

Summary
O-glycan biosynthesis is initiated by the transfer of N-acetylgalactosamine (GalNAc) from a nucleotide-sugar donor (UDP-GalNAc) to Ser/Thr residues of an acceptor substrate. The detailed transfer mechanism, catalyzed by the UDP-GalNAc polypeptide:N-acetyl-α-galactosaminyltransferases (ppGalNAcTs), remains unclear despite structural information available for several isoforms in complex with substrates, at various stages along the catalytic pathway. We used all-atom molecular dynamics (MD) simulations with explicit solvent and counterions to study the conformational dynamics of ppGalNAcT-2 in several enzymatic states along the catalytic pathway. ppGalNAcT-2 is simulated both in the presence and absence of substrates and reaction products to examine the role of conformational changes in ligand binding. In multiple 40 ns–long simulations of more than 600 ns total run time, we studied systems ranging from 45,000 to 95,000 atoms. Our simulations accurately identified dynamically active regions of the protein, as previously revealed by the x-ray structures, and permitted a detailed, atomistic description of the conformational changes of loops near the active site and the characterization of the ensemble of structures adopted by the transferase complex on the transition pathway between the ligand-bound and ligand-free states. In particular, the conformational transition of a functional loop adjacent to the active site from closed (active) to open (inactive) is correlated with the rotameric state of the conserved residue W331. Analysis of water dynamics in the active site revealed that internal water molecules have an important role in enhancing the enzyme flexibility. We also found evidence that charged side chains in the active site rearrange during site opening to facilitate ligand binding. Our results are consistent with the single-displacement transfer mechanism previously proposed for ppGalNAcTs based on x-ray structures and mutagenesis data and provide new evidence for possible functional roles of certain amino acids conserved across several isoforms.
Keywords: glycosyltransferase, functional conformational changes, substrate recognition, active site hydration, molecular dynamics
Introduction
Glycosyltransferases (GTs; EC 2.4.x.y) are a large family of enzymes involved in the biosynthesis of oligosaccharides, polysaccharides, and glycoconjugates.1 GTs catalyze the transfer of the glycosyl moiety from a sugar donor to an acceptor. In most cases, the donor is a nucleoside phosphosugar and the acceptor is a hydroxyl group of another sugar, an amino acid, or a lipid. GTs are classified as either retaining or inverting based upon the stereochemistry of the anomeric carbon atom of the transferred sugar relative to its configuration in the donor substrate.
UDP-N-acetylgalactosamine:polypeptide N-acetylgalactosaminyltransferases (ppGalNAcTs, EC 2.4.1.41, family 27 in CAZy2) are retaining GTs. They initiate synthesis of mucin-type glycoproteins by transferring the GalNAc moiety from the sugar donor UDP-GalNAc to Ser or Thr residues, to form the Tn antigen3 (GalNAcα1-O-Ser/Thr). This family is large, with up to 20 members in humans, and evolutionarily conserved, but the polypeptide substrate preferences of individual isoforms have not been elucidated, despite structural information determined for several isoforms.
The recently solved crystal structures of three isoforms of the ppGalNAcT family capture different stages of the reaction pathway: murine T14 (PDB code 1XHB – apo form), human T25 (PDB code 2FFV – binary complex with UDP, and PDB code 2FFU – ternary complex with UDP and the acceptor peptide EA2); and human T106 (PDB code 2D7I – complex with hydrolyzed UDP-GalNAc and PDB code 2D7R – complex with GalNAcα1-O-Ser). These structures indicate the presence of two domains connected by a flexible linker that permits significant changes in their relative orientations. The catalytic domain belongs to the GT-A fold, whereas the carbohydrate-binding lectin domain (LD) adopts a β-trefoil fold, as classified in the Conserved Domain Database.7 The enzymatic activity of the catalytic domain depends on the presence of a Mn2+ ion coordinated by His and Asp residues, which form the so-called DXH motif of the active site, and by an additional His residue, all conserved throughout the ppGalNAcT family.3
The enzymatic mechanism of retaining GTs is still under debate.3,8,9 However, in the case of ppGalNAcTs a general outline of the process can be proposed, emerging from the available crystal structures as a series of transfer reaction stages listed below.
Stage I
The enzyme in the apo- form, before any ligand is bound, is described by the mT1 structure. Here, the conformation of the loop bridging opposite edges of the active site (shown as loop B in Figure 1, residues R347-T358 in mT1 and R361-S377 in hT2), is not resolved due to crystallographic disorder.
Figure 1.
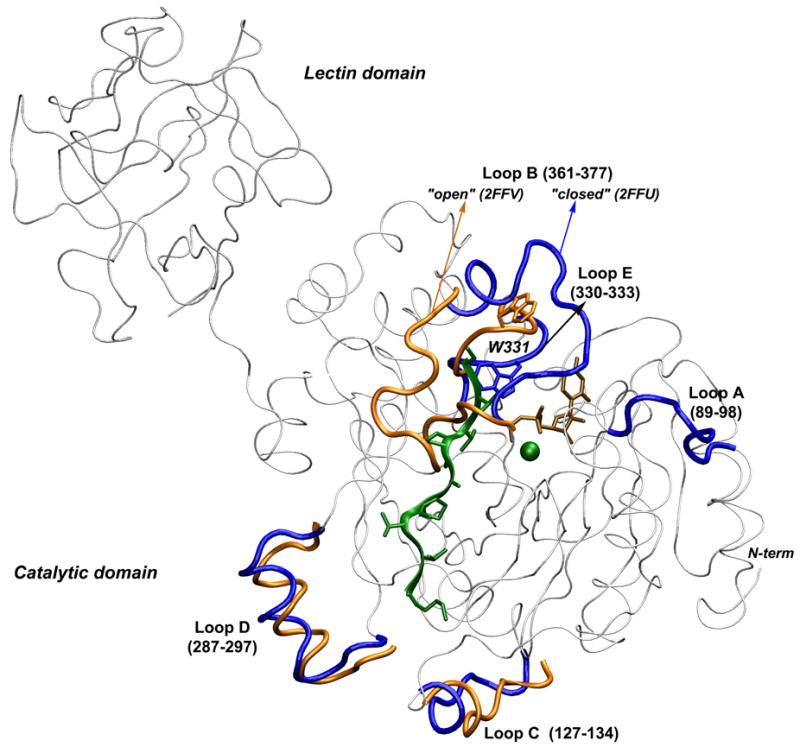
Superposition of x-ray structures of ppGalNAcT2 in the presence (blue) and absence (orange) of the acceptor peptide. Conformational differences of the loops are highlighted. The W331 side chain is represented with sticks. Loop A, which is depicted as blue for the 2FFU structure, is not resolved experimentally in the 2FFV structure. The backbone regions with high conformational similarity are shown in gray. The lectin domain is shown only for the 2FFU structure. UDP is colored in brown, with the peptide and the Mn2+ ion shown in green.
Stage II
The enzyme binds the donor substrate UDP-GalNAc, before peptide binding, according to a proposed ordered-sequential mechanism for retaining GTs.10–12 This is illustrated by the hT10 structure (PDB code 2D7I) containing hydrolyzed UDP-GalNAc. Here, loop R361-S377 (loop B in Figure 1) is stabilized in a “closed” conformation at the edge of the active site, forming a lid over the UDP moiety and leaving the active site groove exposed and able to bind the acceptor peptide.
Stage III
The enzyme, bound with donor and acceptor peptide, is illustrated by the ternary complex of hT2 (PDB code 2FFU). Loop B (Figure 1) maintains its closed conformation, but does not make significant contact with the peptide, as shown by the x-ray structure. Peptide binding is dominated by hydrogen bonds towards the N-terminus and by hydrophobic interactions towards the C-terminus, but the residues mediating these interactions are less conserved, accounting for differences in isoform affinity for different peptides.5 In contrast, UDP-GalNAc interacts with the protein residues mainly via hydrogen bonds, and residues forming its binding pocket are highly conserved among isoforms, accounting for their common preference for UDP-GalNAc as a sugar donor.5
Stage IV
The GalNAc moiety is transferred from the sugar-nucleotide donor to the Ser/Thr residue on the acceptor peptide. A structure containing UDP and a glycosylated peptide in the active site is not available and the residues involved in catalysis have not been clearly identified.
Stage V
The glycosylated peptide is detached, and the reaction product UDP leaves the active site. This step may be illustrated by the binary complex of hT2 and UDP (PDB code 2FFV), since the orientation of the ribose and uridine moieties of UDP are flipped by ~180° relative to the ternary complex. Loop B (R361-S377) is folded back over the active site, exposing UDP to the bulk water molecules. Comparative structural analysis also indicates that in addition to loop B, the N-terminal segment of the catalytic domain, up to residue D97 in hT2, is highly flexible and undergoes significant conformational changes. However, the relevance of these changes for catalysis is unclear. This stretch is not resolved in mT1, and it forms an α-helix in hT10 whose position relative to the catalytic domain is dramatically altered compared to hT2. Consequently, the conformation of loop A (G89-P98, Figure 1) is changed. Although loop A is not in direct contact with the substrates, it interacts with loop B via hydrogen bonds between residues in the hinge regions (N102-R362 and K103-Q364) and possibly influences its conformation.
The transfer reaction steps described above suggest that by undergoing significant conformational changes, loop A and especially loop B may be involved in positioning the substrates into the active site. Similar mechanisms with concerted conformational changes of loops near the active site upon ligand binding have also been reported for members of other GT families such as: β-(1, 4)-Gal-T113,14 in family 7 of inverting GT, α-(1,3)-galactosyltransferase15,16 and human blood group A and B glycosyltransferases17,18 in family 6 of retaining GT, and α-(1, 4)-galactosyltransferase LgtC19 in family 8 of retaining GT. Loop mobility around the active site may thus be a general feature of the GT-A superfamily but further testing of this hypothesis for other members of the superfamily is needed.
The catalytic mechanism of retaining GTs has not been clearly elucidated yet3,8,9 but several scenarios have been proposed. The first is a double-displacement mechanism, by analogy to the well characterized glycosidases. In the double-displacement mechanism, catalysis requires two nucleophilic active site carboxyl groups and proceeds in two steps that involve the formation and subsequent breakdown of a covalent glycosyl–enzyme intermediate: 1) glycosylation: attack of the anomeric center of the donor sugar by one nucleophile, to form a glycosyl–enzyme species, followed by 2) deglycosylation (hydrolysis): deprotonation of the reactive hydroxyl of the acceptor, performed by the other carboxyl, which serves a dual role as acid-base catalyst.
This mechanism was initially proposed for family 6 of retaining GTs based on a low resolution x-ray structure of α-(1,3)-galactosyltransferase20 showing a carboxylic amino acid residue in a suitable position to act as an enzyme nucleophile. However, it was not confirmed by a subsequent high-resolution structure15 and mutagenesis experiments targeting the proposed nucleophile.21 Also, the x-ray structure of family 8 α-(1, 4)-galactosyltransferase LgtC11 showed no carboxylic amino acids (Asp or Glu) close to the anomeric carbon of the donor substrate and raised further doubts about such a mechanism. A Gln residue was thought to be the enzyme nucleophile and an attempt to trap an enzyme-sugar intermediate did result in such a complex, but surprisingly with the sugar substrate covalently attached to an adjacent Asp residue positioned far, at ~8 Å distance from the reaction center, as shown by the x-ray structure.22
In the case of ppGalNAcTs, a covalent enzyme-sugar intermediate was not present in the x-ray structure of isoform T10 where the sugar-phosphate linkage is hydrolyzed. Moreover, the nearest acidic residues that might function as nucleophiles are D224 of the DxH motif binding Mn2+, and E334 of the WGGEN motif. However, the distances from these residues to the β phosphate oxygen are ~7Å in the x-ray structures. As a consequence, a double displacement mechanism would require a large conformational change to bring these residues in proximity of the β-phosphate oxygen.
Failure to provide clear evidence for a double-displacement mechanism has led to the proposal of an alternative mechanism in which the reaction proceeds via a front-side single displacement, also known as SNi mechanism.8,11 In this “concerted one-step” mechanism the nucleophilic hydroxyl group of the acceptor attacks the anomeric carbon at the same side from which the UDP leaving group departs. This mechanistic view has also been extended to other retaining GTs: GT Family 20 trehalose-6-phosphate synthase OtsA,23 GT Family 4 trehalose phosphorylase24 and GT Family 35 glycogen phosphorylase.25
In this study, we probe the mechanistic aspects related to conformational changes of ppGalNAcT-2 during ligand binding and processing by using all-atom molecular dynamics simulations (MD) in conjunction with the recently determined experimental structures.4–6 Previous MD studies have been used to analyze the flexibility of the above mentioned families 7 and 8 of GTs.13,14,19 Here, we perform the first MD simulations for a member of ppGalNAcTs family (human T2), both in the presence and absence of donor and acceptor substrates. Our MD simulations permit a detailed characterization of the ensemble of structures adopted by the transferase complex on the transition pathway between several experimentally determined ligand-bound and ligand-free states. These simulations reveal large and possibly functional conformational changes of the protein loops near the active site during ligand binding. With regard to possible catalytic mechanisms, we find that throughout our simulations, distances between potential nucleophilic residues and the glycosidic oxygen of the sugar-nucleotide ligand remain large (~7 Å), suggesting that the double-displacement mechanism is less likely than other alternative mechanisms such as SNi.
We investigate also the influence of the UDP-GalNAc and peptide substrates on loop dynamics and the effect of these conformational changes on the mechanism of ligand binding/detachment. Based on our analysis, we discuss a possible binding mechanism that suggests a central role of the evolutionarily conserved residue W331. That residue appears to act as a gatekeeper, controlling the conformational transition of the functional loop B adjacent to the active site, from closed (unable to accept ligand) to open (able to accept ligand). We also analyze the dynamics of water molecules in the active site region, and discuss the possible role of water in the catalytic process. We explore the conformational preferences of the bound acceptor peptide and the stability of its specific interactions with the catalytic domain. This information should prove useful in future sequence-scanning and flexible docking experiments to identify preferences of individual isoforms for peptide substrates and for the design of alternative peptide substrates.
Results and Discussion
Figure 1 depicts the x-ray structures of ppGalNAcT-2 with PDB codes 2FFU and 2FFV. A comparison of these two x-ray structures indicates that the conformational changes of the catalytic domain associated to the ligand binding and processing are concentrated in two long loops: loop A (residues 89–98) located relatively far from the active site (modeled in 2FFV as in 2FFU); and loop B (residues 361–377) located at one end of the active site. Loop B undergoes a large movement, shifting by as much as 24 Å from one side of the peptide binding groove to the other. In addition, smaller conformational changes of the protein backbone occur in three other short loops: loop C (residues 127–134), an exposed loop placed on the side opposite of the active site; loop D (residues 287–297) forming a short α-helix, located more than 9 Å away from the peptide binding groove, that moves by approximately 5 Å in the absence of the peptide; and loop E (residues 330–333), showing a 4 Å shift. These loops are shown in Figure 1, colored blue in 2FFU and orange in 2FFV. In analyzing our simulations, we are particularly interested in the flexibility of the loops A, B and E and their possible role in ligand binding.
To assess the dynamical properties of ppGalNacT-2, we performed extensive MD simulations of a series of complexes between the enzyme and its different ligands based upon the x-ray structures described above. With solvent molecules treated explicitly, including Na+ and Cl− counter ions, the simulation systems contained up to 95,000 atoms. Simulations of up to 40 ns per trajectory resulted in a total simulation time of more than 600 ns (Table 1). To study the relative dynamics of the two domains our initial simulations included both the catalytic and lectin domains using the 2FFU crystal structure from which UDP and EA2 had been removed. We also simulated the same system in the presence of the substrates, but with the lectin domain positioned near the catalytic domain, as guided by the mT1 structure. This simulation allowed us to test whether the binding of substrates triggered movement of the lectin domain away from the catalytic domain. A comparison of the results of these simulations indicated that the average flexibility pattern of each domain, as measured by RMSD per residue, is independent of the presence of the other domain or the distance between the domains. Thus, to reduce computational requirements, subsequent simulations focused on the catalytic domain to test the influence of substrates on the dynamics of the enzyme at different steps in the catalytic process. In this the second series of MD trajectories, we simulated the catalytic domain as in the ternary complex in the absence of substrates, in the presence of UDP-GalNAc, in the presence of UDP-GalNAc and EA2 (substrate complex), in the presence of UDP and glycosylated EA2 (product complex), and in the presence of only UDP or UDP and EA2. To evaluate the dynamics of the enzyme after the reaction (structure 2FFV), we also simulated the catalytic domain as in the binary complex, either alone or in the presence of the reaction product UDP.
Table 1.
Summary of MD simulations performed on ppGalNAcT2 and its substrates. The letter “Y” indicates that the corresponding component is included in the simulated system.
| Structure | C | L | UG | UDP | P (EA2) | PG (EA2-GalNAc) | x | Total water | Na+/Cl− | Total #atoms | System Name |
|---|---|---|---|---|---|---|---|---|---|---|---|
| 2FFU | Y | Y | --- | --- | --- | --- | Y | 29093 | 24/31 | 95179 | CLx |
| Y | Y-mT1 | --- | Y | Y | --- | Y | 18794 | 15/20 | 64436 | CL-close | |
| Y | --- | --- | --- | --- | --- | Y | 14707 | 10/17 | 50141 | Cx | |
| YW | --- | --- | --- | --- | --- | Y | 14707 | 10/17 | 50141 | CWx | |
| Y | --- | --- | --- | --- | --- | --- | 13359 | 9/16 | 46095 | C | |
| Y | --- | Y | --- | --- | --- | Y | 13962 | 10/16 | 47902 | C(UG)x | |
| Y | --- | Y | --- | Y | --- | Y | 13943 | 9/16 | 47983 | C(UG)Px | |
| Y | --- | --- | Y | --- | Y | Y | 13064 | 9/16 | 45345 | CU(PG)x | |
| Y | --- | --- | Y | --- | --- | Y | 14705 | 12/16 | 50171 | CUx | |
| Y | --- | --- | Y | --- | --- | --- | 13354 | 11/15 | 46116 | CU | |
| Y | --- | --- | Y | Y | --- | Y | 14678 | 11/16 | 50228 | CUPx | |
| Y | --- | --- | Y | Y | --- | --- | 13315 | 10/15 | 46137 | CUP | |
| --- | Y | --- | --- | --- | --- | Y | 5481 | 6/5 | 18333 | Lx | |
| 2FFV | Y | --- | --- | --- | N/A | N/A | N/A | 13152 | 9/15 | 45514 | Copen |
| Y | --- | --- | Y | N/A | N/A | N/A | 13143 | 10/14 | 45523 | CopenU | |
| Peptide | --- | --- | --- | --- | Y | --- | N/A | 2366 | 2/3 | 7233 | P |
| --- | --- | --- | --- | --- | Y | N/A | 4199 | 4/5 | 12772 | (PG) |
Abbreviations: Catalytic domain (C); lectin domain (L); UDP-GalNAc (UG); number of water molecules from the x-ray structure (x); The names used for each system (last column) are simple combinations of these abbreviations, the brackets indicating covalently connected moieties. Subscripts W indicate systems, in which the W331 side-chain was restrained to a conformation as in the 2FFU x-ray structure.
We compared the simulations of the catalytic domain starting from the ternary complex 2FFU in the presence or absence of UDP-GalNAc donor and peptide acceptor and starting from the binary complex 2FFV in the presence and absence of the reaction product UDP.
Protein flexibility in the presence and absence of the substrates
As a measure of the average flexibility of various regions in the protein, we first calculated for each residue the root-mean-square distance (RMSD) of the alpha carbon (Cα) from the average position and compared it to the corresponding RMSD between the two crystal structures, 2FFU and 2FFV. The results of the simulations done in the presence of UDP-GalNAc and EA2 (C(UG)Px), in the presence of UDP-GalNAc only (C(UG)x) and in the absence of substrates (Cx) are represented in Figure 2. The theoretical and experimental RMSDs agree well and the MD simulations correctly identify the flexible regions in the protein. In particular, the highest peaks in calculated RMSD as a function of residue number correspond to the flexible loops described above.
Figure 2.
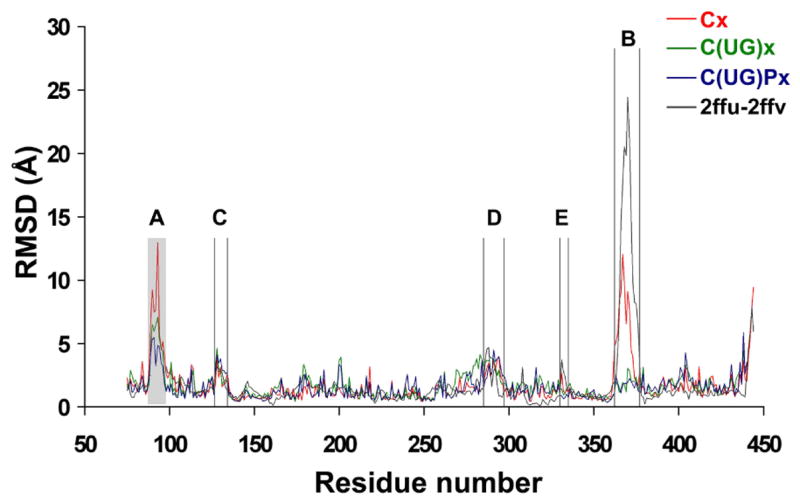
RMSD per residue calculated for the catalytic domain from simulations starting from the x-ray structure 2FFU in the absence of both substrates (red, trajectory Cx, see Table 1 for notation rule), in the presence of UDP-GalNAc (green, C(UG)x), and in the presence of both UDP-GalNAc and peptide (blue, C(UG)Px), with the GalNAc moiety modeled as in the T10 x-ray structure. The RMSD per residue values between the two x-ray structures of hT2 (2FFU and 2FFV, black line) are also shown. The loop unresolved in 2FFV due to crystallographic disorder is indicated as a shaded area.
The highest RMSD (13 Å) is seen in loop A in the absence of ligands. However, when both UDP-GalNAc and the peptide are present, the RMSD of loop A relative to the average position is reduced significantly from 13 Å to 4 Å (Figure 2, peak A). Even though loop A (missing in the 2FFV “open” x-ray structure) was consistently modeled as in the “closed” 2FFU x-ray structure, its dynamics depends not only on the presence of ligands but also the state of loop B. In simulations starting from the structure with loop B in the closed conformation (Figure 3a), loop A appears more stable, because loops A and B interact through a hydrogen bond between the main chain oxygen atom of N102 and the R362 side chain. In the absence of UDP-GalNAc, loop B opens and this interaction is lost. Consequently, loop A becomes more flexible. In simulations starting from the structure with loop B in an open conformation (Figure 3b), this interaction is absent from the beginning and the conformational space sampled by loop A is much larger.
Figure 3.
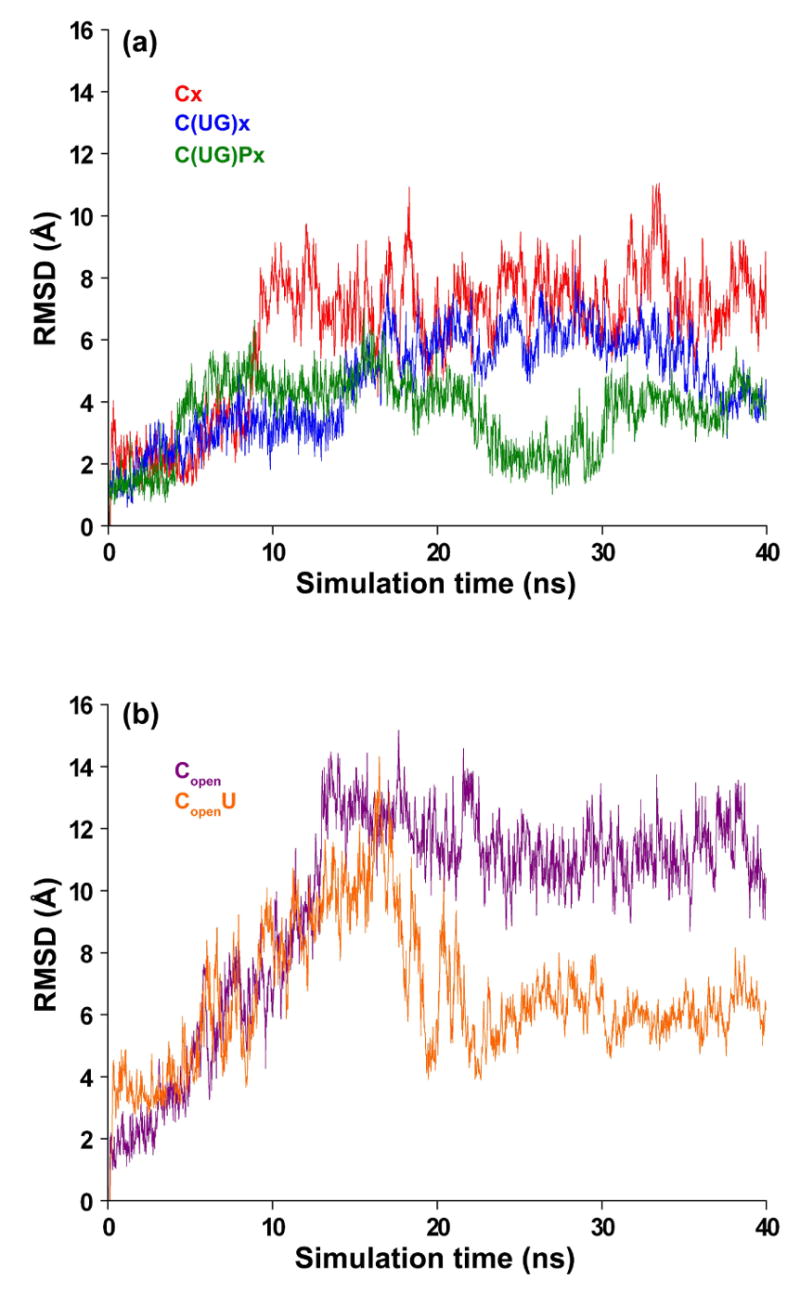
RMSD of loop A as a function of simulation time (ns) calculated using the conformation in the x-ray structure (2FFU) as reference. Results are shown for simulations starting from closed (a) and open (b) conformations of the long loop, both in the presence and absence of substrates (see Table 1 for notations).
The flexibility of loop B is slightly lower than that of loop A in the absence of substrates but is significantly reduced, from 12 Å to less than 4 Å RMSD, upon UDP-GalNAc binding. However, the subsequent binding of the peptide does not provide additional stabilization. Since this loop interacts directly with the donor and acceptor and its dynamics greatly affects the binding of substrates, its motions will be discussed in more detail in the following section.
Access to the active site and mechanism of donor binding: role of W331
In the simulation starting from the closed conformation, but with both substrates removed (Cx), loop B undergoes a conformational transition towards the open state (Figure 4a). This is a slow process that starts almost 1.5 ns after the simulation begins and lasts for about 25 ns. To identify the factors that trigger this large conformational change, we analyzed the residues in the vicinity of loop B. Interacting with its end, we identified W331 (part of the longer, conserved WGGEN motif within loop E) whose side-chain flips by approximately Δχ1~180° around the Cα-Cβ bond after 1.5 ns simulation time. This rearrangement (Figure 4c, d, e) creates an unfavorable steric clash with P366 and Y367, which are both well conserved, and a cluster of bulky residues (H365, F369, F377). The W331 χ1 rotation also appears to trigger the slow movement of loop B towards the opposite side of the peptide binding groove. W331 maintains the flipped conformation until loop B is stabilized in an intermediate conformation at about 8 Å RMSD relative to the completely open state. At this point W331 is no longer interacting with loop B. Thus, its side-chain is free to switch back to the initial conformation (Figure 4e).
Figure 4.
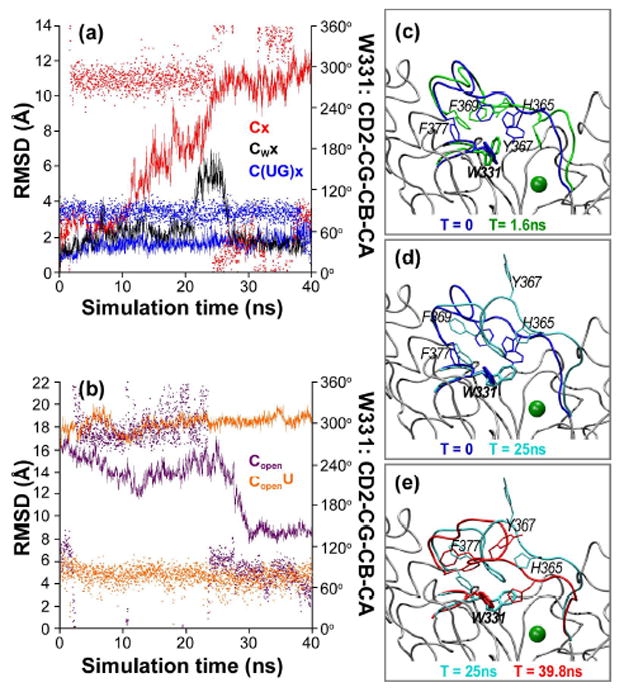
Conformational changes of loop B are correlated with the rotameric state of the W331 side-chain. (a, b) RMSD of loop B (line, left y-axis) relative to the 2FFU conformation, and the side-chain orientation of W331 (χ1 angle, dots, right y-axis), shown as a function of simulation time. (a) Simulations of the catalytic domain in 2FFU in the absence of both substrates (red), with W331 constrained (black), and in the presence of UDP-GalNAc (blue). (b) Simulations of the catalytic domain in 2FFV in the absence (purple) and presence (orange) of UDP. (c, d, e) Snapshots illustrating the transition of loop B correlated with the rotation of W331 side-chain.
To determine whether the opening of loop B observed in the absence of the donor is correlated with the side-chain rotation of W331 or is simply due to the lack of interaction with UDP-GalNAc, we simulated an identical system (i.e., in the absence of the nucleotide-sugar donor) but with the rotameric state of the W331 side chain restrained (Table 1, Cwx). In this simulation, we observe that loop B maintains its original conformation and does not open during the 40 ns-long simulation (Figure 4a), providing further evidence that the change in the W331 side chain orientation is indeed required for loop opening.
Interestingly, the conformational change of the W331 side chain occurs only in the absence of donor UDP-GalNAc. The phosphate groups of either the substrate UDP-GalNAc or the product UDP stabilize the inward orientation of W331 through two hydrogen bonds between their oxygen atoms and nitrogen NE1. In the simulations starting with loop B in its open state and with UDP present (Figure 4b, CopenU), the W331 side chain does not change orientation. In contrast, with the loop initially in the open state but without UDP (Figure 4b, Copen), the side chain behaves similarly to the case with the loop B closed (Figure 4a, Cx). The correlated change in the orientation of the W331 side chain with loop B opening suggests that the W331 amino acid may be facilitating donor access into the active site and product release.
Similar mechanisms, in which access to the active site is controlled by the position of a long loop whose dynamics is in turn correlated with motions in a short and conserved loop, have been described for other families of transferases using different molecules as donors and acceptors. For example, β-(1,4)-Gal-T1 undergoes a transition from an open to a closed conformation upon glycosyl donor substrate binding.26,27 The corresponding Trp residue, W314, is again part of a WGGEN motif within a small flexible loop of that protein. W314 plays an important role in the conformational change of the adjacent long loop, by moving toward the catalytic pocket to interact with the complex of donor substrate and Mn2+ ion upon donor substrate binding.28 This conformational change seems to be essential for the subsequent productive binding of an acceptor sugar substrate, since a W314Y mutation drastically reduced both the binding affinity to the sugar nucleotide and the catalytic activity of the enzyme.29
In molecular dynamics simulations of β-(1,4)-Gal-T1 in both implicit and explicit solution, Gunasekaran et al.13,14 reported tight coupling of motions in a long loop (that would correspond to our loop B) to the structure and motions of the corresponding 314-WGG-316 motif. Mutations of the conserved Trp and the flanking Gly residues to Ala reduced the fluctuations also in the long loop. Overall, the simulation results for β-(1,4)-Gal-T1 are consistent with our results for ppGalNAcT-2. However, in our specific case, the main chain of the WGGEN loop stays relatively rigid and the conformational change appears to be triggered by the side chain of W331 alone. Thus, our simulations suggest a mechanism in which W331 acts as a gatekeeper that controls the transition between the closed (unable to accept ligand) and open (able to accept ligand) conformations of loop B.
Evolution of charge distribution in the active site during loop opening
The accessible surface area of the UDP-GalNAc binding site doubles during loop opening (Figure 5). The newly exposed surface consists mainly of positively charged residues (R201, R362) and hydrophobic residues, while negatively charged residues do not become exposed. The two arginine residues reorient towards solvent, whereas two His residues (H145, H226) do not change orientation but become exposed due to loop repositioning. Both R201 and R362 are highly conserved in the ppGalNAcTs and interact with the uridine ring and phosphate oxygen atoms of UDP-GalNAc. In addition to their favorable electrostatic interactions with the bound UDP, they may also facilitate donor entry into the active site by making weak long-range contacts in the open, highly flexible state of the loop. The observed flexibility of the unstructured regions in the vicinity of the donor binding site may thus be a relevant factor for protein-substrate recognition.
Figure 5.
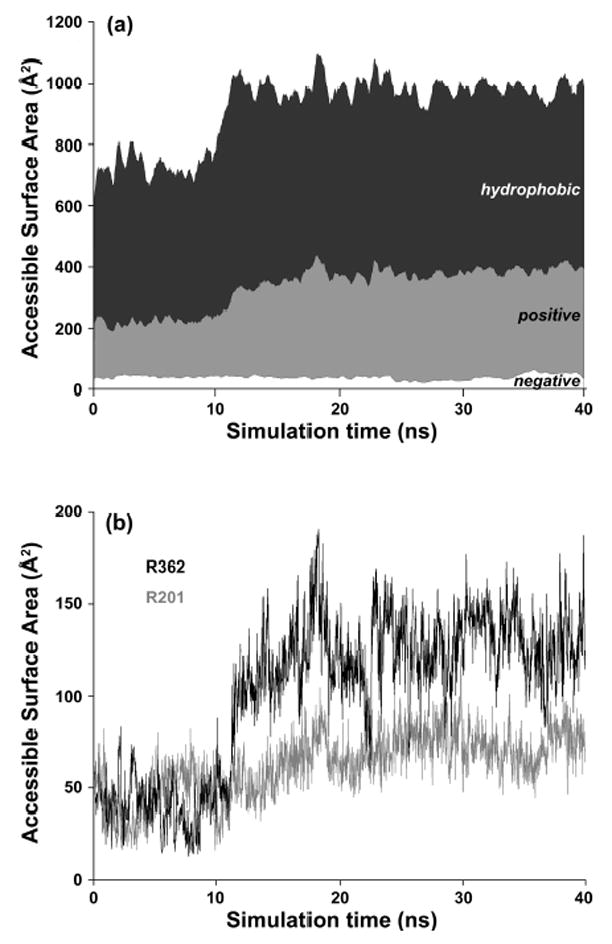
Increase of the accessible surface area (ASA) of the UDP-GalNAc binding site (defined using atoms that are located 5Å around UDP-GalNAc) during loop opening in the Cx simulations. (a) Total ASA and contributions from the hydrophobic (P, G, A, V, L, I, F, W, Y), positively charged (K, R, H) and negatively charged (D, E). (b) ASA for the charged residues R362 and R201.
Hydration of Mn2+ and sugar donor UDP-GalNAc. Implications on enzymatic mechanism
A manganese ion is required for both the stability and activity of ppGalNAcTs and is bound by residues in the active site motif DxH and an additional conserved His.3,5 Mn2+ bridges the protein side chains with the diphosphate group of the donor phosphate. X-ray structures containing UDP-GalNAc or UDP show that Mn2+ bridges three residues in the active site (2 × His, 1 × Asp) and two oxygen atoms in the phosphate groups of UDP. The presence of one water molecule at a distance of ~2.2 Å results in a coordination number 6 of the Mn2+ ion.
We analyzed the stability of Mn2+ interactions in the presence and absence of substrates and the hydration patterns in the active site, to explore possible implications on the enzymatic mechanism of ppGalNAcTs.
Simulations in the presence of UDP-GalNAc indicate that the distance between Mn2+ and the beta-phosphate-oxygen is maintained essentially constant at ~4 Å, in agreement with the x-ray structures. This is also true for simulations containing free UDP, although the oxygen atoms in the terminal phosphate group interchange positions due to rotation of the second phosphate group.
We find that in simulations in the presence of UDP-GalNAc alone or UDP-GalNAc and peptide, a single water molecule with high residence time remains in the first hydration shell of Mn2+ (Figure 6a, b). Even in the simulations performed in the absence of crystal water, a water molecule from the surrounding bulk solvent takes this role, suggesting the importance of hydration for the Mn2+ ion in the active site.
Figure 6.
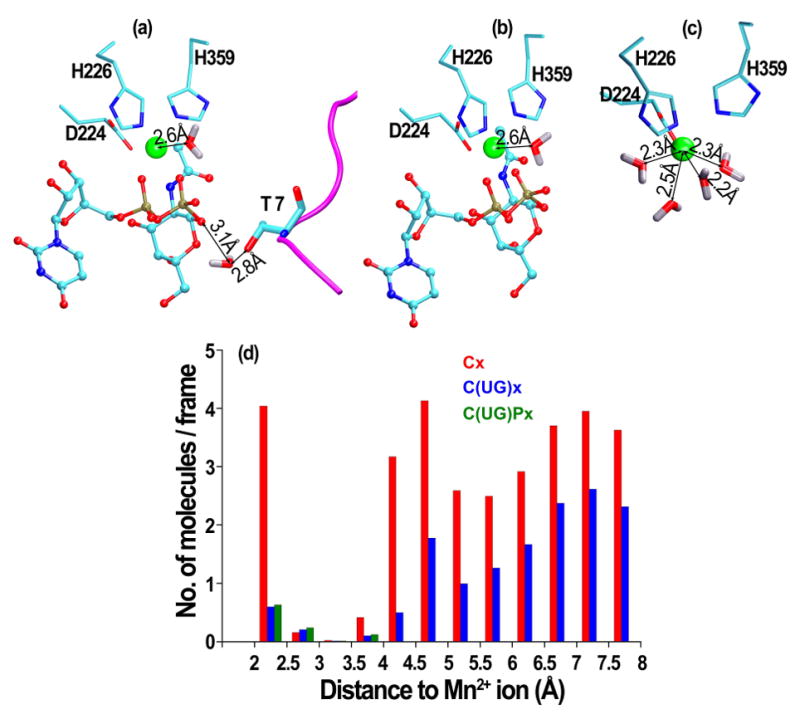
Snapshots of water molecules in the first hydration shell of the Mn2+ ion from simulations (a) in the presence of both UDP-GalNAc and peptide, (b) in the presence of UDP-GalNAc or (c) in the absence of ligands. (d) Histogram of the number of water molecules per simulation frame, as a function of the distance to the Mn2+ ion.
In the presence of UDP-GalNAc, the water molecule in the first hydration layer of Mn2+ is part of a larger water network formed with molecules in the vicinity of glycosidic oxygen. In the presence of both UDP-GalNAc and the EA2 peptide, the glycosidic oxygen interacts with the OG1 atom of the accepting T7 peptide residue via three water molecules: the first bridges only the glycosidic oxygen and OG1, the second bridges in addition the main chain N atom of the T7 side chain, and the third bridges also the O5* atom of the GalNAc moiety. Taken independently, each of these water molecules have a residence time of around 50% of the simulation time. However, taken together, at least one of these water bridges is present during more than 90% of the simulation time. Therefore, the oxygen of the anomeric carbon and the hydroxyl group of the acceptor Thr are nearly continuously bridged by water.
In the simulations of the catalytic domain without substrates, Mn2+ is 7-coordinated, with 3 water molecules replacing the two oxygen atoms of the phosphate groups. The first hydration layer of Mn2+ contains 4 water molecules with high residence time (Figure 6c), as indicated also by the peak at 2.5 Å in the histogram in Figure 6d. The average coordination geometry of Mn2+ is a pentagonal bipyramid; the coordination partners are the water molecules, oxygen atoms of D224 stably coordinating during the entire trajectory, and nitrogen atoms of H226 and H359 competing with additional water molecules for Mn2+ coordination. In the absence of crystal water, molecules from bulk solvent take the role of solvating the Mn2+ ion. The same change of coordination number of the Mn2+ ion has been described also in the case of another retaining glycosyltransferase, α-(1,4)-galactosyltransferase LgtC from Neisseria meningitidis,19 where it was confirmed also by ab-initio quantum mechanics calculations. The increase in the coordination number is correlated with a higher accessibility of the active site in the absence of substrates.
These observations have implications on the enzymatic mechanism. Previously published data do not unequivocally support any of the mechanisms proposed for retaining glycosyltransferases. Our simulations are more consistent with the SNi mechanism than the double displacement mechanism since the large conformational changes of the active site, required by a double-displacement mechanism to bring putative nucleophiles closer to the beta-phosphate-oxygen, did not occur on the timescale of the simulations. However, a double displacement mechanism is still possible if the timescale of the conformational change needed for the enzyme-sugar intermediate is much longer than the time sampled by molecular dynamics. Alternatively, a single displacement mechanism would imply that the nucleophilic attack on C1-O bond is performed by the hydroxy group of the acceptor Ser/Thr. The distance between the glycosidic oxygen and the nucleophilic hydroxy group is about 3 Å and is maintained nearly constant during the simulation, which would at least structurally be consistent with a nucleophile role of the acceptor.
Analysis of tightly bound water molecules
Hydration of the active site is important not only for catalysis, but also for the dynamics and flexibility of loop B controlling ligand access in the active site. Simulations of the system in the presence and absence of crystal water molecules for the catalytic domain alone (Table 1, Cx and C) showed that in the absence of water molecules hydrating deep cavities in the active site, the side chain of W331 is stabilized in the inward orientation, and consequently loop B does not open. Interestingly, loop B is the only region whose dynamics is affected in a major way by the removal of x-ray crystallographic water molecules in the initial structure. We found that in the cavity around W331 two internal water molecules (HOH1160 and 1162) do not get replaced by bulk water. These molecules form hydrogen bonds with W331 and E334 in the conserved loop WGGEN. Without these water molecules, W331 and E334 interact directly, preventing the conformational change of W331 side chain. Therefore, these internal water molecules may have a “lubricating” role in the vicinity of W331. Similar effects of internal water molecules have been reported by calculation of hydration free energies and entropies30 suggesting that the addition of a water molecule can increase protein flexibility through effectively weakened protein-protein hydrogen bonds.
In the presence of UDP (Table 1, CUx and CU) and in the presence of UDP and EA2 (Table 1, CUPx and CUP) the RMSD per residue profiles were not significantly different, explained partly by the fact that large conformational changes do not occur in the presence of ligands, and partly by the observation that bulk water molecules from solution occupy the crystallographic water sites close to the protein surface during the simulation.
Peptide flexibility and interactions
One of the fundamental questions regarding the family of ppGalNAcTs is related to the in vivo peptide substrate preferences of individual isoforms. A detailed understanding of peptide conformational preferences is crucial for the evaluation of peptide affinity in sequence scanning experiments. Moreover, an accurate description of the conformational space sampled by the backbone of the peptide in complex with the enzyme and the identification of patterns of flexibility along peptide sequence is essential for understanding peptide specificity31 and the design of alternative peptide substrates. Previous studies showed that by introducing backbone perturbations in conformations of peptide ligands observed in crystal structures, it was possible to identify a larger number of interacting sequences for a given docking target.32
The analysis of the x-ray ternary complex (PDB code 2FFU5) shows that EA2 binds in an extended conformation into a shallow groove on the surface of hT2. This conformation was maintained throughout our simulation, except for the terminal residues where the backbone residues sample several conformers (Figure 7a). Hydrogen bonds formed between residues at both ends of the peptide (S5 - H365 and K13-G265, Figure 7b, c) are less stable, while interactions near the glycosylation site (T6-R362 and T11-S267) are more stable, consistent with the lower flexibility in the center of the peptide. However, the terminal residues are less flexible when the GalNAc moiety is present, either covalently linked to UDP or to the peptide, compared with the trajectory containing UDP only.
Figure 7.
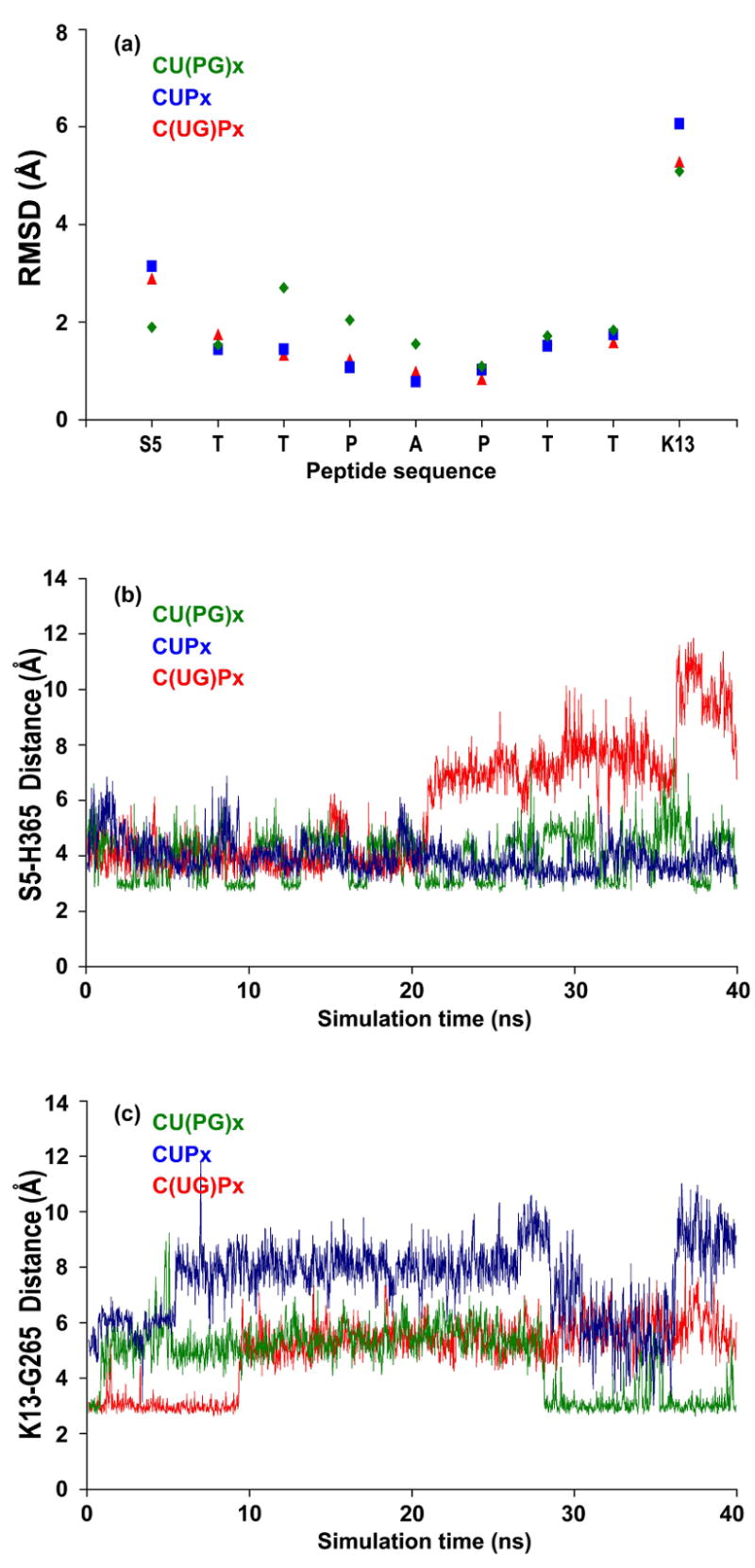
(a) Flexibility of the glycosylated and un-glycosylated peptide in complex with the catalytic domain, measured by the RMSD per residue. (b, c) Evolution of least stable hydrogen bonds between peptide and catalytic domain during simulations before and after catalysis.
A general preference of transferases for peptides adopting extended or random coil conformations has been established both by NMR experiments33,34 and by database analysis,35 and these observations have been used successfully in predictions of mucin-type O-glycosylation sites. The higher backbone flexibility of the peptide termini seen here suggests a less pronounced sequence preference at the ends.
Conclusions
ppGalNAcTases, family 27 of GTs,2 play a crucial role in the first step of O-glycosylation by catalyzing the transfer of the GalNAc moiety from a sugar-nucleotide donor UDP-GalNAc to a Ser/Thr residue in an acceptor peptide. Despite a series of x-ray crystal structures,4–6 details of the catalytic transfer mechanism have remained an open question. Similarly, the functional relevance of large-scale loop rearrangements upon substrate binding, as observed in the crystal structures,5 has been unclear. In this work, we address these questions through multiple molecular dynamics simulations of ppGalNAcT-2, both in the presence and absence of its substrates, based on recently solved crystal structures4–6 of three isoforms of the ppGalNAcT family that capture different stages of the transfer reaction pathway.
Regarding the catalytic transfer mechanism, the results of our study show that potential nucleophilic residues from the catalytic domain do not reach the proximity of the beta-phosphate-oxygen. At the same time, distances between the hydroxy group of the acceptor Ser/Thr side chains and the glycosidic oxygen can be relatively short (~3 Å) during the simulations, which demonstrates the structural possibility for a nucleophilic role of the acceptor. In relation to previously proposed transfer mechanisms, our results are more consistent with a single-displacement mechanism,8,11 as suggested for ppGalNAcTs on the basis of x-ray structures and site-directed mutagenesis data, than a double-displacement transfer mechanism.20 Our simulations also suggest that the partially hydrated Mn2+ ion in the active site may play a role in the enzymatic reaction.
We also observed concerted conformational changes of the loops near the active site. These large-scale loop rearrangements are tied to substrate binding, and thus appear to be crucial for the enzymatic activity. In ppGalNAcT-2, in agreement with the x-ray data, our simulations identify two flexible loops (i.e., A and B, in the vicinity of the active site) as important factors in the binding mechanism. While loop A exhibits a high flexibility both with and without ligands, the dynamics of the longer loop B is significantly reduced when UDP-GalNAc is bound. Analysis of the loop B dynamics revealed a critical interaction with a side chain (W331) located on another less flexible but highly conserved region, loop E. Rotation of the W331 side chain creates unfavorable interactions with residues in loop B. These interactions trigger a conformational change of loop B that exposes the active site, and makes it accessible to ligands. Overall, our analysis suggests a mechanism in which W331 acts as a gatekeeper, controlling the transition of loop B from closed (unable to accept ligand) to open (able to accept ligand) conformations. This structural change is facilitated by internal water molecules, trapped in the structure of the catalytic domain, that prove to be essential for ensuring protein flexibility around the W331 site. Large-scale loop rearrangements have been shown to be important for members of families 6, 7 and 8 of GTs.13–19 In particular, our results, potentially testable through Cys-Cys crosslinking experiments,36,37 are consistent with observations from simulation studies of Beta-1,4-Gal-T113,14 regarding the functional role of a conserved Trp residue (here: W331), suggesting that the observed loop motions and their coupling to local side-chain conformations may also be relevant for other families of GTs.
Our simulations show that the opening of loop B leads to a significant increase in the amount of positively charged surface area in the active site, which can be traced to the exposure of two evolutionarily conserved active-site Arg side chains (R201 and R362). Thus, these residues may play a direct role in the binding of the negatively charged UDP-GalNAc.
Finally, the MD simulations containing the peptide acceptor indicate that its extended conformation is readily maintained when bound to the catalytic domain. The peptide ends exhibit a higher flexibility than the residues near its center, and hydrogen bonds formed by terminal residues are therefore more likely to break than those formed by middle residues. We find that the C-terminal end of the peptide ligand is more dynamic than the N-terminal end, consistent with the observation that the C-terminal region is stabilized only by non-directional hydrophobic interactions. These results are relevant for future studies focused on understanding the binding affinities of peptides to ppGalNAcTs and peptide preferences of individual isoforms.
Methods
Atomically-detailed models of transferase-ligands systems
Table 1 lists the components of the all-atom models that are studied in this paper. The various ppGalNAcT/ligand complexes determined by x-ray crystallography were used as starting conformations. The initial coordinates were taken from the x-ray structures of ppGalNAcT isoform 2, either in ternary complex with UDP and acceptor peptide EA2 (PDB code 2FFU) or in binary complex with UDP (PDB code 2FFV). The missing coordinates in the binary complex, residues G89-P98, were modeled in MODELLER 8v2,38,39 using the ternary complex as a template.
UDP-GalNAc was modeled in the active site of 2FFU using the hT10 structure (PDB code 2D7I) containing hydrolyzed UDP-GalNAc as a template. In one of the systems (CL-close), we positioned the hT2 lectin domain near its catalytic domain using the mT1 x-ray structure (PDB code 1XHB) as a template. The structure containing the EA2 peptide glycosylated at Thr7 was modeled using the Glycam Online Glycoprotein Builder tool.40 (www.glycam.com)
In all cases, starting structures for MD simulations were prepared as follows: (a) hydrogen atoms were added (with acidic residues ionized and His residues neutral, in the absence of experimental evidence for His ionization states) and the protein structure file was generated using the psfgen program in VMD41 (b) the system was explicitly solvated with TIP3P water molecules.42 The dimensions of the rectangular solvent box were chosen so that the minimum distance from the box boundaries to the protein was 10 Å. (c) Na+ and Cl− ions were added in order to maintain electro-neutrality.
Force-field parametrization for UDP-GalNAc
Parameters for the sugar-nucleotide donor UDP-GalNAc were unavailable and were developed as described below. We used the CHARMM27 force field43 parameters and topology for the UDP moiety and the GLYCAM0440 parameters and topology for GalNAc, converted to CHARMM format. Thus, the only missing parameters were for the sugar-phosphate linkage. We tested three types of parameter values: (a) modeled using the AMBER force field parameters for other sugar-nucleotides;44 (b) modeled according to CHARMM2743 parameters for similar atom types; and (c) modeled using a “null model” (i.e., without any additional force constants for bending and torsion, such that the effective angle potentials arise indirectly from other interactions). In a comparison of the energy profiles as a function of the angle, no significant differences were found between these three cases. Therefore, we adopted the simplest “null model” in our simulations.
The energy profile around the sugar-phosphate bond showed a sharp peak at 160 degrees with a half-width of about 30 degrees (corresponding to a conformation in which the N-acetyl group clashes with the oxygen atoms in the second phosphate group), and local minima at 30, 90, 250 and 310 degrees. The conformations in these local minima are extended, with the angle in gauche + and − conformations, in agreement with previous findings from MD simulations and NMR data for UDP-Glucose.45 The UDP-GalNAc moiety modeled using the T10 structure as a template corresponds to the local minimum at 30 degrees. Therefore, this orientation has been used as initial structure in our simulations.
Molecular Dynamics Simulations
Our MD simulations were performed in the NPT ensemble using the NAMD2 package46,47 with the CHARMM2743 force field parameters and topology for the protein and UDP, following simulation and analysis methods used in previous MD studies with explicit solvent of multi-component biomolecular systems.48 For the Mn2+ ion we used a vander-Waals radius of 1.6 Å, ε=−0.014 kcal/mol, similar to the Mg2+ ion and a net charge of +2. The simulation systems were subject to a stepwise equilibration before the production phase, involving initial energy minimization, followed by 10 ps of MD with the protein backbone constrained to the initial coordinates, and 10 ps without any constraints. The systems were gradually heated to 298 K during 20 ps runs at a pressure of 1 atm, first with the protein Cα trace constrained to the crystal coordinates, and then equilibrated for 20 ps with restraints on the Cα atoms, followed by 10 ps without any restraints. After the initial minimization, heating and equilibration stages, the production phase of each MD simulation lasted 40 ns (using a timestep of 2 fs) and the coordinates were saved every 10 ps. The Langevin piston method49,50 was used to maintain a constant pressure of 1 atm. The temperature (298 K) was controlled by using Langevin dynamics with a coupling coefficient of 1 ps−1. We used periodic boundary conditions and the particle mesh Ewald (PME) method51 with a real-space cutoff distance of 10 Å and a grid width of 0.96–1.07 Å. The switching distance for non-bonded electrostatics and van der Waals interactions was 8.5 Å and the integration time-step was 1 fs.
Data visualization and calculation of geometric parameters (RMSD, distances, angles etc) were carried out using VMD.41 For the analysis of water interactions we used in-house code.
Supplementary Material
Acknowledgments
This study utilized the high-performance computational capabilities of the Biowulf Linux cluster at the National Institutes of Health, Bethesda, Md. (http://biowulf.nih.gov). This research was supported by the Intramural Research Program of the NIH, NIDDK. The authors want to thank Dr. Stefano Costanzi, Laboratory of Biological Modeling, NIDDK, NIH for fruitful discussions.
Abbreviations used
- ppGalNAcT
UDP-N-acetylgalactosamine:polypeptide N-acetyl-α-galactosaminyltransferase
- EA2
peptide STTPAPTTK
- GT
Glycosyltransferase
- mT1
x-ray structure of murine T1 (PDB code 1XHB – apo- form)
- hT2
x-ray structure of human T2 (PDB code 2FFV – binary complex with UDP, and PDB code 2FFU – ternary complex with UDP and the acceptor peptide EA2)
- hT10
x-ray structure of human T10 (PDB code 2D7I – complex with hydrolyzed UDP-GalNAc and PDB code 2D7R – complex with GalNAc-Ser)
- RMSD
root-mean-square distance
- ASA
accessible surface area
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Breton C, Snajdrová L, Jeanneau C, Koca J, Imberty A. Structures and mechanisms of glycosyltransferases. Glycobiology. 2006;16:29R–37R. doi: 10.1093/glycob/cwj016. [DOI] [PubMed] [Google Scholar]
- 2.Coutinho PM, Henrissat B. Carbohydrate-active enzymes: an integrated database approach. In: Gilbert HJ, Davies G, Henrissat B, Svensson B, editors. Recent Advances in Carbohydrate Bioengineering. The Royal Society of Chemistry; Cambridge: 1999. pp. 3–12. [Google Scholar]
- 3.Ten Hagen KG, Fritz TA, Tabak LA. All in the family: the UDP-GalNAc:polypeptide N-acetyl-galactosaminyltransferases. Glycobiology. 2003;13:1R–16R. doi: 10.1093/glycob/cwg007. [DOI] [PubMed] [Google Scholar]
- 4.Fritz TA, Hurley JH, Trinh LB, Shiloach J, Tabak LA. The beginnings of mucin biosynthesis: the crystal structure of UDP-GalNAc:polypeptide alpha-N-acetylgalactosaminyltransferase-T1. Proc Natl Acad Sci USA. 2004;101:15307–15312. doi: 10.1073/pnas.0405657101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Fritz TA, Raman J, Tabak LA. Dynamic association between the catalytic and lectin domains of human UDP-GalNAc:polypeptide alpha-N-acetylgalactosaminyltransferase-2. J Biol Chem. 2006;281:8613–8619. doi: 10.1074/jbc.M513590200. [DOI] [PubMed] [Google Scholar]
- 6.Kubota T, Shiba T, Sugioka S, Furukawa S, Sawaki H, Kato R, Wakatsuki S, Narimatsu H. Structural basis of carbohydrate transfer activity by human UDP-GalNAc: polypeptide alpha-N-acetylgalactosaminyltransferase (pp-GalNAc-T10) J Mol Biol. 2006;359:708–727. doi: 10.1016/j.jmb.2006.03.061. [DOI] [PubMed] [Google Scholar]
- 7.Marchler-Bauer A, Bryant SH. CD-Search: protein domain annotations on the fly. Nucleic Acids Res. 2004;32:327–331. doi: 10.1093/nar/gkh454. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Lairson LL, Withers SG. Mechanistic analogies amongst carbohydrate modifying enzymes. Chem Commun. 2004;20:2243–2248. doi: 10.1039/b406490a. [DOI] [PubMed] [Google Scholar]
- 9.Monegal A, Planas A. Chemical rescue of alpha3-galactosyltransferase. Implications in the mechanism of retaining glycosyltransferases. J Am Chem Soc. 2006;128:16030–16031. doi: 10.1021/ja0659931. [DOI] [PubMed] [Google Scholar]
- 10.Boix E, Zhang Y, Swaminathan GJ, Brew K, Acharya KR. Structural basis of ordered binding of donor and acceptor substrates to the retaining glycosyltransferase, alpha-1,3-galactosyltransferase. J Biol Chem. 2002;277:28310–28318. doi: 10.1074/jbc.M202631200. [DOI] [PubMed] [Google Scholar]
- 11.Persson K, Ly HD, Dieckelmann M, Wakarchuk WW, Withers SG, Strynadka NC. Crystal structure of the retaining galactosyltransferase LgtC from Neisseria meningitidis in complex with donor and acceptor sugar analogs. Nat Struct Biol. 2001;8:166–175. doi: 10.1038/84168. [DOI] [PubMed] [Google Scholar]
- 12.Pedersen LC, Dong J, Taniguchi F, Kitagawa H, Krahn JM, Pedersen LG, Sugahara K, Negishi M. Crystal structure of an alpha 1,4-N-acetylhexosaminyltransferase (EXTL2), a member of the exostosin gene family involved in heparan sulfate biosynthesis. J Biol Chem. 2003;278:14420–14428. doi: 10.1074/jbc.M210532200. [DOI] [PubMed] [Google Scholar]
- 13.Gunasekaran K, Ma B, Ramakrishnan B, Qasba PK, Nussinov R. Interdependence of backbone flexibility, residue conservation, and enzyme function: a case study on beta1,4-galactosyltransferase-I. Biochemistry. 2003;42:3674–387. doi: 10.1021/bi034046r. [DOI] [PubMed] [Google Scholar]
- 14.Gunasekaran K, Ma B, Nussinov R. Triggering loops and enzyme function: identification of loops that trigger and modulate movements. J Mol Biol. 2003;332:143–159. doi: 10.1016/s0022-2836(03)00893-3. [DOI] [PubMed] [Google Scholar]
- 15.Boix E, Swaminathan GJ, Zhang Y, Natesh R, Brew K, Acharya KR. Structure of UDP complex of UDP-galactose:beta-galactoside-alpha-1,3-galactosyltransferase at 1.53-A resolution reveals a conformational change in the catalytically important C terminus. J Biol Chem. 2001;276:48608–48614. doi: 10.1074/jbc.M108828200. [DOI] [PubMed] [Google Scholar]
- 16.Boix E, Zhang Y, Swaminathan GJ, Brew K, Acharya KR. Structural basis of ordered binding of donor and acceptor substrates to the retaining glycosyltransferase, alpha-1,3-galactosyltransferase. J Biol Chem. 2002;277:28310–28318. doi: 10.1074/jbc.M202631200. [DOI] [PubMed] [Google Scholar]
- 17.Patenaude SI, Seto NO, Borisova SN, Szpacenko A, Marcus SL, Palcic MM, Evans SV. The structural basis for specificity in human ABO(H) blood group biosynthesis. Nat Struct Biol. 2002;9:685–690. doi: 10.1038/nsb832. [DOI] [PubMed] [Google Scholar]
- 18.Nguyen HP, Seto NO, Cai Y, Leinala EK, Borisova SN, Palcic MM, Evans SV. The influence of an intramolecular hydrogen bond in differential recognition of inhibitory acceptor analogs by human ABO(H) blood group A and B glycosyltransferases. J Biol Chem. 2003;278:49191–49195. doi: 10.1074/jbc.M308770200. [DOI] [PubMed] [Google Scholar]
- 19.Snajdrova L, Kulhanek P, Imberty A, Koca J. Molecular dynamics simulations of glycosyltransferase LgtC. Carbohydr Res. 2004;339:995–1006. doi: 10.1016/j.carres.2003.12.024. [DOI] [PubMed] [Google Scholar]
- 20.Gastinel LN, Bignon C, Misra AK, Hindsgaul O, Shaper JH, Joziasse DH. Bovine alpha1,3-galactosyltransferase catalytic domain structure and its relationship with ABO histo-blood group and glycosphingolipid glycosyltransferases. EMBO J. 2001;20:638–649. doi: 10.1093/emboj/20.4.638. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Zhang Y, Swaminathan GJ, Deshpande A, Boix E, Natesh R, Xie Z, Acharya KR, Brew K. Roles of individual enzyme-substrate interactions by α-1,3-galactosyltransferase in catalysis and specificity. Biochemistry. 2003;42:13512–13521. doi: 10.1021/bi035430r. [DOI] [PubMed] [Google Scholar]
- 22.Lairson LL, Chiu CP, Ly HD, He S, Wakarchuk WW, Strynadka NC, Withers SG. Intermediate trapping on a mutant retaining alpha-galactosyltransferase identifies an unexpected aspartate residue. J Biol Chem. 2004;279:28339–28344. doi: 10.1074/jbc.M400451200. [DOI] [PubMed] [Google Scholar]
- 23.Gibson RP, Turkenburg JP, Charnock SJ, Lloyd R, Davies GJ. Insights into trehalose synthesis provided by the structure of the retaining glucosyltransferase OtsA. Chem Biol. 2002;12:1337–1346. doi: 10.1016/s1074-5521(02)00292-2. [DOI] [PubMed] [Google Scholar]
- 24.Goedl C, Griessler R, Schwarz A, Nidetzky B. Structure-function relationships for Schizophyllum commune trehalose phosphorylase and their implications for the catalytic mechanism of family GT-4 glycosyltransferases. Biochem J. 2006;397:491–500. doi: 10.1042/BJ20060029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Watson KA, McCleverty C, Geremia S, Cottaz S, Driguez H, Johnson LN. Phosphorylase recognition and phosphorolysis of its oligosaccharide substrate: answers to a long outstanding question. EMBO J. 1999;18:4619–32. doi: 10.1093/emboj/18.17.4619. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Gastinel LN, Cambillau C, Bourne Y. Crystal structures of the bovine beta-4-galactosyltransferase catalytic domain and its complex with uridine diphosphogalactose. EMBO J. 1999;18:3546–3557. doi: 10.1093/emboj/18.13.3546. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Ramakrishnan B, Qasba PK. Crystal structure of lactose synthase reveals a large conformational change in its catalytic component, the β 1,4-galactosyltransferase-I. J Mol Biol. 2001;310:205–218. doi: 10.1006/jmbi.2001.4757. [DOI] [PubMed] [Google Scholar]
- 28.Ramakrishnan B, Boeggeman E, Ramasamy V, Qasba PK. Structure and catalytic cycle of beta-1,4-galactosyltransferase. Curr Opin Struct Biol. 2004;14:593–600. doi: 10.1016/j.sbi.2004.09.006. [DOI] [PubMed] [Google Scholar]
- 29.Ramasamy V, Ramakrishnan B, Boeggeman E, Qasba PK. The role of tryptophan 314 in the conformational changes of β-1,4-galactosyltransferase-I. J Mol Biol. 2003;331:1065–1076. doi: 10.1016/s0022-2836(03)00790-3. [DOI] [PubMed] [Google Scholar]
- 30.Olano LR, Rick SW. Hydration free energies and entropies for water in protein interiors. J Am Chem Soc. 2004;126:7991–8000. doi: 10.1021/ja049701c. [DOI] [PubMed] [Google Scholar]
- 31.Sood VD, Baker D. Recapitulation and design of protein binding peptide structures and sequences. J Mol Biol. 2006;357:917–927. doi: 10.1016/j.jmb.2006.01.045. [DOI] [PubMed] [Google Scholar]
- 32.Wollacott AM, Desjarlais JR. Virtual interaction profiles of proteins. J Mol Biol. 2001;313:317–342. doi: 10.1006/jmbi.2001.5035. [DOI] [PubMed] [Google Scholar]
- 33.Kirnarsky L, Nomoto M, Ikematsu Y, Hassan H, Bennett EP, Cerny RL, Clausen H, Hollingsworth MA, Sherman S. Structural analysis of peptide substrates for mucin-type O-glycosylation. Biochemistry. 1998;37:12811–12817. doi: 10.1021/bi981034a. [DOI] [PubMed] [Google Scholar]
- 34.Kirnarsky L, Prakash O, Vogen SM, Nomoto M, Hollingsworth MA, Sherman S. Structural effects of O-glycosylation on a 15-residue peptide from the mucin (MUC1) core protein. Biochemistry. 2000;39:12076–12082. doi: 10.1021/bi0010120. [DOI] [PubMed] [Google Scholar]
- 35.Julenius K, Molgaard A, Gupta R, Brunak S. Prediction, conservation analysis, and structural characterization of mammalian mucin-type O-glycosylation sites. Glycobiology. 2005;15:153–164. doi: 10.1093/glycob/cwh151. [DOI] [PubMed] [Google Scholar]
- 36.Matsumura M, Becktel WJ, Levitt M, Matthews BW. Stabilization of phage T4 lysozyme by engineered disulfide bonds. Proc Natl Acad Sci USA. 1989;86:6562–6566. doi: 10.1073/pnas.86.17.6562. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Ma K, Temiakov D, Anikin M, McAllister WT. Probing conformational changes in T7 RNA polymerase during initiation and termination by using engineered disulfide linkages. Proc Natl Acad Sci USA. 2005;102:17612–17617. doi: 10.1073/pnas.0508865102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Marti-Renom MA, Stuart A, Fiser A, Sánchez R, Melo F, Sali A. Comparative protein structure modeling of genes and genomes. Annu Rev Biophys Biomol Struct. 2000;29:291–325. doi: 10.1146/annurev.biophys.29.1.291. [DOI] [PubMed] [Google Scholar]
- 39.Sali A, Blundell TL. Comparative protein modelling by satisfaction of spatial restraints. J Mol Biol. 1993;234:779–815. doi: 10.1006/jmbi.1993.1626. [DOI] [PubMed] [Google Scholar]
- 40.Woods RJ, Dwek RA, Edge CJ, Fraser-Reid B. Molecular mechanical and molecular dynamical simulations of glycoproteins and oligosaccharides. 1 GLYCAM_93 Parameter Development. J Phys Chem. 1995;99:3832–3846. [Google Scholar]
- 41.Humphrey W, Dalke A, Schulten K. VMD - Visual Molecular Dynamics. J Molec Graphics. 1996;14:33–38. doi: 10.1016/0263-7855(96)00018-5. [DOI] [PubMed] [Google Scholar]
- 42.Jorgensen WL, Chandrasekhar J, Madura JD, Impey RW, Klein ML. Comparison of simple potential functions for simulating liquid water. J Chem Phys. 1983;79:926–935. [Google Scholar]
- 43.MacKerell AD, Jr, Bashford D, Bellott RL, Dunbrack RL, Jr, Evanseck JD, Field MJ, Fischer S, Gao J, Guo H, Ha S, Joseph-McCarthy D, Kuchnir L, Kuczera K, Lau FTK, Mattos C, Michnick S, Ngo T, Nguyen DT, Prodhom B, Reiher WE, III, Roux B, Schlenkrich M, Smith JC, Stote R, Straub J, Watanabe M, Wiorkiewicz-Kuczera J, Yin D, Karplus M. All-atom empirical potential for molecular modeling and dynamics studies of proteins. J Phys Chem B. 1998;102:3586–3616. doi: 10.1021/jp973084f. [DOI] [PubMed] [Google Scholar]
- 44.Petrova P, Koca J, Imberty A. Potential energy hypersurfaces of nucleotide sugars: ab initio calculations, force-field parametrization, and exploration of the flexibility. J Am Chem Soc. 1999;121:5535–5547. [Google Scholar]
- 45.Petrova P, Koca J, Imberty A. Molecular dynamics simulations of solvated UDP-glucose in interaction with Mg2+ cations. Eur J Biochem. 2001;268:5365–5374. doi: 10.1046/j.0014-2956.2001.02469.x. [DOI] [PubMed] [Google Scholar]
- 46.Phillips JC, Braun R, Wang W, Gumbart J, Tajkhorshid E, Villa E, Chipot C, Skeel RD, Kale L, Schulten K. Scalable molecular dynamics with NAMD. J Comput Chem. 2005;26:1781–1802. doi: 10.1002/jcc.20289. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Kale L, Skeel R, Bhandarkar M, Brunner R, Gursoy A, Krawetz N, Phillips J, Shinozaki A, Varadarajan K, Schulten K. NAMD2: greater scalability for parallel molecular dynamics. J Comput Phys. 1999;151:283–312. [Google Scholar]
- 48.Buchete NV, Tycko R, Hummer G. Molecular dynamics simulations of Alzheimer’s beta-amyloid protofilaments. J Mol Biol. 2005;353:804–821. doi: 10.1016/j.jmb.2005.08.066. [DOI] [PubMed] [Google Scholar]
- 49.Martyna GJ, Tobias DJ, Klein ML. Constant-pressure molecular-dynamics algorithms. J Chem Phys. 1994;101:4177–4189. [Google Scholar]
- 50.Feller SE, Zhang YH, Pastor RW, Brooks BR. Constant-pressure molecular-dynamics simulation– the Langevin piston method. J Chem Phys. 1995;103:4613–4621. [Google Scholar]
- 51.Darden T, York D, Pedersen L. Particle Mesh Ewald–an N log(N) method for Ewald sums in large systems. J Chem Phys. 1993;98:10089–10092. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.