

Abstract
Human vestibular dysfunction is an increasing clinical problem. Degeneration or displacement of otoconia is a significant etiology of age-related balance disorders and Benign Positional Vertigo (BPV). In addition, commonly used antibiotics, such as aminoglycoside antibiotics, can lead to disruption of otoconial structure and function. Despite such clinical significance, relatively little information has been compiled about the development and maintenance of otoconia in humans. Recent studies in model organisms and other mammalian organ systems have revealed some of the proteins and processes required for the normal biomineralization of otoconia and otoliths in the inner ear of vertebrates. Orchestration of extracellular biomineralization requires bringing together ionic and proteinaceous components in time and space. Coordination of these events requires the normal formation of the otocyst and sensory maculae, specific secretion and localization of extracellular matrix proteins, as well as tight regulation of the endolymph ionic environment. Disruption of any of these processes can lead to the formation of abnormally shaped, or ectopic, otoconia, or otoconial agenesis. We propose that normal generation of otoconia requires a complex temporal and spatial control of developmental and biochemical events. In this review, we suggest a new hypothetical model for normal otoconial and otolith formation based on matrix vesicle mineralization in bone which we believe to be supported by information from existing mutants, morphants, and biochemical studies.
Keywords: Vestibular development, Otoconia, Otolith, Biomineralization, Matrix vesicle mineralization, Calcium carbonate
1. Introduction
Otoconia are complex calcium carbonate (CaCO3) biominerals in the utricle and saccule of the vertebrate inner ear. Otoconia are embedded in a fibrous extracellular matrix (gelatinous membrane) which couples the force of gravity to cilia of the sensory cells. Bending of the cilia in response to linear accelerations initiates the neuronal response. Otoconia are required for normal balance and the sensation of linear acceleration (gravity). In mammals, the majority of otoconia are generated only in the embryonic ear and must be maintained throughout life. Human otoconia are subject to demineralization and to alterations in structure and composition because of aging, disease, and exposure to common pharmaceutical agents. Ross et al. (1976) reported that temporal bones examined from all patients over 50 years of age exhibited saccular otoconial degeneration, which increased in severity with advancing age (Ross et al., 1976). Typically, the affected otoconia appear pitted or hollowed out, and subsequently break into fragments, which can result in displacement of utricular otoconia into the semicircular canals. In the semicircular canals, ectopic otoconia may cause abnormal sensations of dizziness and loss of balance, a condition referred to as Benign Positional Vertigo (BPV) (House and Honrubia, 2003; Lim, 1984; Tusa, 2001; Welling et al., 1997). Vestibular problems, like BPV, are reported in about 9% of people 65 years of age or older (Oghalai et al., 2000). An NIH report (1991 Annual Report, National Deafness and Other Communication Disorders Advisory Board) states that balance-related falls account for more than half of the accidental deaths in the elderly. Despite such significant morbidity, and even mortality, relatively little is known about the development and maintenance of otoconia, and the genes and processes required to assemble these complex extracellular structures.
Studies of human otoconial development are rare and most commonly associated with syndromic developmental anomalies or other pathologies (Sanchez-Fernandez and Rivera-Pomar, 1984; Sanchez-Fernandez et al., 1989; Wright and Hubbard, 1982; Wright et al., 1979, 1982). The majority of studies on otoconial development to date have focused on the mouse, rat, and chick which all have similar inner ear and otoconial structures as humans. Recent studies suggest that the genes and processes required for inner ear and otoconial development are evolutionarily conserved in the formation of the inner ear and otoliths of the teleost fish, yielding another, easily manipulated, model system to those that can be used for examining vestibular formation and dysfunction.
2. Structure and development of the otoconial and otolithic membranes
The structure and development of the otoconial organs have been best described in the mouse and the rat. The otoconial complex can be divided into 3 layers (Fig. 1A). Beginning in the endolymphatic space, (1) the otoconial layer contains thousands of otoconia, 0.1 to 25 μm biominerals consisting of a glycoprotein/proteoglycan core surrounded by minute crystallites (Figs. 1B, C), and the fibrous proteins that attach them to the underlying matrix, (2) the gelatinous layer is glutinous and amorphous, and (3) the subcupular meshwork (also called the veils) is a dense reticular network of fibrillar proteins that surrounds the processes of sensory hair cells. This complex extracellular structure is established in mid-embryonic development within the aqueous environment of the endolymph.
Fig. 1.
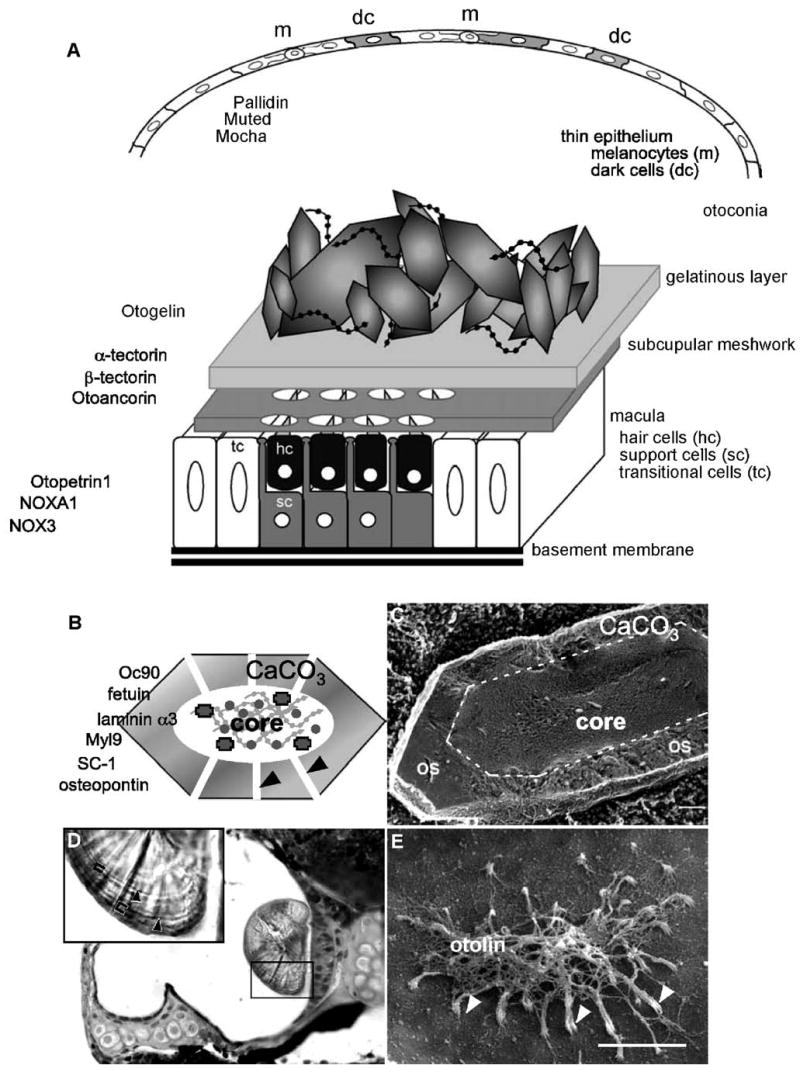
Structure of the otoconial organs in higher vertebrates. (A) Schematic representation of the utricular sensory maculae and associated structures. A thin, nonsensory epithelium is found opposite the sensory macula. This layer has many melanocytes (m), which provide trace elements in melanosomes to the epithelial cells, giving them a dark appearance and a common name of “dark cells” (dc). The macula is composed of sensory hair cells (hc) and supporting cells (sc); transitional cells (tc) border the edge of the maculae. Directly above the macula, the otoconial membrane is composed of the subcupular meshwork, a fibrillar structure which rings the stereociliary projections of each hair cells, and the gelatinous membrane which is amorphous. Otoconia, CaCO3 biominerals precipitated around a proteinaceous core, are embedded in the gelatinous membrane and maintained in place by strands of noncollagenous extracellular matrix proteins that resemble beads on a string. Proteins that have been identified to influence the activity or structure of each of these layers are listed on the left. (B) The otoconial protein core is a rhomboid protein scaffold composed of Oc90 and the minor otoconins. In vertebrates, CaCO3 is precipitated on the otoconial core as tiny crystallites, separated by large channels that are continuous with the protein core. (C) Scanning electron micrograph of a freeze fractured otoconium reveals a complex proteinaceous core surrounded by an outer shell (os) of CaCO3. (D) Plastic section through a 7 dpf zebrafish otop1 morphant otocyst showing a wild type-like otolith attached to the saccular macula. Richardson’s stain identifies concentric rings of darker proteinaceous otolith matrix (black arrowheads in inset) separated by lighter areas of CaCO3 mineralization (40×). The approximate areas covered by a daily growth ring are bracketed. (E) Scanning electron micrograph of an otop1 morphant utricular otolithic membrane at 7 dpf. Otop1 morphant fish formed an otolithic membrane in the absence of the otolith. The otolithic membrane is fibrous and connects the stereocilia of all hair cells (white arrowheads) in the macula and is, in part, made up of the collagen-like protein otolin. Panel C is reprinted from Lins et al. (2000), Journal of Structural Biology, 131(1), 67–78 (Lins et al., 2000) with permission from Elsevier. Panels D and E are reprinted from Hughes and Blasiole et al. (2004), Developmental Biology, 276(2), 391–402 (Hughes et al., 2004) with permission from Elsevier.
Otoconial formation begins over the sensory epithelia of the utricle and saccule when the core proteins (Otoconin 90 (Oc90) and other “minor” otoconial proteins) coalesce into distinct structures, with faint rhombohedral shapes (Ballarino and Howland, 1982) at approximately embryonic day 14 (E14) in the mouse. Calcification of this proteinaceous otoconial precursor is rapid, with maximal mineralization occurring between E15–E16.5 (Anniko, 1980; Anniko et al., 1987). Otoconial growth is most likely mediated through accretion of new CaCO3 crystals at the pointed tips of the calcifying otoconia; this is supported by the localization of the calcium binding protein calbindin d28K to these sites in developing avian otoconia (Balsamo et al., 2000). Otoconia achieve essentially full size by postnatal day 7 (Erway et al., 1986; James et al., 1969; Lim, 1973) and, in mammals, it is presumed that no new otoconia will be added after this point, although turnover of otoconial Ca2+ has been observed at a low rate in the adult rodent (Erway et al., 1986; Preston et al., 1975).
Teleost otoliths have a very different structure as well as some differences in the developmental processes required to generate them. In contrast to the thousands of small otoconial particles in mammals and birds, only three large otoliths form in fish. These “ear rocks” are initially dome-like in structure (Fig. 1D), but will, with age, take on complex shapes consistent with the shape of the underlying sensory maculae, and with the biophysical requirements of each maculae for the sensing of motion and sound (Das, 1994; Lychakov and Rebane, 2000). Otolith formation begins with the aggregation of free-floating protein core particles, identifiable at 18–20 h post-fertilization (hpf). Core particles are directed to either of the two developing sensory maculae through the action of ciliated cells that line the otocyst cavity (Riley et al., 1997) where they adhere to the modified stereocilia on the first hair cells (tether cells) at the anterior and posterior poles of the otocyst. These preotoliths are rapidly mineralized with the aragonitic polymorph of CaCO3 by 24 hpf. The otoliths remain attached to the fibrillar otolithic membrane linked to the hair cells of the sensory macula (Fig. 1E) throughout the morphogenetic movements required to generate the utricular and saccular maculae. A third otolith will begin to form about 11–12 days post-fertilization (dpf) in the lagena (Riley and Moorman, 2000) and is believed to utilize the same matrix components and developmental pathways used during the formation of the utricular and saccular otoliths. Otoliths continue to grow throughout the life of the fish, with daily accretion of layers of extracellular matrix proteins and deposited CaCO3 (Borelli et al., 2003a) (Fig. 1D, inset).
3. Formation of otoconia and otoliths requires a stepwise process
Normal generation of otoconia requires the orchestration of complex temporal and spatial developmental and biochemical events. In the mouse, the first seeding of CaCO3 crystals takes place at E14 and otoconia with defined forms can first be observed at E15–16, but genetic evidence suggests that the processes necessary for normal otoconial development begin much earlier. Normal otoconial formation requires (1) the correct induction and formation of the otocyst; (2) specification and differentiation of the sensory maculae and sensory and supporting cells; (3) establishing the correct ionic environment that allows for normal export and processing of matrix proteins and ions; (4) production and export of the otoconial matrix proteins and gelatinous membrane; (5) assembly of a protein core from free-floating matrix proteins; and (6) locally increasing Ca2+ and carbonate ( ) concentrations to initiate crystal formation on the proteinaceous core. The above elements must occur in a specific order and within specific time points to develop correctly formed otoconia or otoliths. Calcification, once initiated, must occur only above the utricular and saccular sensory maculae to prevent general mineralization in the ear which could severely impair balance and auditory function. The temporal specificity of otoconial development is determined by a combination of localized production of key elements and most likely by removal of excess otoconial proteins through the endolymphatic sac (Ignatova et al., 2004). In addition, the expression of inhibitors of calcification, which prevents mineralization of the rest of the endolymph, is probably complementary to the expression of pro-calcifying proteins, further specifying the time of otoconial/otolith initiation to a tight window during development and restricting locations of mineralization. Support for a critical period of otoconial development comes from dietary studies (Zn2+ rescue of lethal milk mice; Erway and Grider, 1984) and from studies in the fish in which otoconial formation is delayed using laser ablation or gene dosage (Hughes et al., 2004; Riley et al., 1997). Spatial specificity is most likely conferred by specific sets of integrins which localize otoconial matrix proteins to the sensory maculae and by the interactions of mineralizing matrix vesicles with specific extracellular matrix proteins in the developing gelatinous membrane. Disruptions of any of these elements can lead to lack of formation of the CaCO3 biominerals (otoconial/otolith agenesis) or can lead to the formation of inorganic crystals which lack organized matrix.
3.1. Form a normal otocyst
Patterning of the developing otocyst requires interaction of the developing hindbrain with the overlying neurectoderm. Disruptions in inner ear patterning through alterations in the development of the hindbrain rhombomeres have been well documented through the study of naturally occurring mutants and engineered knockout and transgenic mice. In several of these animals, otoconial defects have been described, or can be assumed, based on the severity of the developmental anomalies in inner ear formation.
Targeted deletions of Otx1, one of the earliest expressed transcription factors in the inner ear, lead to impaired otic development, characterized by a continuous utricular and saccular epithelium. Otx1−/− mice do not develop otoconia despite formation of a small, relatively normal sensory epithelium (Morsli et al., 1999). Similarly, Tbx1 mutants do not form the normal structures of the inner ear, and the single macula formed in these mutant ears does not develop otoconia (Vitelli et al., 2003).
The zebrafish valentino (val) mutant ectopically expresses fgf3 in the hindbrain causing alterations in otocyst formation. Otoliths in val mutants are poorly adherent and fail to associate with the sensory maculae (Kwak et al., 2002). Acerebellar (ace) zebrafish mutants, which inactivate the fgf8 gene, typically have a small otocyst, abnormal semicircular canals, and only one otolith (Leger and Brand, 2002). The foxi1 mutant hearsay often forms a single otic vesicle with one otolith (Solomon et al., 2003). Foxi1 is upstream of several transcription factors required for normal otic morphogenesis and regulates the expression of the anion transporter pendrin which is important in endolymph ionic regulation (discussed below) (Table 1).
Table 1.
Otolith-deficient zebrafish
| Strain | Otolith number | Otolith morphology | Other defects | Gene | Authors |
|---|---|---|---|---|---|
| acerebellar | 1 | fgf8 | Leger and Brand (2002) | ||
| backstroke | 0 | Otopetrin1 | Sollner et al. (2004) | ||
| blanched | 2 | Small | PD | ? | Kelsh et al. (1996) |
| bleached | 2 | Small | PD | ? | Kelsh et al. (1996) |
| clorix | 2 | Small | PD | ? | Odenthal et al. (1996a,b |
| colourless | 2 | Tiny | PD, abnl ear dev. | Sox10 | Odenthal et al. (1996a,b |
| delta | 1 | Abnl shape | DeltaA | Riley et al. (1999) | |
| dog-eared | 2 | Small | Abnl jaw, abnl ear dev. | Eya1 | Kozlowski et al. (2005) |
| einstein | 1 | ? | Whitfield et al. (1996) | ||
| earplugs | 2 | Small | Abnl ear dev | ? | Whitfield et al. (1996) |
| halfstoned | 1 | Small | ? | Whitfield et al. (1996) | |
| keinstein | 0/2 | Absent or tiny | ? | Whitfield et al. (1996) | |
| menhir | 1 | ? | Whitfield et al. (1996) | ||
| mindbomb | 2 | CNS, somites, | Ubiquitin E3 ligase | Haddon et al. (1999) | |
| monolith | 1 | ? | Riley and Grunwald (1996) | ||
| nonblond | 2 | Small | PD | ? | Odenthal et al. (1996a,b) |
| pech | 2 | Small | PD | ? | Kelsh et al. (1996) |
| rolling stones | 2 | Loose | ? | Whitfield et al. (1996) | |
| spock | 2 | Tiny | Abnl ear dev. | ? | Whitfield et al. (1996) |
| stein und bein | 1 | No pelvic fins | ? | Whitfield et al. (1996) | |
| stonewashed | Small | PD, abnl ear dev. | ? | Kelsh et al. (1996) | |
| what’s up | 0 | ? | Whitfield et al. (1996) | ||
| valentino | ? | fgf3 | Kwak et al. (2002) | ||
| claudinj MO | 1 | Small | Free otolith matrix material | Hardison et al. (2005) | |
| otolin MO | 2 | Decalcified, unattached | Murayama et al. (2005) | ||
| omp MO | 2 | Slow to grow | Murayama et al. (2005) | ||
| otopetrin MO | 0 | Hughes et al. (2004), Sollner et al. (2004) | |||
| ptena MO | 1 | Abnl shape | Abnl ear dev., small eyes, Curved tail, short body axis | Croushore et al. (2005) |
MO = morpholino; NA = not applicable; R = recessive; SD = semidominant; D = dominant; PD = pigmentation defect; SCL = sensory cell loss; HL = hearing loss; CP = cerebellar phenotype, abnl = abnormal.
3.2. Form normal macular compartments (utricle/saccule sensory regions) and the nonsensory epithelium
The formation of the sensory macula requires the formation of boundaries separating presumptive neural tissue from surrounding nonsensory support structures. At these boundaries, nonsensory transitional cells will form, which have been shown to have an important role in the secretion of the extra-cellular matrix of the otoconial/otolithic membrane and may have a role in the maintenance of otoconia later in life (Murayama et al., 2000; Yamane et al., 1984). While the definition of the sensory region is required for otoconial initiation, the formation of mature sensory hair cells or mature supporting cells does not appear to be necessary. Mouse mutants in Atoh-1/Math1 do not develop sensory hair cells, but have a normal appearing otoconial membrane (Bermingham et al., 1999). Similarly, mature supporting cells are not required for normal otolith formation as the zebrafish mindbomb mutant, which generates only sensory hair cells due to a lack of lateral inhibition, can initiate otolith formation (Haddon et al., 1999). This suggests that initiation of otoconial development is determined by the interaction of Oc90 and activities of the early macular cells before hair cells and supporting cells acquire lineage specific properties. Formation of the precursor cells from which both vestibular hair and supporting cells are derived is both necessary and sufficient (in the presence of Oc90) for otoconial initiation and development, as it has been shown that ectopic vestibular type hair and supporting cells induced by Wnt overexpression in the cochlea can produce small regions of relatively normal looking otoconial membrane and otoconia (Stevens et al., 2003).
Alteration in the size of the sensory maculae causes an associated change in the size of the developing otolith. The dog eared zebrafish mutants have truncation mutations within the transcription factor Eya1 (a homologue of the Drosophila eyes absent 1) and have small utricular and saccular maculae due to increased cell death during early stages of inner ear development. These animals have significantly undersized otoliths, which may correlate with reduced CaCO3 and minor otoconin production by the degenerating maculae (Kozlowski et al., 2005; Whitfield et al., 1996). Similarly, colourless (cls) zebrafish mutants have tiny otoliths that correlate well with the diminished macular size of the utricle and saccule. The cls mutation leads to alteration in the activity of Sox10, a transcription factor required for neural crest differentiation. However, Sox10 activity in the otocyst is independent of its role in neural crest identity (Dutton et al., 2001). Sox10 is believed to play a role in specifying macular fate (Dutton et al., 2001), and its expression is reduced in cls otocyst as is the expression of otx1 and mshC, markers of presumptive macular components (Whitfield et al., 1996).
After the maculae are specified, changes in the relative density of hair cells and supporting cells can also lead to alterations in otolith formation. Otolith formation is delayed until 30 hpf in the zebrafish mutant in deltaA, a component of the Notch lateral inhibition system. DeltaA-deficient fish have malformed otoliths that appear elongated across the macula most likely due to an increase in the number of tether cells, which anchor otolith core proteins to the macula (Riley et al., 1999). Similarly, mindbomb mutants, which also form excess hair cells without supporting cells, fail to enlarge their otoliths by 60 hpf (Haddon et al., 1998). The otoliths in these mutants are poorly adherent and have increased mineralization (Haddon et al., 1999).
3.3. Establish the ionic environment
The endolymph of the inner ear represents a unique extracellular ionic milieu. While measurements of ionic composition have not been done in the embryonic/developing ear, young adult studies suggest that endolymph has concentrations of Ca2+ (~20–100 μM) and sodium (<1 mM), considerably lower than that in the serum (Salt et al., 1989). This unique extracellular fluid is maintained by cells throughout the inner ear including the vestibular dark cells of the thin nonsensory epithelium of the utricle (Fermin et al., 1990; Hsu, 1991; Ichimiya et al., 1994), the stria vascularis (Kambayashi et al., 1982; Kusakari et al., 1978), the fibrocytes of the spiral ligament (Spicer and Schulte, 1991), and the endolymphatic sac and endolymphatic duct (Salt et al., 1989; Thalmann and Thalmann, 1999). Specialized tight junctions and interactions with a complex basement membrane are required to maintain the activities of these compartments. Disruptions in any of these structures could lead to significant alterations in endolymph ionic content and pH and thus alteration in otoconial formation and maintenance.
The dancer mouse has an abnormally high concentration of PO4 in otoconia (Anniko et al., 1988), suggesting a defect in the ionic environment and crystal formation. Dancer mice have subtle abnormalities in inner ear structure that suggests alterations in the development the endolymphatic sac and duct and the spiral ligament (Anniko et al., 1988). This phenotype is the result of an insertion mutation of a portion of the p23 gene into the Tbx10 locus, leading to ectopic and overexpression of the Tbx10 transcript in a near ubiquitous pattern, as opposed to its normal highly restricted expression in rhombomere 4 (Bush et al., 2004). Alterations in ionic content in this case are due to abnormalities of the organization of the inner ear compartments required for endolymph regulation.
Specific loss of individual ion-regulators has also been shown to lead to abnormalities in otoconial formation. The Plasma Membrane Calcium ATPase 2 (PMCA2) has been implicated in the maintenance of endolymph Ca2+ levels. Animals deficient in PMCA2 show an intact macular epithelium and gelatinous membrane but lack otoconia (Kozel et al., 1998). PMCA2 has been proposed as the primary Ca2+ pump for maintenance of endolymph Ca2+ levels, and other Ca2+ pump mutants have not been shown to affect inner ear development (Shull et al., 2003). It may be that the role of PMCA2 in general maintenance of endolymph Ca2+ is required for normal otoconial development; alternatively, PMCA2 may have a more direct role in locally increasing Ca2+ during otoconia formation. Pendrin (Pds) is an anion transporter of the solute carrier 26 family. Loss of Pendrin activity leads to Pendred syndrome, an autosomal recessive syndrome characterized by deafness and goiter. Pendrin specifically transports chloride and iodide and is thought to be important for endolymphatic fluid resorption in the inner ear (Everett et al., 1999). Pds−/− mice lack otoconia or develop giant otoconia (Everett et al., 2001), but also have significant defects in the remainder of the ear, including stereociliary degeneration. Foxi1, a forkhead transcription factor, regulates expression of Pendrin in the developing inner ear. Loss of function of Foxi1 also leads to Pendred syndrome, with expansion of the endolymph compartment leading to auditory and vestibular dysfunction (Hulander et al., 2003).
Claudinj (cldnj) is a component of tight junctions in the inner ear. Zebrafish cldnj mutants develop normal sensory structures within the inner ear, but their otoliths are severely reduced in size and unincorporated otolith core material can be seen throughout the early otic vesicle (Hardison et al., 2005). Claudinj is expressed in the nonsensory epithelium opposite the developing sensory maculae; Hardison et al. (2005) propose that loss of this protein causes a deficiency in some barrier function of the dorsolateral wall of the otocyst for specific ions required in the endolymph for otolith core matrix aggregation (Hardison et al., 2005).
3.4. Make and export the matrix proteins correctly
As with any extracellular matrix, normal protein production, posttranslational modification, packaging, and movement into the extracellular space are required for otoconial and otolith formation. Oc90 accounts for 90% of the total protein of the otoconial core and is expressed by the nonsensory epithelium of the inner ear (Verpy et al., 1999; Wang et al., 1998). Knockout mice for Oc90 initially develop giant otoconia, suggesting that Oc90 is essential for normal otoconial formation (Y. Lundberg, personal communication). The Oc90 protein within otoconia is highly glycosylated, suggesting that mutations or disruption of the activity of glycosylating enzymes would lead to abnormalities in otoconial formation. Indeed, depletion of the divalent cation manganese (Mn2+), an important cofactor for glycosylating enzymes, leads to a significant decrease in the synthesis of sulfated glycoproteins/proteoglycans and agenesis of otoconia in mammals and birds (Erway et al., 1986).
Two of the “minor” otoconins have been identified as the ubiquitous calcium binding proteins osteopontin (Sakagami, 2000; Takemura et al., 1994) and calbindin D28K (in chick and lizard) (Balsamo et al., 2000; Piscopo et al., 2003), which are believed to be produced in the developing sensory macula. Oc90 has 2 EF hands and has been shown to bind Ca2+ (I. Thalmann, unpublished data); osteopontin and calbindin have similar calcium binding motifs. Several other minor constituents of the mammalian otoconial core were identified using microscale protein preparatory techniques and tandem mass spectroscopy and include fetuin, a proposed inhibitor of calcium phosphate-based calcification; myosin regulatory light polypeptide 9, which contains 2 EF-hand domains and binds calcium; osteopontin, a sialoprotein, known to be a component of the matrix of bone and other calcified tissue; SC-1 (also known as “hevin”), a member of the SPARC family of matricellular proteins, which mediate interactions between cells and their extracellular matrices; and laminin α3, a component of basal lamina involved in attaching cells to the extracellular matrix via interactions with integrins (Thalmann et al., in press).
In the teleost fish, three major matrix proteins have been described: starmaker, otolith matrix protein, and otolin. Starmaker is homologous to dental sialophosphoprotein (DSPP) in mammals, which is required for the normal mineralization of teeth (Sollner et al., 2003). DSPP mutations have been shown to result in Dentinogenesis Imperfecta (Sreenath et al., 2003). Morpholino-mediated knockdown of starmaker leads to a delay in otolith formation as well as loss of the core particles and other proteinaceous components of the developing crystal. In animals injected with very high doses of starmaker morpholino, otoliths consist of inorganic CaCO3 and have an altered crystal structure (Sollner et al., 2003), most likely due to an inability to coordinate and organize the CaCO3 into the complex biomineral of the normal otolith. Otolith matrix protein (omp) is the primary matrix protein of the aragonitic fish otolith (Murayama et al., 2000). Omp morphant fish initiate normal otolith formation, but have significantly reduced rates of otolith growth, suggesting that omp is not required for otolith initiation but is required for the daily growth increments (Murayama et al., 2002). Otolin, first identified in trout (Murayama et al., 2002; Takagi and Takahashi, 1999), is expressed by the transitional cells of the developing sacculus and has short collagen like repeats similar to collagen X (Murayama et al., 2002). Otolin is similar to the fish saccular collagen proposed to have a role in the calcification process of the aragonitic otolith (Davis et al., 1995, 1997); this has been supported by the phenotype of otolin morphants. Morphant otoliths lose adherence to the sensory maculae (but not to hair cell kinocilia) and fuse by 72 hpf. Interestingly, otolin morphant otoliths spontaneously decalcify and swell during experimental procedures, suggesting that otolin is required to fully organize the inorganic and organic components required to generate mineralized otoliths in addition to a function as part of the gelatinous membrane anchoring the otolith close to the sensory maculae (Murayama et al., 2005).
The gelatinous membrane and the underlying subcupular meshwork are made up of many proteins and extracellular matrix molecules. The fish otolithic membrane appears much simpler than the mammalian otoconial membrane (Fig. 1C), with a thick fibrillar network that connects each hair cell stereociliary bundle to the otolith, but is predicted to have similar biochemical complexity (Khan and Drescher, 1990). Organic substances, including acidic proteins, glycosaminoglycans (GAGs), and proteoglycans, are essential to regulate crystal growth (Addadi et al., 1989; Khan, 1997) and have been identified in both otoliths (Borelli et al., 2003b) and otoconia (Tachibana and Morioka, 1992). Sulfated GAGs may play a crucial role in locally increasing Ca2+ concentration at the site of biomineralization, as is discussed in more detail below. Proteins of the gelatinous membrane that have been identified include α-tectorin, β-tectorin, otolin (in the fish and probably other vertebrates as well), and otogelin. Knockout mice for α-tectorin had significant abnormalities of the extracellular matrices of the inner ear, including decreased thickness of the gelatinous membrane in the utricle and saccule, a complete lack of utricular otoconia and abnormal or giant otoconia in the saccule (Cohen-Salmon et al., 1997; Legan et al., 2000). This suggests that the integrity of the structure of the gelatinous membrane and subcupular meshwork is required for normal otoconial development.
3.5. Assemble and attach matrix proteins in the right place in the inner ear
Assembly of the otoconial complex in the aqueous endolymphatic space and its maintenance above the sensory maculae require a variety of adhesion molecules. In particular, at least two adhesion molecule complexes appear to be required to first localize the major core protein Oc90 specifically to the otoconial membrane during development and to then maintain adherence of calcified mature otoconia to the gelatinous membrane. The integrins or other molecular recognition and adhesion molecules required for otoconial formation have not yet been identified, though Otogelin and Otoancorin have been shown to be required to maintain adhesion of the mature otoconial membrane to the sensory maculae. The Otogelin (Otog) mutant twister and targeted deletion of Otogelin result in deafness and considerable balance deficits. Otog null mice initially form normal otoconia, but shortly after birth the gelatinous membrane and otoconia detach from both the utricular and saccular maculae, indicating that Otogelin is essential for anchoring the otoconial complex to the sensory epithelium (Cohen-Salmon et al., 1997; Simmler et al., 2000a,b). Otoancorin is expressed by supporting cells and is localized to the point of interaction between the support cells and the acellular otoconial membrane (Zwaenepoel et al., 2002).
Several zebrafish mutants have been described with aberrant otolith attachment that support the hypothesis that two independent adhesion complexes are required for otolith formation and maintenance. The monolith mutant has only one otolith, found on either of the two early sensory epithelia or at an ectopic location in the inner ear. Without the monolith gene signal from supporting cells, the early tether cells were not capable of binding pre-otolith core particles and attaching them to developing sensory maculae (Riley and Grunwald, 1996; Riley and Moorman, 2000). In these animals, a single otolith is formed from the free-floating otolith core particles and attaches later in development to either of the sensory epithelia, depending on orientation of the fish (Riley and Moorman, 2000). Similarly, the einstein and menhir mutants have only one aberrant otolith despite normal macular formation, which suggests that a group of genes is required to attach otolith core particles to the tether cells (Whitfield et al., 1996). The GP96 morphant fish mimic these adherence mutants in that they do not attach otolith core particles to the tether cells. GP96 is a heat shock protein and molecular chaperone which has been described in mouse cells to be involved in the correct processing of Toll-like receptors and integrins (Sumanas et al., 2003). The rolling stones mutant cannot attach otoliths later in life, though they initially attach the core particles to tether cells and initiate normal otolith formation. After mineralization, rolling stones otoliths are free to move about the inner ear. It is only when the stones shift onto a sensory macula that the developing fish can orient and swim normally (Whitfield et al., 1996). Cloning and further characterization of these and other zebrafish otolith adherence mutants may reveal important aspects of otoconial and otolith adhesion that may help to identify factors involved in the etiology of Benign Positional Vertigo.
3.6. Locally increase calcium and carbonate concentrations to initiate otolith/otoconial growth
Once adherence of early otoconial and otolith matrix particles to the maculae occurs, deposition of CaCO3 is rapid. The essential requirement to form CaCO3 is the availability of Ca2+ and ions. The latter depend on the availability of carbonic anhydrase, which is abundant in the inner ear (Lim et al., 1983; Shiao et al., 2005). This has been supported both by lack of otoconial formation in rodent and chick embryos treated with carbonic anhydrase inhibitors (Kido et al., 1991) as well as dietary studies which have shown that severe deficiency of Zn2+, an essential cofactor for carbonic anhydrase activity, leads to otoconial agenesis (Erway and Grider, 1984; Erway et al., 1986). Little is currently known about the source of Ca2+ in otoconial formation. It is believed that PMCA2 has a significant role in establishing the Ca2+ concentration within the endolymph as discussed above, but a role specifically in otoconial formation has not been identified for this protein.
Several mouse mutants have been characterized that appear to have primary defects in otoconial mineralization. Head tilt (het) and head slant (hslt) mutants have complete lack of otoconial mineralization despite the presence of Oc90 in the developing inner ear. The het phenotype results from a recessive mutation within the gene encoding NADPH-Oxidase 3 (NOX3), a reactive oxygen species generating oxidase specific to the inner ear (Banfi et al., 2004; Paffenholz et al., 2004). Hslt mice, with a similar lack of otoconia, carry a mutation in an accessory protein for NOX3 and NOXO1 (Kiss et al., 2006). NOX3 and NOXA1 are localized to the sensory maculae and het and hslt mice are proposed to have significant alterations in the formation of reactive oxygen species (ROS). The role of ROS in otoconial formation has not yet been elucidated, though much work in other model systems has shown that ROS generation is coupled to alterations in mitochondrial and endoplasmic reticulum Ca2+ regulation (Ermak and Davies, 2002), which may be required for concentrating the Ca2+ used for otoconial mineralization (discussed below).
Similar to the phenotypes seen in het and hslt mice, the tilted (tlt), mergulhador (mlh), and inner ear defect (ied) mice carry mutations in the novel multi-transmembrane domain protein, Otopetrin 1 (Otop1), resulting in nonsyndromic otoconial agenesis and a severe balance disorder in mice (Besson et al., 2005; Hurle et al., 2003; Ornitz et al., 1998). The zebrafish otop1 orthologue is a highly conserved gene that is essential for otolith initiation as seen in both morphant otop1 fish and in the backstroke mutant (Hughes et al., 2004; Sollner et al., 2004). Despite lack of otoliths in early development, otop1 morphant fish partially recover otolith formation after 2 days, in a timeframe consistent with dilution of the morpholino and reexpression of otop1, indicating that Otop1 has an essential and conserved role in the timing of formation and the size and shape of the developing otolith. In mouse, Otop1 immunoreactivity localizes to the otoconial membrane in the utricular and saccular maculae; this suggests that Otop1 is localized to extracellular vesicles, called globular substance, that have been suggested to be the site of initiation of otoconial mineralization (Erway et al., 1986; Preston et al., 1975; Ross, 1979). Globular substance vesicles are proposed to bud from the supporting cells (or the precursor cells) of the sensory macula (Suzuki et al., 1995, 1997b). This hypothesis is supported by electron microscopic data (Anniko et al., 1987; Kawamata and Igarashi, 1993; Lim, 1973) that have shown cytoplasmic blebs at the surface of supporting cells or within the otoconial membrane, similar to those seen in chondrocytes before the release of matrix vesicles during bone mineralization.
4. Model of extracellular mineralization in bone suggests mechanism of otoconia development
The formation of calcium phosphate in developing bone is the best described calcification process to date. Bone mineralization begins within differentiating hypertrophic chondrocytes when Ca2+ concentrations increase within mitochondria and the endoplasmic reticulum (Fig. 2A). During the terminal differentiation process, alkaline phosphatase, phospholipase A (PLA), annexins II, V, and VI, Ca2+ ATPase, metalloproteases, calbindin, and chondroitin 6 sulfate are localized to the tips of cytoplasmic extensions. The membranes within these cytoplasmic blebs increase in relative concentrations of the acidic lipids sphingomyelin and phosphatidylserine. Ca2+ levels in mitochondria and the endoplasmic reticulum drop and Ca2+ is concentrated into the developing blebs (Fig. 2B). In an actin- and microtubule-mediated process, these blebs are released into the extracellular environment where they are referred to as matrix vesicles (Fig. 2C). F-actin remains with the vesicles and may be involved in their motility in the extracellular matrix (Hale and Wuthier, 1987). Phosphatidylserine and sphingomyelin concentrate Ca2+ along the surfaces of the matrix vesicle. In the higher extracellular Ca2+, the annexins bind phosphatidylserine in a calcium-dependent manner and act as Ca2+ channels, allowing an influx of Ca2+ into the matrix vesicle. Interactions with Type II and Type X collagen stimulate the activity of annexin V by 2–3-fold (Kirsch et al., 2000; Wang et al., 2003), allowing close regulation of the site of nucleation in the extracellular matrix (Fig. 2D). Alkaline phosphatase and other phosphohydrolases enriched in the matrix vesicles locally increase inorganic phosphate by breakdown of extravesicular ATP to concentrations suitable for nucleation of needle-like calcium phosphate crystals (Fig. 2E). Carbonic anhydrase (also concentrated within matrix vesicles) may increase pH within the matrix vesicles to stabilize the forming crystal. During nucleation, the increase in intravesicular Ca2+ activates phospholipase A, which hydrolyzes diacylphosphatides, leading to changes in membrane fluidity and the breakdown of matrix vesicles (Fig. 2F). Rupture of the matrix vesicle membrane is required for continued growth of the apatite crystal in the extracellular fluid which has a high maintained Ca2+ concentration due to the presence of negatively charged sulfated GAGs that coordinate cations (Fig. 2G) (reviewed extensively in Anderson, 2003).
Fig. 2.
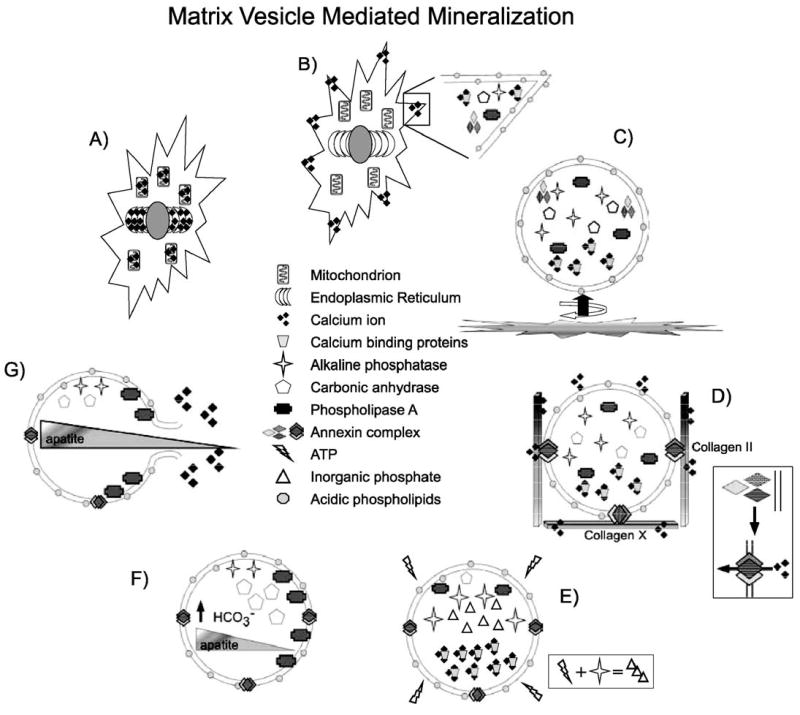
Matrix vesicle mineralization model in bone. (A) Ca2+ concentrations increase in the endoplasmic reticulum and mitochondria of hypertrophic chondrocytes during terminal differentiation. (B) Ca2+, calcium binding proteins, alkaline phosphatase, carbonic anhydrase, and the annexin complex are localized to cytoplasmic blebs, where the composition of the plasma membrane is locally altered to increase the concentration of acidic phospholipids. (C) The cytoplasmic bleb is budded from the plasma membrane by alterations in the cytoskeleton (circular arrow) and is moved into the extracellular space by a tail of polymerizing F-actin (straight arrow). (D) In the extracellular space, the negatively charged acidic phospholipid heads attract and coordinate extracellular Ca2+. Interaction with Collagens II and X leads to membrane insertion of Annexins II, V, and VI and formation of the annexin Ca2+ channel. (E) In the high intravesicular Ca2+, alkaline phosphatase breaks down ATP to form inorganic phosphate for nucleation of calcium phosphate (apatite) crystals. (F) Phospholipase A is activated to begin to break down the vesicular membrane. Carbonic anhydrase activity maintains alkaline pH within vesicles to protect the developing apatite crystal. (G) Phospholipase activity disrupts the vesicular membrane and releases the apatite crystal to grow in the extracellular matrix.
Otoconial formation has several similarities to the processes described above for bone matrix vesicle mineralization (Table 2, Fig. 3). Before the onset of otoconial formation, increases in Ca2+ concentrations in the endoplasmic reticulum and mitochondria have been documented (Fermin and Igarashi, 1985; Harada et al., 1998) (Fig. 3A). High Ca2+ concentrations have also been identified at cytoplasmic blebs at the apical surface of the developing sensory maculae (Anniko et al., 1987; Harada et al., 1998) (Fig. 3B). Large vesicles filled with Ca2+, consistent with apical blebs released into the otoconial membrane, have been identified by electron microscopy during otoconial formation (Anniko, 1980; Nakahara and Bevelander, 1979) (Fig. 3C). Some researchers have termed these vesicles “globular substance” and a role for them in otoconial development has long been advocated. Negatively charged acidic proteins, glycosaminoglycans (GAGs) and proteoglycans, in the early otoconial membrane may locally increase Ca2+ concentrations above the sensory maculae by coordinating endolymph Ca2+. The minor otoconins, some of which are proposed to be secreted from the sensory maculae, and Oc90, may either associate with these vesicles in the extracellular space through Oc90 interaction with lipid membrane through its Phospholipase A2 domains, or may be endocytosed into the maculae and specifically packaged into the globular substance before release (Fig. 3B). Evidence supporting the latter hypothesis includes uptake of the anti-calcifying serum protein fetuin into smooth muscle cells in the walls of blood vessels and its release into matrix vesicles to control ectopic calcification (Reynolds et al., 2005). In addition, a small amount of Oc90 immunoreactivity can often be seen with immunohistochemistry in vestibular maculae at E16.5 (R. Thalmann, unpublished observation), which may indicate a low level of Oc90 uptake into the macula and possible packaging into globular substance vesicles. Carbonic anhydrase activity has been localized to the globular substance (Tateda et al., 1998) and it has been shown in vitro that the calcite form of CaCO3 is preferentially formed in the presence of phosphate (Bachra et al., 1963), suggesting that alkaline phosphatase may also be localized to the gelatinous membrane. Thus, the precipitation of CaCO3 instead of calcium phosphate, as in bone, may simply be due to relative increase in the activity of carbonic anhydrase over alkaline phosphatase (Figs. 3D–F).
Table 2.
Similarities in bone and otoconial development
| Bone | Otoconia |
|---|---|
| Early rise in endoplasmic reticulum and mitochondrial Ca2+ | Early rise in endoplasmic reticulum and mitochondrial Ca2+ |
| Concentration of Ca2+ in cytoplasmic blebs | Concentration of Ca2+ in cytoplasmic blebs |
| Matrix vesicles provide lipid | Globular substance provides lipid |
| Specific collagen interactions | Otolin-1 collagen-like repeats |
| Glycosaminoglycans in the ECM | Glycosaminoglycans in the ECM |
| Ca2+ actively concentrated in extracellular matrix vesicles | Ca2+ actively concentrated in extracellular globular substance |
| Alkaline phosphatase activity | Alkaline phosphatase activity |
| Carbonic anhydrase activity | Carbonic anhydrase activity |
| Annexins concentrate Ca2+ | Otopetrin1 concentrates Ca2+ (?) |
| PLA binds Ca2+ and lipids | Oc90 binds Ca2+ and lipids (?) |
Fig. 3.
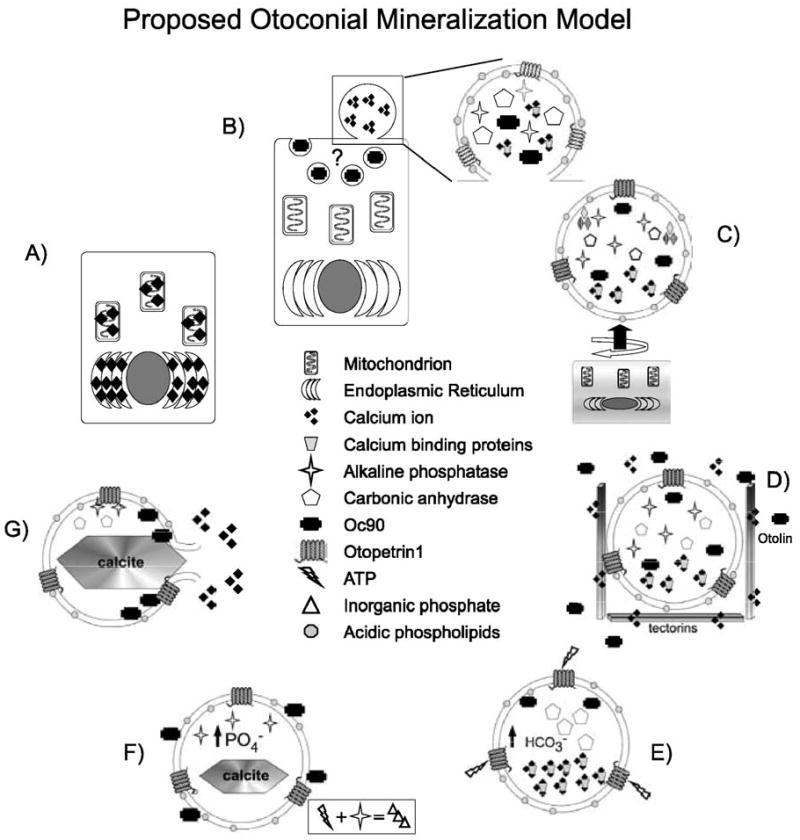
Proposed matrix vesicle/globular substance mineralization model in otoconia. (A) Ca2+ concentrations increase in the endoplasmic reticulum and mitochondria of the precursor cells of the sensory macula. (B) Ca2+, calcium binding proteins, carbonic anhydrase, and the Otopetrin1 are localized to apical cytoplasmic blebs, where the composition of the plasma membrane is locally altered to increase the concentration of acidic phospholipids. Oc90 may be taken up by macular cells through endocytosis and may contribute to the proteins of the apical blebs. (C) The cytoplasmic bleb is budded from the plasma membrane (round arrow) and moves into the otoconial membrane (straight arrow). (D) In the extracellular space, the negatively charged acidic phospholipid heads attract and coordinate extracellular Ca2+. Interaction with otolin, the tectorins, or other otoconial membrane constituents tethers the vesicle and may lead to alterations in the activity of Otop1 or other Ca2+ channels. Alternatively, ATP in the endolymph may activate Otop1 leading to increases in intravesicular Ca2+. Oc90 may be coordinated around the vesicle by interaction with the acidic phospholipids or with Ca2+. (E) Carbonic anhydrase increases HCO3− concentrations, nucleating the calcitic CaCO3 crystal. (F) Alkaline phosphatase breaks down ATP to maintain high intravesicular phosphate to prevent formation of less stable CaCO3 polymorphs. (G) Phospholipase/Oc90 activity disrupts the vesicular membrane and releases the calcium carbonate crystal to be incorporated into the otoconial core particle.
The activity of annexin family members are crucial for initiating increases in intravesicular Ca2+ within the matrix vesicles of developing bone only when the vesicle is in contact with the correct extracellular matrix (collagens II and X) (Anderson, 2003; Kirsch et al., 2000; Wang et al., 2003). Studies of annexin homologues in the zebrafish have not identified expression of these proteins in the inner ear—despite whole-mount in situ hybridization at the appropriate time points (Farber et al., 2003). While a more careful study of annexin expression in the inner ear will be required to rule out their role in otoconial formation, the lack of obvious annexin expression in the inner ear may indicate that a different family of calcium binding proteins or calcium channels is required for concentrating Ca2+ in globular substance vesicles. As with bone matrix vesicle mineralization, specific interactions between proteins on globular substance vesicles and extracellular matrix proteins like the tectorins or the collagen-like otolin and otolin like molecules may be required for localizing CaCO3 crystal development to the otoconial membrane (Fig. 3D). Alternatively, growth factors or extracellular signaling molecules may play a role in initiating increases in Ca2+ concentration during otoconial formation. Suzuki et al. (1997a,b) have shown that the globular substance within isolated guinea pig gelatinous membrane exhibited a rapid and dose-dependent increase in intravesicular Ca2+ in response to ATP (Suzuki et al., 1997a), suggesting that ATP from the sensory maculae, or localized to the otoconial membrane, may play a role in locally increasing Ca2+ required for otoconial mineralization (Fig. 3E). We propose that Otop1, which has the ability to regulate intracellular Ca2+ increases in response to ATP (I. Hughes et al., in preparation), plays an important part in increasing Ca2+ concentrations within the globular substance to initiate nucleation of otoconial CaCO3 crystals. Otop1 may act in place of the annexin family of calcium binding proteins as a regulatable Ca2+ pore in globular substance vesicles or may act within the macular cells to regulate the concentration of Ca2+ within the endoplasmic reticulum that will be destined for the globular substance vesicles.
5. Vesicular trafficking mutants impact several points in otoconial development
Trafficking Ca2+ and otoconial matrix proteins from the cell to the extracellular space require the formation and regulation of a variety of intracellular vesicles. The protein complexes that are required for the formation of intracellular vesicular compartments like lysosomes and melanosomes have been implicated in otoconial formation. Both humans and animal models with mutations in proteins required for the formation of lysosomal-related intracellular vesicles have been shown to have pigmentation defects, as well as a range of abnormalities in platelet and immunologic function; this group of anomalies is clinically referred to as Hermansky Pudlak Syndrome (HPS). As seen in Table 3, the pallid and muted mice, commonly used mouse models of HPS, lack otoconia in the utricle and saccule (Trune and Lim, 1983). The pallid protein (Pallidin), a novel, Syntaxin 13-interacting protein (Huang et al., 1999), and the Muted protein associate in the BLOC-1 (Biogenesis of Lysosome-related Organelles Complex-1) protein complex. BLOC-1 is one of several BLOC or Adaptor Protein (AP) complexes required for the formation of intracellular vesicle compartments (Huizing et al., 2000, 2002; Li et al., 2004). Supplementation with Mn2+ in pregnant pallid mice partially restores otoconial formation in the subsequent generation, suggesting that in part, the pallid phenotype arises from Mn2+ depletion within the inner ear (Erway et al., 1986). Cappuccino encodes a ubiquitously expressed cytoplasmic protein that coassembles with Pallidin and the Muted protein in the BLOC-1 complex (Ciciotte et al., 2003). Otoconial development has not been directly examined in cappuccino mice, though the animals display behavioral abnormalities such as head tilting and poor balance that are suggestive of otoconial agenesis (Gwynn et al., 2000). Mutations in any one of these proteins can affect the distribution or activity of the remainder of the complex, suggesting that it is lack of BLOC-1 activity in general that is responsible for otoconial agenesis in these mice.
Table 3.
Otoconia-deficient mouse mutants
| Strain | Otoconia number | Otoconia morphology | Metal ions in diet | Other defects | Gene | Authors |
|---|---|---|---|---|---|---|
| ames waltzer | Normal | Degenerate with age | HL | Protocadherin 15 | Alagramam et al. (2001) | |
| Cappuccino | Unknown | Unknown | PD | Cappuccino | Ciciotte et al. (2003) | |
| dancer | High PO4 conc | CL and P | Tbx10 | Anniko et al. (1988), Bush et al. (2004) | ||
| head tilt | Absent | NA | None | NOX3 | Paffenholz et al. (2004) | |
| headslant | Absent | NA | NOXO1 | Kiss et al. (2006) | ||
| lethal-milk | Decreased | Abnormal | Zn2+ helps | PD | Znt4 | Huang and Gitschier (1997) |
| mocha | Decreased | Mn2+ helps | HPS | δ subunit AP3 | Kantheti et al. (1998, 2003) | |
| muted | Decreased | HPS | Muted | Falcon-Perez et al. (2002) | ||
| new-mutant | Degenerate | SCL | ? | Kitamura et al. (1991a,b) | ||
| pallid | Decreased | Abnormal | Mn2+ helps | HPS | Pallidin | Huang et al. (1999) |
| tilted/mergulhador/ied | Absent | NA | Otopetrin1 | Hurle et al. (2003), Besson et al. (2005) | ||
| tilted-head | Decreased | Abnormal | Mn2+ helps | None | ? | Rauch (1979) |
| twirler | Decreased | Abnormal | CL and P | ? | Lyon (1958) | |
| wocko | Decreased | Abnormal | SCL | ? | Crenshaw et al. (1991) | |
| Targeted deletions | ||||||
| atoh1/Math1−/− | Absent | Bermingham et al. (1999) | ||||
| foxi1−/− | Absent | Hulander et al. (2003) | ||||
| limp2−/− | Absent | OM reduced | Gamp et al. (2003) | |||
| pendrin−/− | Decreased | Abnormal | HL | Everett et al. (1997) | ||
| pmca2−/− | Absent | NA | HL, CP | Kozel et al. (1998) | ||
| otog−/− | Decreased | Not attached to macula | HL | Simmler et al. (2000a,b) | ||
| otoc−/− | Not attached to macula | HL | Zwaenepoel et al. (2002) | |||
| otx1−/− | Absent | Hindbrain | Morsli et al. (1999) | |||
| tecta−/− | Absent in Ut, giant in Sacc | OM reduced | HL | Legan et al. (2000) |
NA = not applicable; R = recessive; SD = semidominant; D = dominant; PD = pigmentation defect; HPS = Hermansky Pudlak Syndrome; CL and P = cleft lip and palate; SCL = sensory cell loss; HL = hearing loss; CP = cerebellar phenotype.
The mocha mouse also lacks otoconia in the utricle and saccule. The mocha phenotype results from a deletion in the δ subunit of AP3, an adaptor protein complex required for vesicle budding in lysosomes and melanocytes. Supplementation with Mn2+ in pregnant mocha mice can partially correct otoconial development in offspring, supporting the hypothesis that, in some part, the HPS vestibular phenotype in mice arises from Mn2+ depletion within the inner ear (Rolfsen and Erway, 1984). Pearl mice have a mutation in the β subunit of the AP3 complex. Pearl mice have deficiencies/alterations in the localization and distribution of Zn2+, which can also be seen in mocha mutant mice, but pearl mice have normal hearing and balance (Balkema et al., 1983; Feng et al., 1999). This could be due to the subtlety of some vestibular defects, or may be because the pearl mutation alters some splice forms of the β3A protein, but not that expressed in neural tissue. Mutant AP3 may alter the localization of Zn2+ transporters like ZnT-3 and ZnT-4 (protein mutated in lethal milk mouse; Huang and Gitschier, 1997), whereas pallid mice do not have alterations in intracellular Zn2+ (Falcon-Perez et al., 2002).
Lysosomal integral membrane protein-2 (LIMP2) was identified as a component of the lysosomal membrane (Lewis et al., 1985). LIMP2 knockout mice have significant defects in the development of the kidney, ureter, and bladder epithelia. LIMP2 null mice become deaf between 3 and 7 months of age due to atrophy of the stria vascularis, with subsequent loss of outer hair cells, and a reduction in spiral ganglion neurons. In addition, these mice also completely lack otoconia (Gamp et al., 2003). The ureter epithelium has defective membrane trafficking of uroplakin, suggesting that alteration in secretion may lead to abnormalities of the otoconia during development. In support of this, researchers found that the thickness of the otoconial membrane was severely reduced, most likely indicating a loss of tectorins and other proteins secreted from the macular supporting cells. Alternatively, degeneration of the stria vascularis may be sufficient to lead to changes in the endolymph environment and loss of otoconia through acidification later in life along with poor maintenance of the otoconial membrane.
The expression of the HPS proteins described here has yet to be examined in the inner ear, though it has been suggested that they are required in melanocytes in the mammalian inner ear which provide Zn2+ and Mn2+ inside melanosomes for the vestibular dark epithelium, which in turn help to regulate the ionic environment and the posttranslational modification of otoconial proteins. One issue with this hypothesis is that the zebrafish inner ear has no melanocytes, but fish with mutations in genes that are likely to be part of the HPS family of proteins, based on phenotypes that include abnormalities in hemopoeitic system and coloration (blanched, bleached, clorix, nonblond, pech, and stone-washed), are commonly associated with malformation of the inner ear and poor otolith formation (Haffter et al., 1996). This suggests that HPS proteins are required in more cells than melanocytes for otolith/otoconial formation. These proteins may be required for the normal packaging and budding of globular substance. Though the globular substance has not been shown to directly be a lysosomes-related organelle, it has a very important point of similarity with these structures in that it has high intravesicular Ca2+ (Li et al., 2004; Suzuki et al., 1995).
6. Maintenance of otoconia
An increased percentage of abnormal otoconia or a reduction in number has been observed in humans and/or experimental animals following treatment with inhibitors of carbonic anhydrase, prostaglandins, and secondary to intoxication with ethacrynic acid, dilantin, or the aminoglycoside antibiotics streptomycin, neomycin, or gentamicin (Harada and Sugimoto, 1977; Johnsson et al., 1980; Lim, 1984; Minck et al., 1989; Takumida et al., 1997a; Wright et al., 1979, 1982). The majority of these pharmacological agents are believed to lead to significant alterations in the pH of the endolymph (carbonic anhydrase inhibitors) through damage to vestibular dark cells or the death of vestibular hair cells. This suggests that there are two main factors for the maintenance of adult otoconia: (1) the maintenance of a slightly alkaline pH and (2) the maintenance of healthy of vestibular hair cells.
The endolymph of the adult ear is a highly specialized extracellular fluid with high K+, relatively low Ca2+ and Na+ concentrations, and a slightly basic pH. Establishing the high K+ environment unique to the adult inner ear is not necessary for otoconial formation or even otoconial maintenance. Targeted deletions in the Na+-K+-2Cl− cotransporter (NKCC1) (Flagella et al., 1999) and the Kcnq1 K+ channel (Casimiro et al., 2001) both result in a reduction of endolymph volume with age and disrupt the endolymphatic potential, but neither exhibit otoconial defects. Acidification of the endolymph due to asphyxia, or the activity of carbonic anhydrase, has dramatic effects on otoconial morphogenesis and maintenance as CaCO3 is easily solubilized in low pH solutions.
The requirement of hair cells for the maintenance of otoconia is supported by numerous studies. Several alleles of the mouse mutant Ames waltzer have mutations in Protocadherin 15 and lose their sensory epithelium as they age. The otoconia in these mutants degenerate in a similar time course as the loss of the epithelium, though fragments of the gelatinous membrane can still be present (Johnsson et al., 1980; Takumida et al., 1997a,b; Yamane et al., 1984). Young Ames waltzer mice have no obvious defects in vestibular structure (Alagramam et al., 2005), suggesting degeneration, as opposed to altered development, as the cause of otoconial loss. New-mutant mice have age-dependent demineralization of saccular otoconia (Kitamura et al., 1991b) that accompanies the complete degeneration of saccular hair cells. This suggests that the hair cells specifically are required to maintain otoconia mineralization under normal conditions.
Aminoglycosides cause destruction of otoconia and increase the likelihood of otoconia-associated vestibular dysfunction through both hair cell destruction and disruption of endolymph ionic content. Direct treatment of isolated otoconia with high dose aminoglycosides does not appear to alter the surface of otoconia (M. Warchol, unpublished data), but treatment of the inner ear with streptomycin or gentamicin leads to otoconia degeneration, the timing and distribution of which coincide with hair cell degeneration (Johnsson et al., 1980; Takumida et al., 1997a). In addition, streptomycin has been shown to disrupt the activity of cells in the vestibular dark epithelium that are responsible for the maintenance of the endolymphatic ion content (Fujii et al., 1995; Harada and Sugimoto, 1977; Yamane et al., 1984).
7. Otoconial calcium
Otoconial Ca2+ turnover is observed at a low rate in the adult rodent (Erway et al., 1986; Preston et al., 1975), as would be expected in any extracellular mineral. In the frog Rana catesbeiana, the levels of Otoconin22(Oc22) mRNA, the major core matrix protein of aragonitic otoconia (Pote and Ross, 1991; Pote et al., 1993), have been shown to be regulated by the activity of calcitonin (Yaoi et al., 2003), suggesting that, in some animals, otoconia serve as a rapidly ionizable store of Ca2+ that can be (reversibly) demineralized and dispersed. In humans, bone serves as a readily ionizable and regulatable source of Ca2+, but as shown above, many of the systems for the formation and fate of otoconia are well conserved throughout vertebrates. It is unknown if similar mechanisms of calcitonin-regulated expression of proteins required for otoconial mineralization exist in humans, but it may be possible that extreme hyper- or hypo-calcemia may alter the ionic content of otoconia, leading to abnormal otoconial mineralization and degeneration. A recent study has identified a set of patients that exhibit both osteoporosis and a high rate of Benign Positional Vertigo, which may support the coregulation of bone and otoconial mineralization (Vibert et al., 2003).
8. Conclusion
Orchestration of extracellular biomineralization in the inner ear requires bringing together organic and inorganic components in time and space. Coordination of these events requires the normal formation of the otocyst and sensory maculae, specific secretion and localization of extracellular matrix proteins, as well as tight regulation of the local and endolymph ionic environments. Disruption of any of these processes can lead to the formation of abnormally shaped or ectopic otoconia, or otoconial agenesis. We have proposed a new model for normal otoconial and otolith formation based on the matrix vesicle model characterized for bone mineralization. Though speculative, we believe this model is supported by information from existing mutants, morphants, and biochemical studies done in the mouse, chick, and fish model systems. Further characterization of these mutants may help to reveal important clinical clues for the diagnosis, treatment, and prevention of vestibular dysfunction.
Acknowledgments
This work was supported by NIDCD DC06093 (IH), DC02236 (DMO), and DC01414 (IT).
References
- Addadi L, Berman A, Oldak JM, Weiner S. Structural and stereochemical relations between acidic macromolecules of organic matrices and crystals. Connect Tissue Res. 1989;21:127–134. doi: 10.3109/03008208909050003. discussion 135. [DOI] [PubMed] [Google Scholar]
- Alagramam KN, Murcia CL, Kwon HY, Pawlowski KS, Wright CG, Woychik RP. The mouse Ames waltzer hearing-loss mutant is caused by mutation of Pcdh15, a novel protocadherin gene. Nat Genet. 2001;27:99–102. doi: 10.1038/83837. [DOI] [PubMed] [Google Scholar]
- Alagramam KN, Stahl JS, Jones SM, Pawlowski KS, Wright CG. Characterization of vestibular dysfunction in the mouse model for Usher syndrome 1F. J Assoc Res Otolaryngol. 2005;6:106–118. doi: 10.1007/s10162-004-5032-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Anderson HC. Matrix vesicles and calcification. Curr Rheumatol Rep. 2003;5:222–226. doi: 10.1007/s11926-003-0071-z. [DOI] [PubMed] [Google Scholar]
- Anniko M. Development of otoconia. Am J Oto-Laryngol. 1980;1:400–410. doi: 10.1016/s0196-0709(80)80021-4. [DOI] [PubMed] [Google Scholar]
- Anniko M, Wikstrom SO, Wroblewski R. X-ray microanalytic studies on developing otoconia. Acta Oto-Laryngol. 1987;104:285–289. doi: 10.3109/00016488709107330. [DOI] [PubMed] [Google Scholar]
- Anniko M, Wenngren BI, Wroblewski R. Aberrant elemental composition of otoconia in the dancer mouse mutant with a semidominant gene causing a morphogenetic type of inner ear defect. Acta Oto-Laryngol. 1988;106:208–212. doi: 10.3109/00016488809106427. [DOI] [PubMed] [Google Scholar]
- Bachra BN, Trautz ON, Simon SL. Precipitation of calcium carbonates and phosphates: 1. Spontaneous precipitation of calcium carbonates and phosphates under physiological conditions. Arch Biochem Biophys. 1963;103:124–138. doi: 10.1016/0003-9861(63)90018-3. [DOI] [PubMed] [Google Scholar]
- Balkema GW, Mangini NJ, Pinto LH. Discrete visual defects in pearl mutant mice. Science. 1983;219:1085–1087. doi: 10.1126/science.6600521. [DOI] [PubMed] [Google Scholar]
- Ballarino J, Howland HC. Otoconial morphology of the developing chick. Anat Rec. 1982;204:83–87. doi: 10.1002/ar.1092040111. [DOI] [PubMed] [Google Scholar]
- Balsamo G, Avallone B, Del Genio F, Trapani S, Marmo F. Calcification processes in the chick otoconia and calcium binding proteins: patterns of tetracycline incorporation and calbindin-D28K distribution. Hear Res. 2000;148:1–8. doi: 10.1016/s0378-5955(00)00094-0. [DOI] [PubMed] [Google Scholar]
- Banfi B, Malgrange B, Knisz J, Steger K, Dubois-Dauphin M, Krause KH. NOX3, a superoxide-generating NADPH oxidase of the inner ear. J Biol Chem. 2004;279:46065–46072. doi: 10.1074/jbc.M403046200. [DOI] [PubMed] [Google Scholar]
- Bermingham NA, Hassan BA, Price SD, Vollrath MA, Ben-Arie N, Eatock RA, Bellen HJ, Lysakowski A, Zoghbi HY. Math1: an essential gene for the generation of inner ear hair cells. Science. 1999;284:1837–1841. doi: 10.1126/science.284.5421.1837. [DOI] [PubMed] [Google Scholar]
- Besson V, Nalesso V, Herpin A, Bizot JC, Messaddeq N, Romand R, Puech A, Blanquet V, Herault Y. Training and aging modulate the loss-of-balance phenotype observed in a new ENU-induced allele of Otopetrin1. Biol Cell. 2005;97:787–798. doi: 10.1042/BC20040525. [DOI] [PubMed] [Google Scholar]
- Borelli G, Guibbolini ME, Mayer-Gostan N, Priouzeau F, De Pontual H, Allemand D, Puverel S, Tambutte E, Payan P. Daily variations of endolymph composition: relationship with the otolith calcification process in trout. J Exp Biol. 2003a;206:2685–2692. doi: 10.1242/jeb.00479. [DOI] [PubMed] [Google Scholar]
- Borelli G, Mayer-Gostan N, Merle PL, Pontual H, Boeuf G, Allemand D, Payan P. Composition of biomineral organic matrices with special emphasis on turbot (Psetta maxima) otolith and endolymph. Calcif Tissue Int. 2003b;72:717–725. doi: 10.1007/s00223-001-2115-6. [DOI] [PubMed] [Google Scholar]
- Bush JO, Lan Y, Jiang R. The cleft lip and palate defects in Dancer mutant mice result from gain of function of the Tbx10 gene. Proc Natl Acad Sci U S A. 2004;101:7022–7027. doi: 10.1073/pnas.0401025101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Casimiro MC, Knollmann BC, Ebert SN, Vary JC, Jr, Greene AE, Franz MR, Grinberg A, Huang SP, Pfeifer K. Targeted disruption of the Kcnq1 gene produces a mouse model of Jervell and Lange–Nielsen Syndrome. Proc Natl Acad Sci U S A. 2001;98:2526–2531. doi: 10.1073/pnas.041398998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ciciotte SL, Gwynn B, Moriyama K, Huizing M, Gahl WA, Bonifacino JS, Peters LL. Cappuccino, a mouse model of Hermansky–Pudlak syndrome, encodes a novel protein that is part of the pallidin-muted complex (BLOC-1) Blood. 2003;101:4402–4407. doi: 10.1182/blood-2003-01-0020. [DOI] [PubMed] [Google Scholar]
- Cohen-Salmon M, El-Amraoui A, Leibovici M, Petit C. Otogelin: a glycoprotein specific to the acellular membranes of the inner ear. Proc Natl Acad Sci U S A. 1997;94:14450–14455. doi: 10.1073/pnas.94.26.14450. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Crenshaw EB, III, Ryan A, Dillon SR, Kalla K, Rosenfeld MG. Wocko, a neurological mutant generated in a transgenic mouse pedigree. J Neurosci. 1991;11:1524–1530. doi: 10.1523/JNEUROSCI.11-06-01524.1991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Croushore JA, Blasiole B, Riddle RC, Thisse C, Thisse B, Canfield VA, Robertson GP, Cheng KC, Levenson R. Ptena and ptenb genes play distinct roles in zebrafish embryogenesis. Dev Dyn. 2005;234:911–921. doi: 10.1002/dvdy.20576. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Das M. Age determination and longevity in fishes. Gerontology. 1994;40:70–96. doi: 10.1159/000213580. [DOI] [PubMed] [Google Scholar]
- Davis JG, Oberholtzer JC, Burns FR, Greene MI. Molecular cloning and characterization of an inner ear-specific structural protein. Science. 1995;267:1031–1034. doi: 10.1126/science.7863331. [DOI] [PubMed] [Google Scholar]
- Davis JG, Burns FR, Navaratnam D, Lee AM, Ichimiya S, Oberholtzer JC, Greene MI. Identification of a structural constituent and one possible site of postembryonic formation of a teleost otolithic membrane. Proc Natl Acad Sci U S A. 1997;94:707–712. doi: 10.1073/pnas.94.2.707. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dutton KA, Pauliny A, Lopes SS, Elworthy S, Carney TJ, Rauch J, Geisler R, Haffter P, Kelsh RN. Zebrafish colourless encodes sox10 and specifies non-ectomesenchymal neural crest fates. Development. 2001;128:4113–4125. doi: 10.1242/dev.128.21.4113. [DOI] [PubMed] [Google Scholar]
- Ermak G, Davies KJ. Calcium and oxidative stress: from cell signaling to cell death. Mol Immunol. 2002;38:713–721. doi: 10.1016/s0161-5890(01)00108-0. [DOI] [PubMed] [Google Scholar]
- Erway LC, Grider A., Jr Zinc metabolism in lethal-milk mice. Otolith, lactation, and aging effects. J Hered. 1984;75:480–484. doi: 10.1093/oxfordjournals.jhered.a109990. [DOI] [PubMed] [Google Scholar]
- Erway LC, Purichia NA, Netzler ER, D’Amore MA, Esses D, Levine M. Genes, manganese, and zinc in formation of otoconia: labeling, recovery, and maternal effects. Scan Electron Microsc. 1986:1681–1694. [PubMed] [Google Scholar]
- Everett LA, Glaser B, Beck JC, Idol JR, Buchs A, Heyman M, Adawi F, Hazani E, Nassir E, Baxevanis AD, Sheffield VC, Green ED. Pendred syndrome is caused by mutations in a putative sulphate transporter gene (PDS) Nat Genet. 1997;17:411–422. [Google Scholar]
- Everett LA, Morsli H, Wu DK, Green ED. Expression pattern of the mouse ortholog of the Pendred’s syndrome gene (Pds) suggests a key role for pendrin in the inner ear. Proc Natl Acad Sci U S A. 1999;96:9727–9732. doi: 10.1073/pnas.96.17.9727. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Everett LA, Belyantseva IA, Noben-Trauth K, Cantos R, Chen A, Thakkar SI, Hoogstraten-Miller SL, Kachar B, Wu DK, Green ED. Targeted disruption of mouse Pds provides insight about the inner-ear defects encountered in Pendred syndrome. Hum Mol Genet. 2001;10:153–161. doi: 10.1093/hmg/10.2.153. [DOI] [PubMed] [Google Scholar]
- Falcon-Perez JM, Starcevic M, Gautam R, Dell’Angelica EC. BLOC-1, a novel complex containing the pallidin and muted proteins involved in the biogenesis of melanosomes and platelet-dense granules. J Biol Chem. 2002;277:28191–28199. doi: 10.1074/jbc.M204011200. [DOI] [PubMed] [Google Scholar]
- Farber SA, De Rose RA, Olson ES, Halpern ME. The zebrafish annexin gene family. Genome Res. 2003;13:1082–1096. doi: 10.1101/gr.479603. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Feng L, Seymour AB, Jiang S, To A, Peden AA, Novak EK, Zhen L, Rusiniak ME, Eicher EM, Robinson MS, Gorin MB, Swank RT. The beta3A subunit gene (Ap3b1) of the AP-3 adaptor complex is altered in the mouse hypopigmentation mutant pearl, a model for Hermansky–Pudlak syndrome and night blindness. Hum Mol Genet. 1999;8:323–330. doi: 10.1093/hmg/8.2.323. [DOI] [PubMed] [Google Scholar]
- Fermin CD, Igarashi M. Development of otoconia in the embryonic chick (Gallus domesticus) Acta Anat (Basel) 1985;123:148–152. [PubMed] [Google Scholar]
- Fermin CD, Lovett AE, Igarashi M, Dunner K., Jr Immunohistochemistry and histochemistry of the inner ear gelatinous membranes and statoconia of the chick (Gallus domesticus) Acta Anat (Basel) 1990;138:75–83. doi: 10.1159/000146922. [DOI] [PubMed] [Google Scholar]
- Flagella M, Clarke LL, Miller ML, Erway LC, Giannella RA, Andringa A, Gawenis LR, Kramer J, Duffy JJ, Doetschman T, Lorenz JN, Yamoah EN, Cardell EL, Shull GE. Mice lacking the basolateral Na-K-2Cl cotransporter have impaired epithelial chloride secretion and are profoundly deaf. J Biol Chem. 1999;274:26946–26955. doi: 10.1074/jbc.274.38.26946. [DOI] [PubMed] [Google Scholar]
- Fujii M, Harada Y, Hirakawa K, Takumida M. Otoconia on the vestibular dark cells of the ampullar areas. Acta Oto-Laryngol. 1995;Suppl. 519:140–142. doi: 10.3109/00016489509121889. [DOI] [PubMed] [Google Scholar]
- Gamp AC, Tanaka Y, Lullmann-Rauch R, Wittke D, D’Hooge R, De Deyn PP, Moser T, Maier H, Hartmann D, Reiss K, Illert AL, von Figura K, Saftig P. LIMP-2/LGP85 deficiency causes ureteric pelvic junction obstruction, deafness and peripheral neuropathy in mice. Hum Mol Genet. 2003;12:631–646. [PubMed] [Google Scholar]
- Gwynn B, Ciciotte SL, Hunter SJ, Washburn LL, Smith RS, Andersen SG, Swank RT, Dell’Angelica EC, Bonifacino JS, Eicher EM, Peters LL. Defects in the cappuccino (cno) gene on mouse chromosome 5 and human 4p cause Hermansky–Pudlak syndrome by an AP-3-independent mechanism. Blood. 2000;96:4227–4235. [PubMed] [Google Scholar]
- Haddon C, Jiang YJ, Smithers L, Lewis J. Delta–Notch signalling and the patterning of sensory cell differentiation in the zebrafish ear: evidence from the mind bomb mutant. Development. 1998;125:4637–4644. doi: 10.1242/dev.125.23.4637. [DOI] [PubMed] [Google Scholar]
- Haddon C, Mowbray C, Whitfield T, Jones D, Gschmeissner S, Lewis J. Hair cells without supporting cells: further studies in the ear of the zebrafish mind bomb mutant. J Neurocytol. 1999;28:837–850. doi: 10.1023/a:1007013904913. [DOI] [PubMed] [Google Scholar]
- Haffter P, Granato M, Brand M, Mullins MC, Hammerschmidt M, Kane DA, Odenthal J, van Eeden FJ, Jiang YJ, Heisenberg CP, Kelsh RN, Furutani-Seiki M, Vogelsang E, Beuchle D, Schach U, Fabian C, Nusslein-Volhard C. The identification of genes with unique and essential functions in the development of the zebrafish, Danio rerio. Development. 1996;123:1–36. doi: 10.1242/dev.123.1.1. [DOI] [PubMed] [Google Scholar]
- Hale JE, Wuthier RE. The mechanism of matrix vesicle formation. Studies on the composition of chondrocyte microvilli and on the effects of microfilament-perturbing agents on cellular vesiculation. J Biol Chem. 1987;262:1916–1925. [PubMed] [Google Scholar]
- Harada Y, Sugimoto Y. Metabolic disorder of otoconia after streptomycin intoxication. Acta Oto-Laryngol. 1977;84:65–71. doi: 10.3109/00016487709123943. [DOI] [PubMed] [Google Scholar]
- Harada Y, Kasuga S, Mori N. The process of otoconia formation in guinea pig utricular supporting cells. Acta Oto-Laryngol. 1998;118:74–79. doi: 10.1080/00016489850155161. [DOI] [PubMed] [Google Scholar]
- Hardison AL, Lichten L, Banerjee-Basu S, Becker TS, Burgess SM. The zebrafish gene claudinj is essential for normal ear function and important for the formation of the otoliths. Mech Dev. 2005;122:949–958. doi: 10.1016/j.mod.2005.03.009. [DOI] [PubMed] [Google Scholar]
- House MG, Honrubia V. Theoretical models for the mechanisms of benign paroxysmal positional vertigo. Audiol Neuro-Otol. 2003;8:91–99. doi: 10.1159/000068998. [DOI] [PubMed] [Google Scholar]
- Hsu CJ. Ultrastructural study of cytochemical localization of carbonic anhydrase in the inner ear. Acta Oto-Laryngol. 1991;111:75–84. doi: 10.3109/00016489109137357. [DOI] [PubMed] [Google Scholar]
- Huang L, Gitschier J. A novel gene involved in zinc transport is deficient in the lethal milk mouse. Nat Genet. 1997;17:292–297. doi: 10.1038/ng1197-292. [DOI] [PubMed] [Google Scholar]
- Huang L, Kuo YM, Gitschier J. The pallid gene encodes a novel, syntaxin 13-interacting protein involved in platelet storage pool deficiency. Nat Genet. 1999;23:329–332. doi: 10.1038/15507. [DOI] [PubMed] [Google Scholar]
- Hughes I, Blasiole B, Huss D, Warchol ME, Rath NP, Hurle B, Ignatova E, Dickman JD, Thalmann R, Levenson R, Ornitz DM. Otopetrin 1 is required for otolith formation in the zebrafish Danio rerio. Dev Biol. 2004;276:391–402. doi: 10.1016/j.ydbio.2004.09.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hughes I, Speck JD, Saito M, Schlesinger PH, Warchol ME, Ornitz DM. Otopetrin 1: a novel purinergic nucleotide-gated regulator of intracellular calcium in preparation. [Google Scholar]
- Huizing M, Anikster Y, Gahl WA. Hermansky–Pudlak syndrome and related disorders of organelle formation. Traffic. 2000;1:823–835. doi: 10.1034/j.1600-0854.2000.011103.x. [DOI] [PubMed] [Google Scholar]
- Huizing M, Boissy RE, Gahl WA. Hermansky–Pudlak syndrome: vesicle formation from yeast to man. Pigm Cell Res. 2002;15:405–419. doi: 10.1034/j.1600-0749.2002.02074.x. [DOI] [PubMed] [Google Scholar]
- Hulander M, Kiernan AE, Blomqvist SR, Carlsson P, Samuelsson EJ, Johansson BR, Steel KP, Enerback S. Lack of pendrin expression leads to deafness and expansion of the endolymphatic compartment in inner ears of Foxi1 null mutant mice. Development. 2003;130:2013–2025. doi: 10.1242/dev.00376. [DOI] [PubMed] [Google Scholar]
- Hurle B, Ignatova E, Massironi SM, Mashimo T, Rios X, Thalmann I, Thalmann R, Ornitz DM. Non-syndromic vestibular disorder with otoconial agenesis in tilted/mergulhador mice caused by mutations in Otopetrin 1. Hum Mol Genet. 2003;12:777–789. doi: 10.1093/hmg/ddg087. [DOI] [PubMed] [Google Scholar]
- Ichimiya I, Adams JC, Kimura RS. Immunolocalization of Na+, K(+)–ATPase, Ca(++)–ATPase, calcium-binding proteins, and carbonic anhydrase in the guinea pig inner ear. Acta Oto-Laryngol. 1994;114:167–176. doi: 10.3109/00016489409126037. [DOI] [PubMed] [Google Scholar]
- Ignatova EG, Thalmann I, Xu B, Ornitz DM, Thalmann R. Molecular mechanisms underlying ectopic otoconia-like particles in the endolymphatic sac of embryonic mice. Hear Res. 2004;194:65–72. doi: 10.1016/j.heares.2004.03.019. [DOI] [PubMed] [Google Scholar]
- James J, Schellens JP, Veenhof VB. Electron microscopy of formation of statoconia. Experientia. 1969;25:1173–1174. doi: 10.1007/BF01900258. [DOI] [PubMed] [Google Scholar]
- Johnsson LG, Wright CG, Preston RE, Henry PJ. Streptomycin-induced defects of the otoconial membrane. Acta Oto-Laryngol. 1980;89:401–406. doi: 10.3109/00016488009127155. [DOI] [PubMed] [Google Scholar]
- Kambayashi J, Kobayashi T, DeMott JE, Marcus NY, Thalmann I, Thalmann R. Effect of substrate-free vascular perfusion upon cochlear potentials and glycogen of the stria vascularis. Hear Res. 1982;6:223–240. doi: 10.1016/0378-5955(82)90056-9. [DOI] [PubMed] [Google Scholar]
- Kantheti P, Qiao X, Diaz ME, Peden AA, Meyer GE, Carskadon SL, Kapfhamer D, Sufalko D, Robinson MS, Noebels JL, Burmeister M. Mutation in AP-3 delta in the mocha mouse links endosomal transport to storage deficiency in platelets, melanosomes, and synaptic vesicles. Neuron. 1998;21:111–122. doi: 10.1016/s0896-6273(00)80519-x. [DOI] [PubMed] [Google Scholar]
- Kantheti P, Diaz ME, Peden AE, Seong EE, Dolan DF, Robinson MS, Noebels JL, Burmeister ML. Genetic and phenotypic analysis of the mouse mutant mh2J, an Ap3d allele caused by IAP element insertion. Mamm Genome. 2003;14:157–167. doi: 10.1007/s00335-002-2238-8. [DOI] [PubMed] [Google Scholar]
- Kawamata S, Igarashi Y. The fine structure of the developing otolithic organs of the rat. Acta Oto-Laryngol. 1993;Suppl. 504:30–37. doi: 10.3109/00016489309128118. [DOI] [PubMed] [Google Scholar]
- Kelsh RN, Brand M, Jiang YJ, Heisenberg CP, Lin S, Haffter P, Odenthal J, Mullins MC, van Eeden FJ, Furutani-Seiki M, Granato M, Hammerschmidt M, Kane DA, Warga RM, Beuchle D, Vogelsang L, Nusslein-Volhard C. Zebrafish pigmentation mutations and the processes of neural crest development. Development. 1996;123:369–389. doi: 10.1242/dev.123.1.369. [DOI] [PubMed] [Google Scholar]
- Khan SR. Interactions between stone-forming calcific crystals and macromolecules. Urol Int. 1997;59:59–71. doi: 10.1159/000283025. [DOI] [PubMed] [Google Scholar]
- Khan KM, Drescher DG. Proteins of the gelatinous layer of the trout saccular otolithic membrane. Hear Res. 1990;43:149–158. doi: 10.1016/0378-5955(90)90224-d. [DOI] [PubMed] [Google Scholar]
- Kido T, Sekitani T, Yamashita H, Endo S, Masumitsu Y, Shimogori H. Effects of carbonic anhydrase inhibitor on the otolithic organs of developing chick embryos. Am J Otolaryngol. 1991;12:191–195. doi: 10.1016/0196-0709(91)90119-z. [DOI] [PubMed] [Google Scholar]
- Kirsch T, Harrison G, Golub EE, Nah HD. The roles of annexins and types II and X collagen in matrix vesicle-mediated mineralization of growth plate cartilage. J Biol Chem. 2000;275:35577–35583. doi: 10.1074/jbc.M005648200. [DOI] [PubMed] [Google Scholar]
- Kiss PJ, Knisz J, Zhang Y, Baltrusaitis J, Sigmund CD, Thalmann R, Smith RJ, Verpy E, Baufi B. Inactivation of NADPH oxidase organizer 1 results in severe imbalance. Curr Biol. 2006;16:208–213. doi: 10.1016/j.cub.2005.12.025. [DOI] [PubMed] [Google Scholar]
- Kitamura K, Nomura Y, Yagi M, Yoshikawa Y, Ochikubo F. Morphological changes of cochlea in a strain of new-mutant mice. Acta Oto-Laryngol. 1991a;111:61–69. doi: 10.3109/00016489109137355. [DOI] [PubMed] [Google Scholar]
- Kitamura K, Yoshikawa Y, Ochikubo F. An ultrastructural study on vestibular sensory cells in a new-mutant mouse. Acta Oto-Laryngol. 1991b;111:1013–1020. doi: 10.3109/00016489109100750. [DOI] [PubMed] [Google Scholar]
- Kozel PJ, Friedman RA, Erway LC, Yamoah EN, Liu LH, Riddle T, Duffy JJ, Doetschman T, Miller ML, Cardell EL, Shull GE. Balance and hearing deficits in mice with a null mutation in the gene encoding plasma membrane Ca2+-ATPase isoform 2. J Biol Chem. 1998;273:18693–18696. doi: 10.1074/jbc.273.30.18693. [DOI] [PubMed] [Google Scholar]
- Kozlowski DJ, Whitfield TT, Hukriede NA, Lam WK, Weinberg ES. The zebrafish dog-eared mutation disrupts eya1, a gene required for cell survival and differentiation in the inner ear and lateral line. Dev Biol. 2005;277:27–41. doi: 10.1016/j.ydbio.2004.08.033. [DOI] [PubMed] [Google Scholar]
- Kusakari J, Ise I, Comegys TH, Thalmann I, Thalmann R. Effect of ethacrynic acid, furosemide, and ouabain upon the endolymphatic potential and upon high energy phosphates of the stria vascularis. Laryngoscope. 1978;88:12–37. doi: 10.1002/lary.1978.88.1.12. [DOI] [PubMed] [Google Scholar]
- Kwak SJ, Phillips BT, Heck R, Riley BB. An expanded domain of fgf3 expression in the hindbrain of zebrafish valentino mutants results in mis-patterning of the otic vesicle. Development. 2002;129:5279–5287. doi: 10.1242/dev.129.22.5279. [DOI] [PubMed] [Google Scholar]
- Legan PK, Lukashkina VA, Goodyear RJ, Kossi M, Russell IJ, Richardson GP. A targeted deletion in alpha-tectorin reveals that the tectorial membrane is required for the gain and timing of cochlear feedback. Neuron. 2000;28:273–285. doi: 10.1016/s0896-6273(00)00102-1. [DOI] [PubMed] [Google Scholar]
- Leger S, Brand M. Fgf8 and Fgf3 are required for zebrafish ear placode induction, maintenance and inner ear patterning. Mech Dev. 2002;119:91–108. doi: 10.1016/s0925-4773(02)00343-x. [DOI] [PubMed] [Google Scholar]
- Lewis V, Green SA, Marsh M, Vihko P, Helenius A, Mellman I. Glycoproteins of the lysosomal membrane. J Cell Biol. 1985;100:1839–1847. doi: 10.1083/jcb.100.6.1839. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li W, Rusiniak ME, Chintala S, Gautam R, Novak EK, Swank RT. Murine Hermansky–Pudlak syndrome genes: regulators of lysosome-related organelles. Bioessays. 2004;26:616–628. doi: 10.1002/bies.20042. [DOI] [PubMed] [Google Scholar]
- Lim DJ. Formation and fate of the otoconia. Scanning and transmission electron microscopy. Ann Otol Rhinol Laryngol. 1973;82:23–35. doi: 10.1177/000348947308200109. [DOI] [PubMed] [Google Scholar]
- Lim DJ. Otoconia in health and disease. A review. Ann Otol Rhinol Laryngol. 1984;Suppl. 112:17–24. doi: 10.1177/00034894840930s404. [DOI] [PubMed] [Google Scholar]
- Lim DJ, Karabinas C, Trune DR. Histochemical localization of carbonic anhydrase in the inner ear. Am J Otolaryngol. 1983;4:33–42. doi: 10.1016/s0196-0709(83)80005-2. [DOI] [PubMed] [Google Scholar]
- Lins U, Farina M, Kurc M, Riordan G, Thalmann R, Thalmann I, Kachar B. The otoconia of the guinea pig utricle: internal structure, surface exposure, and interactions with the filament matrix. J Struct Biol. 2000;131:67–78. doi: 10.1006/jsbi.2000.4260. [DOI] [PubMed] [Google Scholar]
- Lychakov DV, Rebane YT. Otolith regularities. Hear Res. 2000;143:83–102. doi: 10.1016/s0378-5955(00)00026-5. [DOI] [PubMed] [Google Scholar]
- Lyon MF. Twirler: a mutant affecting the inner ear of the house mouse. J Embryol Exp Morphol. 1958;6:105–116. [PubMed] [Google Scholar]
- Minck DR, Erway LC, Vorhees CV. Preliminary findings of a reduction of otoconia in the inner ear of adult rats prenatally exposed to phenytoin. Neurotoxicol Teratol. 1989;11:307–311. doi: 10.1016/0892-0362(89)90073-1. [DOI] [PubMed] [Google Scholar]
- Morsli H, Tuorto F, Choo D, Postiglione MP, Simeone A, Wu DK. Otx1 and Otx2 activities are required for the normal development of the mouse inner ear. Development. 1999;126:2335–2343. doi: 10.1242/dev.126.11.2335. [DOI] [PubMed] [Google Scholar]
- Murayama E, Okuno A, Ohira T, Takagi Y, Nagasawa H. Molecular cloning and expression of an otolith matrix protein cDNA from the rainbow trout, Oncorhynchus mykiss. Comp Biochem Physiol Part B: Biochem Mol Biol. 2000;126:511–520. doi: 10.1016/s0305-0491(00)00223-6. [DOI] [PubMed] [Google Scholar]
- Murayama E, Takagi Y, Ohira T, Davis JG, Greene MI, Nagasawa H. Fish otolith contains a unique structural protein, otolin-1. Eur J Biochem. 2002;269:688–696. doi: 10.1046/j.0014-2956.2001.02701.x. [DOI] [PubMed] [Google Scholar]
- Murayama E, Herbomel P, Kawakami A, Takeda H, Nagasawa H. Otolith matrix proteins OMP-1 and Otolin-1 are necessary for normal otolith growth and their correct anchoring onto the sensory maculae. Mech Dev. 2005;122:791–803. doi: 10.1016/j.mod.2005.03.002. [DOI] [PubMed] [Google Scholar]
- Nakahara H, Bevelander G. An electron microscope study of crystal calcium carbonate formation in the mouse otolith. Anat Rec. 1979;193:233–241. doi: 10.1002/ar.1091930205. [DOI] [PubMed] [Google Scholar]
- Odenthal J, Haffter P, Vogelsang E, Brand M, van Eeden FJ, Furutani-Seiki M, Granato M, Hammerschmidt M, Heisenberg CP, Jiang YJ, Kane DA, Kelsh RN, Mullins MC, Warga RM, Allende ML, Weinberg ES, Nusslein-Volhard C. Mutations affecting the formation of the notochord in the zebrafish, Danio rerio. Development. 1996a;123:103–115. doi: 10.1242/dev.123.1.103. [DOI] [PubMed] [Google Scholar]
- Odenthal J, Rossnagel K, Haffter P, Kelsh RN, Vogelsang E, Brand M, van Eeden FJ, Furutani-Seiki M, Granato M, Hammerschmidt M, Heisenberg CP, Jiang YJ, Kane DA, Mullins MC, Nusslein-Volhard C. Mutations affecting xanthophore pigmentation in the zebrafish, Danio rerio. Development. 1996b;123:391–398. doi: 10.1242/dev.123.1.391. [DOI] [PubMed] [Google Scholar]
- Oghalai JS, Manolidis S, Barth JL, Stewart MG, Jenkins HA. Unrecognized benign paroxysmal positional vertigo in elderly patients. Otolaryngol-Head Neck Surg. 2000;122:630–634. doi: 10.1016/S0194-5998(00)70187-2. [DOI] [PubMed] [Google Scholar]
- Ornitz DM, Bohne BA, Thalmann I, Harding GW, Thalmann R. Otoconial agenesis in tilted mutant mice. Hear Res. 1998;122:60–70. doi: 10.1016/s0378-5955(98)00080-x. [DOI] [PubMed] [Google Scholar]
- Paffenholz R, Bergstrom RA, Pasutto F, Wabnitz P, Munroe RJ, Jagla W, Heinzmann U, Marquardt A, Bareiss A, Laufs J, Russ A, Stumm G, Schimenti JC, Bergstrom DE. Vestibular defects in head-tilt mice result from mutations in Nox3, encoding an NADPH oxidase. Genes Dev. 2004;18:486–491. doi: 10.1101/gad.1172504. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Piscopo M, Balsamo G, Mutone R, Avallone B, Marmo F. Calbindin D28K is a component of the organic matrix of lizard Podarcis sicula otoconia. Hear Res. 2003;178:89–94. doi: 10.1016/s0378-5955(03)00053-4. [DOI] [PubMed] [Google Scholar]
- Pote KG, Ross MD. Each otoconia polymorph has a protein unique to that polymorph. Comp Biochem Physiol Part B: Biochem Mol Biol. 1991;98:287–295. doi: 10.1016/0305-0491(91)90181-c. [DOI] [PubMed] [Google Scholar]
- Pote KG, Hauer CR, III, Michel H, Shabanowitz J, Hunt DF, Kretsinger RH. Otoconin-22, the major protein of aragonitic frog otoconia, is a homolog of phospholipase A2. Biochemistry. 1993;32:5017–5024. doi: 10.1021/bi00070a007. [DOI] [PubMed] [Google Scholar]
- Preston RE, Johnsson LG, Hill JH, Schacht J. Incorporation of radioactive calcium into otolithic membranes and middle ear ossicles of the gerbil. Acta Oto-Laryngol. 1975;80:269–275. doi: 10.3109/00016487509121327. [DOI] [PubMed] [Google Scholar]
- Rauch TM. The additive effects of two mutant genes on otolith formation in mice: an animal model to assess otolith function. J Aud Res. 1979;19:259–265. [PubMed] [Google Scholar]
- Reynolds JL, Skepper JN, McNair R, Kasama T, Gupta K, Weissberg PL, Jahnen-Dechent W, Shanahan CM. Multifunctional roles for serum protein fetuin-a in inhibition of human vascular smooth muscle cell calcification. J Am Soc Nephrol. 2005;16:2920–2930. doi: 10.1681/ASN.2004100895. [DOI] [PubMed] [Google Scholar]
- Riley BB, Grunwald DJ. A mutation in zebrafish affecting a localized cellular function required for normal ear development. Dev Biol. 1996;179:427–435. doi: 10.1006/dbio.1996.0272. [DOI] [PubMed] [Google Scholar]
- Riley BB, Moorman SJ. Development of utricular otoliths, but not saccular otoliths, is necessary for vestibular function and survival in zebrafish. J Neurobiol. 2000;43:329–337. doi: 10.1002/1097-4695(20000615)43:4<329::aid-neu2>3.0.co;2-h. [DOI] [PubMed] [Google Scholar]
- Riley BB, Zhu C, Janetopoulos C, Aufderheide KJ. A critical period of ear development controlled by distinct populations of ciliated cells in the zebrafish. Dev Biol. 1997;191:191–201. doi: 10.1006/dbio.1997.8736. [DOI] [PubMed] [Google Scholar]
- Riley BB, Chiang M, Farmer L, Heck R. The deltaA gene of zebrafish mediates lateral inhibition of hair cells in the inner ear and is regulated by pax2.1. Development. 1999;126:5669–5678. doi: 10.1242/dev.126.24.5669. [DOI] [PubMed] [Google Scholar]
- Rolfsen RM, Erway LC. Trace metals and otolith defects in mocha mice. J Hered. 1984;75:159–162. [PubMed] [Google Scholar]
- Ross MD. Calcium ion uptake and exchange in otoconia. Adv Oto-Rhino-Laryngol. 1979;25:26–33. doi: 10.1159/000402913. [DOI] [PubMed] [Google Scholar]
- Ross MD, Peacor D, Johnsson LG, Allard LF. Observations on normal and degenerating human otoconia. Ann Otol Rhinol Laryngol. 1976;85:310–326. doi: 10.1177/000348947608500302. [DOI] [PubMed] [Google Scholar]
- Sakagami M. Role of osteopontin in the rodent inner ear as revealed by in situ hybridization. Med Electron Microsc. 2000;33:3–10. doi: 10.1007/s007950000001. [DOI] [PubMed] [Google Scholar]
- Salt AN, Inamura N, Thalmann R, Vora A. Calcium gradients in inner ear endolymph. Am J Otolaryngol. 1989;10:371–375. doi: 10.1016/0196-0709(89)90030-6. [DOI] [PubMed] [Google Scholar]
- Sanchez-Fernandez JM, Rivera-Pomar JM. A scanning electron microscopy study on human otoconia genesis. Acta Oto-Laryngol. 1984;97:479–488. doi: 10.3109/00016488409132925. [DOI] [PubMed] [Google Scholar]
- Sanchez-Fernandez JM, Rivera-Pomar JM, Tello MJ. Human otoconial crystal growth. An approach from morphological and morphometric data. ORL J Otorhinolaryngol Relat Spec. 1989;51:108–115. doi: 10.1159/000276042. [DOI] [PubMed] [Google Scholar]
- Shiao JC, Lin LY, Horng JL, Hwang PP, Kaneko T. How can teleostean inner ear hair cells maintain the proper association with the accreting otolith? J Comp Neurol. 2005;488:331–341. doi: 10.1002/cne.20578. [DOI] [PubMed] [Google Scholar]
- Shull GE, Okunade G, Liu LH, Kozel P, Periasamy M, Lorenz JN, Prasad V. Physiological functions of plasma membrane and intracellular Ca2+ pumps revealed by analysis of null mutants. Ann N Y Acad Sci. 2003;986:453–460. doi: 10.1111/j.1749-6632.2003.tb07229.x. [DOI] [PubMed] [Google Scholar]
- Simmler MC, Cohen-Salmon M, El-Amraoui A, Guillaud L, Benichou JC, Petit C, Panthier JJ. Targeted disruption of otog results in deafness and severe imbalance. Nat Genet. 2000a;24:139–143. doi: 10.1038/72793. [DOI] [PubMed] [Google Scholar]
- Simmler MC, Zwaenepoel II, Verpy E, Guillaud L, Elbaz C, Petit C, Panthier JJ. Twister mutant mice are defective for otogelin, a component specific to inner ear acellular membranes. Mamm Genome. 2000b;11:961–966. doi: 10.1007/s003350010197. [DOI] [PubMed] [Google Scholar]
- Sollner C, Burghammer M, Busch-Nentwich E, Berger J, Schwarz H, Riekel C, Nicolson T. Control of crystal size and lattice formation by starmaker in otolith biomineralization. Science. 2003;302:282–286. doi: 10.1126/science.1088443. [DOI] [PubMed] [Google Scholar]
- Sollner C, Schwarz H, Geisler R, Nicolson T. Mutated otopetrin 1 affects the genesis of otoliths and the localization of Starmaker in zebrafish. Dev Genes Evol. 2004;214:582–590. doi: 10.1007/s00427-004-0440-2. [DOI] [PubMed] [Google Scholar]
- Solomon KS, Kudoh T, Dawid IB, Fritz A. Zebrafish foxi1 mediates otic placode formation and jaw development. Development. 2003;130:929–940. doi: 10.1242/dev.00308. [DOI] [PubMed] [Google Scholar]
- Spicer SS, Schulte BA. Differentiation of inner ear fibrocytes according to their ion transport related activity. Hear Res. 1991;56:53–64. doi: 10.1016/0378-5955(91)90153-z. [DOI] [PubMed] [Google Scholar]
- Sreenath T, Thyagarajan T, Hall B, Longenecker G, D’Souza R, Hong S, Wright JT, MacDougall M, Sauk J, Kulkarni AB. Dentin sialophosphoprotein knockout mouse teeth display widened predentin zone and develop defective dentin mineralization similar to human dentinogenesis imperfecta type III. J Biol Chem. 2003;278:24874–24880. doi: 10.1074/jbc.M303908200. [DOI] [PubMed] [Google Scholar]
- Stevens CB, Davies AL, Battista S, Lewis JH, Fekete DM. Forced activation of Wnt signaling alters morphogenesis and sensory organ identity in the chicken inner ear. Dev Biol. 2003;261:149–164. doi: 10.1016/s0012-1606(03)00297-5. [DOI] [PubMed] [Google Scholar]
- Sumanas S, Larson JD, Miller Bever M. Zebrafish chaperone protein GP96 is required for otolith formation during ear development. Dev Biol. 2003;261:443–455. doi: 10.1016/s0012-1606(03)00322-1. [DOI] [PubMed] [Google Scholar]
- Suzuki H, Ikeda K, Takasaka T. Biological characteristics of the globular substance in the otoconial membrane of the guinea pig. Hear Res. 1995;90:212–218. doi: 10.1016/0378-5955(95)00168-7. [DOI] [PubMed] [Google Scholar]
- Suzuki H, Ikeda K, Furukawa M, Takasaka T. P2 purinoceptor of the globular substance in the otoconial membrane of the guinea pig inner ear. Am J Physiol. 1997a;273:C1533–C1540. doi: 10.1152/ajpcell.1997.273.5.C1533. [DOI] [PubMed] [Google Scholar]
- Suzuki H, Ikeda K, Takasaka T. Age-related changes of the globular substance in the otoconial membrane of mice. Laryngoscope. 1997b;107:378–381. doi: 10.1097/00005537-199703000-00019. [DOI] [PubMed] [Google Scholar]
- Tachibana M, Morioka H. Glucuronic acid-containing glycosaminoglycans occur in otoconia: cytochemical evidence by hyaluronidase-gold labeling. Hear Res. 1992;62:11–15. doi: 10.1016/0378-5955(92)90198-v. [DOI] [PubMed] [Google Scholar]
- Takagi Y, Takahashi A. Characterization of ootolith soluble-matrix producing cells in the saccular epithelium of rainbow trout (Oncorhynchus mykiss) inner ear. Anat Rec. 1999;254:322–329. doi: 10.1002/(SICI)1097-0185(19990301)254:3<322::AID-AR2>3.0.CO;2-Q. [DOI] [PubMed] [Google Scholar]
- Takemura T, Sakagami M, Nakase T, Kubo T, Kitamura Y, Nomura S. Localization of osteopontin in the otoconial organs of adult rats. Hear Res. 1994;79:99–104. doi: 10.1016/0378-5955(94)90131-7. [DOI] [PubMed] [Google Scholar]
- Takumida M, Zhang DM, Yajin K, Harada Y. Effect of streptomycin on the otoconial layer of the guinea pig. ORL J Otorhinolaryngol Relat Spec. 1997a;59:263–268. doi: 10.1159/000276950. [DOI] [PubMed] [Google Scholar]
- Takumida M, Zhang DM, Yajin K, Harada Y. Formation and fate of giant otoconia of the guinea pig following streptomycin intoxication. Acta Oto-Laryngol. 1997b;117:538–544. doi: 10.3109/00016489709113434. [DOI] [PubMed] [Google Scholar]
- Tateda M, Suzuki H, Ikeda K, Takasaka T. pH regulation of the globular substance in the otoconial membrane of the guinea-pig inner ear. Hear Res. 1998;124:91–98. doi: 10.1016/s0378-5955(98)00115-4. [DOI] [PubMed] [Google Scholar]
- Thalmann R, Thalmann I. Source and role of endolymph macromolecules. Acta Oto-Laryngol. 1999;119:293–296. doi: 10.1080/00016489950181260. [DOI] [PubMed] [Google Scholar]
- Trune DR, Lim DJ. The behavior and vestibular nuclear morphology of otoconia-deficient pallid mutant mice. J Neurogenet. 1983;1:53–69. doi: 10.3109/01677068309107072. [DOI] [PubMed] [Google Scholar]
- Thalmann I, Hughes I, Tong B, Thalmann R. Microscale analysis of proteins in inner ear tissues and fluids with emphasis on endolymphatic sac, otoconia and organ of Corti, Electrophoresis. doi: 10.1002/elps.200500768. in press. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tusa RJ. Benign paroxysmal positional vertigo. Curr Neurol Neurosci Rep. 2001;1:478–485. doi: 10.1007/s11910-001-0110-y. [DOI] [PubMed] [Google Scholar]
- Verpy E, Leibovici M, Petit C. Characterization of otoconin-95, the major protein of murine otoconia, provides insights into the formation of these inner ear biominerals. Proc Natl Acad Sci U S A. 1999;96:529–534. doi: 10.1073/pnas.96.2.529. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vibert D, Kompis M, Hausler R. Benign paroxysmal positional vertigo in older women may be related to osteoporosis and osteopenia. Ann Otol Rhinol Laryngol. 2003;112:885–889. doi: 10.1177/000348940311201010. [DOI] [PubMed] [Google Scholar]
- Vitelli F, Viola A, Morishima M, Pramparo T, Baldini A, Lindsay E. TBX1 is required for inner ear morphogenesis. Hum Mol Genet. 2003;12:2041–2048. doi: 10.1093/hmg/ddg216. [DOI] [PubMed] [Google Scholar]
- Wang Y, Kowalski PE, Thalmann I, Ornitz DM, Mager DL, Thalmann R. Otoconin-90, the mammalian otoconial matrix protein, contains two domains of homology to secretory phospholipase A2. Proc Natl Acad Sci U S A. 1998;95:15345–15350. doi: 10.1073/pnas.95.26.15345. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang W, Xu J, Kirsch T. Annexin-mediated Ca2+ influx regulates growth plate chondrocyte maturation and apoptosis. J Biol Chem. 2003;278:3762–3769. doi: 10.1074/jbc.M208868200. [DOI] [PubMed] [Google Scholar]
- Welling DB, Parnes LS, O’Brien B, Bakaletz LO, Brackmann DE, Hinojosa R. Particulate matter in the posterior semicircular canal. Laryngoscope. 1997;107:90–94. doi: 10.1097/00005537-199701000-00018. [DOI] [PubMed] [Google Scholar]
- Whitfield TT, Granato M, van Eeden FJ, Schach U, Brand M, Furutani-Seiki M, Haffter P, Hammerschmidt M, Heisenberg CP, Jiang YJ, Kane DA, Kelsh RN, Mullins MC, Odenthal J, Nusslein-Volhard C. Mutations affecting development of the zebrafish inner ear and lateral line. Development. 1996;123:241–254. doi: 10.1242/dev.123.1.241. [DOI] [PubMed] [Google Scholar]
- Wright CG, Hubbard DG. SEM observations on development of human otoconia during the first trimester of gestation. Acta Oto-Laryngol. 1982;94:7–18. doi: 10.3109/00016488209128884. [DOI] [PubMed] [Google Scholar]
- Wright CG, Hubbard DG, Graham JW. Absence of otoconia in a human infant. Ann Otol Rhinol Laryngol. 1979;88:779–783. doi: 10.1177/000348947908800607. [DOI] [PubMed] [Google Scholar]
- Wright CG, Rouse RC, Johnsson LG, Weinberg AG, Hubbard DG. Vaterite otoconia in two cases of otoconial membrane dysplasia. Ann Otol Rhinol Laryngol. 1982;91:193–199. doi: 10.1177/000348948209100215. [DOI] [PubMed] [Google Scholar]
- Yamane H, Imoto T, Nakai Y, Igarashi M, Rask-Andersen H. Otoconia degradation. Acta Oto-Laryngol. 1984;Suppl. 406:263–270. doi: 10.3109/00016488309123047. [DOI] [PubMed] [Google Scholar]
- Yaoi Y, Suzuki M, Tomura H, Sasayama Y, Kikuyama S, Tanaka S. Molecular cloning of otoconin-22 complementary deoxyribonucleic acid in the bullfrog endolymphatic Sac: effect of calcitonin on otoconin-22 messenger ribonucleic acid levels. Endocrinology. 2003;144:3287–3296. doi: 10.1210/en.2002-0181. [DOI] [PubMed] [Google Scholar]
- Zwaenepoel I, Mustapha M, Leibovici M, Verpy E, Goodyear R, Liu XZ, Nouaille S, Nance WE, Kanaan M, Avraham KB, Tekaia F, Loiselet J, Lathrop M, Richardson G, Petit C. Otoancorin, an inner ear protein restricted to the interface between the apical surface of sensory epithelia and their overlying acellular gels, is defective in autosomal recessive deafness DFNB22. Proc Natl Acad Sci U S A. 2002;99:6240–6245. doi: 10.1073/pnas.082515999. [DOI] [PMC free article] [PubMed] [Google Scholar]