses caused an approximate 8-fold increase of MyD-88 expression when compared to sham animals while Ly96 expression did not change in all three experimental groups (Figure 7). Hepatic TLR-2 expression increased more than 30-fold in all mice after CLP and LPS-treatment compared to sham animals, whereas TLR-4 expression was only slightly elevated in the LPS groups. Hepatic TLR-4 expression did not change in the CLP group when compared to sham-operated mice. The observed changes in MyD-88 and TLR2 were not significant, most likely due to small numbers/group.
Figure 7.

Fold changes in hepatic mRNA expression of MyD-88, Ly96, TLR-2 and TLR-4, measured using RT-PCR in LPS-sensitive mice following sham-surgery, CLP, or administration of either 50 or 500 μg/kg LPS.
Discussion
A recent study performed in our laboratory indicates that the liver microvasculature of C57Bl/6 mice is profoundly affected by experimental sepsis induced by CLP. The hepatic microvascular responses to CLP include l) the accumulation of leukocytes and platelets in THV and hepatic sinusoids, and 2) an increase in the number of nonperfused sinusoids. (21) In the present study, similar responses of the hepatic microvasculature to CLP were observed in C3HeB/FeJ (LPS-sensitive) mice. Furthermore, we observed that administration of exogenous LPS to C3HeB/FeJ mice elicits qualitatively and quantitatively (at 500 μg/kg) similar responses to CLP in the hepatic microvasculature. While these similarities between LPS- and CLP-induced sepsis suggest that LPS mediates this response, a comparison of the hepatic microvascular responses to CLP between LPS-sensitive and LPS-insensitive mice does not support this possibility.
A comparison of the responses in the hepatic microvasculature to different doses of LPS in sensitive and insensitive mice confirms the view that these mice can distinguish between LPS-dependent and LPS-independent pathways. The mechanism underlying the LPS-insensitivity in C3H/HeJ mice involves a defect in the TLR-4 receptor. (24) This receptor is known to engage LPS produced by Gram-negative bacteria by forming a molecular protein complex with CD14 and the secreted protein Ly96. (25) Our data indicate that the LPS-induced blood cell/endothelial cell interactions and sinusoidal malperfusion in the liver are not manifested in the absence of a functional TLR-4. This observation with exogenous LPS is consistent with a previous report demonstrating a more profound E-selectin upregulation on endothelial cells in LPS-sensitive mice exposed to LPS, compared to the largely absent response observed in LPS-insensitive mice. (14) Other cell types are also likely to participate in the blunted responses observed in C3H/HeJ mice since macrophages derived from these mice fail to induce inflammatory cytokines such as TNF, IL-1 and IL-6 in response to LPS challenge and their splenic B-cells do not proliferate after LPS-exposure. (26)
In order to optimize our comparison of the CLP and LPS models of experimental sepsis, both experimental procedures were kept as similar as possible, e.g., both groups were anesthetized, and the animals received the same fluid resuscitation. There are several lines of evidence derived from our work that support the view that LPS (and consequentially TLR-4) is not important in eliciting the hepatic microvascular responses to the CLP model of polymicrobial sepsis. First, induction of sepsis using CLP caused similar alterations of the hepatic microvasculature, i.e. leukocyte and platelet adhesion in venules, blood cell recruitment in sinusoids and number of nonperfused sinusoids, in both LPS-insensitive and LPS-sensitive mice. Second, a dose of 500 μg/kg LPS was required to produce quantitatively similar responses of the hepatic microcirculation to CLP in LPS-sensitive mice. Even at this high dose of LPS, we were unable to elicit any of the hepatic microvascular alterations in LPS-insensitive mice, which is consistent to previous reports that describe a hyporesponsiveness of the hepatic microcirculation (leukocyte adhesion to sinusoids and central venules and sinusoidal perfusion) in LPS-insensitive mice challenged with LPS. (27) Third, injection of both doses of LPS tended to reduce blood leukocyte count in LPS-sensitive mice, while CLP had no effect on this variable in the same mouse strain (see Table 1). Fourth, injection of LPS at a dose of 500 μg/kg caused a significant elevation in ALT levels in LPS-sensitive mice, while CLP did not alter serum ALT at the same time (6 hrs) after induction of the inflammatory state.
Another finding that argues against a role for LPS as a mediator of the responses elicited by CLP is the absence of detectable LPS in portal and vena caval blood after CLP, while the bacterial product was detected in blood after LPS administration. Our results contrast others that show a significant increase of LPS in blood samples obtained from the hearts of mice after CLP. (28) However, the levels of LPS detected by these authors in mice following CLP were about one-tenth the values detected in our experimental group treated with 50 μg/kg LPS and about 380-fold lower than the values detected in our 500 μg/kg LPS group.
The increase in hepatic gene expression of TLR-2 and the unchanged expression of TLR-4 mRNA in LPS-sensitive animals following two doses of LPS in our study are consistent with previous reports. Murine splenic macrophages respond to treatment with LPS with an increased TLR-2 mRNA while TLR-4 gene expression remains constant. (29) Similarly, it has been shown in mice that LPS treatment upregulates hepatic TLR-2 mRNA and does not affect TLR-4 gene expression in the liver. (27) Our results in the CLP group (upregulation of TLR-2 and unchanged TLR-4) are in contrast to a report showing increases of both TLR-2 and TLR-4 in the livers of CLP treated ICR/HSD mice. (30) This difference may relate to the different mouse strains studied.
Ly96 is a binding protein that is required for the function of TLR-4 and several reports have identified the TLR-4/Ly96 complex as the major signaling receptor for LPS in mammals. (26, (31) Therefore, it is not unexpected that Ly96 mRNA levels in mouse liver do not change after either CLP or LPS treatment, showing the same pattern as TLR-4 mRNA expression. MyD-88 has been identified as a cytosolic adaptor protein for several TLRs including both TLR-2 and TLR-4. (32) Thus, the upregulation of TLR-2 noted in all of our experimental groups may be closely linked to the increased expression of MyD-88. It has been shown that a deficiency of MyD-88, but not TLR-2 or TLR-4, renders mice more resistant to the lethal effects of polymicrobial sepsis. (32) Our results, coupled to the findings of the latter report, argue against a role of TLR-4 in mediating the hepatic responses to polymicrobial sepsis.
While our results argue against a role for LPS as a mediator of the hepatic microvascular responses to CLP, a contribution of other bacterial products cannot be discounted, as supported by studies showing a significantly improved survival of CLP-mice treated with antibiotics. (33) Furthermore, the phagocytic function of neutrophils appears to be important in the control of sepsis. Depletion of neutrophils before the induction of CLP greatly enhanced the bacteremic response to this insult (34) and a recent study from our laboratory strongly implicates neutrophils in the hepatic microvascular alterations induced by CLP. (21) However, it remains unclear whether the presence of whole bacteria per se and/or immune responses related to the presence of bacteria in the blood stream (eg. complement activation) contributes to the blood cell recruitment and sinusoidal malperfusion that result from CLP-induced sepsis.
Acknowledgments
Supported by a grant from the National Institute of Diabetes and Digestive and Kidney Diseases (P01 DK43785)
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Angus DC, Linde-Zwirble WT, Lidicker J, Clermont G, Carcillo J, Pinsky MR. Epidemiology of severe sepsis in the United States: analysis of incidence, outcome, and associated costs of care. Crit Care Med. 2001;29:1303–1310. doi: 10.1097/00003246-200107000-00002. [DOI] [PubMed] [Google Scholar]
- 2.Fry DE, Pearlstein L, Fulton RL, Polk HC., Jr Multiple system organ failure. The role of uncontrolled infection. Arch Surg. 1980;115:136–140. doi: 10.1001/archsurg.1980.01380020006003. [DOI] [PubMed] [Google Scholar]
- 3.Beal AL, Cerra FB. Multiple organ failure syndrome in the 1990s. Systemic inflammatory response and organ dysfunction. Jama. 1994;271:226–233. [PubMed] [Google Scholar]
- 4.Eipel C, Bordel R, Nickels RM, Menger MD, Vollmar B. Impact of leukocytes and platelets in mediating hepatocyte apoptosis in a rat model of systemic endotoxemia. Am J Physiol Gastrointest Liver Physiol. 2004;286:G769–G776. doi: 10.1152/ajpgi.00275.2003. [DOI] [PubMed] [Google Scholar]
- 5.Salkowski CA, Detore G, Franks A, Falk MC, Vogel SN. Pulmonary and hepatic gene expression following cecal ligation and puncture: monophosphoryl lipid A prophylaxis attenuates sepsis-induced cytokine and chemokine expression and neutrophil infiltration. Infect Immun. 1998;66:3569–3578. doi: 10.1128/iai.66.8.3569-3578.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Russwurm S, Vickers J, Meier-Hellmann A, Spangenberg P, Bredle D, Reinhart K, et al. Platelet and leukocyte activation correlate with the severity of septic organ dysfunction. Shock. 2002;17:263–268. doi: 10.1097/00024382-200204000-00004. [DOI] [PubMed] [Google Scholar]
- 7.Garrido AG, Poli de Figueiredo LF, Rocha e Silva M. Experimental models of sepsis and septic shock: an overview. Acta Cirurgica Brasileira. 2004;19:82–88. [Google Scholar]
- 8.Remick DG, Newcomb DE, Bolgos GL, Call DR. Comparison of the mortality and inflammatory response of two models of sepsis: lipopolysaccharide vs. cecal ligation and puncture. Shock. 2000;13:110–116. doi: 10.1097/00024382-200013020-00004. [DOI] [PubMed] [Google Scholar]
- 9.Villa P, Sartor G, Angelini M, Sironi M, Conni M, Gnocchi P, et al. Pattern of cytokines and pharmacomodulation in sepsis induced by cecal ligation and puncture compared with that induced by endotoxin. Clin Diagn Lab Immunol. 1995;2:549–553. doi: 10.1128/cdli.2.5.549-553.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Croner RS, Hoerer E, Kulu Y, Hackert T, Gebhard MM, Herfarth C, et al. Hepatic platelet and leukocyte adherence during endotoxemia. Crit Care. 2006;10:R15. doi: 10.1186/cc3968. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Lush CW, Kvietys PR. Microvascular dysfunction in sepsis. Microcirculation. 2000;7:83–101. doi: 10.1038/sj.mn.7300096. [DOI] [PubMed] [Google Scholar]
- 12.Sorkine P, Szold O, Halpern P, Gutman M, Greemland M, Rudick V, et al. Gut decontamination reduces bowel ischemia-induced lung injury in rats. Chest. 1997;112:491–495. doi: 10.1378/chest.112.2.491. [DOI] [PubMed] [Google Scholar]
- 13.Hiramatsu M, Hotchkiss RS, Karl IE, Buchman TG. Cecal ligation and puncture (CLP) induces apoptosis in thymus, spleen, lung, and gut by an endotoxin and TNF-independent pathway. Shock. 1997;7:247–253. doi: 10.1097/00024382-199704000-00002. [DOI] [PubMed] [Google Scholar]
- 14.Bauer P, Russell JM, Granger DN. Role of endotoxin in intestinal reperfusion-induced expression of E-selectin. Am J Physiol. 1999;276:G479–G484. doi: 10.1152/ajpgi.1999.276.2.G479. [DOI] [PubMed] [Google Scholar]
- 15.Medzhitov R. Toll-like receptors and innate immunity. Nat Rev Immunol. 2001;1:135–145. doi: 10.1038/35100529. [DOI] [PubMed] [Google Scholar]
- 16.Miller SI, Ernst RK, Bader MW. LPS, TLR4 and infectious disease diversity. Nat Rev Microbiol. 2005;3:36–46. doi: 10.1038/nrmicro1068. [DOI] [PubMed] [Google Scholar]
- 17.Gangloff M, Gay NJ. MD-2: the Toll 'gatekeeper' in endotoxin signalling. Trends Biochem Sci. 2004;29:294–300. doi: 10.1016/j.tibs.2004.04.008. [DOI] [PubMed] [Google Scholar]
- 18.Nagai Y, Akashi S, Nagafuku M, Ogata M, Iwakura Y, Akira S, et al. Essential role of MD-2 in LPS responsiveness and TLR4 distribution. Nat Immunol. 2002;3:667–672. doi: 10.1038/ni809. [DOI] [PubMed] [Google Scholar]
- 19.Bauer P, Lush CW, Kvietys PR, Russell JM, Granger DN. Role of endotoxin in the expression of endothelial selectins after cecal ligation and perforation. Am J Physiol Regul Integr Comp Physiol. 2000;278:R1140–R1147. doi: 10.1152/ajpregu.2000.278.5.R1140. [DOI] [PubMed] [Google Scholar]
- 20.Tailor A, Lefer DJ, Granger DN. HMG-CoA reductase inhibitor attenuates platelet adhesion in intestinal venules of hypercholesterolemic mice. Am J Physiol Heart Circ Physiol. 2004;286:H1402–H1407. doi: 10.1152/ajpheart.00993.2003. [DOI] [PubMed] [Google Scholar]
- 21.Singer G, Urakami H, Specian RD, Stokes KY, Granger DN. Platelet recruitment in the murine hepatic microvasculature during experimental sepsis: role of neutrophils. Microcirculation. 2006;13:89–97. doi: 10.1080/10739680500466343. [DOI] [PubMed] [Google Scholar]
- 22.Massberg S, Eisenmenger S, Enders G, Krombach F, Messmer K. Quantitative analysis of small intestinal microcirculation in the mouse. Res Exp Med (Berl) 1998;198:23–35. doi: 10.1007/s004330050086. [DOI] [PubMed] [Google Scholar]
- 23.Rivera CA, Thurman RG. Tips for measuring endotoxin in plasma. Alcohol Clin Exp Res. 1998;22:2192–2194. doi: 10.1111/j.1530-0277.1998.tb05933.x. [DOI] [PubMed] [Google Scholar]
- 24.Palsson-McDermott EM, O'Neill LA. Signal transduction by the lipopolysaccharide receptor, Toll-like receptor-4. Immunology. 2004;113:153–162. doi: 10.1111/j.1365-2567.2004.01976.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Akashi S, Shimazu R, Ogata H, Nagai Y, Takeda K, Kimoto M, et al. Cutting edge: cell surface expression and lipopolysaccharide signaling via the toll-like receptor 4-MD-2 complex on mouse peritoneal macrophages. J Immunol. 2000;164:3471–3475. doi: 10.4049/jimmunol.164.7.3471. [DOI] [PubMed] [Google Scholar]
- 26.Hoshino K, Takeuchi O, Kawai T, Sanjo H, Ogawa T, Takeda Y, et al. Cutting edge: Toll-like receptor 4 (TLR4)-deficient mice are hyporesponsive to lipopolysaccharide: evidence for TLR4 as the Lps gene product. J Immunol. 1999;162:3749–3752. [PubMed] [Google Scholar]
- 27.Matsumoto Y, Ito Y, Hayashi I, Majima M, Ishii K, Katagiri H, et al. Effect of FR167653, a novel inhibitor of tumor necrosis factor-alpha and interleukin-1beta synthesis on lipopolysaccharide-induced hepatic microvascular dysfunction in mice. Shock. 2002;17:411–415. doi: 10.1097/00024382-200205000-00012. [DOI] [PubMed] [Google Scholar]
- 28.Vianna RC, Gomes RN, Bozza FA, Amancio RT, Bozza PT, David CM, et al. Antibiotic treatment in a murine model of sepsis: impact on cytokines and endotoxin release. Shock. 2004;21:115–120. doi: 10.1097/01.shk.0000111828.07309.21. [DOI] [PubMed] [Google Scholar]
- 29.Matsuguchi T, Musikacharoen T, Ogawa T, Yoshikai Y. Gene expressions of Toll-like receptor 2, but not Toll-like receptor 4, is induced by LPS and inflammatory cytokines in mouse macrophages. J Immunol. 2000;165:5767–5772. doi: 10.4049/jimmunol.165.10.5767. [DOI] [PubMed] [Google Scholar]
- 30.Williams DL, Ha T, Li C, Kalbfleisch JH, Schweitzer J, Vogt W, et al. Modulation of tissue Toll-like receptor 2 and 4 during the early phases of polymicrobial sepsis correlates with mortality. Crit Care Med. 2003;31:1808–1818. doi: 10.1097/01.CCM.0000069343.27691.F3. [DOI] [PubMed] [Google Scholar]
- 31.Shimazu R, Akashi S, Ogata H, Nagai Y, Fukudome K, Miyake K, et al. MD-2, a molecule that confers lipopolysaccharide responsiveness on Toll-like receptor 4. J Exp Med. 1999;189:1777–1782. doi: 10.1084/jem.189.11.1777. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Weighardt H, Kaiser-Moore S, Vabulas RM, Kirschning CJ, Wagner H, Holzmann B. Cutting edge: myeloid differentiation factor 88 deficiency improves resistance against sepsis caused by polymicrobial infection. J Immunol. 2002;169:2823–2827. doi: 10.4049/jimmunol.169.6.2823. [DOI] [PubMed] [Google Scholar]
- 33.Echtenacher B, Weigl K, Lehn N, Mannel DN. Tumor necrosis factor-dependent adhesions as a major protective mechanism early in septic peritonitis in mice. Infect Immun. 2001;69:3550–3555. doi: 10.1128/IAI.69.6.3550-3555.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Hoesel M, Neff TA, Neff SB, Younger JG, Olle EW, Gao H, et al. Harmful and protective roles of neutrophils in sepsis. Shock. 2005;24:40–47. doi: 10.1097/01.shk.0000170353.80318.d5. [DOI] [PubMed] [Google Scholar]