

Abstract
The characterization of protein-folding kinetics with increasing chain length under various thermodynamic conditions is addressed using the capillarity picture in which distinct spatial regions of the protein are imagined to be folded or trapped and separated by interfaces. The quantitative capillarity theory is based on the nucleation theory of first-order transitions and the droplet analysis of glasses and random magnets. The concepts of folding funnels and rugged energy landscapes are shown to be applicable in the large size limit just as for smaller proteins. An ideal asymptotic free-energy profile as a function of a reaction coordinate measuring progress down the funnel is shown to be quite broad. This renders traditional transition state theory generally inapplicable but allows a diffusive picture with a transition-state region to be used. The analysis unifies several scaling arguments proposed earlier. The importance of fluctuational fine structure both to the free-energy profile and to the glassy dynamics is highlighted. The fluctuation effects lead to a very broad trapping-time distribution. Considerations necessary for understanding the crossover between the mean field and capillarity pictures of the energy landscapes are discussed. A variety of mechanisms that may roughen the interfaces and may lead to a complex structure of the transition-state ensemble are proposed.
Proteins can be thought of as mesoscopic systems; that is, they are large in atomistic terms but are too small to be completely analyzed by macroscopic reasoning alone. The energy landscape theory of protein folding describes folding kinetics through a statistical characterization of the energies of different conformations (1, 2). This makes a bridge with the theory of phase transitions in disordered systems. The theory suggests that rapidly foldable proteins have an energy landscape dominated by a funnel to a large basin of attraction, the native state, and many small rugged features that give rise to trapping in local minima (3–5). Reliable folding requires the guiding forces of the funnel to be strong enough to overcome the entropy of the unfolded states. This must occur at a temperature such that trapping (also favored at low temperature) is not so strong that flow down the funnel is too slow. Much of the qualitative content of this picture has been confirmed through the computer simulation of small lattice models of proteins (5–8). Quantitatively, the analytical theories of folding landscapes have often made use of mean field descriptions that should be most accurate when the protein is small enough that we can think of each rearranging subunit of the chain as being able to interact with a fair fraction of the others at any time (1, 2, 9–13). This is true of the smallest lattice models. If the rearranging units are appropriately taken to be small segments of helices rather than individual residues, it is also true for the smallest globular proteins in nature, which have about 70 residues (4).
This paper discusses how the folding dynamics of larger proteins can be described in terms of funnels and landscapes also in the opposite limit, where the protein is much larger than the range of the interresidue forces and the local correlations. In this limit we can define an interface or front between the folded and unfolded parts of a protein. Similar fronts, which are relatively free to move, exist between segments of the chain that are trapped in incommensurate misfolded configurations. To obtain a complete picture of the problem, these two aspects must be combined. The resulting capillarity model seeks to address the asymptotic behavior of folding kinetics with increasing protein size. As for the funneled aspects of the landscape, the capillarity picture is just like that of a first-order phase transition in a cluster. Regarding the rugged features of the landscape, the capillarity picture of the energy landscape is based on the droplet model of glasses, spin glasses, and random ferromagnets (14–19). For at least a decade, despite many simulation studies and much analysis, it has remained controversial at what size, if any, such disordered systems exhibit a crossover from the mean field behavior to the one envisioned by droplet arguments. Experiments on mesoscopic spin glasses show features in common with both limits (20). One apparent difference between the predictions of mean field and droplet theories is the way kinetics changes with size. But it is hard to convincingly debate this issue in the biophysical context because natural proteins, being the result of evolution, are not truly scaleable at the experimenter’s discretion. Instead, large proteins typically have identifiable domains. Although the mean field theories of folding and trapping give barriers scaling linearly with chain length, these barriers still are predicted to be modest for proteins in the size range of a typical naturally occurring domain (11). Weaker scaling with chain length than mean field theory has already been suggested several times (21–23). Finkelstein and Badredtinov (21) obtain a barrier scaling like N2/3, using the capillarity picture in an elegant way that, however, neglects ruggedness of the landscape. Thirumalai (22) focuses on the rugged features of the landscape and uses the scaling theory for glasses (18, 19) to obtain a barrier scaling as N1/2. Simulations of proteins selected from a random pool to be fast folders or designed using the minimal frustration principle suggest a logarithmic scaling of the barrier height at the temperature of fastest folding but linear scaling with N of slow-folding events at low temperature (23). The general treatment here unifies these disparate arguments and observations by highlighting the importance of fluctuations and of how the barriers depend on thermodynamic state as well as chain length. The treatment also emphasizes that the dynamics in the capillarity picture is totally consistent with the funneled landscape pictures used phenomenologically.
The organization of this paper is as follows: In the first section the funneled aspects of the landscape are discussed within the capillarity approximation. The similarity with the kinetic description usually discussed via mean field theory is made clear. In the next section the glassy landscape ruggedness is discussed using an explicit capillarity-based picture generalizing Thirumalai’s scaling arguments. These aspects are combined to discuss the “typical” behavior of folding times for a large protein. The “fine structure” of the free-energy profile and other fluctuation effects are discussed in the following section. This analysis shows the delicacy required of computer simulations addressing size scaling issues and why fluctuation effects need to be considered in interpreting recent simulations. Further questions raised, especially involving the crossover between the mean field and capillarity approximations to the landscape, are discussed in the final section.
Capillarity Description of a Folding Funnel.
Bryngelson and Wolynes (9) examined folding barriers with capillarity ideas in 1990. Following classical nucleation theory for first-order transitions, they wrote the free energy in terms of a progress coordinate, the number of residues folded, Nf. Their expression contains a linear “bulk” term and an interfacial term scaling like Nf2/3:
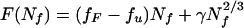 |
1 |
The bulk term depends on the free-energy difference per particle Δf between folded (fF) and unfolded (fu) protein; the small value of Δf = fF − fu under folding conditions reflects the near cancellation of the entropy of unfolded state and the stabilizing energy of the native structure. The “interface” term γ was taken to be largely energetic. Putting in the typical stabilization of proteins under physiological conditions (10kBT), they concluded that of the order ≈100 residues need to be ordered at the folding transition state, a number comparable to a protein domain size. At this level, that a biopolymer is being studied is largely immaterial and, in fact, nearly the same description was used somewhat earlier by Reiss et al. to describe mesoscopic cluster freezing transitions. In that context they made an observation that is important for proteins as well: the bulk transition temperature at which Δf vanishes does not coincide with the transition temperature of the cluster TF. The temperature at which the two global free-energy minima have the same free energy is depressed by the surface contribution, as has long been known for the boiling of drops (25). Following Reiss et al. (24), we can then rewrite Eq. 1 by showing explicitly the temperature dependence of the free energy referenced to the transition temperature
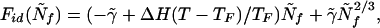 |
2 |
where we have written γ̃ = γN2/3 and scaled Nf by the chain length N. At TF, this is a crudely universal form for the “ideal” free-energy profile since Fid(0) must equal Fid(1). The temperature dependence of the stability depends on the enthalpy of unfolding ΔH. In this expression we see the normalized folded fraction Ñf = Nf/N can be used as a progress coordinate for the unfolding reaction here just as reaction coordinates highlighted in other theories of the funnel. The specific numeric coefficients above (which are used for concreteness in this paper) assume the protein or cluster is nearly spherical, so the curvature of the front is equally limited by all dimensions of the protein (see Fig. 1).
Figure 1.

A schematic view, according to the capillarity picture of structure of a protein (lysozyme is shown for illustration) once it is advanced partially down the folding funnel. When several phases of the protein are possible, they can lead to a rather complex interface, as illustrated here. Thermal- and disorder-induced roughening of the interfaces can smooth out the transition region seriously. The figure shows several kinds of ordering that are possible: a region in which side chains are completely ordered (blue), a transition zone with topologically correct folding but no side-chain order (red), a molten-disordered phase (yellow), and the random coil (green). Having such a gradual progression of phases can lower the activation energy but broadens the interface between completely folded and unfolded regions. The front is shown, for clarity, as progressing from one end of the protein to the other. The most well ordered part may be buried in the core, depending on the relative surface tensions between the phases and the solvent.
At TF the free-energy profile is a rather broad curve as shown in Fig. 2. The maximum occurs at Nf≠ = 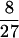 N. The barrier can be ascribed to the interface term and is given by ΔF≠ =
N. The barrier can be ascribed to the interface term and is given by ΔF≠ = 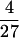 γN2/3. This is the barrier scaling obtained by Finkelstein and Badredtinov (21), using a more elaborate treatment of the interface contributions and a more careful treatment of the protein shape. These effects can be important; for example, if the protein is cylindrical, the front will orient orthogonal to the long axis and the barrier will not depend on the total cylinder length, a case reminiscent of coiled coils such as gcn4, where folding was recently studied (26).
γN2/3. This is the barrier scaling obtained by Finkelstein and Badredtinov (21), using a more elaborate treatment of the interface contributions and a more careful treatment of the protein shape. These effects can be important; for example, if the protein is cylindrical, the front will orient orthogonal to the long axis and the barrier will not depend on the total cylinder length, a case reminiscent of coiled coils such as gcn4, where folding was recently studied (26).
Figure 2.

The funnel free-energy profile in the capillarity approximation. The solid curve is the ideal profile of the free energy versus the folded fraction, assuming no heterogeneity of stabilization energy. An actual profile differs by a fluctuation that is random and Gaussianly distributed due to this heterogeneity. This is the “fine structure” of the folding funnel. A specific such profile is indicated by a dotted line. Notice the cusp-like minima occur at precisely Ñf = 0 and Ñf = 1, because the capillarity theory is an asymptotic result for large NTOT. Simulations and mean field theory show smooth minima at nonextreme values of the order parameters.
The breadth of the free-energy profile has consequences for the kinetic description and is reflected in the size of the transition state region δNTST, defined as the range over which F(N) changes by kBT. This range in the fractional progress coordinate is δÑTST = 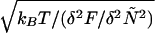 . Using Eq. 2 at TF, the number of residues displaced in moving over the transition region is approximately δNTST ≅ 2.
. Using Eq. 2 at TF, the number of residues displaced in moving over the transition region is approximately δNTST ≅ 2.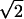 /3N2/3
/3N2/3 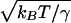 , showing the barrier becomes broader in terms of displaced residues with increasing chain length. An elementary move in folding dynamics is thought to involve displacing a loop (27) whose length scales like N1/3. Thus, crossing this region will take many elementary moves and will be expected to be at least at the border of diffusive behavior. Even in the small-size regime, Socci et al. (28) have shown that, for the 27-mer lattice model, the diffusive dynamics works quite well. Thus, we see at TF, the capillarity argument, like that of the mean field, suggests that Ñf can be taken as a reaction coordinate but must be treated as diffusive and traditional transition state theory should not be used. The only difference from the mean field funnel description is that capillarity theory assumes most contacts made in the partially folded protein are contiguous in physical space, whereas mean field estimates allow them to spread out. The capillarity model, therefore, obtains a different scaling of the thermodynamic barrier with chain length, but the phenomenological analysis is unchanged. It is worth noting that predictions of mean field barrier heights (11) at TF actually are smaller than Finkelstein’s (21) specific estimates for barriers even for N ≈ 100, suggesting that the contiguous configurations assumed by capillarity need not dominate in this size range over a set of more diffuse arrangements of contacts with greater diversity. Eventually as temperature decreases when T ≪ TF, the curvature of the fronts will be large and the barrier to folding will be nearly independent of chain length. Polymeric effects of chain connectivity should give a weak logarithmic dependence on size for the purely thermodynamic barrier (22).
, showing the barrier becomes broader in terms of displaced residues with increasing chain length. An elementary move in folding dynamics is thought to involve displacing a loop (27) whose length scales like N1/3. Thus, crossing this region will take many elementary moves and will be expected to be at least at the border of diffusive behavior. Even in the small-size regime, Socci et al. (28) have shown that, for the 27-mer lattice model, the diffusive dynamics works quite well. Thus, we see at TF, the capillarity argument, like that of the mean field, suggests that Ñf can be taken as a reaction coordinate but must be treated as diffusive and traditional transition state theory should not be used. The only difference from the mean field funnel description is that capillarity theory assumes most contacts made in the partially folded protein are contiguous in physical space, whereas mean field estimates allow them to spread out. The capillarity model, therefore, obtains a different scaling of the thermodynamic barrier with chain length, but the phenomenological analysis is unchanged. It is worth noting that predictions of mean field barrier heights (11) at TF actually are smaller than Finkelstein’s (21) specific estimates for barriers even for N ≈ 100, suggesting that the contiguous configurations assumed by capillarity need not dominate in this size range over a set of more diffuse arrangements of contacts with greater diversity. Eventually as temperature decreases when T ≪ TF, the curvature of the fronts will be large and the barrier to folding will be nearly independent of chain length. Polymeric effects of chain connectivity should give a weak logarithmic dependence on size for the purely thermodynamic barrier (22).
In using capillarity arguments we note there are two reasons proteins may have quite a complicated structure for the interface between “folded” and “unfolded” regions. First, thermal fluctuations and heterogeneity of native contacts roughen the interface. Indeed, because of the reduction in the number of neighbors when going from three dimensions to the two dimensions of the interface, the roughening temperature of the interface could be quite a bit lower than the folding transition (29). Another source of interfacial width is the possible existence of what can be thought of as multiple phases of the protein in bulk. Even when the equilibrium unfolded state is expanded, a metastable compact liquid or even liquid crystalline state, or one topologically correct without side chain ordering may exist and have been theoretically suggested. Being intermediate in structure, these phases normally will partially wet the interface between completely folded and unfolded states, as shown in Fig. 1. This reduces the interface energy γ and the thermodynamic activation barrier. Thus, for instance, although in the interior of the folding phase side chains may be ordered, they need not be ordered in the interface. If the interface is very rough and wide enough to be comparable to the diameter of the protein, the capillarity picture will be inappropriate, but one crosses over to a more mean field-like description with contacts spread everywhere.
Because of the diffusive nature of the barrier crossing at TF, the time for folding will involve both the thermodynamic barrier ΔF≠ and the reconfiguration times, which themselves can involve an activated barrier crossing. The description using diffusive theory (2, 8) gives a simple dependence on the typical reconfiguration time in the unfolded state, τ0RC:
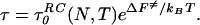 |
3 |
In the case where T ≪ TF, with its resulting “small” folding nucleus, the free-energy profile will be sharper at the transition-state region, allowing the barrier crossing to be less diffusive and more direct as in transition-state theory. If the inherent thermodynamic barrier is small, however, the subsequent growth of the folded region for a large enough protein can be rate limiting. The time for this thermodynamically downhill step will also be proportional to τ0RC. Within the capillarity picture, as in mean field theory, the reconfiguration times depend on escaping from traps and on the ruggedness of the landscape. These effects have been neglected in earlier work by focusing primarily on the case where the unfolded part of the chain is not compact (21). This requires TF to exceed the random heteropolymer collapse transition, TC, otherwise an additional barrier for expanding the chain, scaling with N, is introduced. TC is a fortiori even higher than the glass transition temperature, so ruggedness effects can be neglected for such a well designed, minimally frustrated protein. It is not yet clear whether proteins are in fact frustrated so little. If the stability gap is not so large as to allow TF to exceed TC, the unfolded segment will be collapsed and possibly transiently misfolded. We must then consider trapping in determining the N and T dependence of the trap escape processes that enter τ0RC.
Capillarity Description of Reconfiguration Dynamics.
The capillarity description of first-order phase transitions is well established by experiments documenting the reduction of freezing temperature with size. The size dependence of glassy phenomena, the sort needed to understand trapping, is hardly explored. In mesoscopic samples of magnetic spin glasses, elegant experiments of Weissman (20) show behavior reminiscent of mean field theories, although in some cases droplet-like excitations seem to be involved, too. Experiments on glass transitions of fluids confined to pores also sometimes show a reduction in TG, but there are complications due to surface anchoring (30). Nevertheless, it is worth examining where theory leads.
The glass transition of random heteropolymers is believed to be a “random first-order transition” involving a discontinuous change in heat capacity and an entropy crisis similar to the supposed ideal glass transition of supercooled liquids (31, 32). The scaling theory of dynamics of liquids approaching such an ideal glass transition (18–19) has been formulated partially in terms of capillarity arguments. We can explicitly adapt those arguments to discuss the finite size effects appropriate to proteins. Above the thermodynamic transition a system that will undergo a random first-order transition can be thought of as a system that has an exponentially large number of metastable phases. In the heteropolymer context these are the trapping states. Above a dynamic transition temperature TA, which is greater than TG, each of those phases becomes even locally unstable to thermal motions. Above TA, the chain dynamics is slowed down by caging but is much like a free chain. Below this temperature, on the other hand, when the interactions are short ranged, reconfigurational movements become slow and activated. The driving force for escape from one to another is the configurational entropy density. When the system is trapped in a given minimum it costs energy to move into another one, but there are so many ways to do this that escape becomes favorable. If a region of size Ne monomers to reconfigure the free-energy cost is
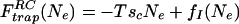 |
4 |
where sc is the configurational entropy per residue and fI(Ne) is an interfacial energy term representing the mismatch between the configurations, each one of which is a local minimum, naively the interface energy fI would scale as a surface term Ne2/3. A detailed approximate calculation for the random heteropolymer yields this result (33) for the interface energy and an approximate quantification of trapping barriers. To understand the scaling we must recognize also that the existence of many “phases,” which represent proteins in globally different local minima, however, reduces this surface energy by means of a wetting phenomenon such as the one we discussed for a finite number of phases at the end of section two of this paper.
As discussed earlier for specific, qualitatively different phases, a series of different phases also can decorate the interface, making it very broad and lowering the barrier. For highly curved interfaces fI will behave like a surface term, but as the reconfiguring region increases in size, the effective surface tension will diminish through wetting as the interface straightens. A detailed argument much like the one for the random field Ising magnet (16, 18) finally gives a contribution that scales like αΔENe1/2, where ΔE is the root mean square of the energy variance per particle of the trapped states. In more elaborated theories of random systems there can be a more anomalous scaling such as Ne1/2+x. The maximum of F(Ne) gives the barrier for reconfiguration, ΔFtrap≠RC and we can write τ0RC = τ0expΔFtrapRC/kBT, as the typical reconfiguration time. For an infinite system the critical size corresponding to the maximum is Ne≠ = (αΔE/Tsc)2 and the barrier is ΔFtrap≠RC = α2ΔE2/2Tsc. The barrier is the same as the empirical Vogel–Fulcher law and depends on the configurational entropy density just as in the Adam–Gibbs description of glassy dynamics (31), but the number of units in the reconfiguring region is quite a bit larger than the Adam–Gibbs result for the critical size that scales only like the sc−1 rather than sc−2 predicted by random first-order transition scaling, where the so-called correlation length exponent is ν = 2/d.
The capillarity argument shows that well above TG the reconfiguration barriers are finite (i.e., they do not scale with chain length) just as the thermodynamic barriers to folding are chain length-independent far below TF. Nevertheless there is a connection of the capillarity result to the famous Levinthal estimate, which arises in the mean field theory. We can see that at any temperature, the reconfiguration barrier is ΔFtrap≠RC = ½scNe≠ = ½S(Ne≠). This is one-half of a renormalized Levinthal entropy for the critical sized drop S(Ne≠). The barrier now depends on the actual configurational entropy of the critical drop, not the infinite temperature value of the entropy of the entire system So. The barrier thus grows as TG approaches, but more slowly than in the mean field theory, because the random wetting phenomenon allows the entropy to be gained in stages. At TG the energy landscape for an infinite random heteropolymer is self-similar for different size regions. There are minifunnels within minifunnels.
Once Ne≠ grows to a size such that the rearranging region is comparable to the protein diameter, the interface cannot get any larger. Within the capillarity approximation, the driving force term can now be neglected, and again the barrier involves moving a front between different trap states across the molecule, giving a barrier ΔFtrap≠RC = αΔEN1/2. This is the result obtained more elliptically by Thirumalai (22). We now see it is an asymptotic result valid near TG.
Reconfiguration dynamics then gives a contribution to the apparent folding barrier that is independent of N at high T and scales like N1/2 for T near TG.
The same wetting phenomenon operating for traps can change the scaling of the thermodynamic barrier, too. If the randomness is large enough, the native state is hardly distinguishable from any other trap in energetics. The resulting multiple-phase wetting resembles disorder-induced roughening of the interface.
When we combine the results for the thermodynamic barriers and the typical configurational diffusion barriers in the diffusive barrier-crossing expression, we obtain various different scalings of folding time for a “typical” protein with chain length under different thermodynamic conditions, as shown in Fig. 3. For very well designed proteins that can be made to stably fold well below TF but above TG, the asymptotic scaling of folding time is polynomial in chain length. There is no contradiction with well known NP completeness theorems (34) about predicting global minima of a random heteropolymer because well designed proteins are selected, minimally frustrated systems. If still above TG but near TF, the scaling for the time grows exponentially in size but the barrier rises sublinearly as N2/3 in Finkelstein’s (21) calculation. Near to TG, the scaling even for a well designed protein again would be exponential but with a different power, N1/2, for the barrier, as Thirumalai (22) suggests. This is also the behavior of the typical folding time for an unselected random heteropolymer.
Figure 3.

The various regimes of scaling behavior for the typical folding time with length are indicated for different values of TF/T, the folding temperature-to-ambient ratio, and TG/T, the glass temperature-to-ambient ratio. At high temperatures, folding is always thermodynamic uphill and the barrier grows like N. At low temperatures, the rate is trap-dominated, and the typical barrier behaves like N1/2, whereas the slowest escape (not indicated in the figure) will have bigger barriers, scaling like N. Fastest folding occurs for T ≪ TF but T ≫ TG where polynomial behavior of the folding time in N is to be found. This occurs for “well designed” proteins. The figure also shows the boundary between trap escape and quasi-free chain dynamics, as indicated by the mean field dynamic transition temperature TA. A realistic value of TF/TG is 1.6. In this figure we assume temperatures are all less than the collapse temperature TC. If T is bigger than TC, ruggedness effects can be neglected.
Fine Structure of the Funnel Free-Energy Profile and of Reconfiguration Dynamics.
The simplest phenomenological funnel description of folding either in mean field or capillarity approximations maps folding kinetics near TF onto the diffusion of a one-dimensional progress coordinate. More coordinates are needed if different phases are taken into account (e.g., secondary structure formed versus disordered, collapsed versus noncollapsed, etc.), but another effect is also important. At any given value of the progress coordinate, in a specific protein, different contacts will be made to varying extents, depending on their energetics. Although the ideal funnel has a fairly predictable shape in either approximation, the heterogeneity of contact energies and local propensities modifies the free-energy profile for any specific protein, as shown in Fig. 2. This heterogeneity can be probed experimentally by protein engineering (35) and by elegant NMR experiments (B. A. Shulman, P. S. Kim, C. M. Dobson, and C. Redfield, personal communication). For specific proteins the resulting structural correlations can be discussed using free-energy functionals like those used for liquids or random magnets (37).
We now examine what the capillarity arguments give for the fluctuation effects on folding and trapping free-energy profiles. Let us assume the fluctuations in stabilization energy for different residues are small and independent, with a typical value being Δeo. We expect Δeo to be somewhat smaller than the ruggedness energy scale for compact states, ΔE. The ratio Δeo/ΔE depends on the flexibility of the sequence code used for encoding the protein. At TF, if the interface is sharp, a specific protein free-energy profile will differ from the ideal profile by a random contribution δFrand(Ñf) so that F(Ñf) = Fis(Ñf) + δFrand(Ñf). δFrand(Ñf) will be characteristic of a diffusion process in Nf that must return at the end of its journey to the origin while never crossing the origin, if folding a single domain. Thus, δF(Ñf) is typically Δeo N1/2(Ñf(1 − Ñf))1/4. At TF the transition state of the ideal funnel is nearly midway. Thus, the fluctuation in barrier height is ∼½ΔeoN1/2. The thermodynamic activation barrier will be approximately Gaussianly distributed with this width. Taking the rather large value of Δeo ≈1kBT, one finds fluctuations as big as 5kBT for a 100-mer. In fact, if δΔF≠ is sufficiently large to overcome ΔFideal≠, the folding event will be broken up into two parts (i.e., a kinetic intermediate will exist). The probability of finding a sequence that folds 100 times faster than typical is about one-third for such a large Δeo. It becomes necessary in computer simulations to examine the distribution of measured rates rather than, for example, selecting (after the fact) the fastest folders, if the model is to be compared with analytical work.
There are many different trapping configurations; simply knowing their typical behavior is not enough. Fine structure effects on the distribution of trapping escape times are predicted to be especially large (and more subtle than on the thermodynamic folding profile) if the capillarity argument is followed. Adding a Gaussian random contribution to Eq. 4, which represents fluctuations in energy of the trapped states contacts just as was done for the folding profile, the typical trap escape free-energy profile becomes widely distributed because now the renormalized interfacial energy term scales in exactly the same way with chain length as the randomness. The mean and variance of the escape barriers are comparable. Equivalently said, the assumed scaling exponent, x = 0, is marginal for a random system’s transition (39).† Although escape from the typical and, more importantly, the deepest traps will significantly slow as TG is approached, there is now a significant chance that trapping can be avoided altogether through motions involving one of these low barrier traps, even for long chains. The wide distribution suggests that trapping dynamics will be strongly multiexponential as TG is approached. Even when the overall thermodynamic barrier still is large enough to give single exponential kinetics, the widely distributed trapping times can modify the transmission coefficient computed using the pure diffusion description of the barrier crossing; instead, a frequency-dependent Kramers theory should be used. This acts to diminish the influence of trapping on the rate, since the weaker traps will be used in the crossing. At low temperatures, once the thermodynamic barrier is small (“downhill kinetics”), the entire folding kinetics will be nonexponential and reflect the trap distribution directly. As in the kinetic partitioning phenomenology (40, 41), a small fraction of protein molecules can fold on a fast track, while the bulk of them will be trapped in misfolded states.
Although the heterogeneity effects on trapping can allow a small fraction of fast track folders, they also lead to a finite fraction of very slow folding molecules. The capillarity picture yields a roughly Gaussian form (except near ΔF = 0) for the distribution of escape barriers
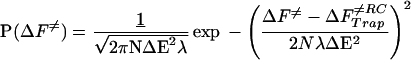 |
5 |
so the distribution of long escape times is roughly log normal. At TG, the most probable escape time scales as N1/2 as discussed in the previous section, but the mean folding time is dominated by the deepest traps. Averaging the escape time, which depends exponentially on the barrier over the log normal distribution, the apparent activation energy for the slowest events then scales linearly in N, just as it does from mean field theory. Bigger molecules can sample less and less typical, deeper traps. The linear dependence of the longest escape time was noted in recent simulations and may also be due to topological constraints on motion (23). This wide variance of escape times also was predicted at all temperatures on the basis of the random energy model (2). It is interesting that it persists below TG in the capillarity description as well. The effects of native contact heterogeneity near TF and trapping due to glassy dynamics near TG are very similar and scale in the same way with N. The fluctuations away from the ideal funnel free-energy profile will typically act to slow the folding from the ideal profile result, because it is the big barriers that count for a long, random walk in a one-dimensional random potential (42). Below TG, folding times, even of well designed proteins, will be quite sensitive to sequence.
Both the fluctuational fine structure of the free-energy profile and the broad distribution of trapping times from the glassy dynamics suggest the largest proteins will fold near TG by a highly intermittent, partially progressive escape from traps, much like a Levy flight (42). This may be studied using single-molecule detection techniques.
Discussion.
The analysis presented here shows that the energy landscape description of protein folding has much the same mathematical structure within the capillarity approximation as when mean field theories are used. Just as in mean field approaches, a folding funnel with a diffusive progress coordinate can be defined. Folding kinetics reflects both the free-energy profile of this coordinate and the reconfigurational motions that depend on the nature of glassy dynamics representing escape from local traps. The questions arise then of what determines which limit, mean field or capillarity based, is more appropriate for describing real proteins, and how can we find this out?
The crossover between mean field-like and capillarity behavior for the funnel characteristics depends on the breadth of the interface compared with the protein size. Even in the simplest analyses, this breadth will be proportional to the range of the interactions. For hydrophobic forces this range may be as small as a water molecule’s diameter or as large as a residue’s diameter. It still is longer for electrostatic forces. Using the residue size range, much of a small protein (≈60 residues) is at the surface, arguing for the mean field picture. The breadth of the interface also increases with the heterogeneity of contact energies, eventually leading to disorder-induced roughening of the interface. There is also the possibility of interpolated, partially ordered phases (e.g., molten globule) broadening the interface. The magnitude of these effects is hard to estimate currently. One difficulty is that database contact potentials still are rather too crude to be sure of the size of heterogeneity effects. Also, the energetics of detailed side-chain packing, which determines whether a protein “sublimes” or “melts,” remain uncertain.
Experimentally, recent studies using NMR of the progressive denaturation of lactalbumin (≈140 residues) by urea do seem to indicate the disordering of the molecule by means of a front moving across the structure (B. A. Shulman, P. S. Kim, C. M. Dobson, and C. Redfield, personal communication). This suggests there is a crossover to capillarity behavior, but greater precision will be needed to pin down the interface width. According to the one-dimensional funnel picture, the equilibrium front should be closely tied to the landscape relevant to kinetics, but if additional order parameters are involved, this is less clear. The existence of a sharp, contiguous front could be probed kinetically by protein engineering using simultaneous mutations of contiguous sites, which could affect the fine structure of the funnel.
The existence of fine structure fluctuations in the capillarity picture has an impact on understanding the constraints faced in the evolution of protein-folding energy landscapes. Although the minimal frustration principle is still needed for reliable folding without traps, the fine structure effects on trapping apparently may allow a small fraction of the population of a molecule with a long, random sequence to fold quickly, even when the bulk of the population does not. Thus, the minimal frustration constraint is more appropriately thought of as one on yield rather than speed per se. Conversely, to achieve very fast folding may require, in addition to the minimal frustration constraint, that very stable contacts be judiciously placed in the structure so that even the ideal capillarity barrier is reduced. Such a feature may be correlated with the existence of foldons or modules in the larger proteins (36).
Acknowledgments
I thank C. Dobson, W. Eaton, M. Oliveberg, J. Onuchic, S. Plotkin, A. Szabo, and J. Wang for enjoyable discussions, and Ben Shoemaker for his assistance with the figures. The work at the University of Illinois was supported by National Institutes of Health Grant PHS R01 GM44557. This paper was written while I was a Scholar-in-Residence at the Fogarty International Center for Advanced Study in the Health Sciences of the National Institutes of Health (Bethesda, MD).
Footnotes
For concreteness, the exponent phenomenology for a random first-order glass transition has been followed in this paper. More involved phenomenologies in which the interface energy, randomness contributions, and their distributions and even activation barriers have anomalous exponents for size scaling are possible (see refs. 15 and 16). These possibilities that allow the glass state to be a “chaotic” phase (39) would give rise to considerations similar to those here but with quantitative modification.
References
- 1.Bryngelson J D, Wolynes P G. Proc Natl Acad Sci USA. 1987;84:7524–7528. doi: 10.1073/pnas.84.21.7524. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Bryngelson J D, Wolynes P G. J Phys Chem. 1989;93:6902–6915. [Google Scholar]
- 3.Leopold P E, Montal M, Onuchic J N. Proc Natl Acad Sci USA. 1992;89:8721–8725. doi: 10.1073/pnas.89.18.8721. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Onuchic J N, Wolynes P G, Luthey-Schulten Z, Socci N D. Proc Natl Acad Sci USA. 1995;92:3626–3630. doi: 10.1073/pnas.92.8.3626. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Bryngelson J D, Onuchic J N, Socci N D, Wolynes P G. Proteins. 1995;21:167–195. doi: 10.1002/prot.340210302. [DOI] [PubMed] [Google Scholar]
- 6.Dill K A, Bromberg S, Yue K, Fiebig K M, Yee D P, Thomas P D, Chan H S. Protein Science. 1995;4:561–601. doi: 10.1002/pro.5560040401. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Mirny L A, Abkevich V, Shakhnovich E I. Folding Des. 1996;1:103–116. doi: 10.1016/S1359-0278(96)00019-3. [DOI] [PubMed] [Google Scholar]
- 8.Socci N D, Onuchic J N. J Chem Phys. 1995;103:4732–4744. [Google Scholar]
- 9.Bryngelson J D, Wolynes P G. Biopolymers. 1990;30:177–188. [Google Scholar]
- 10.Plotkin S S, Wang J, Wolynes P G. Phys Rev E. 1996;53:6271–6296. doi: 10.1103/physreve.53.6271. [DOI] [PubMed] [Google Scholar]
- 11.Plotkin S S, Wang J, Wolynes P G. J Chem Phys. 1997;106:2932–2948. [Google Scholar]
- 12.Plotkin S S, Wang J, Wolynes P G. J Physique I. 1997;7:395–421. [Google Scholar]
- 13.Takada S, Wolynes P G. Phys Rev E. 1997;55:4562–4577. [Google Scholar]
- 14.McMillan W L. Phys Rev B. 1985;31:340–341. doi: 10.1103/physrevb.31.340. [DOI] [PubMed] [Google Scholar]
- 15.Fisher D S, Huse D A. Phys Rev B. 1988;38:373–385. doi: 10.1103/physrevb.38.373. [DOI] [PubMed] [Google Scholar]
- 16.Villain J. J Phys (Orsay, Fr) 1985;46:1843–1852. [Google Scholar]
- 17.Kirkpatrick T R, Wolynes P G. Phys Rev B. 1987;36:8552–8564. doi: 10.1103/physrevb.36.8552. [DOI] [PubMed] [Google Scholar]
- 18.Kirkpatrick T R, Thirumalai D, Wolynes P G. Phys Rev A. 1989;40:1045–1054. doi: 10.1103/physreva.40.1045. [DOI] [PubMed] [Google Scholar]
- 19.Wolynes P G. In: Proceedings International Symposium on Frontiers in Science. Chan S, DeBrunner P G, editors. Hans Frauenfelder Festschrift; 1988. (Am. Inst. Physics, New York), pp. 39–65. [Google Scholar]
- 20.Weissman M. Rev Mod Phys. 1988;60:537. [Google Scholar]
- 21.Finkelstein A V, Badredtinov A Y. Fold Des. 1997;2:115–121. doi: 10.1016/s1359-0278(97)00016-3. [DOI] [PubMed] [Google Scholar]
- 22.Thirumalai D. J Phys (Orsay, Fr) 1995;15:1457–1467. [Google Scholar]
- 23.Gutin A M, Abkevich V, Shakhnovich E I. Phys Rev Lett. 1996;77:5433–5436. doi: 10.1103/PhysRevLett.77.5433. [DOI] [PubMed] [Google Scholar]
- 24.Reiss H, Mirabel P, Whetten R. J Phys Chem. 1988;92:7241–7250. [Google Scholar]
- 25.Rowlinson J T, Widom B. Molecular Theory of Capillarity. Oxford: Clarendon Press; 1982. [Google Scholar]
- 26.Sosnick T, Jackson S, Wilk R R, Englander S W, De Grado W F. Proteins Struct Funct Genet. 1996;24:427–432. doi: 10.1002/(SICI)1097-0134(199604)24:4<427::AID-PROT2>3.0.CO;2-B. [DOI] [PubMed] [Google Scholar]
- 27.Hagen S J, Hofrichter J, Szabo A, Eaton W A. Proc Natl Acad Sci USA. 1996;93:11615–11617. doi: 10.1073/pnas.93.21.11615. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Socci N, Onuchic J, Wolynes P G. J Chem Phys. 1996;104:5860–5868. [Google Scholar]
- 29.Weeks J D, Gilmer G H, Leary H J. Phys Rev Lett. 1973;39:549–553. [Google Scholar]
- 30.Zhang J, Liu G, Jonas J. J Phys Chem. 1992;96:3478–3480. [Google Scholar]
- 31.Kauzmann W. Chem Rev. 1948;43:219–256. [Google Scholar]
- 32.Adam G, Gibbs J H. J Chem Phys. 1965;43:139–146. [Google Scholar]
- 33.Takada, S. & Wolynes, P. G. (1996) Prog. Theor. Phys. Suppl., in press.
- 34.Ngo J T, Marks J. Protein Eng. 1984;5:313–321. doi: 10.1093/protein/5.4.313. [DOI] [PubMed] [Google Scholar]
- 35.Fersht A R. Curr Opin Struct Biol. 1997;7:3–9. doi: 10.1016/s0959-440x(97)80002-4. [DOI] [PubMed] [Google Scholar]
- 36.Panchenko A R, Luthey-Schulten Z, Wolynes P G. Proc Natl Acad Sci USA. 1996;93:2008–2013. doi: 10.1073/pnas.93.5.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Shoemaker B J, Wang J, Wolynes P G. Proc Natl Acad Sci USA. 1997;94:777–782. doi: 10.1073/pnas.94.3.777. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Chayes J T, Chayes L, Fisher D S, Spencer T. Phys Rev Lett. 1986;57:1999–2002. doi: 10.1103/PhysRevLett.57.2999. [DOI] [PubMed] [Google Scholar]
- 39.Bray A J, Moore M A. Phys Rev Lett. 1987;58:57–61. doi: 10.1103/PhysRevLett.58.57. [DOI] [PubMed] [Google Scholar]
- 40.Thirumalai D, Woodson S A. Acc Chem Res. 1996;29:433–439. [Google Scholar]
- 41.Kiefhaber T. Proc Natl Acad Sci USA. 1995;92:9029–9033. doi: 10.1073/pnas.92.20.9029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Bouchaud, J. & Georges, A. (1990) Phys. Rep. 127–293.