

Abstract
STAT3 has been indirectly implicated in numerous fundamental cellular processes, including proliferation, survival and differentiation. We provide genetic evidence from studies of STAT3-null cells that STAT3 is dispensable for normal growth of mouse fibroblasts in culture. STAT3 contributed to the full induction of some (typified by c-fos) but not all (typified by c-myc) immediate early gene expression, but STAT3-independent processes were sufficient to support full cell growth and survival. However, STAT3 was required to manifest a transformed state following expression of v-src, and STAT3-null cells were impaired for anchorage independent growth as colonies in soft agar and as tumors in mice. The data suggest that STAT3 mediates the maintenance of FAK activity in the absence of cell adhesion by suppressing the action of an inhibitory phosphatase.
Signal transducers and activators of transcription (STATs) are latent transcription factors that are activated by cytokines and growth factors. Tyrosine phosphorylation leads to STAT dimerization, nuclear translocation and activation of gene expression (1, 2). STAT3 is phosphorylated by various tyrosine kinases such as Janus Kinases (JAKs) and src-family kinases that are associated with and activated by cytokine and growth factor receptors. STAT3 activation has been implicated in various biological responses including proliferation, survival, differentiation and transformation (2). In addition, genetic ablation of STAT3 in mice leads to embryonic lethality, suggesting that STAT3 is essential for basic processes.
An important role for STAT3 in src signaling has been suggested by a variety of experimental data, both as a mediator of proliferation downstream of c-src and as an essential arbiter of cell transformation by the v-src oncogene. For instance, STAT3 is activated by c-src downstream of growth factor receptors, such as EGF and PDGF receptors. Indeed, activated STAT3 was first detected as a DNA binding activity that associated with the c-fos promoter in PDGF-stimulated cells (3). Activation of STAT3 by c-src downstream of the PDGF receptor has also been associated with c-myc transcriptional induction and proliferation of fibroblasts (4), suggesting a direct role for STAT3 in cell growth.
Following the seminal observation that STAT3 is a phosphorylated substrate of v-src (5), increasing evidence has accumulated for a role for STAT3 in the oncogenic action of v-src. Interference with STAT3 signaling by ectopic expression of STAT3 mutants that act as dominant interfering molecules impairs cell transformation (6, 7). Moreover, a constitutively dimerized form of STAT3 that displays unregulated transactivation potential can promote fibroblast and epithelial cell transformation in the absence of v-src, implying that STAT3 is a potential oncogene on its own (8, 9). This notion is supported by the observation that STAT3 is often abnormally expressed and activated in diverse human tumors, and interference with STAT3 function can impair the viability of transformed cells. Such data provide compelling evidence for a role for STAT3 in tumorigenesis involving src and additional oncogenes (10-12).
Roles for c-src in cell proliferation and malignant transformation remain incompletely understood. During normal cell growth, c-src can be activated by growth factor stimulation and integrin engagement to affect cell proliferation, adhesion and migration (13). Many of the effects of src on cell adhesion, cell spreading and migration are thought to be mediated by activation of focal adhesion kinase (FAK), a tyrosine kinase that contributes to the oncogenic action of v-src (14) and is activated by integrins (15). Integrin clustering recruits FAK into focal adhesions where it is activated by autophosphorylation. FAK activation leads not only to proliferation and survival but also regulates cell spreading and migration, most clearly demonstrated by the defects in FAK null cells (16). Down-regulation of FAK activity can occur through mechanisms involving a naturally occurring dominant negative form of FAK, proteolytic cleavage, or dephosphorylation by phosphatases (14, 17). FAK downregulation is important for the turnover of focal adhesions during cell migration and is a major target for v-src in transformed cells, contributing to altered adhesion, migration and invasion (18).
In this study, we took a genetic approach to studying STAT3 signaling, by conditional gene ablation in primary and immortal mouse fibroblasts. We show that STAT3 is required for some immediate early gene induction downstream of c-src, activated by growth factor receptors such as PDGF receptor, and we identify a novel role for STAT3 in regulation of cell adhesion, through control of FAK phosphorylation in response to v-src. Although we provide definitive evidence for STAT3 involvement in v-src transformation, we found no evidence for an obligatory role for STAT3 in regulation of c-myc expression or for normal fibroblast proliferation.
Experimental Procedures
Antibodies, DNA constructs, and chemicals
The following antibodies were obtained from commercial sources: STAT3 (clone 5G7, Zymed), P-STAT3 (Cell Signaling), pp60-src (Upstate), tubulin (Sigma), c-Fos, FAK (Santa Cruz), P-FAK397 (Biosource). PDGF-BB was purchased from Becton Dickinson; SU6656 from Calbiochem; and staurosporine from LC Laboratories.
Expression construct for v-src (pMv-src) has been described (19). STAT3 constructs were generated by PCR-directed mutagenesis, and cloned into the bicistronic GFP retroviral vector Pallino (20). STAT3Y705F carries a mutation of tyr 705 to phe, and STAT3DBD carries a five amino acid substitution in the DNA binding domain (21).
Cell culture
Primary mouse embryo fibroblasts (MEF) were prepared by standard techniques (22) from E14-15 d STAT3flox/flox or STAT3flox/- embryos and used for experiments prior to passage 5. Immortalized cell lines were generated by the 3T3 protocol (23). The lines used had sustained inactivating mutations of the tumor suppressor p53 (data not shown).
Protein analysis
Total cell extracts were prepared in 300 mM NaCl, 50 mM HEPES, pH 7.6, 1.5 mM MgCl2, 10% glycerol, 1% Triton X-100, 10 mM NaPyrPO4, 20 mM NaF, 1 mM EGTA, 0.1 mM EDTA, 1 mM DTT, 1 mM Na3VO4, and Protease Inhibitor Cocktail (Sigma). Proteins were analyzed by SDS-PAGE and immunoblotting, using HRP-labeled secondary antibodies (Pierce), as previously described (24).
Retroviral infection
Recombinant retroviruses were packaged using Phoenix cells, and infections were carried out using standard protocols (25). In brief, Phoenix cells were transfected in the presence of 25 μM chloroquine using the calcium phosphate method, 48 h supernatants were used to infect 1×105 cells in the presence of 40 μg/ml polybrene and infected cells were selected by drug resistance. STAT3 constructs also expressed GFP from the same RNA, and GFP expression was used to select infected cells displaying similar expression levels by flow cytometry. Levels of v-src protein in transformed cells were verified by antibody blots.
RNA quantification
Abundance of specific RNA species was measured by real-time fluorescent RT-PCR using Syber green dye (Molecular Probes), as previously described (26). Each measurement was performed in triplicate, quantified by comparison to a standard dilution series, normalized to the abundance of GAPDH or ribosomal protein L32, and presented as the mean and standard error. For RNase protection, primary MEF were serum-starved 48 h in DMEM containing 0.5% serum, stimulated with 40 ng/ml PDGF-BB or 100 ng/ml EGF, and RNA was quantified using the RiboQuant system from BD-Pharmingen using multi-probe sets for mFos/Jun and mMyc.
Colony formation in soft agar
Cells were seeded in 0.35% low-melting agarose, prepared in growth medium. Colonies that reached 0.2 mm2 or larger were enumerated after 10-14 d (27).
Focus formation assay
Confluent cells were incubated in medium containing 0.1% serum for 7 d and foci were enumerated by light microscopy.
Subcutaneous tumor formation
Balb/c-nu/nu mice (Taconic Labs) were injected with 100 mg/kg cyclophosphamide in PBS 3 d prior to inoculation with 1×104 cells. Tumor growth was monitored every 3-4 d, and tumor volume was calculated as 4/3 of the product of length, height, and depth, measured with calipers.
Cell proliferation
Cells were plated on 18 mm2 glass cover slips, serum starved, growth factor stimulated, and labeled with 10 μM BrdU (Zymed) for 2 h. Fixed cells were stained with FITC-labeled anti-BrdU (Becton Dickinson), counter-stained with 0.1 μg/ml propidium iodide, and counted by fluorescence microscopy.
Adhesion assay
Non-treated plastic dishes were coated with 25 μg/ml collagen I or 10 μg/ml fibronectin. Cells were detached in 0.5 mM EDTA, washed and plated in triplicate at 5×105/ml in DMEM containing 0.5% BSA, 1 mM MgCl2 and 0.2 mM MnCl2. When cells exhibited first signs of attachment, but not yet spread, non-attached cells were removed by rinsing with PBS until no cells remained in control wells (BSA coated). Attached cells were stained with 0.4% crystal violet in methanol, extracted in 10% acetic acid and quantified by absorbance at 590 nm.
Results
Generation of STAT3-null fibroblasts
Analysis of STAT3 function in cell proliferation and transformation has largely relied on data generated using dominant interfering mutants over-expressed in cells. To study the role of STAT3 in a more physiologically relevant environment, STAT3-null fibroblasts were generated from STAT3 conditional knockout mice (28, 29). Primary MEF from STAT3 flox/- or flox/flox embryos (referred to as STAT3WT) were subsequently infected with a control or a retrovirus encoding Cre-recombinase (30) to facilitate deletion of exons 16-21 of the STAT3 gene (referred to as STAT3KO). STAT3 gene ablation and protein loss were monitored by PCR, EMSA, and western blotting. Near 100% gene deletion was obtained, accompanied by complete loss of detectable STAT3 protein and DNA binding activity (Suppl. Fig. 1 and data not shown). In addition, immortal STAT3WT cell lines were generated using the 3T3 protocol (23), and STAT3KO cells were derived by Cre infection. Primary MEF were used in basic gene expression and proliferation studies, to avoid the potential complication of secondary mutations introduced during immortalization. 3T3 cells were used for transformation studies.
Proliferation and gene expression in primary STAT3-null fibroblasts
Given that STAT3 deletion in mice is embryonic lethal, and that some of the proposed downstream targets for STAT3 are direct regulators of cell proliferation (e.g., c-myc, c-fos and cyclin D1), deletion of STAT3 in cells could be expected to inhibit proliferation (2). Surprisingly, no effects on proliferation were observed in STAT3KO MEF compared with STAT3WT MEF, as measured by steady-state growth, saturation density, or response to growth factors such as EGF, PDGF or serum (see below and data not shown). However, since STAT3 has been implicated in induction gene expression, specifically of c-myc in response to PDGF (4), immediate-early gene induction was compared in STAT3WT and STAT3KO MEF. Interestingly, c-myc RNA levels were not significantly different between STAT3WT and STAT3KO MEF when corrected for levels of the housekeeping RNA, ribosomal protein L32 (Fig. 1A, left). However, c-fos induction was significantly and reproducibly reduced in STAT3KO MEF, most notably in serum-stimulated cells, (Fig. 1A, right). STAT3WT 3T3 fibroblasts expressed a low but measurable level of c-fos protein that was partially reduced in STAT3KO 3T3 fibroblasts (Fig. 2B), reflecting the partial reduction in c-fos mRNA accumulation observed in growth-stimulated primary cells. Levels of c-fos protein were dependent on transcriptionally active STAT3, since reconstitution of STAT3KO cells with recombinant wild type but not transcriptionally inactive STAT3 mutants (STAT3Y705F or STAT3DBD) restored c-fos protein (Fig. 1B) and RNA levels (Suppl. Fig. 2). Interestingly, c-Fos protein levels in cells reconstituted with STAT3 mutants were slightly lower than in STAT3KO cells, suggesting that over-expression of transcriptionally inactive STAT3 may interfere with endogenous transcription of c-fos, perhaps by interfering with upstream signaling pathways or co-activator recruitment to the c-fos promoter. Levels of c-myc were not affected by the presence or absence of STAT3 (not shown).
Fig. 1.
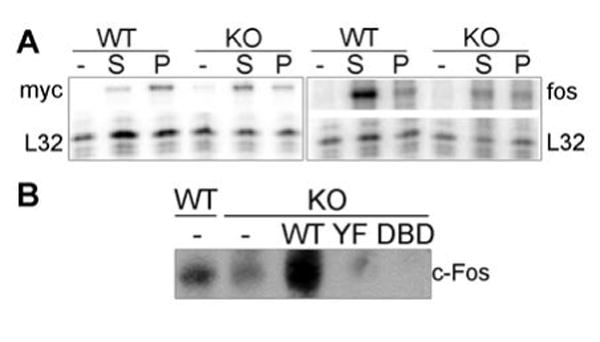
STAT3 is required for c-fos but not c-myc induction. (A) STAT3WT or STAT3KO MEF were starved for 48 h in 0.5% serum and re-stimulated with 20% serum (S) or 40 ng/ml PDGF-BB (P) for 30 and 120 min. Total RNA was prepared and c-fos or c-myc transcript levels were measured using RNase protection assays. c-fos was measured in samples from cells stimulated for 30 min while c-myc was measured in RNA samples from cells stimulated for 120 min. Representative images are shown for c-myc (left) and c-fos (right). L32 served as internal control.
(B) STAT3KO 3T3 cells were infected with wild type or mutant STAT3, together with v-src to phosphorylate STAT3. c-Fos protein levels were measured by immunoblotting.
Fig. 2.
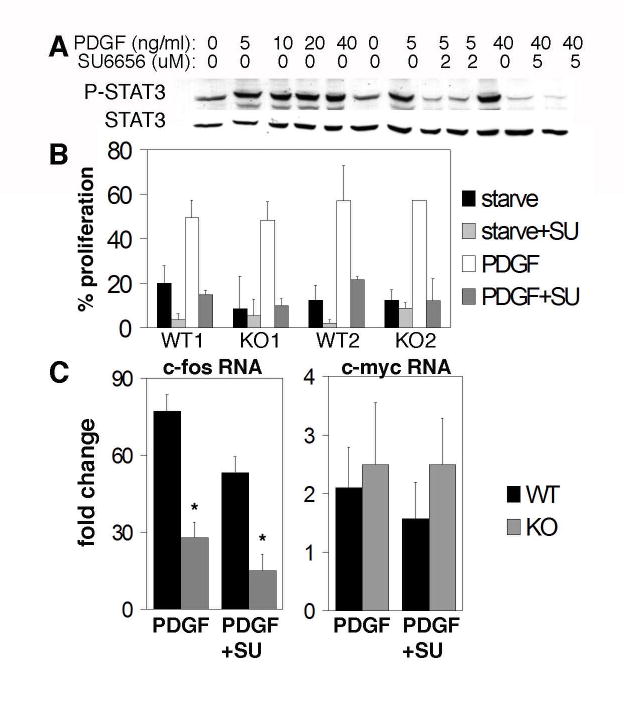
PDGF-induced proliferation and src/STAT3 activation are separate pathways. (A) STAT3WT MEF were starved in 0.5% serum for 48 h and re-stimulated with the indicated amounts of PDGF-BB in the presence of increasing concentrations of the c-src inhibitor, SU6656. Cell lysates were probed for phospho-STAT3 (Y705) and total STAT3. (B) Proliferation of two independent MEF lines, WT or KO (infected with control or Cre retrovirus). BrdU incorporation following stimulation with 40 ng/ml PDGF-BB, with or without 5 μM SU6656, was measured. Results are presented as percent cells stained by anti-BrdU.(C) c-myc and c-fos transcripts were measured by real-time RT-PCR in STAT3WT and STAT3KO MEF, starved and re-stimulated as described in (A). Results (averages of 5 independent experiments) are presented as fold induction over non-stimulated cells. *Student t-test of STAT3KO cells with or without SU6656 showed no significant difference between samples (P=0.14).
Src and STAT3 dependent and independent c-fos induction
STAT3 has been implicated in several signaling events downstream of c-src and v-src. For instance, inhibition of c-src activity by treatment of NIH3T3 cells with the c-src specific inhibitor SU6656 inhibited STAT3 phosphorylation, c-myc induction, and cell proliferation in response to PDGF (31). To test if STAT3 was required in this response, wild type and mutant cells were treated with SU6656 and stimulated with PDGF. In STAT3WT cells increasing concentrations of SU6656 abolished STAT3 phosphorylation (Fig. 2A), as previously observed (31). Proliferation under these conditions was also inhibited, confirming that PDGF-induced proliferation was dependent on the activity of c-src (Fig. 2B). Interestingly, proliferation of STAT3KO cells was comparable to that of STAT3WT cells, and this proliferation was equally sensitive to inhibition by SU6656. Therefore, although SU6656 inhibited STAT3 phosphorylation and blocked proliferation, these two processes are not interdependent in that STAT3 is not required for PDGF-induced proliferation.
Since c-fos gene expression was in part dependent on STAT3 (Fig. 1), we tested whether c-fos induction by PDGF also required c-src (Fig. 2C, left). Induction of c-fos mRNA was partially inhibited by SU6656 in STAT3WT cells. However, SU6656 only minimally inhibited the already reduced level of c-fos induction observed in STAT3KO cells, and this inhibition was not statistically significant (Fig. 2C). These results suggest that PDGF stimulates c-fos induction through two parallel pathways, one that requires c-src and STAT3 and a second that is c-src and STAT3 independent, signaling most likely through the Ras/MAPK pathway (31-33). Importantly, the partial reduction of c-fos expression in the absence of STAT3 was not reflected by altered growth responses to PDGF (Fig. 2B) or to EGF or IGF-I (data not shown), suggesting that the still substantial STAT3-independent induction of c-fos is sufficient to promote full cell proliferation, making STAT3 dispensable for cell proliferation. In contrast to c-fos, no significant effect was observed on the induction of c-myc RNA following c-src inhibition in cells of either genotype (Fig. 2C, right), supporting our initial finding that c-myc induction by PDGF was independent of STAT3. Absence of STAT3 led to a more profound impairment of c-fos induction than did inhibition of c-src activity in STAT3WT cells, suggesting either that inhibition of c-src by SU6656 was incomplete (note the low levels of phosphorylated STAT3 remaining in SU6656-treated cells, Fig. 2A) or that STAT3 can induce c-fos independent of c-src. In either case, the contribution of c-src toward c-fos induction would appear to be predominately if not exclusively mediated by STAT3.
STAT3 is required for v-src transformation of fibroblasts
Previous studies identified STAT3 as a v-src target, and interference with STAT3 signaling by over-expression of dominant-interfering mutants of STAT3 in v-src transformed fibroblasts significantly reduced transformation (6, 7). To examine the requirement for STAT3 in cell transformation mediated by v-src, STAT3WT and STAT3KO 3T3 fibroblasts were infected with a v-src retrovirus, and transformation was measured by quantifying focus formation on dishes and colony formation in soft agar (Fig. 3A, B). v-src transformation of STAT3KO cells was significantly reduced in both assays, providing direct evidence that STAT3 contributes to v-src transformation. Moreover, the continued presence of STAT3 was required for cell transformation, as deleting STAT3 from cells already transformed by v-src led to a similar reduction in colony formation, demonstrating that STAT3 is required for both initiation and maintenance of the transformed phenotype (Fig. 3B, right). Interestingly, when STAT3 was depleted after v-src introduction, the reduction in colony numbers was less profound, suggesting STAT3 may be preferentially required during early stages of transformation in addition to its role in maintenance of transformation.
Fig. 3.
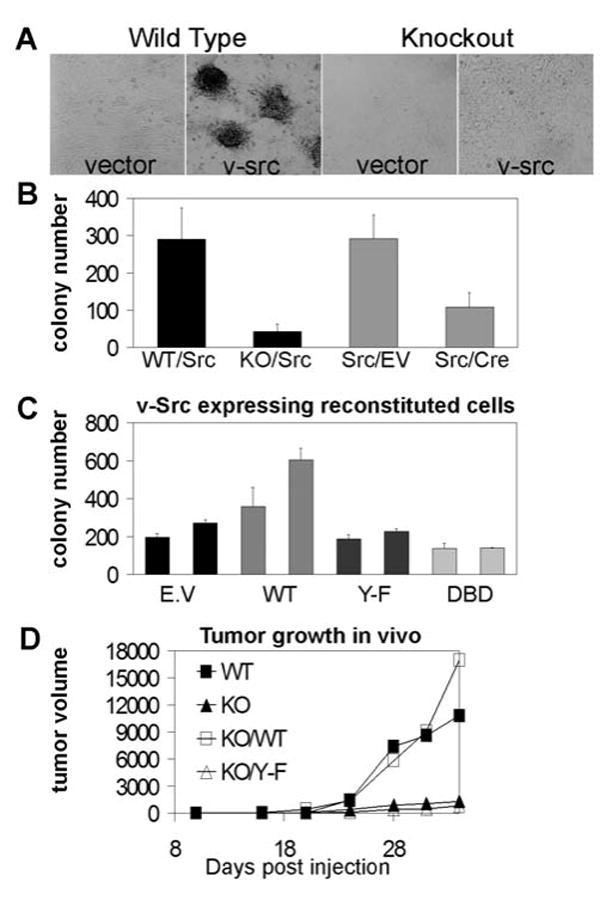
Transcriptionally competent STAT3 is required for v-src transformation in vitro and in vivo. (A) STAT3WT and STAT3KO 3T3 cells, expressing v-src or vector control, were grown until confluent and maintained in low-serum media (0.5%) until foci were visible. Images were taken at 32x magnification. (B) 1×105 cells were plated in 6 cm dishes in complete media with low-melting agarose (0.35%). After 14 d, colonies larger than 0.2 mm in diameter were counted. Experiment was performed in triplicate, and average colony numbers (+/− SEM) are presented. STAT3 was deleted prior to v-src (left) or after (right) v-src transformation. Stable v-src expression and complete STAT3 deletion were confirmed by immunoblotting and PCR, respectively.
(C) STAT3 KO cells expressing v-src were infected with a bicistronic retrovirus containing GFP and STAT3 or control. Cells were sorted by FACS for GFP levels and expanded into “high” and “low” pools. Soft agar colony growth was scored, using 5,000 cells due to higher transformation efficiency of these pure pools. Chart represents two pools isolated for each genotype based on increasing order of GFP (STAT3) expression. E.V, empty vector control (i.e., STAT3 KO cells); WT, Y-F, and DBD, STAT3-null cells reconstituted with wild type STAT3, STAT3Y705F and STAT3DBD, respectively.
(D) STAT3WT, STAT3KO or STAT3KO cells reconstituted with wild type or mutant STAT3 were injected subcutaneously into nude mice, and resulting tumor growth is presented.
To confirm that the defect in transformation was directly due to the absence of STAT3, v-src expressing STAT3KO cells were reconstituted with STAT3 cDNA. In addition to wild type, two transcriptionally-inactive mutants of STAT3 were tested, STAT3Y705F or DNA binding mutant STAT3DBD (Fig. 3C). Reconstitution with wild type STAT3 fully restored transformation by v-src, in a dose-dependent manner. However, reconstitution with either transcriptionally incompetent mutant failed to rescue transformation, reinforcing the notion that gene expression by STAT3 is required for v-src transformation.
The role of STAT3 in v-src transformation was further examined in vivo. Transformed STAT3WT, STAT3KO and STAT3KO cells reconstituted with wild type or mutant STAT3 were inoculated subcutaneously in nude mice (Fig. 3D). Clear impairment of tumor formation was observed for STAT3KO cells, confirming the in vitro results. Tumors from STAT3WT cells developed much more rapidly and reached greater masses compared to tumors derived from STAT3KO cells. Furthermore, cells reconstituted with wild type STAT3 formed large tumor masses equivalent to those formed by the original STAT3WT cells, while cells reconstituted with mutant STAT3 formed minimal tumors similar to those formed by STAT3KO cells.
STAT3 is required for reduced adhesion and for FAK activation by v-src
In normal cells, growth and survival are dependent on proliferative and anti-apoptotic signals delivered both from growth factor receptors and adhesion receptors, such as integrins. Oncogenes circumvent these requirements, disconnecting cell growth from external signals. The finding that STAT3 is required for v-src transformation but not for growth factor induced proliferation (Fig. 1-3) suggested a possible role for STAT3 in regulation of adhesion signals. Adhesion of STAT3WT and STAT3KO cells to the integrin substrates collagen and fibronectin was measured (Fig. 4). While non-transformed cells showed no differences in adhesion to the substrates tested, v-src expressing cells exhibited reduced adhesion to collagen I (approximately 60% of non-transformed cells) that was statistically significant. However, oncogene-induced reduction in adhesion was not observed in the absence of STAT3, indicating that loss of adhesion induced by v-src required STAT3. Similar results were observed following different times of adhesion (not shown). Adhesion to fibronectin was not significantly affected by the absence of STAT3, suggesting that STAT3 is required for signaling of specific integrin subsets and does not affect adhesion non-specifically.
Fig. 4.
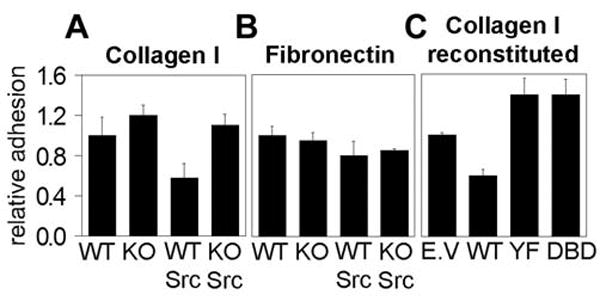
STAT3 is required for v-src induced loss of adhesion. Adhesion of non-transformed and v-src transformed STAT3WT and STAT3KO cells to collagen I (A) or fibronectin (B). (C) Adhesion to collagen I of v-src transformed STAT3KO cells reconstituted with wild type or mutant STAT3. At least 4 independent experiments were performed, each in triplicate.
To confirm the requirement of STAT3 for integrin-mediated adhesion, STAT3KO cells were reconstituted with wild type or mutant STAT3 and their adhesion to ECM substrates was tested. Adhesion of STAT3KO cells reconstituted with wild type STAT3 was again reduced by 60%, similar to the effect observed in STAT3WT cells (Fig. 4). In contrast, neither STAT3 mutant mediated the v-Src-dependent decrease in adhesion, suggesting that transcriptionally active STAT3 is required to modulate adhesion.
Impaired cellular adhesion is associated with a decrease in focal adhesion complex turnover, which is dependent on FAK activity. In addition to regulating focal adhesion complexes, FAK has been implicated in cell proliferation and survival (14, 15, 34). Therefore, it was possible that STAT3 regulated FAK, contributing to the reduced cell adhesion and increased survival observed in suspension culture. To characterize FAK activation in STAT3KO cells, v-src transformed cells were plated in suspension and FAK autophosphorylation on Tyr 397 was measured (Fig. 5A). STAT3WT cells maintained high levels of FAK phosphorylation when grown in suspension, although there was some degree of variability over time. In contrast, STAT3KO cells exhibited a marked reduction in FAK phosphorylation within 24 h. Loss of FAK phosphorylation correlates with the inability of STAT3KO cells to establish colonies in soft agar, suggesting that in addition to mediating loss of cell adhesion, STAT3 contributes to the enhanced FAK phosphorylation observed in v-src-transformed cells.
Fig. 5.
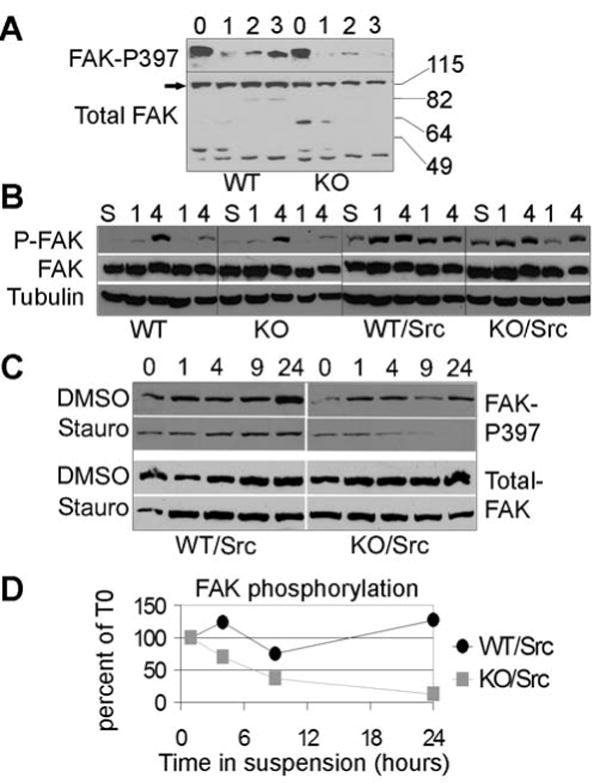
STAT3 is required for v-src-mediated FAK phosphorylation in suspension. (A) STAT3WT and STAT3KO cells in suspension for the indicated number of days were assayed for phospho-FAK (Y397) and total FAK by immunoblotting. (B) WT and KO cells, with and without v-src as indicated, were grown under adherent conditions in low serum (0.1%) for 24 h, detached in EDTA, washed, and kept in suspension for 1 h and subsequently replated on dishes pre-coated with collagen-I (second and third lanes of each set) or fibronectin (fourth and fifth lanes of each set), and collected after 1 or 4 h as indicated. Prior to plating a fraction was reserved to serve as starved/suspension control (S). Phospho-FAK, total FAK, and tubulin levels were detected by immunoblotting, as indicated. (C) Cells were pretreated with 0.5 μM staurosporin or solvent (DMSO) for 1 hr and either lysed immediately (adherent) or plated in suspension for the indicated number of hours, in media containing staurosporin or solvent. (D) Results from (C) were quantified using ImageQuant™ and presented as a ratio between phospho-FAK and total FAK at each time point. Average of two experiments is shown.
To test whether FAK was appropriately activated by integrins, FAK autophosphorylation was measured in starved cells replated on ECM (Fig. 5B). Non-transformed cells placed in suspension lost FAK phosphorylation, which was fully restored by 4 h of replating in both STAT3WT and STAT3KO cells (Fig. 5B, left), suggesting that FAK is activated in a STAT3 independent manner in the absence of v-src. In transformed cells, the kinetics and extent of FAK activation were different (Fig. 5B, right). First, FAK phosphorylation was not completely abolished in suspension, and second, FAK re-activation was maximal already by 1 h. Importantly, STAT3WT and STAT3KO cells behaved similarly, suggesting that activation of FAK by integrins occurs normally in both genotypes. Coimmunoprecipitation experiments of FAK with v-src showed no significant differences between STAT3WT and STAT3KO cells, supporting the notion that upstream activation of FAK by either integrin engagement or v-src is not affected by the absence of STAT3 (not shown).
FAK phosphorylation levels are the consequence of phosphorylation by upstream kinases and dephosphorylation by phosphatases. To measure the rate of FAK dephosphorylation, cells were grown in the presence of the kinase inhibitor staurosporine to block upstream kinase activity. The rate of decay of phosphorylated FAK was subsequently measured over a period of 24 h (Fig. 5C). When STAT3WT cells were plated in suspension with no inhibitor present FAK phosphorylation was maintained or increased over time. In contrast, STAT3KO cells were unable to maintain FAK phosphorylation, as shown previously (Fig. 5A). In the presence of staurosporine, FAK phosphorylation in STAT3WT cells was still maintained, albeit at somewhat lower levels. However, in STAT3KO cells FAK phosphorylation decayed rapidly, and was completely lost by 24 h, without loss of FAK protein levels. Quantitation of two independent experiments showed the half-life of phosphorylated FAK in STAT3KO cells to be approximately 6 h, in contrast to a half-life of greater than 24 h in wild type cells (Fig. 5D). The same experiment performed in the presence of the src inhibitor SU6656 showed a similar although less complete loss of FAK phosphorylation (data not shown), consistent with the Y397 phosphorylation site being an autophosphorylation target and only indirectly dependent on v-src. We conclude that STAT3 contributes to the maintenance of high levels of phosphorylated FAK downstream of v-src through inhibition of FAK dephosphorylation.
Discussion
Numerous reports have suggested a role for STAT3 in cell proliferation, survival, differentiation, and transformation. Much of this previous evidence was derived from studies involving expression of constitutively active or dominant-negative versions of STAT3 (2). In this report, we have directly examined the requirement for STAT3 in fibroblasts by using gene ablation. Given initial gene ablation studies showing a requirement for survival of early embryos (35), it was surprising to find that STAT3 was dispensable for normal growth of fibroblasts in vitro. Wild type and STAT3-null primary fibroblasts and immortalized 3T3 cells grew at equivalent rates, reached comparable saturation densities, and displayed similar serum dependence. Moreover, no gene product was found to be absolutely dependent on STAT3 for expression, including such previously reported STAT3 target genes as cyclin D, Bcl-X (data not shown), c-fos, or c-myc (Fig. 1).
In spite of no genes being absolutely dependent on STAT3 for expression, induction of c-fos in response to mitogens was substantially impaired. This impairment presumably reflects the recruitment of STAT3 to its cognate binding site (SIE) in the c-fos promoter (3, 36). Although this enhancer element is dispensable in transient transfection assays, it clearly plays a substantial role in the regulation of c-fos gene expression in its normal chromosomal context, as has been also noted in transgenic mice (37). Induction of c-fos by PDGF was partially dependent on c-src activity, as revealed by its impairment by the src inhibitor SU6656 (Fig. 2). A greater inhibition of c-fos expression was observed in the absence of STAT3 than in the absence of c-src activity. Either additional enzymes are involved in STAT3 activity or the SU6656 was unable to completely inhibit c-src activity. In either case, most if not all of the induction of c-fos by c-src was mediated by STAT3, since inhibition of c-src had an insignificant effect on c-fos expression in STAT3-null cells. c-fos gene expression was also impaired in src-transformed cells in the absence of STAT3, confirming the ability of src to phosphorylate STAT3. This partial loss of c-fos gene expression may have contributed to the impaired transformation of STAT3 null cells, since reconstitution of STAT3-null cells with constitutively expressed c-fos partially rescued the transformation defect (data not shown).
A different pattern was observed for c-myc expression. Although c-myc has been suggested to depend on STAT3 in response to PDGF (4), no significant differences in c-myc mRNA levels were detected in the absence of STAT3. Moreover, inhibition of c-src activity also did not impair c-myc expression, in spite of its ability to block PDGF-induced cell proliferation (Fig. 2). The lack of a requirement for STAT3 for c-myc expression was observed both in primary MEF (Fig. 2), as well as in established cells lines (data not shown). It has been postulated that transcription factor requirements for c-myc expression can be affected by the activity of p53 or SV40 T antigen (38). However, this explanation cannot account for lack of a STAT3 requirement observed in these cells, since the primary MEF were wild type for p53 and neither primary nor immortalized lines expressed SV40 proteins (data not shown).
These results suggest that STAT3 contributes to the full induction of c-fos in response to mitogens, largely through a c-src-dependent pathway. However, residual STAT3-independent induction of c-fos is sufficient to drive full cell proliferation in vitro. On the other hand, STAT3 is dispensable for the induction of c-myc expression, which is driven through a c-src-independent pathway. This model differs from previous conclusions drawn from data using NIH 3T3 cells (39), in which over-expression of c-myc rescued the cell cycle block imposed by inhibition of c-src. This disparity may lie in inherent differences between normal primary fibroblasts and immortalized NIH 3T3 cells that lack ARF, a c-myc target gene (40), or may result from the higher levels of myc achieved in those experiments by ectopic expression. In either case, normal primary fibroblasts are capable of maintaining sufficient immediate early gene expression for cell growth in the absence of STAT3.
A different picture emerged for v-src-induced transformation. Here, as previously suggested from experiments involving dominant-negative forms of STAT3 (5, 7), STAT3 played an essential role in the full transforming potential of v-src. STAT3-null cells expressing v-src were impaired in their ability to form foci in culture, to grow as colonies in soft agar, and to develop as tumors in nude mice. This requirement for STAT3 reflected its role as a transcription factor, since only wild type STAT3 and not transcriptionally compromised mutants was capable of restoring the transforming potential of v-src.
The step in cell transformation compromised by loss of STAT3 involved cell-substratum interactions. While growth of v-src-expressing cells in adherent culture was equivalent in the presence or absence of STAT3 (data not shown), the reduction in cell adhesion to integrin substrates typical of src-transformed cells was abrogated (Fig. 4) and a biochemical correlate of this defect was an inability to maintain phosphorylated FAK in the absence of integrin signaling (Fig. 5). Direct phosphorylation of FAK did not appear to depend on STAT3, either downstream of c-src activated by integrins during adhesion or following expression of v-src. However, the ability to maintain FAK phosphorylation was impaired, even in the presence of v-src, which appeared to result from more rapid dephosphorylation. Regulation of the rate of FAK dephosphorylation required STAT3 target genes, since only wild type and not transcriptionally compromised STAT3 mediated this process. Although several phosphatases have been proposed to dephosphorylate FAK, e.g., PTP1B and SHP2, (41, 42), we did not detect changes in protein levels of these phosphatases in STAT3KO cells (data not shown). Nevertheless, it is possible that STAT3 regulates the expression of another phosphatase. Alternatively, STAT3-dependent genes might affect phosphatase or regulatory protein recruitment to focal adhesion complexes.
The contrast between a requirement for STAT3 during cell transformation and its dispensable role during cell growth suggests that STAT3 possesses a transformation-specific function, consistent with its postulated role as an oncogene (9). No abnormalities were noted for adherent cell growth in the absence of STAT3, implying normal integrin function is independent of STAT3. However, transformation by dominant oncogenes, such as v-src, overrides the proliferation requirement for integrin engagement through a process at least partially dependent on FAK activation (43), and this process was STAT3 dependent. Therefore, while it might have been imagined that what v-src does during transformation is to constitutively activate normal growth signaling pathways, it appears in this case to supplant an integrin-dependent signal by a STAT3-dependent process. Thus, while STAT3 is not required for normal cell proliferation, either downstream of growth factor receptors or adhesion receptors, it is necessary for v-src to render cells adhesion-independent. This concept suggests that the activation of STAT3 that is characteristic of many human tumors may contribute to metastatic growth and tumor invasion. It will be important to identify the critical STAT3 target genes that mediate this process.
Supplementary Material
Supplementary Fig. 1. STAT3 is efficiently deleted in fibroblasts generating completely null cells. The top panel represents results of semi-quantitative PCR for STAT3 genotyping (29). The top band is a product of the null allele while the bottom is a product of the flox allele. The bottom panel shows an immunoblot for STAT3 protein. Shown are one STAT3WT (vector infected cell line) and two STAT3KO (Cre infected) cell lines.
Supplementary Fig. 2. STAT3 is required for c-fos but not c-myc induction. STAT3KO 3T3 cells were infected with wild type or mutant STAT3, together with v-src to phosphorylate STAT3. c-fos RNA levels were measured by real-time RT-PCR. Levels of c-myc did not change (not shown).
Acknowledgments
We thank P. Brooks for help with adhesion assays, J. Hirst for expert flow cytometry, M. Resh, I. Gellman, C. Horvath, J. Jonkers, A. Loonstra, and G. Nolan for the kind gifts of vectors and reagents, Yaming Wang and Isabelle Marié for help with figures, and members of the lab for helpful discussions. This work was supported by grants from the National Institutes of Health.
References
- 1.O'Shea JJ, Gadina M, Schreiber RD. Cell. 2002;109:S121–S131. doi: 10.1016/s0092-8674(02)00701-8. [DOI] [PubMed] [Google Scholar]
- 2.Levy DE, Lee CK. J Clin Invest. 2002;109:1143–1148. doi: 10.1172/JCI15650. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Hayes TE, Kitchen AM, Cochran BH. Proc Natl Acad Sci USA. 1987;84:1272–1276. doi: 10.1073/pnas.84.5.1272. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Bowman T, Broome MA, Sinibaldi D, Wharton W, Pledger WJ, Sedivy JM, Irby R, Yeatman T, Courtneidge SA, Jove R. Proc Natl Acad Sci USA. 2001;98:7319–7324. doi: 10.1073/pnas.131568898. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Yu CL, Meyer DJ, Campbell GS, Larner AC, Carter-Su C, Schwartz J, Jove R. Science. 1995;269:81–83. doi: 10.1126/science.7541555. [DOI] [PubMed] [Google Scholar]
- 6.Turkson J, Bowman T, Garcia R, Caldenhoven E, De Groot RP, Jove R. Mol Cell Biol. 1998;18:2545–2552. doi: 10.1128/mcb.18.5.2545. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Bromberg JF, Horvath CM, Besser D, Lathem WW, Darnell JE., Jr Mol Cell Biol. 1998;18:2553–2558. doi: 10.1128/mcb.18.5.2553. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Dechow TN, Pedranzini L, Leitch A, Leslie K, Gerald WL, Linkov I, Bromberg JF. Proc Natl Acad Sci USA. 2004;101:10602–10607. doi: 10.1073/pnas.0404100101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Bromberg JF, Wrzeszczynska MH, Devgan G, Zhao Y, Pestell RG, Albanese C, Darnell JE., Jr Cell. 1999;98:295–303. doi: 10.1016/s0092-8674(00)81959-5. [DOI] [PubMed] [Google Scholar]
- 10.Bromberg J. J Clin Invest. 2002;109:1139–1142. doi: 10.1172/JCI15617. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Mora LB, Buettner R, Seigne J, Diaz J, Ahmad N, Garcia R, Bowman T, Falcone R, Fairclough R, Cantor A, Muro-Cacho C, Livingston S, Karras J, Pow-Sang J, Jove R. Cancer Res. 2002;62:6659–6666. [PubMed] [Google Scholar]
- 12.Niu G, Bowman T, Huang M, Shivers S, Reintgen D, Daud A, Chang A, Kraker A, Jove R, Yu H. Oncogene. 2002;21:7001–7010. doi: 10.1038/sj.onc.1205859. [DOI] [PubMed] [Google Scholar]
- 13.Martin GS. Nat Rev Mol Cell Biol. 2001;2:467–475. doi: 10.1038/35073094. [DOI] [PubMed] [Google Scholar]
- 14.Jones RJ, Brunton VG, Frame MC. Eur J Cancer. 2000;36:1595–1606. doi: 10.1016/s0959-8049(00)00153-2. [DOI] [PubMed] [Google Scholar]
- 15.Schaller MD. Biochim Biophys Acta. 2001;1540:1–21. doi: 10.1016/s0167-4889(01)00123-9. [DOI] [PubMed] [Google Scholar]
- 16.Ilic D, Furuta Y, Kanazawa S, Takeda N, Sobue K, Nakatsuji N, Nomura S, Fujimoto J, Okada M, Yamamoto T. Nature. 1995;377:539–544. doi: 10.1038/377539a0. [DOI] [PubMed] [Google Scholar]
- 17.Carragher NO, Westhoff MA, Riley D, Potter DA, Dutt P, Elce JS, Greer PA, Frame MC. Mol Cell Biol. 2002;22:257–269. doi: 10.1128/MCB.22.1.257-269.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Guo W, Giancotti FG. Nat Rev Mol Cell Biol. 2004;5:816–826. doi: 10.1038/nrm1490. [DOI] [PubMed] [Google Scholar]
- 19.Johnson PJ, C PM, Danko AV, Shalloway D. Mol Cell Biol. 1985;5:1073–1083. doi: 10.1128/mcb.5.5.1073. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Grignani F, K T, Mencarelli A, Valtieri M, Riganelli D, Grignani F, Lanfrancone L, Peschle C, Nolan GP, Pelicci PG. Cancer Res. 1998;58:14–19. [PubMed] [Google Scholar]
- 21.Horvath CM, Wen Z, Darnell JE. Genes Dev. 1995;9:984–994. doi: 10.1101/gad.9.8.984. [DOI] [PubMed] [Google Scholar]
- 22.Hogan B, Beddington R, Costantini F, Lacy E. Manipulating the Mouse Embryo. A Laboratory Manual. Cold Spring Harbor Laboratory Press; Cold Spring Harbor: 1994. [Google Scholar]
- 23.Todaro GJ, Green H. J Cell Biol. 1963;17:299–313. doi: 10.1083/jcb.17.2.299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Paulson M, Pisharody S, Pan L, Guadagno S, Mui AL, Levy DE. J Biol Chem. 1999;274:25343–25349. doi: 10.1074/jbc.274.36.25343. [DOI] [PubMed] [Google Scholar]
- 25.Pear W, Nolan G, Scott M, Baltimore D. Proc Natl Acad Sci USA. 1993;90:8392–8396. doi: 10.1073/pnas.90.18.8392. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Chang HM, Paulson M, Holko M, Rice CM, Williams BR, Marie I, Levy DE. Proc Natl Acad Sci USA. 2004;101:9578–9583. doi: 10.1073/pnas.0400567101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Jankun J, Matsumura F, Kaneko H, Trosko JE, Pellicer A, Greenberg AH. Mol Toxicol. 1989;2:177–186. [PubMed] [Google Scholar]
- 28.Raz R, Lee CK, Cannizzaro LA, d'Eustachio P, Levy DE. Proc Natl Acad Sci USA. 1999;96:2846–2851. doi: 10.1073/pnas.96.6.2846. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Lee Ck, Raz R, Gimeno R, Gertner R, Wistinghausen B, Takeshita K, DePinho RA, Levy DE. Immunity. 2002;17:63–72. doi: 10.1016/s1074-7613(02)00336-9. [DOI] [PubMed] [Google Scholar]
- 30.Zou YR, Muller W, Gu H, Rajewsky K. Curr Biol. 1994;4:1099–1103. doi: 10.1016/s0960-9822(00)00248-7. [DOI] [PubMed] [Google Scholar]
- 31.Blake RA, Broome MA, Liu X, Wu J, Gishizky M, Sun L, Courtneidge SA. Mol Cell Biol. 2000;20:9018–9027. doi: 10.1128/mcb.20.23.9018-9027.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Heldin CH, Ostman A, Ronnstrand L. Biochim Biophys Acta. 1998;1378:F79–113. doi: 10.1016/s0304-419x(98)00015-8. [DOI] [PubMed] [Google Scholar]
- 33.Karpova AY, Abe MK, Li J, Liu PT, Rhee JM, Kuo WL, Hershenson MB. Am J Physiol. 1997;272:L558–565. doi: 10.1152/ajplung.1997.272.3.L558. [DOI] [PubMed] [Google Scholar]
- 34.Westhoff MA, Serrels B, Fincham VJ, Frame MC, Carragher NO. Mol Cell Biol. 2004;24:8113–8133. doi: 10.1128/MCB.24.18.8113-8133.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Takeda K, Noguchi K, Shi W, Tanaka T, Matsumoto M, Yoshida N, Kishimoto T, Akira S. Proc Natl Acad Sci USA. 1997;94:3801–3804. doi: 10.1073/pnas.94.8.3801. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Yang E, Lerner L, Besser D, Darnell JE., Jr J Biol Chem. 2003;278:15794–15799. doi: 10.1074/jbc.M213073200. [DOI] [PubMed] [Google Scholar]
- 37.Robertson LM, Kerppola TK, Vendrell M, Luk D, Smeyne RJ, Bocchiaro C, Morgan JI, Curran T. Neuron. 1995;14:241–252. doi: 10.1016/0896-6273(95)90282-1. [DOI] [PubMed] [Google Scholar]
- 38.Broome MA, Courtneidge SA. Oncogene. 2000;19:2867–2869. doi: 10.1038/sj.onc.1203608. [DOI] [PubMed] [Google Scholar]
- 39.Barone MV, Courtneidge SA. Nature. 1995;378:509–512. doi: 10.1038/378509a0. [DOI] [PubMed] [Google Scholar]
- 40.Zindy F, Eischen CM, Randle DH, Kamijo T, Cleveland JL, Sherr CJ, Roussel MF. Genes Dev. 1998;12:2424–2433. doi: 10.1101/gad.12.15.2424. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Liu F, Sells MA, Chernoff J. Curr Biol. 1998;8:173–176. doi: 10.1016/s0960-9822(98)70066-1. [DOI] [PubMed] [Google Scholar]
- 42.Yu DH, Qu CK, Henegariu O, Lu X, Feng GS. J Biol Chem. 1998;273:21125–21131. doi: 10.1074/jbc.273.33.21125. [DOI] [PubMed] [Google Scholar]
- 43.Guan JL, Shalloway D. Nature. 1992;358:690–692. doi: 10.1038/358690a0. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary Fig. 1. STAT3 is efficiently deleted in fibroblasts generating completely null cells. The top panel represents results of semi-quantitative PCR for STAT3 genotyping (29). The top band is a product of the null allele while the bottom is a product of the flox allele. The bottom panel shows an immunoblot for STAT3 protein. Shown are one STAT3WT (vector infected cell line) and two STAT3KO (Cre infected) cell lines.
Supplementary Fig. 2. STAT3 is required for c-fos but not c-myc induction. STAT3KO 3T3 cells were infected with wild type or mutant STAT3, together with v-src to phosphorylate STAT3. c-fos RNA levels were measured by real-time RT-PCR. Levels of c-myc did not change (not shown).