

Abstract
Mammalian selenocysteine-containing thioredoxin reductase (TR) isolated from HeLa cells and from human lung adenocarcinoma cells was separated into two major enzyme species by heparin-agarose affinity chromatography. The low-affinity enzyme forms that were not retained on heparin agarose showed strong crossreactivity in immunoblot assays with anti-rat liver TR polyclonal antibodies, whereas the high-affinity enzyme forms that were retained by the heparin column were not detected. Both low and high heparin-affinity enzyme forms contained FAD, were indistinguishable on SDS/PAGE analysis, and exhibited similar catalytic activities in the NADPH-dependent DTNB [5,5′-dithiobis(2-nitrobenzoate)] assay. The C-terminal amino acid sequences of 75Se-labeled tryptic peptides from lung adenocarcinoma low- and high heparin-affinity enzyme forms were identical to the predicted C-terminal sequence of human placental TR. These two determined peptide sequences were -Ser-Gly-Ala-Ser-Ile-Leu-Gln-Ala-Gly-Cys-Secys-(Gly). Occurrence of the Se-carboxymethyl derivative of radioactive selenocysteine in the position corresponding to TGA in the gene confirmed that UGA is translated as selenocysteine. The presence of cysteine followed by a reactive selenocysteine residue in this C-terminal region of the protein may explain some of the unusual properties of the mammalian TRs.
Keywords: heparin affinity, selenocysteine/NADPH-linked reductase/flavoprotein
The occurrence of an in-frame TGA codon in a mutant form, hIIB3, of the human lung cytochrome P450 CYP2B7 gene has been reported (1) and a radioactive selenoprotein of the expected mass (57 kDa) that contained selenocysteine, was isolated from a human lung adenocarcinoma cell line, NCI-H441, cultured in media containing 75Se-selenite (2). However, this selenoprotein was not a cytochrome P450 and instead proved to be a subunit of mammalian thioredoxin reductase (TR). The selenoprotein, a homodimer of approximately 57 kDa subunits, is a flavoprotein containing bound FAD that uses NADPH as electron donor (2). It reduces 5,5′-dithiobis(2-nitrobenzoic acid) (DTNB) and catalyzes the thioredoxin-dependent reduction of insulin, properties that are typical of mammalian TR (3). The pure selenoprotein, however, failed to react with polyclonal antibodies elicited to rat liver TR (2), whereas a purified TR from human Jurkat T cells was recognized by the same rat liver TR antibodies (4). The rat liver TR used for antibody production and the T cell enzyme had been purified by a standard procedure that included a 2′,5′-ADP agarose-affinity matrix chromatographic step, which is selective for NADPH-dependent redox enzymes (5). A different isolation procedure, devised for purification of the 75Se-labeled protein from the lung adenocarcinoma cells prior to its identification as TR, included a heparin-agarose-affinity matrix adsorption step. The resulting homogeneous enzyme preparation was obtained after less than 150-fold enrichment (2), and similar high levels of TR in other transformed cells are indicated by the reported 500-fold purification required for isolation of the enzyme from HeLa cells (6) and 300-fold for Novikoff rat tumor TR (7). In contrast, the levels of TR in rat liver (5) and T cells (4) are very low and required extensive enrichment (3,000- to 5,000-fold) to obtain pure enzyme. Isolation of TR from various other normal tissues also has been reported to require enrichment of 4,000-fold for calf liver (3), 3,200-fold for bovine liver (8), and 4,100-fold for human placenta (9). Whether the differences in immunoreactivity of the TR preparations from rat liver, human T cells, and transformed lung cells are the result of a posttranslational modification that is less efficient in the rapidly growing transformed cells as compared with normal cells, the presence of more than one TR gene product, or other reasons is unknown. It has been reported by several investigators that their TR preparations contain enzyme forms with pKa values varying from 4.7–4.9 (10), 5.2–5.3 (2), 5.2–5.4 (4), and 5.5–5.8 (8). The basis of these differences has not been determined.
As shown in this study, TR preparations isolated from transformed cell lines by the standardized procedures that include chromatography on a 2′,5′-ADP-affinity matrix actually contain two forms of the enzyme that differ in immunoreactivity and also in affinity for heparin agarose. The isolated low heparin-affinity enzymes from the human lung adenocarcinoma cells and also from HeLa cells reacted with rat liver TR polyclonal antibodies in immunoblot assays, whereas the isolated high heparin-affinity enzymes did not. These results are consistent with the original observation (2) that human lung adenocarcinoma TR, which had been purified by heparin-affinity chromatography, failed to react with antibodies elicited to rat liver TR.
MATERIALS AND METHODS
Materials.
[75Se]Selenite (1,000 Ci/mmol; 1 Ci = 37 GBq) was purchased from the Research Reactor Facility (University of Missouri, Columbia). Heparin agarose was from Sigma, a heparin-5PW Progel-TSK HPLC column was from Supelco, and 2′,5′-ADP Sepharose-4B, octyl-Sepharose CL-4B, phenyl-Sepharose CL-4B, butyl-Sepharose-4B, and isoelectrofocusing standards were from Pharmacia LKB. Precast polyacrylamide gels and molecular weight standards were purchased from NOVEX (San Diego). Rabbit antiserum raised against rat liver TR was a gift from H.-J. Kim, H. Z. Chae, and Sue Goo Rhee (National Institutes of Health). Rabbit anti-goat IgG (heavy and light chains), goat anti-rabbit IgG (heavy and light chains), and the BCIP/NBT phosphatase substrate visualization system were purchased from Kirkegaard & Perry Laboratories. Heat-inactivated fetal bovine serum, antibiotic-antimycotic solution (×100), trypsin–EDTA solution (×1), and RPMI 1640 medium were purchased from Mediatech (Herndon, VA).
Cell Culture and Preparation of Extracts.
Human lung adenocarcinoma cells, NCI-H441, from the American Type Tissue Collection, were cultured in monolayers and labeled with 75Se as described (2). The same procedures were used for culture and labeling of HeLa cells. Cells were harvested, washed, and stored as frozen pellets at −20°C until needed. Extracts were prepared and proteins, after precipitation by addition of ammonium sulfate to 80% of saturation and dialysis, were used as enzyme source (2). Purification of 75Se-labeled TR by DEAE-cellulose chromatography followed by affinity chromatography on 2′,5′-ADP Sepharose-4B and then chromatography on phenyl-Sepharose CL-4B was carried out using minor variations of established procedures (2, 3, 5). Radioactivity of protein fractions was determined in a Beckman model 5500 γ counter.
Other Methods.
SDS and native PAGE analyses, immunoblot analysis, and protein determinations were performed as described (2). For detection of 75Se in gels, a PhosphorImager (Molecular Dynamics) was used. TR activity was determined by measuring DTNB reduction or the thioredoxin-dependent reduction of insulin by NADPH (2).
Affinity of the enzyme purified from lung adenocarcinoma cells or from HeLa cells for heparin was determined using either heparin agarose buffered with 50 mM sodium phosphate (pH 7.3) containing 2 mM DTT and 1 mM MgEDTA (buffer A) or a heparin HPLC column equilibrated with the same buffer. The heparin HPLC column was eluted with a 50-min linear gradient of 0–0.5 M NaCl in buffer A at a flow rate of 1 ml/min. For regeneration, the column was washed with 1 M NaCl in buffer A from 51–55 min.
RESULTS
Purified 75Se-labeled TR isolated from human lung adenocarcinoma cell extracts by DEAE-Sepharose chromatography followed by 2′,5′-ADP-affinity chromatography was shown to consist of a mixture of two enzyme forms when subjected to chromatography on heparin agarose (Fig. 1). The protein in the major radioactive protein peak, which was retained on the column and eluted with about 250 mM NaCl, corresponds to the form originally described by Tamura and Stadtman (2). A smaller radioactive protein peak from the lung cell extract was eluted earlier from the column by buffer lacking NaCl. When this low-affinity enzyme fraction was rechromatographed on the heparin column under the same conditions, there was no change in its elution position confirming its low binding property. An enzyme preparation from HeLa cell extract that had been purified by the same procedure using chromatography on DEAE Sepharose and 2′,5′-ADP agarose also was separated into two protein components by passage over the heparin-agarose-affinity column (Fig. 1), but in this case most of the protein was eluted in the radioactive peak that emerged before application of the NaCl gradient and only a small amount of enzyme was found in the salt gradient fractions. In control experiments in which the high heparin-affinity lung cell enzyme was chromatographed on a Bio-Gel A1.5m agarose gel filtration column, the protein appeared in the effluent fraction showing that the matrix alone was not responsible for retention of the enzyme on the heparin-agarose-affinity column. As can be seen in both elution profiles of Fig. 1, the low-affinity protein peaks are nonsymmetrical, indicating the presence of multiple enzyme forms.
Figure 1.

Separation of two forms of TR from transformed cell lines on a heparin-5PW Progel-TSK HPLC column. The enzyme preparations applied to the column had been purified by chromatography on DEAE Sepharose followed by 2′,5′-ADP agarose-affinity chromatography.
Properties of the lung cell enzyme forms shown in Fig. 1 that exhibited high and low affinities for heparin are summarized in Table 1. The 75Se that copurified with both forms of the enzyme is present in the form of selenocysteine residues in the proteins. This selenoamino acid was identified earlier (2) in the high heparin-affinity enzyme purified from lung cells and shown to account satisfactorily for the 75Se content of the protein. In this study, the location of 75Se in a radioactive tryptic peptide derived from both high and low heparin-affinity enzyme forms corresponded to selenocysteine, which was predicted by a TGA codon in the gene (4, 11). Based on migration on SDS/PAGE gels, a subunit molecular weight of about 57,000 is indicated for both enzyme forms (Fig. 2), and protein bands derived from the two forms comigrate when mixed (lane 4) as judged by staining (Fig. 2 Upper) and by detection of radioactivity (Fig. 2 Lower). Catalytic activities of the two enzyme forms, measured by reduction of DTNB with NADPH, are similar (Table 1) and tightly bound FAD is present in both as was reported earlier for the high heparin-affinity enzyme (2). Unlike the high heparin-affinity TR from lung adenocarcinoma cells that failed to react in immunoblot assays with polyclonal antibodies elicited to rat liver TR (2), the low-affinity enzyme from the lung cell line and also from HeLa cells showed strong positive reactions with the same antibody preparation (Fig. 3). The high heparin-affinity HeLa enzyme, like the high-affinity lung enzyme, failed to react with these antibodies in the immunoblot assay under the same conditions (data not shown).
Table 1.
TR from human lung adenocarcinoma cells exhibits differing affinities to heparin
| Low affinity form | High affinity form | |
|---|---|---|
| Selenocysteine in C-terminal | ||
| tryptic peptide | + | + |
| Subunit mass | ||
| by SDS/PAGE | 57 kDa | 57 kDa |
| NADPH linked | + | + |
| Specific activity (DTNB) | 26 units/mg | 32 units/mg |
| Bound cofactor | FAD | FAD |
| Heparin binding | − | + |
| Reaction with rat liver | ||
| polyclonal antibody | + | − |
Figure 2.

SDS/PAGE analysis of low heparin and high heparin-affinity enzyme forms on a 12% polyacrylamide gel. Protein bands stained with Coomassie brilliant blue (Upper) and 75Se-labeled protein bands detected by PhosphorImager (Lower). Lane 1, prestained standard proteins (Novex) in 250, 98, 64, 50, 36, 30, 16, and 6 kDa; lane 2, 1 μg of high heparin-affinity HeLa cell enzyme (2,100 cpm); lane 3, 1 μg of low heparin-affinity human lung adenocarcinoma cell enzyme (1,200 cpm); lane 4, mixture of 1 μg of each (3,300 cpm).
Figure 3.

Immunoblot analysis of low heparin-affinity enzyme forms. TR isolated from human lung adenocarcinoma cells and from HeLa cells was chromatographed on heparin agarose and 1 μg samples of enzyme fractions were subjected to electrophoresis and transferred to a poly(vinylidene difluoride) membrane followed by immunoblotting with a polyclonal anti-rat liver TR antibody (1:1,000 dilution). Goat anti-rabbit IgG (H+L), 5-bromo-4-chloro-3-indolyl phosphate, and nitroblue tetrazolium were used for visualization of reactive protein bands. Transferred standards: 98 kDa at 3.2 cm and 64 kDa at 3.9 cm (lane 1), low heparin-affinity enzyme from lung cells (lane 2) and from HeLa cells (lane 3). The high heparin-affinity enzyme forms were not detected.
To determine the location of the selenocysteine residue in the TR preparations that exhibit differing binding affinities to heparin, both of the 75Se-labeled enzyme forms isolated from the human lung adenocarcinoma cell line were reduced, carboxymethylated, and subjected to trypsin digestion as described (2, 4). A radioactive selenopeptide isolated from each of these enzyme preparations by C18 reverse-phase HPLC was subjected to automated Edman degradation on Hewlett–Packard Protein Sequencer G100A (4).
Sequence data for the tryptic peptides derived from the carboxymethylated heparin high-affinity and the heparin low-affinity enzymes are shown in Fig. 4. The amino acid identified in each cycle is identical to the predicted amino acid sequence of the C-terminal region of human placental TR (11). The peak of radioactivity eluted from both peptides at cycle 11 corresponds to the position of selenocysteine deduced from the presence of a TGA codon in the gene sequence of human placental TR (11). Furthermore, identification of dehydroalanine in cycle 11 of the analytical profile of the peptide from heparin low-affinity enzyme provided additional evidence of the occurrence of the unstable selenocysteine derivative at the corresponding position. Cysteine was identified in cycle 10 of both peptide sequences as the carboxymethyl derivative. Except for the C-terminal glycine residue, which was not identified in cycle 12 in either sample, the amino acids identified in cycles 1–11 correspond to the predicted gene sequence of a C-terminal tryptic peptide produced by cleavage after an arginine residue (Fig. 4). In this deduced sequence, reported originally for human placental TR, the TGA codon was interpreted as a stop codon (11), whereas it was shown to correspond actually to the elution position of 75Se present in a tryptic peptide derived from human T cell TR (4). The C-terminal residue of the T cell peptide, although obtained in low yield, was identified as glycine, which corresponds to GGT in the gene sequence and the actual stop codon therefore is TAA (Fig. 4).
Figure 4.

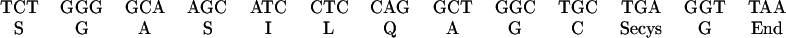 |
DISCUSSION
Our recent unexpected finding that TR from mammalian cells contains a selenocysteine residue preceded by cysteine near the C terminus of each subunit indicates that in enzymes of this class there is one more redox center in addition to the pair of redox-active cysteine residues located in the amino-terminal domain. This serves to further differentiate the mammalian enzyme from the smaller TRs of Escherichia coli and yeast origin, which are not selenoenzymes. In the 116-kDa mammalian TRs, the redox-active cysteines are separated by four amino acid residues in a motif within a putative FAD-binding domain of the amino-terminal region of each 55-kDa subunit. In human placenta TR this motif is -Cys-59 X X X X Cys-64- (11). In contrast, the E. coli and yeast enzymes are 70-kDa homodimers of 34-kDa subunits, each of which contains two redox-active cysteines separated by two amino acid residues in a -Cys X X Cys- motif (12). In the E. coli enzyme the redox-active disulfide is located within the NADPH-binding domain in the amino-terminal half of each subunit. Both types of TRs are flavoproteins that contain tightly bound FAD, use NADPH as electron donor, and normally catalyze the reduction of the disulfide forms of thioredoxins.
The occurrence of a selenocysteine residue in the C-terminal sequence (-Cys-497 Secys-498 Gly 499) of mammalian TR (4, 11) introduces entirely new possibilities concerning the mechanism of action of this redox enzyme. The involvement of the selenocysteine and possibly the adjacent cysteine as an additional redox center in the overall electron transport pathway and interaction of this center with FAD and the redox-active cysteine residues in the amino-terminal domain of the enzyme are clearly suggested by the similar occurrence in mercuric ion reductase (13) of a pair of cysteines, residues 558 and 559, near the C terminus, in addition to the active-site redox-active disulfide and FAD. The fact that the double mutant Cys/558/Ala, Cys/559/Ala exhibited greatly reduced mercury ion reductase activity in vitro and increased sensitivity to HgCl2 in vivo showed that at least one of these cysteine residues is essential for optimal enzyme function. From a number of lines of evidence it has been suggested that the Cys/558-Cys/559 pair are positioned so that they are actually part of the active site together with the previously determined redox-active disulfide, Cys/135 and Cys/140 (14).
As pointed out in the Introduction, an initial search (2) for a putative selenocysteine containing cytochrome P450 in a human lung adenocarcinoma cell line led to the discovery that mammalian TR is a selenoprotein. Although the monomeric cytochrome P450 is similar in mass to the mammalian TR subunit, the native proteins are readily distinguishable on the basis of size and identities of their bound chromophores. Another mammalian selenoprotein, selenoprotein P, which contains up to 10 selenocysteine residues, occurs as a 57-kDa monomer in its glycosylated form (15). When labeled with 75Se and monitored on SDS/PAGE gels, selenoprotein P could be difficult to distinguish from TR based on mobility alone. Thus, in the earlier experiments (2), the 75Se-labeled unknown protein isolated from human lung adenocarcinoma cells by heparin-affinity chromatography was subjected to deglycosylation tests as described by Read et al. (15). No glycosyl groups were detected on the radioactive protein and, in addition, it failed to crossreact with anti-human selenoprotein P polyclonal antibodies. Thus, two selenoproteins, the putative cytochrome P450 and selenoprotein P, that might occur in lung cells, were clearly distinguished from the unknown radioactive isolated protein. Although limited amounts of this protein were available, its identity as TR could be established on the basis of cofactor content, catalytic activity, and other properties (2). In some instances, glycoproteins exhibit appreciable affinity for heparin, but the failure to detect glycosyl groups on the high heparin-affinity form of lung TR using N-glycosidase tests, periodate oxidation, and the dansyl hydrazine staining assay (2) indicates that glycosylation is not a likely determinant in binding of this TR to heparin. Lack of glycosyl groups on the low heparin-affinity form of lung TR also is indicated by the fact that it is similar in size, as judged by its comigration on SDS/PAGE, with the high heparin-affinity form as shown in Fig. 2. From these data it appears that mammalian TR is not a glycoprotein, although this has been suggested as an explanation of the discrepancy in molecular mass values determined by gel filtration chromatography and sucrose density centrifugation in the case of the human placenta enzyme (9).
The differences in affinity for heparin demonstrated by the native forms of TR isolated from the human lung adenocarcinoma and HeLa transformed cell lines could be due to variable amounts of covalent derivatives or tightly bound compounds. The failure of the high heparin-affinity form of the enzyme to react with the polyclonal antibodies that crossreact with the low-affinity form of the enzyme, even after SDS/PAGE analysis and electrotransfer to membranes, implies a difference based on the presence or absence of a somewhat stable modified form. The nonreactive form of the enzyme originally isolated from the lung cells by heparin-affinity chromatography was blocked at the amino terminus (2), as was also the enzyme isolated from human T cells that did crossreact with the same polyclonal antibodies (4). Thus, the presence or absence of a ligand on the amino group of the N-terminal residue of the protein should not be the variable. Furthermore, the presence of identical 12-residue selenocysteine-containing peptides in the C-terminal domains of both enzyme forms show that this potentially labile region of the protein is not modified. Lack of some relatively stable modification that prevents binding to heparin, but is essential for antibody recognition in the immunoblot assay, is one possible explanation of the phenomenon observed. In normal cells that contain low levels of mammalian TR, the efficiency of such a putative modifying system could be sufficient to fully derivatize the enzyme, but in rapidly growing transformed cells the enzyme could be incompletely modified resulting in a mixture of enzyme species. Thus, the low heparin-affinity enzyme from transformed cells would be the modified form and the high heparin-affinity enzyme form would lack the adduct. As pointed out in the Introduction, the wide variation in isoelectric points reported for mammalian TRs, and in several cases the presence in a single enzyme preparation of multiple species with differing pI values, suggests a normal heterogeneity in these enzyme populations.
ABBREVIATIONS
- TR
thioredoxin reductase
- DTNB
5,5′-dithiobis(2-nitrobenzoic acid)
References
- 1.Yamano S, Nhamburo P T, Aoyama T, Meyer U A, Inaba T, Kalow W, Gelboin H V, McBride O W, Gonzalez F J. Biochemistry. 1989;28:7340–7348. doi: 10.1021/bi00444a029. [DOI] [PubMed] [Google Scholar]
- 2.Tamura T, Stadtman T C. Proc Natl Acad Sci USA. 1996;93:1006–1011. doi: 10.1073/pnas.93.3.1006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Holmgren A. J Biol Chem. 1977;252:4600–4606. [PubMed] [Google Scholar]
- 4.Gladyshev V N, Jeang K-T, Stadtman T C. Proc Natl Acad Sci USA. 1996;93:6146–6151. doi: 10.1073/pnas.93.12.6146. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Luthgren M, Holmgren S. Biochemistry. 1982;21:6628–6633. doi: 10.1021/bi00269a003. [DOI] [PubMed] [Google Scholar]
- 6.Tsang M L-S, Weatherbee J A. Proc Natl Acad Sci USA. 1981;78:7478–7482. doi: 10.1073/pnas.78.12.7478. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Chen C-C, Borns-McCall B L, Moore E C. Prep Biochem. 1977;7:165–177. doi: 10.1080/00327487708061633. [DOI] [PubMed] [Google Scholar]
- 8.Martinez-Galisteo E, Padilla C A, Garcia-Alfonso C, Lopez-Barea J, Barcena J A. Biochimie. 1993;75:803–809. doi: 10.1016/0300-9084(93)90131-b. [DOI] [PubMed] [Google Scholar]
- 9.Oblong J E, Gasdaska P Y, Sherrill K, Powis G. Biochemistry. 1993;32:7271–7277. doi: 10.1021/bi00079a025. [DOI] [PubMed] [Google Scholar]
- 10.Chen C-C, Moore E C, Borns-McCall B L. Cancer Res. 1978;38:1885–1888. [PubMed] [Google Scholar]
- 11.Gasdaska P Y, Gasdaska J R, Cochran S, Powis G. FEBS Lett. 1995;373:5–9. doi: 10.1016/0014-5793(95)01003-w. [DOI] [PubMed] [Google Scholar]
- 12.Holmgren A. Annu Rev Biochem. 1985;54:237–271. doi: 10.1146/annurev.bi.54.070185.001321. [DOI] [PubMed] [Google Scholar]
- 13.Moore M J, Walsh C T. Biochemistry. 1989;28:1183–1194. doi: 10.1021/bi00429a036. [DOI] [PubMed] [Google Scholar]
- 14.Miller S M, Moore M J, Massey V, Williams C H, Jr, Distefano M D, Ballou D P, Walsh C T. Biochemistry. 1989;28:1194–1205. doi: 10.1021/bi00429a037. [DOI] [PubMed] [Google Scholar]
- 15.Read R, Bellew T, Yang J-G, Hill K E, Palmer I S, Burk R F. J Biol Chem. 1990;265:17899–17905. [PubMed] [Google Scholar]