

Abstract
It has been questioned recently whether populated intermediates are important for the protein folding process or are artefacts trapped in nonproductive pathways. We report here that the rapidly formed intermediate of the spliceosomal protein U1A is an off-pathway artefact caused by transient aggregation of denatured protein under native conditions. Transient aggregates are easily mistaken for structured monomers and could be a general problem in time-resolved folding studies.
Keywords: U1A spliceosomal protein, aggregation artefacts, off-pathway intermediates
In time-resolved refolding experiments, some unfolded proteins (D) collapse rapidly (<ms) into compact and partly structured denatured species prior to the formation of the native structure (N) (1–6). The nature of these folding intermediates† has attracted much attention because they are believed to reveal key information about the folding process. However, populated intermediates are not necessary for folding. Several small proteins (<90 residues) fold rapidly in a two-state process directly from the unfolded state (7–12). Two-state folding is seen also with larger proteins which may or may not accumulate intermediates depending on the conditions: intermediates that are populated under physiological conditions become destabilized and disappear in the presence of denaturant (3, 13) or at elevated temperatures (14). The findings have triggered a lively debate about the role of populated intermediates, and it has been argued that they could even be misfolds trapped in nonproductive pathways (15–19). Here we report that folding intermediates can also be artefacts of rapid aggregation of denatured protein.
Materials and Methods
The protein examined in this study is human spliceosomal protein U1A (20) (102 residues and no cysteines) in which the semiburied phenylalanine-56 was replaced with a tryptophan, a variant that occurs naturally in U1A from potato (21). The substitution produces large fluorescence changes upon denaturation and thereby facilitates time-resolved studies at very low protein concentrations.
Protein stability was obtained by standard linear free-energy assumptions and titration with guanidine⋅hydrochloride (Gdn⋅HCl) (22) using a Perkin–Elmer LS 50 luminescence spectrometer.
Unfolding and refolding kinetics were monitored using a SX.18MV stopped-flow instrument from Applied Photophysics set up for 1:10 volumes mixing. Excitation was at 280 nm and detection was with a 320-nm cut-off filter. All experiments were done at 25°C and the buffer was 50 mM Mes at pH 6.3.
The Folding Intermediate of U1A Is an Artefact Caused by Rapid (ms) Aggregation
A common way to classify folding behavior is to test if the experimental data obey the two-state relation, N ⇄ D (3, 7, 17),
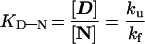 |
1 |
where KD–N is the equilibrium constant for unfolding, determined here by Gdn⋅HCl denaturation (22) (Table 1), and ku and kf are the rate constants from the unfolding and refolding kinetics, respectively. With U1A, the observed refolding kinetics, log kf, deviates from the nearly linear Gdn⋅HCl dependence predicted by Eq. 1 at [Gdn⋅HCl] lower than 1.5 M (Fig. 1). The behavior is seen also with other proteins, where it has been suggested to result from a rapid collapse of the unfolded protein into an intermediate at low concentrations of denaturant (3, 23). Since this collapse occurs in the dead-time of the stopped-flow instrument, the observed reaction takes place from a stabilized form of the polypeptide which folds slower (3). Closer analysis of the U1A time course, however, reveals also a faster reaction preceding the predominant refolding phase (Fig. 2). Interestingly, the rate constant for this fast reaction (kffast) agrees precisely with that expected for two-state folding according to Eq. 1 (Fig. 1). We conclude, therefore, that this fast reaction represents two-state folding directly from the denatured state.
Table 1.
Comparison of stability and kinetic data shows that the fast folding reaction of U1A takes place directly from the denatured state.
| Measurement | ΔGD–NH2O, kcal/mol | Gdn⋅HCl1/2, M | mD–N |
|---|---|---|---|
| Equilibrium unfolding | 9.3 ± 0.2* | 4.07 ± 0.02* | −2.3 ± 0.1* |
| Kinetics | 9.1 ± 0.2† | 4.08§ | −2.2 ± 0.1¶ |
As expected from a two-state process, the free energy of unfolding in pure water (ΔGD–NH2O) derived from the kinetics is the same as from equilibrium denaturation experiments (Eq. 1). Gdn⋅HCl1/2 is the midpoint for the unfolding transition and mD–N is the Gdn⋅HCl dependence of ΔGD–N (22).
The value is determined by standard Gdn⋅HCl denaturation experiments (22).
ΔGD–NH2O = −2.3RTlog KD–N = −2.3RT(log ku − log kffast) at [Gdn⋅HCl] = 0 M, Eq. 1. The derivation does not take into account the cis–trans equilibria of the prolines in the denatured state, which contribute to a small underestimate of ΔGD–NH2O (7). The difference is within the experimental error.
Obtained from the intersect between the fits in Fig. 1, where log kf = log ku.
Figure 1.

Gdn⋅HCl dependence of the rate constants for folding and unfolding of U1A. The rate constants are in units of s−1. The left arm of the V-shaped plot shows the refolding rate constant (○, kf, [U1A] = 3.1 μM; and ▾, kffast, [U1A] = 1 μM) following 1:10 dilution (stopped-flow) of denatured U1A (in 5.1 M Gdn⋅HCl) into lower [Gdn⋅HCl], and the right arm shows the unfolding rate constant (•, ku, [U1A] = 3.1 μM) upon 1:10 mixing of native protein (in water) into high [Gdn⋅HCl]. The curves are polynomial fits which precisely obey Eq. 1 and, hence, represent two-state folding directly from the denatured state—i.e., log kf = log ku − log KD–N. The deviation from two-state folding observed at low [Gdn⋅HCl] (○) is found also for other proteins and is usually believed to result from accumulation of an intermediate. With U1A, the deviation is caused by transient aggregation of denatured protein under refolding conditions. At low protein concentrations the denatured protein remains monomeric during the refolding process and the rate constant (kffast) follows Eq. 1, but at higher protein concentrations the denatured protein aggregates in the dead-time of the stopped-flow instrument, giving rise to a retardation of the refolding rate. Conditions where aggregation occurs are marked gray.
Figure 2.

(A) Time course for refolding of U1A at different protein concentrations. Final [Gdn⋅HCl] = 0.46 M. At moderate to high protein concentrations (>5 μM), the time course is dominated by the slow phase, but at low protein concentrations folding occurs mainly by the fast reaction. (B) The rate constant of the slow phase decreases slightly with increasing protein concentration, whereas the fast reaction appears independent of protein concentration. The negative concentration dependence of the slow phase is inconsistent with formation of aggregates, since this process would become faster at high protein concentrations. Hence, it is likely that the slow phase represents a dissociation process—i.e., folding from an aggregate. Data from the first 6 ms were excluded from the fits. Control experiments were conducted with free tryptophan and with U1A contained in the dilution buffer.
Similar results were obtained recently with lysozyme, where a small population of unfolded protein folds fast in parallel with the slower conversions of various intermediates (17). The author points out that kinetic partitioning into fast and slow pathways (compare Fig. 2) is inconsistent with rapid interconversion between the unfolded state and other species in the pre-equilibrium, and he concludes that the intermediate, despite its rapid formation, is trapped in a nonproductive conformation that equilibrates slowly with the unfolded protein.
With U1A, the extent of fast refolding varies with protein concentration (Figs. 2 and 3). At low concentrations (<1 μM) U1A folds mainly by the fast pathway, but at higher concentrations the slow reaction becomes predominant. The behavior is typical of high-order reactions such as complexation and shows that the slow conversion into the native state takes place from aggregates which form in the dead-time of the stop-flow instrument (<5 ms). It remains to establish whether the fraction of monomer folding is determined by a rapid pre-equilibration of denatured monomers and aggregates or results from kinetic competition between monomer folding and aggregation. The kinetic partitioning favors the latter explanation. If poor solubility under native conditions is a general property of denatured proteins (24), the competition scenario predicts that slowly folding proteins will fold with substantial formation of transient aggregates, whereas more rapidly folding proteins will tend to be two-state. At the Gdn⋅HCl concentration where aggregation occurs, log kf vs. [Gdn⋅HCl] bends down and displays a positive slope (Fig. 1). From mass-action, this indicates that the protein expands upon activation—i.e. the transition state exhibits more Gdn⋅HCl-binding sites than the dead-time species. This is another clear indication of the dead-time species being off-pathway, since it has to unfold (or dissociate) before it adopts the native conformation.
Figure 3.

Fraction of monomer folding at different concentrations of U1A, expressed as the ratio of the amplitudes of the fast and slow refolding phase (compare Fig. 2A). In fits where [U1A] ≥ 3.1 μM the rate constant for the fast phase was locked to 200 s−1. Since refolding is usually monitored at relatively high concentrations of protein, the proportion of monomer folding may be very small and undetected. For example, standard stopped-flow (≈10 μM), stopped-flow CD (10–50 μM), and quench-flow NMR (>100 μM). Hence, tests of concentration dependence in these regions may not reveal aggregation artefacts.
Accordingly, U1A may fold either rapidly in a two-state process (D → N) or more slowly from aggregates, depending on solvent conditions and protein concentration. Since the aggregation artefact is revealed only at very low protein concentrations, it may well have been missed in earlier rapid-mixing studies done at much higher protein concentrations (compare Figs. 2 and 3).
The curved plots of log kf and log ku under two-state conditions suggest that U1A features a nonlinear Gdn⋅HCl dependence of the activation energy. The behavior has been examined in detail and is suggested to be caused by movements of the transition state along the top of a very broad and flat activation barrier upon destabilization by Gdn⋅HCl (unpublished results).
Could Concealed Aggregation Be a General Problem in Folding Studies?
Dead-time aggregation is found also with the small and rapidly folding chymotrypsin inhibitor 2 (CI2), but only at protein concentrations above ≈200 μM (compared with ≈1 μM for U1A) (M.O., Y.-J. Tan, and A. R. Fersht, unpublished results). It must be noticed, however, that previous results with CI2 were obtained at protein concentrations well below 200 μM, where aggregation can be excluded. We cannot detect any aggregation during folding of the ribonuclease barnase. The difference may be that barnase collapses rapidly into a folding intermediate (3) which is more soluble than the unfolded protein. Aggregation is observed also during refolding of larger proteins—e.g., bovine growth hormone (25), reduced lysozyme (26), and phosphoglycerate kinase (27). Since these proteins fold much more slowly than U1A there is ample of time for aggregation to occur, and the implications of the results for rapidly folding proteins are uncertain. It is now important to find out if artefacts of transient aggregation are a widespread problem in time-resolved folding studies.
Acknowledgments
We thank Dr. Kiyoshi Nagai and coworkers for generously providing the U1A clones and Dr. Alan Fersht and his group for help with expression systems. Our work is supported by the Swedish Natural Science Research Council and the Sven and Lilly Lawski Foundation for Natural Science.
ABBREVIATION
- Gdn⋅HCl
guanidine⋅hydrochloride
Footnotes
The intermediates discussed in this study are those that accumulate transiently in the refolding reaction of small proteins without being restricted by nonnative disulfide links or nonnative proline isomerizations.
References
- 1.Udgaonkar J B, Baldwin R L. Nature (London) 1988;335:700–704. [Google Scholar]
- 2.Roder H, Elöve G A, Englander S W. Nature (London) 1988;335:694–699. doi: 10.1038/335700a0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Matouschek A, Kellis J T, Serrano L, Bycroft M, Fersht A R. Nature (London) 1990;346:440–445. doi: 10.1038/346440a0. [DOI] [PubMed] [Google Scholar]
- 4.Kim P S, Baldwin R L. Annu Rev Biochem. 1990;59:631–660. doi: 10.1146/annurev.bi.59.070190.003215. [DOI] [PubMed] [Google Scholar]
- 5.Radford S E, Dobson C M, Evans P A. Nature (London) 1992;358:302–307. doi: 10.1038/358302a0. [DOI] [PubMed] [Google Scholar]
- 6.Bai Y, Sosnick T R, Mayne L, Englander S W. Science. 1995;269:192–197. doi: 10.1126/science.7618079. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Jackson S E, Fersht A R. Biochemistry. 1991;30:10428–10435. doi: 10.1021/bi00107a010. [DOI] [PubMed] [Google Scholar]
- 8.Alexander P, Orban J, Bryan P. Biochemistry. 1992;31:7243–7248. doi: 10.1021/bi00147a006. [DOI] [PubMed] [Google Scholar]
- 9.Viguera A R, Martinez J C, Filimonov V V, Mateo P L, Serrano L. Biochemistry. 1994;33:2142–2150. doi: 10.1021/bi00174a022. [DOI] [PubMed] [Google Scholar]
- 10.Kuszewski J, Clore G M, Gronenborn A M. Protein Sci. 1994;3:1945–1952. doi: 10.1002/pro.5560031106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Kragelund B B, Robinson C V, Knudsen V, Dobson C M, Poulsen F M. Biochemistry. 1995;34:7217–7224. doi: 10.1021/bi00021a037. [DOI] [PubMed] [Google Scholar]
- 12.Schindler T S, Herrier M, Marahiel M A, Schmid F X. Nat Struct Biol. 1995;2:663–673. doi: 10.1038/nsb0895-663. [DOI] [PubMed] [Google Scholar]
- 13.Khorasanizadeh S, Peters I D, Butt T R, Roder H. Biochemistry. 1993;32:7054–7063. doi: 10.1021/bi00078a034. [DOI] [PubMed] [Google Scholar]
- 14.Oliveberg M, Tan Y J, Fersht A R. Proc Natl Acad Sci USA. 1995;92:8926–8929. doi: 10.1073/pnas.92.19.8926. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Shakhnovich E I, Finkelstein A V. Biopolymers. 1989;28:1667–1680. doi: 10.1002/bip.360281003. [DOI] [PubMed] [Google Scholar]
- 16.Chan H S, Dill K A. J Chem Phys. 1994;100:9238–9275. [Google Scholar]
- 17.Kiefhaber T. Proc Natl Acad Sci USA. 1995;92:9029–9033. doi: 10.1073/pnas.92.20.9029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Fersht A R. Proc Natl Acad Sci USA. 1995;92:10869–10873. doi: 10.1073/pnas.92.24.10869. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Mirny L A, Abkevich V, Shakhnovich E I. Folding and Design. 1996;1:103–116. doi: 10.1016/S1359-0278(96)00019-3. [DOI] [PubMed] [Google Scholar]
- 20.Nagai K, Oubridge C, Jessen T-H, Li J, Evans P R. Nature (London) 1990;348:515–520. doi: 10.1038/348515a0. [DOI] [PubMed] [Google Scholar]
- 21.Simpson G G, Clark G P, Rothnie H M, Boelens W, van Venrooij W, Brown J W S. EMBO J. 1995;14:4540–4550. doi: 10.1002/j.1460-2075.1995.tb00133.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Pace C N. Methods Enzymol. 1986;131:266–279. doi: 10.1016/0076-6879(86)31045-0. [DOI] [PubMed] [Google Scholar]
- 23.Muñoz V, Lopez E M, Jager M, Serrano L. Biochemistry. 1994;33:5858–5866. doi: 10.1021/bi00185a025. [DOI] [PubMed] [Google Scholar]
- 24.Dill K A, Shortle D. Annu Rev Biochem. 1991;60:795–825. doi: 10.1146/annurev.bi.60.070191.004051. [DOI] [PubMed] [Google Scholar]
- 25.Brems D N. Biochemistry. 1988;27:4541–4546. [Google Scholar]
- 26.Goldberg M E, Rudolph R, Jaenicke R. Biochemistry. 1991;30:2790–2797. doi: 10.1021/bi00225a008. [DOI] [PubMed] [Google Scholar]
- 27.Pecorari F, Minard P, Desmadril M, Yon J M. J Biol Chem. 1996;271:5270–5276. doi: 10.1074/jbc.271.9.5270. [DOI] [PubMed] [Google Scholar]